

ABSTRACT
Varicella-zoster virus (VZV) causes chickenpox upon primary infection and establishes latency in ganglia. Reactivation from latency causes herpes zoster, which may be complicated by postherpetic neuralgia. Innate immunity mediated by interferon and proinflammatory cytokines represents the first line of immune defense upon infection and reactivation. VZV is known to interfere with multiple innate immune signaling pathways, including the central transcription factor NF-κB. However, the role of these inhibitory mechanisms in vivo is unknown. Simian varicella virus (SVV) infection of rhesus macaques recapitulates key aspects of VZV pathogenesis, and this model thus permits examination of the role of immune evasion mechanisms in vivo. Here, we compare SVV and VZV with respect to interference with NF-κB activation. We demonstrate that both viruses prevent ubiquitination of the NF-κB inhibitor IκBα, whereas SVV additionally prevents IκBα phosphorylation. We show that the ORF61 proteins of VZV and SVV are sufficient to prevent IκBα ubiquitination upon ectopic expression. We further demonstrate that SVV ORF61 interacts with β-TrCP, a subunit of the SCF ubiquitin ligase complex that mediates the degradation of IκBα. This interaction seems to inactivate SCF-mediated protein degradation in general, since the unrelated β-TrCP target Snail is also stabilized by ORF61. In addition to ORF61, SVV seems to encode additional inhibitors of the NF-κB pathway, since SVV with ORF61 deleted still prevented IκBα phosphorylation and degradation. Taken together, our data demonstrate that SVV interferes with tumor necrosis factor alpha (TNF-α)-induced NF-κB activation at multiple levels, which is consistent with the importance of these countermechanisms for varicella virus infection.
IMPORTANCE The role of innate immunity during the establishment of primary infection, latency, and reactivation by varicella-zoster virus (VZV) is incompletely understood. Since infection of rhesus macaques by simian varicella virus (SVV) is used as an animal model of VZV infection, we characterized the molecular mechanism by which SVV interferes with innate immune activation. Specifically, we studied how SVV prevents activation of the transcription factor NF-κB, a central factor in eliciting proinflammatory responses. The identification of molecular mechanisms that counteract innate immunity might ultimately lead to better vaccines and treatments for VZV, since overcoming these mechanisms, either by small-molecule inhibition or by genetic modification of vaccine strains, is expected to reduce the pathogenic potential of VZV. Moreover, using SVV infection of rhesus macaques, it will be possible to study how increasing the vulnerability of varicella viruses to innate immunity will impact viral pathogenesis.
INTRODUCTION
Varicella-zoster virus (VZV) is a member of the subfamily Alphaherpesvirinae and is the causative agent of chickenpox and herpes zoster (HZ). Following primary infection, VZV establishes latency in ganglia. Reactivation from latency, which typically occurs later in life due to a weakened or compromised immune system, causes HZ, or shingles. HZ is characterized by a painful itching rash that typically appears on the trunk of the body along a thoracic dermatome. The occurrence of HZ is associated with serious debilitating complications, which include postherpetic neuralgia (PHN), blindness, paralysis, and hearing loss. PHN is characterized by pain, or allodynia, that remains after the HZ rash has subsided (1–3). In vivo research on VZV has been constrained in the past due to the lack of an adequate animal model. Simian varicella virus (SVV) is organizationally and genetically similar to VZV, sharing about 75% DNA homology and a colinear genome (4). Recently, SVV infection of rhesus macaques (RM) has been shown to recapitulate many features of VZV pathogenesis, including a varicella-like rash that disappeared around 3 weeks postinfection. Latency was confirmed by detection of viral DNA in neuronal ganglia months after primary infection (5). Thus, SVV infection of RM can be used as a model for VZV infection.
Nuclear factor kappa B (NF-κB) signaling plays a critical role in the establishment of antiviral immune responses (6). NF-κB signaling drives the expression of many proteins that aid in blocking viral replication and stimulate the development of specific adaptive immune responses. These factors include proinflammatory cytokines, regulators of apoptosis, and chemokines (7–10). NF-κB signaling is initiated by pattern recognition receptors (PRRs) that recognize pathogen-associated molecular patterns (PAMPs), as well as cytokine receptors. They include Toll-like receptor 3 (TLR-3), TLR-4, interleukin 1 receptor (IL-1R), and tumor necrosis factor receptor 1 (TNFR1), which are activated by double-stranded RNA, lipopolysaccharide (LPS), IL-1, and tumor necrosis factor alpha (TNF-α), respectively (10–13). Signaling through PRRs leads to the phosphorylation of the inhibitor of NF-κB kinase (IKK) complex, which is composed of the subunits IKKα, IKKβ, and IKKγ, or NEMO. The activated IKK complex phosphorylates the inhibitor of NF-κB (IκBα), which keeps the NF-κB subunits RelA, or p65, and p50 inactive. Phosphorylation of IκBα leads to its rapid degradation and the release of the NF-κB subunits, which initiates the expression of numerous genes (10, 14). NF-κB–IκBα subunits have been shown to shuttle between the nucleus and cytoplasm (15). Degradation of IκBα and activation of NF-κB occur predominantly in the cytoplasm but have been observed in the nucleus, as well (15, 16). IκBα degradation is mediated by an E3 ubiquitin ligase complex that consists of the F-box protein β-TrCP, Skp1, Cullin1 (Cul1), and the adaptor protein Roc1, or Rbx1 (the SCFβ-TrCP complex). Targets of this complex contain a phosphodegron domain (DSGΦXS, where Φ indicates a hydrophobic domain), which is recognized by β-TrCP upon phosphorylation of the two serines. This is followed by the ubiquitination of the target protein and subsequent degradation by the proteasome (17, 18).
