

ABSTRACT
Hepatitis B virus (HBV) infects hundreds of millions of people worldwide and causes acute and chronic hepatitis, cirrhosis, and hepatocellular carcinoma. HBV is an enveloped virus with a relaxed circular (RC) DNA genome. In the nuclei of infected human hepatocytes, conversion of RC DNA from the incoming virion or cytoplasmic mature nucleocapsid (NC) to the covalently closed circular (CCC) DNA, which serves as the template for producing all viral transcripts, is essential to establish and sustain viral replication. For reasons yet to be understood, HBV is apparently unable to make CCC DNA in normal mouse hepatocytes in the liver. We report here that HBV CCC DNA was formed efficiently in an immortalized mouse hepatocyte cell line, AML12HBV10, and this is associated with destabilization of mature NCs in these cells. These results suggest that destabilization of mature HBV NCs in AML12HBV10 cells facilitates efficient NC uncoating and subsequent CCC DNA formation. They further implicate NC uncoating as an important step in CCC DNA formation that is subject to host regulation and potentially a critical determinant of host range and/or cell tropism of HBV.
IMPORTANCE Persistent infection by hepatitis B virus (HBV), afflicting hundreds of millions worldwide, is sustained by the episomal viral covalently closed circular (CCC) DNA in the nuclei of infected hepatocytes. CCC DNA is converted from the viral genomic (precursor) DNA contained in cytoplasmic viral nucleocapsids. The conversion process remains ill defined, but host cell factors are thought to play an essential role. In particular, HBV fails to make CCC DNA in normal mouse hepatocytes despite the presence of large amounts of nucleocapsids containing the precursor viral DNA. We have found that in an immortalized mouse hepatocyte cell line, HBV is able to make abundant amounts of CCC DNA. This ability correlates with increased instability of viral nucleocapsids in these cells, which likely facilitates nucleocapsid disassembly (uncoating) to release the genomic DNA for conversion to CCC DNA. Our studies have thus revealed a novel mechanism of controlling viral persistence via regulating nucleocapsid disassembly.
INTRODUCTION
Hepatitis B virus (HBV) has infected approximately 2 billion people worldwide, with ca. 350 million of those becoming chronically infected (1). Annually, 1 million fatalities are attributed to acute and chronic hepatitis, cirrhosis, and hepatocellular carcinoma (HCC) caused by HBV. HBV is a small, enveloped DNA virus that contains a 3.2-kb, partially double-stranded (DS), relaxed circular (RC) DNA genome and replicates via an RNA intermediate, the pregenomic RNA (pgRNA). HBV genome replication starts with the assembly of a replication-competent but immature nucleocapsid (NC) by multiple copies of a single viral protein, the HBV core (HBc) protein, incorporating pgRNA and the viral reverse transcriptase (RT). Within the immature NCs, RT converts pgRNA first to a single-stranded (SS) (minus-strand) DNA and then to the DS RC DNA, and the immature NCs are converted to mature NCs (i.e., containing RC DNA) (2, 3). In contrast to conventional retroviruses, the HBV RT initiates reverse transcription using itself as a protein primer, resulting in the covalent attachment of RT to the 5′ end of the minus-strand DNA (4, 5).
Upon infection, HBV RC DNA is converted to the covalently closed circular (CCC) DNA in the nuclei of infected human hepatocytes, which serves as the transcriptional template for all viral RNAs and thus is essential for establishing and maintaining viral infection (6, 7). CCC DNA can also be produced from RC DNA synthesized de novo in mature cytoplasmic NCs, which is recycled back to the nucleus via an intracellular amplification pathway (5, 8–10). To date, studies on the mechanism of HBV CCC DNA formation are hampered by the lack of convenient in vitro or in vivo model systems that support efficient CCC DNA formation. In particular, normal mouse hepatocytes in vivo are unable to support HBV CCC DNA formation despite accumulating abundant RC DNA (11). Established human hepatoma cell lines also have limited ability to support HBV CCC DNA formation (10, 12). These hepatoma cells instead accumulate high levels of a processed form of RC DNA called protein-free RC (PF-RC) or deproteinated RC (dp-RC) DNA, from which RT has been removed (10, 12).
Regarding viral factors, the envelope proteins, especially the largest one, L, negatively regulate CCC DNA formation (10, 12–15). In addition, as NC uncoating and RC DNA release must precede CCC DNA formation, the maturation-associated NC destabilization, which we have reported recently (16), is likely a prerequisite for CCC DNA formation. Indeed, mature HBV NCs can be differentiated into at least three different subpopulations, M1 to M3, with increasing destabilization evidenced by enhanced susceptibility to exogenous protease and nuclease digestion. In particular, the susceptibility of their interior RC DNA content to exogenous nuclease indicates an increased porosity of the M3 mature NCs (16). These subpopulations also display distinct migration characteristics on a native agarose gel and sediment differentially upon sucrose gradient ultracentrifugation (16). The C-terminal domain (CTD) of HBc has also been implicated in regulating CCC DNA formation potentially by influencing NC nuclear import or stability (17–19).
An immortalized mouse hepatocyte cell line, AML12HBV10, was developed recently that supports high levels of HBV replication in a tetracycline (Tet)-regulated manner (20). In the current study, we found that AML12HBV10 cells, in contrast to normal mouse hepatocytes and another immortalized mouse hepatocyte cell line, could support efficient HBV CCC DNA formation. Furthermore, we found that mature HBV NCs were hyperdestabilized in AML12HBV10 cells, which probably facilitated NC uncoating and subsequent CCC DNA formation.
MATERIALS AND METHODS
Cell cultures.