Numerous viruses code for immune evasion mechanisms that target the NF-κB signaling pathway, illustrating the prominent antiviral role of NF-κB-mediated protein expression (19). For VZV, it was shown that the virus interferes with TNF-α-induced NF-κB promoter activity by preventing the degradation of IκBα (20). This interference seems to be at least partially mediated by ORF61, since Sloan et al. showed that ORF61 expression led to the stabilization of IκBα in TNF-α-treated HEK 293T cells (21). However, Zhu et al. reported that overexpression of ORF61 had only a minor effect on Sendai virus-induced NF-κB signaling, indicating a pathway-specific countermechanism (22). Instead, they showed that ORF61 interacts with phosphorylated IRF3 and induces degradation of the protein, thereby preventing the induction of beta interferon (IFN-β) expression (22). VZV ORF61 is expressed with immediate-early kinetics and is highly homologous to the herpes simplex virus 1 protein ICP0 (23), which has also been shown to be involved in preventing innate immune activation (24). In addition, ORF61 was shown to trans-activate or trans-repress the transcription of other VZV proteins, including its own promoter (25–29). The ORF61 protein contains an N-terminal RING domain, typically found in ubiquitin ligases and known to be required for its gene-regulatory functions (28), as well as the degradation of IRF3 (22). The isolated RING domain displayed ubiquitin ligase activity in vitro (30), and VZV ORF61 was shown to regulate its own stability via autoubiquitination (31).
In the present study, we demonstrate that, similar to VZV, infection with SVV leads to the stabilization of IκBα (20, 21). We further demonstrate that SVV ORF61, which shares 42.8% amino acid identity with VZV ORF61 (4) and regulates viral gene expression (32), also prevents IκBα degradation. Interestingly, IκBα phosphorylation was allowed but degradation was prevented in ORF61-expressing cells stimulated with TNF-α, resulting in the accumulation of phosphorylated IκBα (pIκBα). Inhibition of degradation is likely the consequence of SVV ORF61 forming a complex with the ubiquitin ligase subunit β-TrCP, thereby preventing TNF-α-induced ubiquitination of IκBα by the SCFβ-TrCP complex. This interaction seems to broadly interfere with SCFβ-TrCP function, since SVV ORF61 also affected the turnover of the β-TrCP target Snail. Similarly, VZV ORF61 prevented β-TrCP-mediated IκBα ubiquitination, indicating that this molecular mechanism is conserved between the two viruses. We show that, in addition to inhibiting IκBα ubiquitination, SVV, but not VZV, prevents the phosphorylation of IκBα, suggesting that SVV codes for at least one additional protein that contributes to the evasion of NF-κB signaling.
MATERIALS AND METHODS
Cell lines and recombinant viruses.
Rhesus fibroblasts had their lives extended through stable transduction of constitutively expressed human telomerase reverse transcriptase encoded by the lentivector pBABE and selected using 400 μg/ml G418, generally as described previously (33). The telomerized rhesus fibroblasts (TRFs) were then stably transduced with replication-incompetent lentiviruses containing luciferase-coding sequences from the firefly Photinus pyralis and from Renilla reniformis that were, respectively, inserted downstream of an NF-κB-dependent promoter or the cytomegalovirus (CMV) promoter (SA Biosciences). Transduced cells were selected using 3 μg/ml puromycin. The reverse Tet transactivator (rtTA) was stably introduced into telomerized human fibroblasts (THF) by inserting the coding region into the retrovector pCFG5-IEGZ and transducing the cells with derivative replication-incompetent virus as described previously (34). THF rtTA cells stably expressing inducible VZV ORF61 and simian immunodeficiency virus (SIV) GAG were generated using the pLVX lentivector system (Clontech) by cotransfecting pLVX, along with vectors encoding vesicular stomatitis virus G (VSV-G) (pMD2.G; Addgene 12259) and Gag/Pol (psPAX2; Addgene 12260), into HEK 293T cells using Lipofectamine LTX (Life Technologies) according to the manufacturer's protocol. Supernatant containing lentivirus was harvested from the transfected cells 48 h posttransfection, passed through 0.45-μm filters, and used to transduce THF rtTA in the presence of 5 μg/ml Polybrene (hexadimethrine bromide; Sigma-Aldrich). This process was repeated 24 h later, and the resulting cell lines were grown in the presence of 3 μg/ml puromycin to select for cells that expressed the viral genes. TRFs, TRF NF-κB (see below), THF rtTA, human embryonic kidney (HEK) 293T cells (ATCC), and the human fibroblast cell line MRC-5 (ATCC) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 140 IU of penicillin, and 140 μg of streptomycin per ml of culture medium.
TRFs were infected with the SVV Delta strain, which expresses enhanced green fluorescent protein (eGFP) (SVV.eGFP), which was inserted between US2 and US3 using homologous recombination (35), or with SVVΔ61 (see below). A monolayer of TRFs was infected by cocultivation with previously infected cells at the indicated ratios in DMEM supplemented with 2% FBS. Complete infection with SVV.eGFP was verified by fluorescence microscopy. VZV infections with the recombinant VZV Oka strain, in which eGFP was fused to the N terminus of ORF66 (VZV.eGFP) (generously provided by P. R. Kinchington, University of Pittsburgh, Pittsburgh, PA) (36), in MRC5 cells were performed in the same way as SVV infections.
Reagents and antibodies.
Rhesus (Rh) and human (Hu) TNF-α and IL-1β were obtained from R&D systems. Phorbol 12-myristate 13-acetate (PMA) (Enzo Life Sciences) was dissolved in dimethyl sulfoxide (DMSO) and used at the indicated concentrations. Poly(I·C) and LPS were acquired from Sigma-Aldrich and used at the indicated concentrations. MG132 (Z-Leu-Leu-Leu-al; Fisher Scientific) was dissolved in DMSO and used at 50 μM for 3 h. For detection of cellular and viral proteins in Western blots, we used the following antibodies: anti-IκBα sc-203 (Santa Cruz Biotechnology), anti-IκBα 10B (kindly provided by R. T. Hay, University of Dundee, Dundee, Scotland) (37), anti-phospho-IκBα Ser32/36 (Cell Signaling Technology), anti-FLAG M2 (peroxidase) (Sigma-Aldrich), anti-Snail C15D3 (Cell Signaling Technology), anti-β-TrCP sc-33213 (Santa Cruz Biotechnology), and anti-hemagglutinin (HA) HA-7 (Sigma-Aldrich). The monoclonal antibodies specific for SVV and VZV ORF31 (clone 31C_8) and ORF63 (clone 63_6) have been described previously (38). VZV ORF61 was detected using a rabbit polyclonal that was described previously and was kindly provided by P. R. Kinchington (University of Pittsburgh, Pittsburgh, PA) (39). Primary-antibody binding was visualized using horseradish peroxidase (HRP)-conjugated secondary antibodies specific for mouse (Santa Cruz) or rabbit (Thermo Scientific) IgG. The anti-FLAG M2 and anti-NF-κB p65 sc-372 (Santa Cruz Biotechnology) antibodies were used in immunofluorescence microscopy (immunofluorescence assay [IFA]). Secondary antibodies used for IFA were Alexa Fluor 488 goat anti-mouse IgG and Alexa Fluor 594 goat anti-rabbit IgG (Life Technologies).
Plasmids.
The HA–β-TrCP and FLAG-tagged vaccinia virus (VACV) A49 (FL-A49) expression plasmids were kindly provided by G. L. Smith (University of Cambridge, Cambridge, United Kingdom) and have been previously described (40). For the recombinant adenoviruses (Ad), we cloned SVV ORF61 from DNA isolated from TRFs infected with SVV.eGFP using the DNeasy blood and tissue kit (Qiagen) with the following primers: 5′-CACCGAATTCACCATGAACCCCCCGGCGTATACC-3′ (ORF61 FW) or 5′-CACCGAATTCACCATGGACTACAAGGATGACGACGATAAGAACCCCCCGGCGTATAC-3′ (ORF61 FW-FLAG) and 5′-AATAAAGGATCCTTATTTTCTCCGTACCTTTTTAGTTAACATTTCAATGCG-3′ (ORF61 Rev). The resulting PCR products were inserted in the adenovirus shuttle vector using EcoRI/BamHI sites. The RING mutant of ORF61 was generated using the shuttle vector with FLAG-ORF61 as a template and using the primers 5′-CCACCGGGAACTCCGCTATATGCATGAGC-3′ (QC FW) and 5′-GCTCATGCATATAGCGGAGTTCCCGGTGG-3′ (QC Rev).
For the transfection vectors, we used the same DNA as a template for PCR amplification of SVV ORF63. PCR was performed using the following primers: 5′-AATAAAGAATTCGCCACCATGGACTACAAGGATGACGACGATAAGCAGGCGCCCCGAG-3′ (ORF63 FW-FLAG) and 5′-AATAAAGGATCCTTATGTATTGTGTACAGACTCTCGTAACTCCGTG-3′ (ORF63 Rev). SVV ORF61 was amplified from the adenovirus shuttle vectors using the primers 5′-GCCACCATGGACTACAAGGATGACGACGATAAG-3′ (FW-FLAG) and 5′-TTATTTTCTCCGTACCTTTTTAGTTAACATTTCAATGCG-3′ (ORF61 Rev). These PCR-generated products were inserted into pcDNA3-IRES-nlsGFP, creating pcDNA3 FL-ORF63 and pcDNA3 FL-ORF61. VZV DNA was purified from MRC5 cells infected with VZV.eGFP and used as a template for PCR amplification of VZV ORF61. The primers used were 5′-AATAAAGAATTCGCCACCATGGACTACAAGGATGACGACGATAAGGATACCATATTAGCGGGCGGTAGC-3′ (ORF61VZV FW-FLAG) and 5′-AATAAAGCGGCCGCCTAGGACTTCTTCATCTTGTTTGGAATACC-3′ (ORF61VZV Rev). The PCR-generated product was inserted into the pLVX-Tight-Puro vector (Clonetech Laboratories). pLVX SIV GAG was created using DNA isolated from TRFs infected with a recombinant rhesus cytomegalovirus that expresses the protein (41) using the primers 5′-CACCGAATTCACCATGGGCGTGAGAAACTCCGTCTTG-3′ (GAG FW) and 5′-AATAAAGGATCCCTACTGGTCTCCTCCAAAGAGAGAATTGAG-3′ (GAG Rev).
All PCRs were performed with either the Expand High Fidelity PCR system (Roche) or AccuPrime Taq DNA Polymerase High Fidelity (Life Technologies), and all sequences were verified.