The AML12HBV10 cell line (20), derived from an immortalized murine hepatocyte line (AML12) (21), and the HepAD38 cell line (a gift from Christoph Seeger at the Fox Chase Cancer Center) (22), derived from a human hepatoblastoma cell line (HepG2), were maintained in Dulbecco modified Eagle F-12 medium supplemented with 10% fetal bovine serum (FBS), 50 μg/ml of penicillin-streptomycin, 400 μg/ml G418 (Gibco), and 5 μg/ml of tetracycline (Tet) until HBV replication induction. Both cell lines are induced to replicate HBV upon removal of Tet from the culture medium. HBV-Met.4 cells (23) (a gift of Stefan Wieland and Francis V. Chisari, The Scripps Research Institute), derived from the HBV and Met double transgenic mouse, were maintained in RPMI 1640 medium supplemented with 10% FBS, 55 ng/ml of epidermal growth factor (EGF), 10 μg/ml of insulin, 16 ng of insulin-like growth factor (IGF), and 50 μg/ml of penicillin-streptomycin. The cells were differentiated with 2% dimethyl sulfoxide (DMSO) up to 15 days to express HBV. Alternatively, in the absence of DMSO, HBV-Met.4 cells were transiently transfected with 4 μg of pCMV-HBV/WT per 6-cm dish to engender HBV replication, or pCMV-HBV/POL− (10), which is deficient in viral DNA synthesis (used as a negative control), using Lipofectamine 2000 (Invitrogen). Transiently transfected cells were harvested 5 days posttransfection.
Isolation of viral DNA.
Viral DNAs were isolated from induced HepAD38 or AML12HBV10 cells as previously described (10, 20, 24). In brief, for isolation of core DNA (NC-associated viral DNA), cells were lysed in NP-40 lysis buffer (50 mM Tris-HCl [pH 8.0], 1 mM EDTA, 1% NP-40) containing a protease inhibitor cocktail (Roche) and briefly centrifuged at 14,000 rpm to remove the nuclei and cell debris. The resulting cytoplasmic lysate was incubated with micrococcal nuclease (MNase) (Roche) (150 units/ml) and CaCl2 (5 mM) at 37°C for 90 min to degrade the nucleic acids outside NCs. The MNase was then inactivated by addition of 10 mM EDTA. Subsequently, the NCs were precipitated with polyethylene glycol (PEG), disrupted by 0.5% sodium dodecyl sulfate (SDS), and digested with 0.6 mg/ml of proteinase K (PK) at 37°C for 1 h. Alternatively, the MNase and PK treatments were omitted for detecting cytoplasmic PF DNA (10). The viral DNAs were then recovered by phenol-chloroform extraction and ethanol precipitation. Total PF DNAs, including CCC DNA, were isolated by modified Hirt extraction (10). In brief, cells were lysed with 1 ml of SDS lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 10 mM EDTA, and 1% SDS) and incubated at room temperature for 10 min. For isolation of PF DNA from the nuclei, the nuclear pellet from the NP-40 lysis described above was resuspended in the SDS lysis buffer after extensive washing using the NP-40 lysis buffer. The lysates were then mixed with 250 μl of 2.5 M KCl and incubated at 4°C overnight with gentle rotation. The mixture was subsequently centrifuged at 14,000 rpm for 30 min, and the supernatant was extracted by multiple rounds of phenol and once with chloroform. The DNAs were then recovered with ethanol precipitation and resuspended in TE (10 mM Tris-HCl, 1 mM EDTA [pH 8.0]). Aliquots (1/10 of total) of extracted core and PF DNA were resolved on a 1.2% agarose gel and detected by standard Southern blot analysis using a 32P-labeled HBV DNA probe. To verify the identity of CCC DNA, PF DNA was heat denatured at 100°C for 10 min, and the resulting DNA was then digested with or without EcoRI to linearize the CCC DNA, which was detected by Southern blotting. For PF DNA extracted from transiently transfected HBV-Met.4 cells, DpnI treatment was applied to digest the transfected plasmid DNA prior to heat denaturation.
Isolation of NCs by linear sucrose gradient ultracentrifugation.
Cytoplasmic lysates, without treatment by MNase, were layered on a 15% to 30% linear sucrose gradient in HCB2 buffer (20 mM Tris-HCl [pH 7.5], 50 mM NaCl, 1 mM EDTA, 0.01% [wt/vol] Triton X-100, 1× protease inhibitor cocktail [Roche], and 0.1% β-mercaptoethanol) and centrifuged at 27,000 rpm for 4 h at 4°C in a Beckman SW32 rotor (16, 25). Twenty-three fractions were then collected (from top to bottom). To detect the NC-associated DNA species, individual fractions (fractions 6 to 12, 10 μl each) were first disrupted with 0.1% SDS and then resolved on a 1.2% agarose gel containing 0.1% SDS (16). To detect intact NCs, the same amount of the above fractions was resolved on a 1.2% agarose gel. The viral DNAs were detected by standard Southern blotting.
EPR.
NCs (10 μl of the indicated sucrose fraction) were incubated with 100 μM (each) dATP, dGTP, dCTP, and TTP; an EDTA-free protease inhibitor cocktail (Roche); and the endogenous polymerase reaction (EPR) buffer (50 mM Tris-HCl [pH 7.5], 10 mM MgCl2, 0.1% NP-40, 0.1% 2-mercaptoethanol) for 16 h at 37°C in a final volume of 20 μl (16, 26), whereby the viral RT packaged within NCs synthesizes DNA using the endogenous viral RNA and DNA templates packaged within the NCs.
Treatment of NCs by PK and nuclease.
To study the stability of NCs, the NP-40 cytoplasmic lysate (10 μl) was treated with PK (Invitrogen) (1 mg/ml) in the EPR buffer in a total volume of 20 μl at 37°C for 1 h. The intact NCs and any viral DNA that was released by PK were resolved on a 1.2% agarose gel and detected by Southern blotting (16). Similarly, for nuclease treatment (16), DNase I (Roche) (1 mg/ml) was added and the reaction mixture was incubated at 37°C for 1 h. The nuclease was then inactivated by addition of EDTA to 15 mM. The NCs were disrupted by 0.5% SDS and incubated with 0.6 mg/ml of PK. Then, the mixture was resolved on a 1.2% agarose gel and detected by Southern blotting.
RESULTS
An immortalized mouse hepatocyte cell line produced high levels of HBV CCC DNA.