Luciferase reporter assay.
TRF NF-κB infected with SVV.eGFP were seeded in a black 96-well plate (Corning Inc.) at 24 h postinfection (p.i.). At 42 h p.i., the cells were incubated with the indicated NF-κB activators for 6 h to induce expression of NF-κB-driven firefly luciferase. Firefly and Renilla luciferases were measured using the Dual-Glo luciferase assay system (Promega), and luminescence was measured on a Veritas microplate luminometer (Promega). Data are presented as the ratio of firefly luciferase expression to Renilla luciferase expression.
Semiquantitative PCR.
Total cellular RNA was harvested using the NucleoSpin RNA isolation kit (Macherey-Nagel) in accordance with the supplied protocol. The collected RNA concentration was measured with the NanoDrop 1000 Spectrophotometer (Thermo Scientific), and single-stranded cDNA was made using Maxima Reverse Transcriptase (Thermo Scientific) with the manufacturer's protocol and random hexamers (TaKaRa). RANTES mRNA induction following TNF-α treatment was analyzed by semiquantitative real-time PCR (qPCR) using SYBR green PCR core reagents and Platinum Taq DNA polymerase (Invitrogen). Reactions were performed using the Applied Biosystems StepOnePlus real-time PCR system (Life Technologies). The primers used were RANTES Fw, 5′-CCTCGCTGTCATCCTCG-3′, and RANTES Rev, 5′-GCACACACTTGGCGATC-3′.The GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene was used as a housekeeping gene (GAPDH Fw, 5′-GCACCACCAACTGCTTAGCAC-3′, and GAPDH Rev, 5′-TCTTCTGGGTGGCAGTGATG-3′). The relative expression of RANTES was calculated using the method described by Livak and Schmittgen (42).
Immunofluorescence microscopy.
TRFs were seeded onto glass coverslips and infected the following day with the indicated viruses. Following infection and cytokine treatment, the cells were washed twice with phosphate-buffered saline (PBS) and fixed with 3.7% formaldehyde (Fisher Scientific) in PBS for 40 min at room temperature (RT). The cells were washed again in PBS and incubated with 50 mM ammonium chloride (NH4Cl) for 10 min to reduce nonspecific background. The cells were then permeabilized with 0.1% Triton X-100 for 4 min at RT and washed/blocked with 2% bovine serum albumin (Fisher Scientific) in PBS (PBA). The fixed cells were incubated for 1 h at 37°C with primary antibody diluted in PBA, washed in PBA, and incubated with secondary antibodies diluted in PBA for 1 h at 37°C. The cells were washed with PBA, followed by a PBS rinse, and the coverslips were then mounted on slides using ProLong Gold Anti-fade Reagent with DAPI (4′,6-diamidino-2-phenylindole) (Cell Signaling). Images were captured with an Axioskop 2 Plus fluorescence microscope and AxioVision v4.6 software (Zeiss).
Immunoprecipitations, tandem-repeated ubiquitin binding entity (TUBE) pulldown, and Western blotting.
For immunoprecipitation studies, HEK 293T cells were transfected with 3 μg of the indicated plasmids using the Lipofectamine 2000 reagent (Life Technologies) according to the manufacturer's protocol. Forty-eight hours posttransfection, the cells were lysed in a buffer containing 0.5% Nonidet P-40 (NP-40), 50 mM Tris-HCl (pH 7.5), and 5 mM MgCl2. The lysates were precleared for 1 h using protein A/G Plus-agarose beads and normal mouse serum sc-45051 (Santa Cruz Biotechnology). The lysates were transferred to a new tube with an anti-FLAG antibody and protein A/G Plus-agarose beads and incubated overnight at 4°C. The beads were washed with ice-cold Tris-buffered saline (20 mM Tris-HCl [pH 8.0], 150 mM NaCl, and 0.1% Tween 20), and the precipitated immune complexes were eluted from the beads by resuspension in Laemmli sample buffer (100 mM Tris-HCl [pH 8.0], 4% SDS, 20% glycerol, 10% 2-mercaptoethanol, bromophenol blue) and boiling the samples for 5 min at 95°C.
For the ubiquitination experiment, cells were washed once with ice-cold PBS and were subsequently lysed in a lysis buffer containing 1% NP-40, 50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 10% glycerol, Halt protease inhibitor cocktail, Halt phosphatase inhibitor cocktail (Thermo Scientific), and 15 μM N-ethylmaleimide. The lysates were incubated with protein A/G Plus-agarose beads for 30 min at 4°C to remove any nonspecific protein binding. The lysates were transferred to new tubes and incubated with agarose-TUBE 2 (Lifesensors) overnight at 4°C. After 3 washes with ice-cold Tris-buffered saline (20 mM Tris-HCl [pH 8.0], 150 mM NaCl, and 0.1% Tween 20), proteins were eluted with Laemmli sample buffer.
For all other Western blot analyses, cells were directly lysed in Laemmli sample buffer. Immunoprecipitated complexes and lysates were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Thermo Scientific). The membranes were incubated with primary antibodies, followed by HRP-conjugated secondary antibodies. Bound HRP-labeled antibodies were visualized using either SuperSignal West Pico Chemiluminescent Substrate, Pico substrate mixed with Western blot signal enhancer, SuperSignal West Femto Maximum Sensitivity Substrate, or ECL2 Western blotting substrate (Thermo Scientific).
Recombinant adenovirus production and infection.
The generation of infectious recombinant adenovirus expressing SVV ORF61, FLAG-tagged SVV ORF61 (FL-ORF61), SVV FL-ORF61 C19G, and green fluorescent protein (GFP) was previously described (43, 44). The vectors contain a tetracycline-responsive promoter and require the addition of a tetracycline-regulated transactivator (tTA) (45), which was provided by cotransduction with an adenovirus expressing the tetracycline transactivator (AdTA). TRFs were transduced in six-well clusters with the purified adenoviruses and AdTA at the indicated multiplicity of infection (MOI) in 0.5 ml of serum-free DMEM. After 2 h of rocking at 37°C, 1.5 ml of DMEM supplemented with 10% FBS was added, and incubation was continued for a total of 48 h.
Statistical analysis.
P values were determined using unpaired Student's t tests.
SVV BAC mutagenesis.
The SVV bacterial artificial chromosome (BAC) pSVV-FrDX in Escherichia coli strain DH10B was kindly provided by W. L. Gray (University of Arkansas for Medical Sciences, Little Rock, AR) (46). To create a ΔORF61 deletion mutant based on this wild type (wt) by homologous recombination, primers containing 50-bp homology to regions flanking ORF61 (Fw, 5′-AAAACAGTATTACAGTAAAATAACATGTAACTATGTGAATGTACATTGCT-3′, and Rev, 5′-CTAACGGCATATTTGGGCCTGGTTTTTGGGGACATCTATCTTCCACAGTC-3′) were used to amplify a kanamycin resistance (Kanr) cassette from plasmid pCP015 (47). The pCP015 forward primer binding site (5′-GTAAAACGACGGCCAGT) and reverse primer binding site (5′-GAAACAGCTATGACCATG) were added to the 3′ ends of the mutagenesis primers. Purified SVV BAC DNA was transformed into the E. coli strain SW105, which has heat-inducible λ recombination genes and an arabinose-inducible FLP recombinase (48). Bacterial cultures were grown in LB medium at 30°C until an optical density at 600 nm (OD600) of 0.6 was reached, and the λ-recombinant genes were heat induced by shaking at 42°C in a water bath for 15 min. The bacteria were subsequently chilled on ice for 10 min and made electrocompetent by washing 4 times with cold deionized water. For recombination, the generated competent E. coli cells were electroporated with the PCR product using a MicroPulser (Bio-Rad) and selected for Kan and chloramphenicol (Cm) resistance at 30°C on LB agar for 36 h. Cm/Kan-resistant colonies were grown in LB medium, and BAC DNA was isolated from the bacteria. Restriction digestion was performed using EcoRI, and the resulting DNA fragments were separated by electrophoresis on a 0.75% agarose gel in 0.5× Tris-borate-EDTA (TBE) buffer. The restriction patterns of the generated clones were compared to that of the parental wt and to an in silico restriction analysis.
To induce the FLP recombinase excising the Kanr cassette, clones were grown in LB medium with Cm until they reached an OD600 of 0.5 and incubated with 1 mg/ml arabinose for 1 h. The bacteria were streaked out on an LB plate with Cm selection using an inoculation loop and incubated overnight at 30°C. After colonies were visible, the clones were replica plated first on Cm/Kan LB agar, followed by Cm LB agar, and colonies that had lost Kanr were selected and characterized by restriction digestion and partial sequencing. To reconstitute the virus, BAC DNA was prepared from the bacteria and transfected into Vero cells using Lipofectamine 2000 (Life Technologies) according to the manufacturer's protocol. Viral plaques appeared approximately 7 days posttransfection.
RESULTS
SVV blocks NF-κB activation at or downstream of IKK activation.
Previous studies have shown that VZV infection inhibits TNF-α-induced activation of the NF-κB pathway (20, 21). To determine whether SVV similarly interferes with NF-κB signaling, we studied TNF-α-induced NF-κB activation in SVV-infected TRFs that stably express firefly luciferase under the control of an NF-κB promoter and constitutively express Renilla luciferase (TRF NF-κB). We infected TRF NF-κB with SVV.eGFP by coincubating uninfected and SVV-infected cells at a 5:1 ratio. After 42 h, the cells were incubated with increasing concentrations of RhTNF-α or HuTNF-α for 6 h. Immunofluorescence microscopy for GFP confirmed that nearly all the cells were infected (data not shown). NF-κB activity was calculated as the ratio between induced firefly and constitutive Renilla luciferase expression. The latter was measured to control for cell death resulting from viral infection. Mock-infected cells showed a dose-dependent increase in both RhTNF-α- and HuTNF-α-induced NF-κB activation, whereas NF-κB promoter activity was significantly reduced in SVV-infected cells (Fig. 1A).
FIG 1.

SVV inhibits NF-κB activation induced by various stimuli. TRFs stably expressing firefly luciferase under an NF-κB promoter and constitutively active Renilla luciferase (TRF-NFκB) were mock infected or infected with SVV.eGFP at a ratio of 5:1. At 42 h p.i., the cells were stimulated with the indicated increasing concentrations of RhTNF-α and HuTNF-α (A) or poly(I·C), PMA, LPS, and IL-1β (B) for 6 h. Firefly and Renilla luciferase expression was measured using a dual-luciferase reporter assay, and NF-κB activity was determined by normalizing the firefly signal to the Renilla signal. The results from one out of three (TNF-α) or two (other stimulants) independent experiments are shown. The error bars indicate standard deviations.