In an attempt to understand the block to HBV CCC DNA formation in mouse hepatocytes, we compared the HBV replication characteristics in the mouse hepatocyte cell line AML12HBV10, which was immortalized by stable expression of the human transforming growth factor alpha (TGF-α), to those in the well-characterized HepAD38 cell line, which was derived from the human hepatoma cell line HepG2. In both cell lines, HBV replication can be induced by removal of Tet from the culture medium, which is used to suppress the transcription of the HBV pgRNA under a Tet-repressed promoter (20, 22). Accordingly, upon Tet removal, high levels of viral replicative DNA intermediates associated with NCs (core DNA) were detected by Southern blotting in both cell lines (Fig. 1A, lanes 3 and 4, and C, lanes 1 and 3). As we reported earlier (10), CCC DNA and PF-RC DNA were detected in induced HepAD38 cells (Fig. 1C, lane 4). The PF-RC DNA was also detected in AML12HBV10 cells (Fig. 1A, lanes 6 and 7; B, lane 1; and C, lane 2). Surprisingly, HBV CCC DNA was also readily detectable in AML12HBV10 cells at levels similar to or even higher than those in HepAD38 cells (Fig. 1A, lanes 6 and 7; B, lane 1; and C and D, lanes 2 and 4), in contrast to normal hepatocytes in the mouse liver, where CCC DNA is undetectable (11). The identity of the CCC DNA in AML12HBV10 cells was further verified by its resistance to heat denaturation and linearization into a unit-length DNA species with the single-cut restriction enzyme EcoRI (Fig. 1B, lanes 2 and 3). As in the human cells, the PF-RC and CCC DNAs were predominantly detected in the nucleus of the mouse cells (Fig. 1D). The PF DNA extracted from AML12HBV10 cells also contained variable amounts of immature DNA species, including SS DNA, such that it displayed a smearing pattern in some but not all cases (Fig. 1A, lanes 6 and 7; B, lane 1; C, lane 2; and D, lanes 1 and 2) (see section below also).
FIG 1.

Analysis of core and PF DNA in AML12HBV10 cells and HepAD38 cells. AML12HBV10 or HepAD38 cells were induced to replicate HBV by removal of Tet from the culture medium. (A) At the indicated days after Tet removal, viral core (lanes 2 to 4) and PF (lanes 5 to 7) DNAs from AML12HBV10 cells were isolated and detected by Southern blotting. (B) PF DNA extracted from AML12HBV10 cells induced for 7 days (lane 1) was treated by heat denaturation (100°C), which denatured RC and double-stranded linear DNA to SS DNA without denaturing CCC DNA (lane 2), or by heating to 100°C followed by EcoRI digestion, which linearized CCC DNA (lane 3). (C) Core (lanes 1 and 3) and PF (lanes 2 and 4) DNAs were isolated from induced AML12HBV10 (lanes 1 and 2) or HepAD38 (lanes 3 and 4) cells, resolved on an agarose gel, and analyzed by Southern blotting. (D) PF DNA was isolated from either nuclei (N; lanes 1 and 3) or whole cells (W; lanes 2 and 4) of AML12HBV10 (lanes 1 and 2) or HepAD38 (lanes 3 and 4) cells, resolved on an agarose gel, and detected by Southern blotting. RC, relaxed circular; DSL, double stranded linear; CCC, covalently closed circular; CCC-L, covalently closed circular linearized by EcoRI digestion; SS, single stranded; M, DNA molecular mass marker, indicated in kilobase pairs to the left of panel A.
To determine if HBV CCC DNA could also be formed in other mouse hepatocyte-derived cell lines, we analyzed the viral DNA in the HBV-Met.4 cell line, which is a mouse hepatocyte cell line immortalized by the Met oncogene and harbors stably integrated HBV DNA. HBV-Met.4 cells can be induced to express the viral genome upon differentiation by treatment with DMSO (23). Alternatively, undifferentiated HBV-Met.4 cells can be transiently transfected with HBV DNA to initiate viral gene expression and replication (27). As shown in Fig. 2, both differentiated and undifferentiated (transiently transfected) HBV-Met.4 cells could produce high levels of core DNA as reported previously (23, 27). However, HBV-Met.4 cells produced much lower (less than 2%) levels of PF-RC DNA than did AML12HBV10 cells, as judged by the ratio of the PF-RC DNA to core RC DNA (Fig. 2 versus Fig. 1). HBV CCC DNA was produced by HBV-Met.4 cells at even lower levels, being barely detectable by Southern blotting (Fig. 2A and B, lanes 8). Thus, the efficiency of HBV CCC DNA formation varied dramatically in different immortalized mouse hepatocyte cell lines in culture.
FIG 2.

Analysis of HBV DNA synthesis in HBV-Met.4 cells. (A) HBV-Met.4 cells were differentiated with 2% DMSO up to 15 days to induce HBV gene expression and replication. The viral core DNA (lanes 1 to 3) and PF DNA (lanes 4 to 6) were isolated every 5 days and detected by Southern blotting. The PF DNA from day 15 postinduction was further heat treated (100°C) to denature non-CCC DNA species, which was followed by EcoRI digestion to linearize CCC DNA (lane 8). Lane 7 was a much longer exposure than lane 6 and had the same exposure time as lane 8. (B) HBV-Met.4 cells, without DMSO induction, were transiently transfected with pCMV-HBV/WT (lanes 2, 4, and 6) or pCMV-HBV/Pol− (lanes 1, 3, and 5). Five days later, the viral core DNA (lanes 1 and 2) and PF DNA (lanes 3 and 4) were isolated and detected by Southern blotting. DpnI was used to digest transfected plasmid DNA that was coisolated with HBV PF DNA prior to Southern blot analysis of all PF DNA. The PF DNA was further heat denatured (100°C), followed by EcoRI digestion to linearize the CCC DNA (lanes 5 and 6). Lanes 7 and 8 represent a longer exposure of lanes 5 and 6. RC, relaxed circular; CCC, covalently closed circular; SS, single stranded; DpnI fragments, plasmid DNA fragments generated by DpnI digestion.