NF-κB is activated by many different signaling pathways that are induced by PAMPs, such as microbial RNA or DNA and LPS, or by proinflammatory cytokines, such as TNF-α and IL-1β. While NF-κB-terminal signaling employs multiple alternative upstream factors, they all converge at the IKK complex (49). To determine if SVV inhibits a common event in NF-κB activation, we stimulated mock- and SVV.eGFP-infected TRF NF-κB with increasing concentrations of poly(I·C) (an activator of the MDA5/RIG-I pathway [50]), phorbol 12-myristate 12-acetate (PMA) (which activates NF-κB via protein kinase C [51]), LPS (an activator of TRIF signaling [52]), and IL-1β for 6 h. At 42 h p.i., mock-infected cells showed a dose-dependent increase in firefly luciferase expression for all stimuli, which is indicative of NF-κB activation (Fig. 1B). In contrast, NF-κB activation by all stimuli was significantly reduced in SVV-infected cells (Fig. 1B). These data show that SVV-induced inhibition is not limited to the TNF-α-specific pathway and suggest that SVV targets a common event in the signaling cascade at the level of or downstream from IKK activation.
SVV inhibits NF-κB signaling by preventing the phosphorylation and degradation of IκBα.
To confirm that SVV inhibits NF-κB-driven cytokine expression, we studied RANTES mRNA induction by qPCR in TRFs that were mock or SVV.eGFP infected for 48 h and stimulated with TNF-α during the final 1, 3, or 6 h of infection. The mock-infected cells showed a time-dependent increase in RANTES mRNA expression, which was remarkably diminished in SVV-infected cells (Fig. 2A). To determine the signal transduction step at which SVV interferes with NF-κB activation, we studied the nuclear localization of the NF-κB complex in mock- and SVV-infected cells. TRFs were infected with SVV.eGFP for 48 h and stimulated with TNF-α for 45 min to activate NF-κB. TNF-α treatment resulted in nuclear localization of the NF-κB subunit p65 in uninfected cells, but not in SVV-infected cells (Fig. 2B, green [eGFP]). Since the translocation of the p50/p65 heterodimer from the cytoplasm into the nucleus is dependent on IκBα degradation, which is preceded by IKK-mediated phosphorylation, we analyzed the phosphorylation and degradation of IκBα in SVV-infected cells. TRFs were mock or SVV.eGFP infected for 48 h and subsequently stimulated with TNF-α for up to 60 min. In mock-infected cells, phosphorylated IκBα appeared after 5 min of TNF-α stimulation, and reduced IκBα levels were observed as early as 15 min after cytokine addition. After 60 min of stimulation, IκBα reappeared in mock-infected cells as a result of new synthesis (Fig. 2C). In contrast, in SVV-infected cells, only very low levels of IκBα phosphorylation were observed regardless of TNF-α stimulation (Fig. 2C, long exposure). Furthermore, IκBα was not degraded in SVV-infected cells and was detected at similar levels at all time points of TNF-α stimulation (Fig. 2C). These data suggest that SVV interferes with IκBα phosphorylation, thereby stabilizing IκBα in TNF-α-treated cells.
FIG 2.

SVV inhibits NF-κB-induced cytokine production by preventing IκBα activation. (A) TRFs were mock or SVV.eGFP infected (5:1 ratio) and stimulated with 100 ng/ml RhTNF-α at 42 h p.i. for the indicated times. TNF-α-induced RANTES mRNA expression was measured by qPCR using specific primers. The data were normalized to GAPDH mRNA expression in each sample and are shown as relative fold changes. Shown are the means and standard deviations of the results of two independent experiments with three replicates per sample in each experiment. (B) TRFs were mock or SVV.eGFP infected (10:1 ratio) for 48 h and stimulated with 100 ng/ml RhTNF-α for 45 min. Nuclear localization of the NF-κB subunit p65 was analyzed by immunofluorescence microscopy using a specific antibody (red). SVV.eGFP-infected cells appear green. (C) TRFs were mock or SVV.eGFP infected (5:1 ratio) for 48 h and stimulated with 100 ng/ml RhTNF-α for the indicated times. Lysates of the cells were analyzed for IκBα and phosphorylated IκBα by SDS-PAGE and Western blotting using specific antibodies. ORF31 expression was analyzed to confirm SVV infection, and GAPDH was used as a protein-loading control. The results of one representative experiment out of three independent experiments are shown.
SVV ORF61 inhibits NF-κB activation by preventing the degradation, but not the phosphorylation, of IκBα.
The inhibition of NF-κB activation observed in VZV-infected cells has been attributed to the ORF61 protein (21). VZV and SVV ORF61 proteins share 42.8% overall amino acid identity (4), and both proteins are implicated in the transactivation of expression of other viral genes (26, 32). To assess whether the ORF61 protein was responsible for the inhibition of NF-κB signaling in SVV-infected cells, we used recombinant adenovectors to ectopically express the protein. TRFs were transduced with an adenovector encoding SVV ORF61 (AdORF61, or AdFL-ORF61, in which ORF61 is N-terminally tagged with FLAG). Expression of the gene is dependent on the tetracycline-regulated transactivator, which is provided by cotransducing with AdTA. As a control, we used TRFs transduced with AdORF61 or AdFL-ORF61 only. At 48 h p.i., we studied TNF-α-induced RANTES expression by qPCR. In the control cells, we observed a time-dependent increase in RANTES expression, but when ORF61 was expressed, this response was strongly reduced (Fig. 3A). Similar results were obtained for TRFs expressing the FLAG-tagged ORF61 (data not shown). Thus, SVV ORF61 inhibits TNF-α-induced NF-κB signaling. Next, we determined whether ORF61 inhibits the nuclear translocation of NF-κB as observed in SVV-infected cells. In TRFs transduced with AdFL-ORF61 alone, treatment with TNF-α for 45 min resulted in the nuclear import of the NF-κB subunit p65 (Fig. 3B). In contrast, in TRFs that expressed ORF61 through cotransduction with AdTA, nuclear accumulation of p65 was not observed (Fig. 3B). FLAG-ORF61 expression was confirmed by staining with a FLAG-specific antibody (Fig. 3B). To study whether this block in nuclear translocation of NF-κB resulted from inhibited IκBα activation, we stimulated FLAG-ORF61-expressing cells with TNF-α at the indicated time points. In the absence of ORF61 expression, IκBα was phosphorylated after 5 min and degraded after 15 min and reappeared after 60 min of treatment with TNF-α (Fig. 3C). Conversely, IκBα degradation was not induced in the presence of ORF61 (Fig. 3C), suggesting that ORF61 inhibited NF-κB by preventing IκBα degradation. Interestingly, ORF61 did not affect cytokine-induced phosphorylation of IκBα (Fig. 3C). Thus, ORF61 appears not to be responsible for the inhibition of IκBα phosphorylation observed in SVV-infected cells, but rather, inhibits IκBα degradation at a step that follows phosphorylation.
FIG 3.

SVV ORF61 inhibits NF-κB-induced cytokine production by preventing IκBα degradation. TRFs were coinfected with a recombinant adenovirus expressing SVV ORF61 (AdORF61) or FLAG-tagged ORF61 (AdFL-ORF61) at an MOI of 15 and an adenovirus expressing the tetracycline transactivator (AdTA) at an MOI of 7. TRFs infected with AdORF61 or AdFL-ORF61 only were used as a control. (A) TRFs were infected with AdORF61 only or with AdORF61 and AdTA and stimulated with 100 ng/ml RhTNF-α for the indicated times at 48 h p.i. TNF-α-induced RANTES mRNA expression was measured by reverse transcription-PCR and qPCR using specific primers. The data were normalized to the level of GAPDH mRNA expression measured in each sample and are shown as the relative fold change. Shown are the means and standard deviations of the results of two independent experiments with three replicates per sample in each experiment. (B) TRFs infected with AdFL-ORF61 (MOI, 15) only or AdFL-ORF61 (MOI, 15) and AdTA (MOI, 7) for 48 h were incubated with 100 ng/ml RhTNF-α for 45 min, after which nuclear localization of the NF-κB subunit p65 was analyzed by immunofluorescence microscopy using a specific antibody (red). The cells were stained with a FLAG-specific antibody to visualize FLAG-ORF61 expression (green). (C) TRFs infected with AdFL-ORF61 (MOI, 15) only or AdFL-ORF61 (MOI, 15) and AdTA (MOI, 7) for 48 h were incubated with 100 ng/ml RhTNF-α for the indicated times. Lysates of the cells were analyzed for IκBα and phosphorylated IκBα by SDS-PAGE and Western blotting using the indicated antibodies. ORF61 expression was confirmed using a FLAG-specific antibody, and GAPDH was used as a protein-loading control. The results from one out of three independent experiments are shown.
SVV ORF61 inhibits TNF-α-induced ubiquitination of IκBα by targeting β-TrCP.
Upon being phosphorylated, IκBα is recognized by the F-box protein β-TrCP, which is associated with the proteins Skp1 and Cul1 and the Ring protein Roc1/Rbx1, together forming the E3 ligase complex SCFβ-TrCP. SCFβ-TrCP adds polyubiquitin chains to IκBα, which leads to the proteasomal degradation of IκBα and the subsequent release of the NF-κB subunits to the nucleus (17). The results described above suggest that ORF61 inhibits NF-κB signaling by preventing IκBα degradation postphosphorylation. Conceivably, ORF61 could either interfere with the ubiquitination of IκBα or prevent the degradation of the ubiquitinated protein by the proteasome. To determine if IκBα ubiquitination is affected in ORF61-expressing cells, we used agarose-conjugated TUBEs to isolate polyubiquitinated forms of IκBα (53). TRFs were cotransduced with AdORF61 and AdTA, and at 44 h p.i., the cells were treated with MG132 for 3 h to block the proteasome. The cells were subsequently stimulated with TNF-α for 1 h to initiate NF-κB activation. Lysates of the cells were incubated with TUBEs, and the resulting complexes were analyzed for the presence of IκBα by Western blotting. In stimulated control cells (AdORF61 only), higher-molecular-weight forms of IκBα were detected, corresponding to the polyubiquitinated form of the protein (Fig. 4A, lane 2 from left). In contrast, ubiquitinated forms of IκBα were almost undetectable in ORF61-expressing cells treated with TNF-α (Fig. 4A, lane 4). These results imply that ORF61 blocks the SCFβ-TrCP-mediated addition of ubiquitin chains to IκBα, thereby interfering with the degradation of the protein.
FIG 4.