HBV NCs were unstable in AML12HBV10 cells.
Inspection of the HBV core and PF DNA pattern showed that there was little mature RC DNA associated with NCs in AML12HBV10 cells, which contained mostly SS DNA and immature DS DNA intermediates (Fig. 1A, lanes 3 and 4, and C, lane 1), whereas the levels of mature RC DNA were always higher or at least equal to the SS DNA in HepAD38 cells (Fig. 1C, lane 3) (10) or HBV-Met.4 cells (Fig. 2A, lanes 1 to 3, and B, lane 2) (23, 28, 29). On the other hand, relative to the levels of core RC DNA, there was much more PF-RC DNA (by at least 10-fold) in AML12HBV10 cells than in HepAD38 cells or HBV-Met.4 cells (Fig. 1A, lanes 6 and 7 versus lanes 3 and 4, and 2A, lanes 4 to 6 versus lanes 1 to 3, and B, lane 4 versus lane 2). These results suggested that RC DNA in mature NCs in AML12HBV10 cells was rapidly converted to PF-RC and CCC DNA, accounting for both the abundance of PF-RC and CCC DNA and the depletion of their precursor, the core RC DNA. As the conversion of core RC DNA to PF-RC DNA (i.e., removal of the covalently linked RT protein from core RC DNA or deproteination) or to CCC DNA likely occurs following at least partial disassembly (uncoating) of NCs (10, 12, 16, 30), we hypothesized that mature NCs were destabilized in AML12HBV10 cells.
To test this hypothesis, we estimated the stability of NCs in AML12HBV10 cells by determining their sensitivity to protease and nuclease digestion in comparison to NCs harvested from the well-characterized HepAD38 cells, using procedures that we recently developed (16). As anticipated, most (>60%) of the RC DNA in the cytoplasmic NCs from AML12HBV10 cells was eliminated by exogenous DNase I (Fig. 3B, lane 2 versus lane 1), in contrast to those in HepAD38 cells, where only a minority (less than 30%) of mature NCs failed to protect their RC DNA content from exogenous nuclease (Fig. 3B, lane 4 versus lane 3), as we reported recently (16). This nuclease sensitivity could thus explain the near absence of mature RC DNA extracted from cytoplasmic NCs in AML12HBV10 cells following MNase predigestion (Fig. 1A, lanes 3 and 4, and C, lane 1), which is used routinely during core DNA extraction to remove extraneous DNA/RNA not packaged within NCs. Almost all (80% or more) RC DNA could also be released from mature NCs in AML12HBV10 cells following exogenous protease digestion (Fig. 4B, lane 3 versus lane 1). This was in contrast to mature NCs from HepAD38 cells, in which only a small fraction (ca. 20%) of mature NCs released their RC DNA content following the same protease digestion (Fig. 4B, lane 6 versus lane 4), as we reported recently (16). In addition, a portion of immature NC from AML12HBV10 cells also appeared to be sensitive to protease digestion, leading to the release of small amounts (ca. 10%) of SS DNA (Fig. 4B, lane 3), whereas no release of SS DNA from immature HepAD38 NCs by protease digestion was ever detected (Fig. 4B, lane 6) as we reported (16). Protease digestion of both the AML12HBV10 and HepAD38 lysate led to an apparent increase of the NC signal running as a distinct band near the bottom of the gel (Fig. 4B, lanes 3 and 6). This was due to the fact that protease digestion caused those protease-resistant NCs that migrated as a smear up the lane before protease treatment to migrate to the distinct NC band, possibly by removing cellular proteins associated loosely with the outside of the NCs, as we observed before (16).
FIG 3.
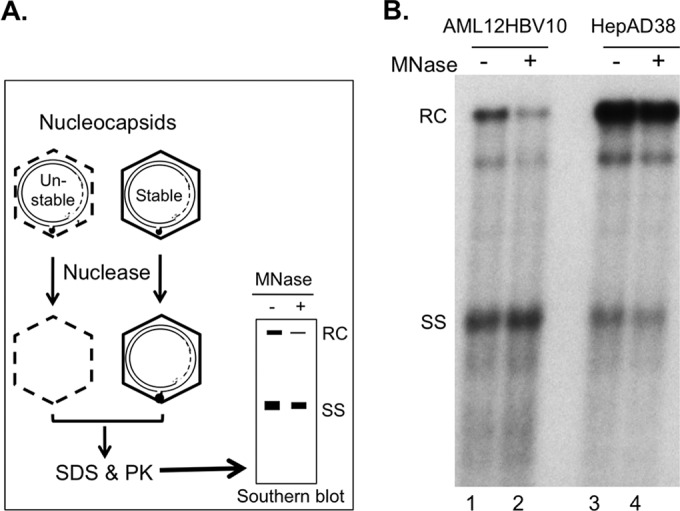
Isolation of core DNA with or without prior nuclease treatment of NCs. (A) Schematic diagram of nuclease treatment of HBV NCs. MNase was used to degrade viral DNA inside the unstable NC. Following inactivation of the nuclease, nuclease-resistant, NC-protected viral DNA was isolated and detected by Southern blotting. (B) Core DNA extracted from cytoplasmic lysate of AML12HBV10 (lanes 1 and 2) or HepAD38 (lanes 3 and 4) cells, with (lanes 2 and 4) or without (lanes 1 and 3) prior MNase digestion, was detected by Southern blotting. RC, relaxed circular; SS, single stranded.
FIG 4.

Analysis of NC stability by protease digestion and extraction of PF DNA. (A) Schematic diagram of protease treatment of HBV NCs. Proteinase K (PK) was used to disrupt the unstable NC to release its DNA content. Released viral DNA and any remaining intact NC could be resolved on an agarose gel and detected by Southern blotting. (B) Aliquots (10 μl) of cytoplasmic lysate from AML12HBV10 (lanes 1 to 3) or HepAD38 (lanes 4 to 6) cells were resolved on an agarose gel, with (lanes 3 and 6) or without (lanes 2 and 5) prior PK digestion (1 mg/ml, 1 h at 37°C). Viral DNA released from unstable NCs following PK digestion as well as DNA associated with remaining intact NCs was resolved on an agarose gel and detected by Southern blotting. HBV DNAs released from all NCs by treatment with 0.5% SDS and 0.6 mg/ml PK were loaded as reference (lanes 1 and 4). (C) Detection of cytoplasmic PF DNA in AML12HBV10 but not HepAD38 cells. Induced AML12HBV10 (lanes 1 and 2) or HepAD38 (lanes 3 and 4) cells were lysed with the NP-40 lysis buffer and centrifuged at 14,000 rpm for 1 min to remove the nuclei and cell debris. The PF DNA was then isolated from the resulting cytoplasmic lysates by modified Hirt extraction with (lanes 1 and 3) or without (lanes 2 and 4) proteinase K (PK) pretreatment and detected by Southern blotting as described in Materials and Methods. RC, relaxed circular; SS, single stranded; NC, nucleocapsid.
Moreover, some PF DNAs (RC and SS) could also be isolated from the cytoplasm of AML12HBV10 cells but not from HepAD38 cells (Fig. 4C, lane 2 versus lane 4), and the total PF-RC DNA extracted from AML12HBV10 cells showed a heterogeneous pattern with variable amounts of SS DNA and other immature DNA species migrating above and even below the SS DNA (Fig. 1A, lanes 6 and 7; B, lane 1; and C, lane 2). These results thus indicated that most mature NCs from AML12HBV10 cells were indeed destabilized. Some immature NCs in these cells may also be destabilized, although their destabilization was to a smaller degree and more variable.
Mature NCs from AML12HBV10 cells displayed altered profiles upon sucrose gradient ultracentrifugation and native agarose gel electrophoresis.
Detailed analyses of mature NCs from HepAD38 cells revealed significant heterogeneity among the mature NCs that is characterized by distinct sedimentation on a sucrose gradient and migration rates on a native agarose gel, in addition to differential protease and nuclease sensitivity (16). To characterize the NCs further in AML12HBV10 cells, we thus performed sucrose gradient centrifugation and native agarose gel analysis of AML12HBV10 NCs in comparison to those from HepAD38 cells. The viral DNA released from the NCs in the different fractions was analyzed by Southern blotting (Fig. 5A). In parallel, native NCs were resolved by agarose gel electrophoresis and viral DNA associated with the NCs was detected by Southern blotting (Fig. 5B). As we reported recently (16), the least stable (sensitive to both exogenous nuclease and protease) M3 mature NC subpopulation from HepAD38 cells sedimented to fractions 6 to 9 on the sucrose gradient (Fig. 5A, lanes 8 to 11), slower than the SS DNA-containing immature NCs (peaked at fraction 10 [Fig. 5A, lane 12]). They also displayed much lower mobility than immature NCs on the agarose gel, comigrating with or migrating just above the isolated RC DNA (Fig. 5B, lanes 8 to 11). When NCs from AML12HBV10 cells were subjected to similar analyses, the canonical M3 mature NCs could not be detected, i.e., no RC DNA was detected in fractions 6 to 9 and no slow-migrating NCs were detected at the RC DNA position (Fig. 5A and B, lanes 1 to 4). The nuclease-sensitive mature NCs in AML12HBV10 cells detected in Fig. 3 above apparently were thus not associated with the typical M3 NCs defined in HepAD38 cells, judging from their sedimentation on the sucrose gradient and mobility on the agarose gel. Rather, they might be in an alternative, nuclease-sensitive structure(s) that could not be resolved by current analyses.
FIG 5.
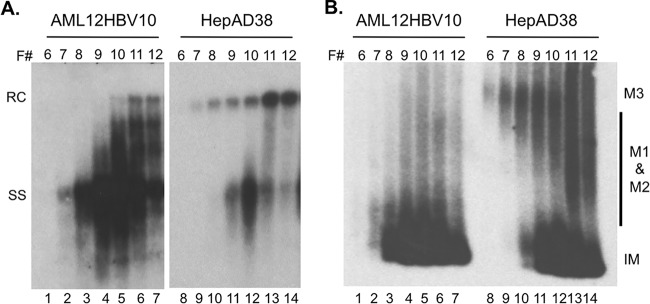
Analysis of HBV NCs by sucrose gradient ultracentrifugation. HBV NCs with different maturities were isolated from cytoplasmic lysates of AML12HBV10 (A and B, lanes 1 to 7) and HepAD38 (A and B, lanes 8 to 14) cells by sucrose gradient ultracentrifugation. (A) To analyze the NC-associated viral DNA from fractions (F#) 6 to 12, 10 μl of each fraction was first incubated with 0.1% SDS to disrupt the NC and release the viral DNA, which was then resolved on a 1.2% agarose gel containing 0.1% SDS and detected by Southern blotting. (B) To detect native NCs from the same fractions, 10 μl of each fraction was resolved directly on a 1.2% agarose gel and detected by Southern blotting. RC, relaxed circular; SS, single stranded; M1 and M2, nuclease-resistant mature NCs; M3, nuclease-sensitive mature NCs; IM, immature NC.
The other two mature NC subpopulations, M1 and M2, which are more stable than M3, with M1 being resistant and M2 being sensitive to protease digestion and both being resistant to exogenous nuclease (16), from HepAD38 cells peaked at fraction 11 (i.e., sedimenting faster than immature NCs) and migrated as a smear between the fast-migrating immature NCs and the RC DNA (Fig. 5A and B, lanes 13 and 14), as we reported earlier (16). The mature, RC DNA-containing NCs from AML12HBV10 cells all sedimented to fractions 10 to 12 (Fig. 5A, lanes 5 to 7), but it was difficult to clearly discern their mobility on the agarose gel due to the presence of some smeary signal above the immature NCs in those fractions and the low abundance of mature NCs in the AML12HBV10 cells (Fig. 5B, lanes 5 to 7). Thus, the M1/M2 subpopulations from AML12HBV10 cells shared sedimentation profiles similar to those from HepAD38 cells, but their migration on the agarose gel was difficult to ascertain. Given that almost all mature NCs in AML12HBV10 cells were sensitive to proteinase K digestion (Fig. 4B, above), these results suggested that AML12HBV10 cells accumulated little of the M1 subpopulation, which would be resistant to exogenous protease, and most or all of the mature NCs in these cells displayed properties of both M2 (protease sensitive) and M3 (nuclease sensitive) found in HepAD38 cells but were also distinct from those species in their sedimentation on sucrose gradients and migration on agarose gels.
NCs from AML12HBV10 cells that were matured in vitro were also unstable.
To help us understand how NCs in AML12HBV10 cells might be destabilized, we tested whether NCs from AML12HBV10 cells that were matured in vitro by EPR, which allows the production of mature RC DNA from more immature DNA species in isolated NCs in vitro (16, 26), were also sensitive to exogenous nuclease. To generate mature NCs in vitro, sucrose fractions that contained mostly immature NCs were subjected to EPR followed by DNase I treatment. Similar to native mature NCs harvested from the AML12HBV10 cells, the majority (60% or more) of the in vitro-generated mature NCs from AML12HBV10 cells (Fig. 6, lane 4 versus lane 3) also failed to protect their RC DNA from exogenous nuclease. In contrast, most of the mature NCs generated in vitro using NCs from HepAD38 cells were resistant to nuclease digestion (Fig. 6, lane 8 versus lane 7). Thus, mature NCs generated in vitro using immature NCs derived from AML12HBV10 cells were also unstable, suggesting that the increased instability of mature NCs from AML12HBV10 cells was an intrinsic structural property of the NCs themselves.
FIG 6.
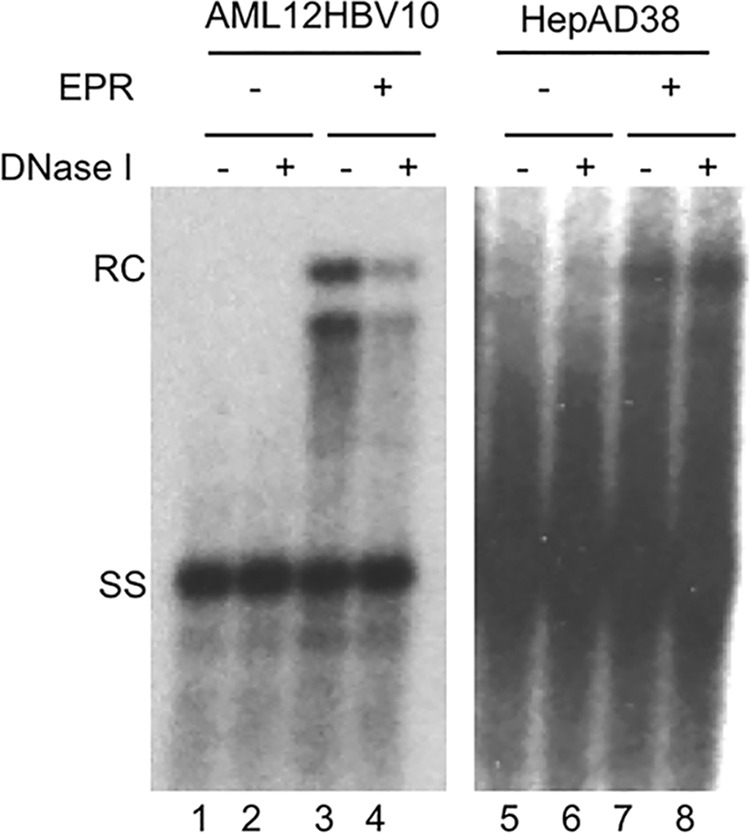
Nuclease sensitivity of mature NCs generated in vitro via EPR. Immature NCs containing mostly SS DNA isolated from AML12HBV10 (lanes 1 to 4) or HepAD38 (lanes 5 to 8) cells by sucrose gradient fractionation were used for the endogenous polymerase reaction (EPR) (lanes 3, 4, 7, and 8) or analyzed directly without EPR (lanes 1, 2, 5, and 6). Subsequent to EPR, the NCs were treated with (lanes 2, 4, 6, and 8) or not treated with (lanes 1, 3, 5, and 7) DNase I (1 mg/ml, 37°C for 1 h), followed by addition of 15 mM EDTA to inactivate the DNase. The reaction mixtures were then incubated with 0.5% SDS and 0.6 mg/ml of PK, and viral DNA was detected by Southern blotting. RC, relaxed circular; SS, single stranded.
DISCUSSION
In contrast to normal mouse hepatocytes in vivo that support little to no CCC DNA formation during HBV replication, we found here that the immortalized mouse hepatocyte cell line AML12HBV10 supports efficient CCC DNA formation. This could be correlated with the accumulation of high levels of PF-RC DNA and little NC-associated RC DNA, suggesting efficient uncoating of mature HBV NCs and subsequent deproteination of their RC DNA content. The extent of destabilization of mature NCs was dramatically enhanced in AML12HBV10 cells compared to human hepatoma cells, in which some mature, as opposed to immature, HBV NCs are also preferentially destabilized. This heightened destabilization of mature NCs is likely responsible, at least in part, for the efficient CCC DNA formation in these immortalized mouse hepatocytes.
We have recently shown that mature HBV NCs in human hepatoma cells are heterogeneous, displaying distinct stability and structural properties (as measured by sensitivity to exogenous protease and nuclease digestion, sedimentation on sucrose gradients, and migration on agarose gels) (16). The mature HBV NCs in the mouse AML12HBV10 cells appeared to differ from those in the human hepatoma cells in that most mature NCs were sensitive to protease and nuclease digestion and their sedimentation and migration profiles on the sucrose gradients and agarose gels were not the same as those of the mature NCs from human hepatoma cells. The absence in the AML12HBV10 cells of the M3 mature NC subpopulation, which are the most unstable mature NCs, thought to be partially uncoated already in the human hepatoma cells (16), suggests that these unstable NCs may be even more rapidly uncoated and their RC DNA content exposed for deproteination and CCC DNA formation in these mouse cells. This is consistent with the high levels of PF-RC DNA (which was much more abundant than RC DNA associated with mature NCs) and CCC DNA detected in the AML12HBV10 cells. This notion is also supported by the detection of PF-RC DNA in the cytoplasm of these cells. The most stable mature NC population, M1, which is resistant to protease and nuclease digestion (16), also failed to accumulate to significant levels in the AML12HBV10 cells, again indicative of rapid destabilization of mature NCs. Judging from the sedimentation and migration properties on sucrose gradients and agarose gels, respectively, the nuclease- and protease-sensitive mature NCs that accumulated in AML12HBV10 cells are apparently in a different structure than are those in HepAD38 cells, which remains to be better characterized.
The mechanisms of mature NC hyperdestabilization remain to be determined. Indeed, the uncoating (disassembly) of HBV NCs, an essential step in viral infection and replication and a prerequisite to CCC DNA formation, represents one of the least understood aspects of its replication cycle, as is true for most other viruses. One potential mechanism of NC hyperdestabilization may be related to altered posttranslational modifications of the NCs. For the related duck hepatitis B virus (DHBV), we have shown previously that altering the state of phosphorylation of the viral core protein, which is dynamic during, and regulates, NC maturation, can indeed lead to preferential destabilization of mature NCs (18, 25). The observation that mature NCs generated in vitro through EPR from immature NCs in AML12HBV10 cells were also destabilized is consistent with an intrinsic biochemical or structural alteration of the NCs themselves and further suggests that the putative biochemical or structural alteration of HBV NCs in AML12HBV10 cells may occur early and yet manifest itself only later as the NCs mature. Due to the low abundance of mature NCs in the background of a large excess of immature NCs and empty capsids which do not participate in maturation at all (16, 31, 32), it remains at present a serious challenge to directly analyze biochemical or structural changes associated with HBV NC maturation.
Our results implicate host regulation of viral NC stability, but the putative host factors that mediate the enhanced destabilization of mature NCs in AML12HBV10 cells remain to be identified. As discussed above, if biochemical modifications, such as the phosphorylation state of HBc, are involved, host factors mediating HBc phosphorylation, including the cyclin-dependent protein kinase 2 (33) and protein kinase C (34), and yet to be identified host phosphatases that mediate HBc dephosphorylation (17, 25) could be implicated. The fact that mouse hepatocytes in the hepatocyte nuclear factor 1α (HNF1α)-knockout animals, in contrast to the wild-type animals, can also support HBV CCC DNA synthesis, albeit at greatly reduced levels compared to AML12HBV10 cells (35), is also consistent with the notion that cellular factors involved in HBV CCC DNA formation may be modulated by the physiological status of the hepatocytes.
It remains unclear why normal mouse hepatocytes in the liver fail to support HBV CCC DNA formation. Among other possibilities, our results suggest that a putative defect in inducing mature NC uncoating may contribute to the deficiency of CCC DNA formation in normal mouse hepatocytes. Identification of mouse hepatocyte factors involved in regulating HBV CCC DNA formation should facilitate the development of a mouse model permissive for HBV infection and replication. Ectopic expression of the recently identified HBV cell surface receptor sodium taurocholate cotransporting polypeptide (36, 37) is insufficient on its own to render mouse hepatocytes susceptible to HBV infection (36–38), a result not entirely surprising given the known inability of normal mouse hepatocytes to support CCC DNA formation.
ACKNOWLEDGMENTS
We thank Frank Chisari at The Scripps Research Institute for providing the HBV-Met.4 cell line and Frank Chisari and Stefan Wieland for a critical reading of the manuscript.
This work was supported by a Public Health Service grant (R01 AI074982 to J.H.) from the National Institutes of Health. Ju-Tao Guo is supported by NIH grant R01 AI113267 and the Hepatitis B Foundation through an appropriation from the Commonwealth of Pennsylvania.
REFERENCES
- 1.Trepo C, Chan HL, Lok A. 2014. Hepatitis B virus infection. Lancet 384:2053–2063. doi: 10.1016/S0140-6736(14)60220-8. [DOI] [PubMed] [Google Scholar]
- 2.Summers J, Mason WS. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403–415. doi: 10.1016/0092-8674(82)90157-X. [DOI] [PubMed] [Google Scholar]
- 3.Seeger C, Zoulim F, Mason WS. 2013. Hepadnaviruses, p 2185–2221. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Wang GH, Seeger C. 1992. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 71:663–670. doi: 10.1016/0092-8674(92)90599-8. [DOI] [PubMed] [Google Scholar]
- 5.Hu J, Seeger C. 2015. Hepadnavirus genome replication and persistence. Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a021386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mason WS, Halpern MS, England JM, Seal G, Egan J, Coates L, Aldrich C, Summers J. 1983. Experimental transmission of duck hepatitis B virus. Virology 131:375–384. doi: 10.1016/0042-6822(83)90505-6. [DOI] [PubMed] [Google Scholar]
- 7.Miller RH, Robinson WS. 1984. Hepatitis B virus DNA forms in nuclear and cytoplasmic fractions of infected human liver. Virology 137:390–399. doi: 10.1016/0042-6822(84)90231-9. [DOI] [PubMed] [Google Scholar]
- 8.Tuttleman JS, Pourcel C, Summers J. 1986. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 47:451–460. doi: 10.1016/0092-8674(86)90602-1. [DOI] [PubMed] [Google Scholar]
- 9.Wu TT, Coates L, Aldrich CE, Summers J, Mason WS. 1990. In hepatocytes infected with duck hepatitis B virus, the template for viral RNA synthesis is amplified by an intracellular pathway. Virology 175:255–261. doi: 10.1016/0042-6822(90)90206-7. [DOI] [PubMed] [Google Scholar]
- 10.Gao W, Hu J. 2007. Formation of hepatitis B virus covalently closed circular DNA: removal of genome-linked protein. J Virol 81:6164–6174. doi: 10.1128/JVI.02721-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guidotti LG, Matzke B, Schaller H, Chisari FV. 1995. High-level hepatitis B virus replication in transgenic mice. J Virol 69:6158–6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81:12472–12484. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lentz TB, Loeb DD. 2011. Roles of the envelope proteins in the amplification of covalently closed circular DNA and completion of synthesis of the plus-strand DNA in hepatitis B virus. J Virol 85:11916–11927. doi: 10.1128/JVI.05373-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Summers J, Smith PM, Horwich AL. 1990. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J Virol 64:2819–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Summers J, Smith PM, Huang MJ, Yu MS. 1991. Morphogenetic and regulatory effects of mutations in the envelope proteins of an avian hepadnavirus. J Virol 65:1310–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cui X, Ludgate L, Ning X, Hu J. 2013. Maturation-associated destabilization of hepatitis B virus nucleocapsid. J Virol 87:11494–11503. doi: 10.1128/JVI.01912-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu K, Ludgate L, Yuan Z, Hu J. 2015. Regulation of multiple stages of hepadnavirus replication by the carboxyl-terminal domain of viral core protein in trans. J Virol 89:2918–2930. doi: 10.1128/JVI.03116-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Basagoudanavar SH, Perlman DH, Hu J. 2007. Regulation of hepadnavirus reverse transcription by dynamic nucleocapsid phosphorylation. J Virol 81:1641–1649. doi: 10.1128/JVI.01671-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rabe B, Vlachou A, Pante N, Helenius A, Kann M. 2003. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc Natl Acad Sci U S A 100:9849–9854. doi: 10.1073/pnas.1730940100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu C, Guo H, Pan XB, Mao R, Yu W, Xu X, Wei L, Chang J, Block TM, Guo JT. 2010. Interferons accelerate decay of replication-competent nucleocapsids of hepatitis B virus. J Virol 84:9332–9340. doi: 10.1128/JVI.00918-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu JC, Merlino G, Fausto N. 1994. Establishment and characterization of differentiated, nontransformed hepatocyte cell lines derived from mice transgenic for transforming growth factor alpha. Proc Natl Acad Sci U S A 91:674–678. doi: 10.1073/pnas.91.2.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother 41:1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pasquetto V, Wieland SF, Uprichard SL, Tripodi M, Chisari FV. 2002. Cytokine-sensitive replication of hepatitis B virus in immortalized mouse hepatocyte cultures. J Virol 76:5646–5653. doi: 10.1128/JVI.76.11.5646-5653.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu J, Flores D, Toft D, Wang X, Nguyen D. 2004. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J Virol 78:13122–13131. doi: 10.1128/JVI.78.23.13122-13131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perlman DH, Berg EA, O'Connor PB, Costello CE, Hu J. 2005. Reverse transcription-associated dephosphorylation of hepadnavirus nucleocapsids. Proc Natl Acad Sci U S A 102:9020–9025. doi: 10.1073/pnas.0502138102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen DH, Gummuluru S, Hu J. 2007. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J Virol 81:4465–4472. doi: 10.1128/JVI.02510-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia ML, Byfield R, Robek MD. 2009. Hepatitis B virus replication and release are independent of core lysine ubiquitination. J Virol 83:4923–4933. doi: 10.1128/JVI.02644-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pagliaccetti NE, Chu EN, Bolen CR, Kleinstein SH, Robek MD. 2010. Lambda and alpha interferons inhibit hepatitis B virus replication through a common molecular mechanism but with different in vivo activities. Virology 401:197–206. doi: 10.1016/j.virol.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robek MD, Boyd BS, Chisari FV. 2005. Lambda interferon inhibits hepatitis B and C virus replication. J Virol 79:3851–3854. doi: 10.1128/JVI.79.6.3851-3854.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo H, Mao R, Block TM, Guo JT. 2010. Production and function of the cytoplasmic deproteinized relaxed circular DNA of hepadnaviruses. J Virol 84:387–396. doi: 10.1128/JVI.01921-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ning X, Nguyen D, Mentzer L, Adams C, Lee H, Ashley R, Hafenstein S, Hu J. 2011. Secretion of genome-free hepatitis B virus-single strand blocking model for virion morphogenesis of para-retrovirus. PLoS Pathog 7:e1002255. doi: 10.1371/journal.ppat.1002255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luckenbaugh L, Kitrinos KM, Delaney WE, Hu J. 2015. Genome-free hepatitis B virion levels in patient sera as a potential marker to monitor response to antiviral therapy. J Viral Hepat 22:561–570. doi: 10.1111/jvh.12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ludgate L, Adams C, Hu J. 2011. Phosphorylation state-dependent interactions of hepadnavirus core protein with host factors. PLoS One 6:e29566. doi: 10.1371/journal.pone.0029566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kann M, Gerlich WH. 1994. Effect of core protein phosphorylation by protein kinase C on encapsidation of RNA within core particles of hepatitis B virus. J Virol 68:7993–8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raney AK, Eggers CM, Kline EF, Guidotti LG, Pontoglio M, Yaniv M, McLachlan A. 2001. Nuclear covalently closed circular viral genomic DNA in the liver of hepatocyte nuclear factor 1 alpha-null hepatitis B virus transgenic mice. J Virol 75:2900–2911. doi: 10.1128/JVI.75.6.2900-2911.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ni Y, Lempp FA, Mehrle S, Nkongolo S, Kaufman C, Falth M, Stindt J, Koniger C, Nassal M, Kubitz R, Sultmann H, Urban S. 2014. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 146:1070–1083. doi: 10.1053/j.gastro.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 37.Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, Li W. 2012. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He W, Ren B, Mao F, Jing Z, Li Y, Liu Y, Peng B, Yan H, Qi Y, Sun Y, Guo JT, Sui J, Wang F, Li W. 2015. Hepatitis D virus infection of mice expressing human sodium taurocholate co-transporting polypeptide. PLoS Pathog 11:e1004840. doi: 10.1371/journal.ppat.1004840. [DOI] [PMC free article] [PubMed] [Google Scholar]