SVV ORF61 interferes with ubiquitination by SCFβ-TrCP. (A) TRFs were infected with AdORF61 (MOI, 15) only or with AdORF61 (MOI, 15) and AdTA (MOI, 7). At 44 h p.i., the TRFs were incubated with 50 μM MG132 for 3 h, followed by stimulation with 100 ng/ml RhTNF-α for 1 h. Agarose-conjugated TUBEs were used for the immunoprecipitation (IP) of ubiquitinated IκBα. Whole lysates and the immunoprecipitated complexes were then analyzed by SDS-PAGE and Western blotting using an IκBα-specific antibody. Input lysates were analyzed for GAPDH as a protein-loading control. ORF61 expression was confirmed using reverse transcription-PCR and PCR with ORF61-specific primers (data not shown). (B) TRFs were infected with AdORF61 (MOI, 15) only, with AdORF61 (MOI, 15) and AdTA (MOI, 7), or with AdGFP (MOI, 15) and AdTA (MOI, 7). At 48 h p.i., whole-cell lysates were analyzed for Snail, FLAG, and GFP expression by SDS-PAGE and Western blotting using specific antibodies. GAPDH was used as a protein-loading control. (C) HEK 293T cells were cotransfected with HA-tagged β-TrCP and FL-A49, FL-ORF61, or FL-ORF63. At 48 posttransfection, the cells were lysed, and viral proteins were immunoprecipitated using a FLAG-specific antibody. The input lysates and immunoprecipitated complexes were analyzed for the presence of HA–β-TrCP by SDS-PAGE and Western blotting using a specific antibody. Viral protein expression was confirmed in the input lysates using the FLAG-specific antibody, and GAPDH was used as a loading control. Shown are the results of one representative experiment out of three independent experiments.
To determine whether ORF61 generally interferes with SCFβ-TrCP function, we examined the turnover of Snail, a well-known target of this ubiquitin ligase complex. Snail is phosphorylated by glycogen synthase kinase 3β (GSK-3β), creating the recognition motif or degron for β-TrCP (54). GSK-3β is constitutively active in resting cells, and therefore, Snail is continuously ubiquitinated by SCFβ-TrCP and subsequently degraded by the proteasome, resulting in a very short half-life (54). TRFs were transduced with AdFL-ORF61 in the absence or presence of AdTA. As an additional control, we used cells cotransduced with an adenovirus expressing GFP in a transactivator-dependent manner (AdGFP) and AdTA. At 48 h p.i., low levels of Snail were detected in TRFs infected with AdFL-ORF61 only or with AdGFP/AdTA (Fig. 4B). In contrast, when FLAG-ORF61 was expressed, we observed an accumulation of Snail, indicating that degradation of the protein by the SCFβ-TrCP complex was inhibited by ORF61 (Fig. 4B). Therefore, we conclude that ORF61 affects multiple SCFβ-TrCP target proteins in addition to IκBα.
To assess whether inhibition of SCF-mediated ubiquitin ligation involved a direct interaction between β-TrCP and ORF61, we performed coimmunoprecipitation experiments. As a viral control protein known to interact with β-TrCP, we included the VACV protein A49 previously reported to block the SCFβ-TrCP-mediated degradation of IκBα by interacting with β-TrCP (40). We cotransfected HEK 293T cells with expression plasmids encoding HA-tagged β-TrCP and FL-A49, FL-ORF61, or FLAG-tagged SVV ORF63 (FL-ORF63) as a negative control. Cells were harvested 48 h posttransfection, and the viral proteins were isolated by immunoprecipitation with a FLAG-specific antibody. β-TrCP was detected by Western blotting using an HA antibody in immunoprecipitates of FL-A49 and FL-ORF61, but not FL-ORF63 (Fig. 4C). Therefore, we conclude that, similar to the poxvirus protein A49, ORF61 specifically forms a complex with β-TrCP, thereby preventing SCFβ-TrCP-mediated ubiquitination of target proteins, such as IκBα and Snail.
The RING domain of SVV ORF61 is necessary for IκBα inhibition.
The RING domain of VZV ORF61 has E3 ubiquitin ligase activity in vitro (30, 31). This domain was found to be essential for the degradation of phosphorylated IRF3 (22) and for the inhibition of TNF-α-induced NF-κB signaling (21). Mutation of the cysteine at position 19 to a glycine residue (C19G) was shown to disrupt the RING domain of ORF61 and the protein's E3 ubiquitin ligase activity (28, 31). We introduced this mutation in the SVV ORF61-expressing adenovirus (AdFL-ORF61 C19G) and studied whether the protein was still able to inhibit NF-κB signaling. TRFs were transduced with AdTA alone or cotransduced with AdTA and AdGFP, AdFL-ORF61, or AdFL-ORF61 C19G for 48 h, followed by stimulation with TNF-α for 6 h to induce cytokine expression. Cells transduced with AdTA alone or AdTA/AdGFP showed RANTES expression upon TNF-α stimulation, which was inhibited in AdTA/AdFL-ORF61-infected cells (Fig. 5A). In contrast, RANTES induction was not inhibited in ORF61 C19G-expressing cells (Fig. 5A) despite comparable expression levels of ORF61 and ORF61 C19G (Fig. 5B). We also analyzed IκBα degradation in these cells using Western blotting. As shown in Fig. 5B, GFP-expressing cells displayed diminished IκBα levels after 20 min and restored IκBα levels after 60 min of treatment with TNF-α. As observed in Fig. 3C, ORF61 expression stabilized IκBα, but this was not observed for ORF61 C19G (Fig. 5B). These data indicate that the inhibition of the SCFβ-TrCP ubiquitin ligase complex by SVV ORF61 requires an intact RING domain and thus a functional ubiquitin ligase activity of ORF61.
FIG 5.
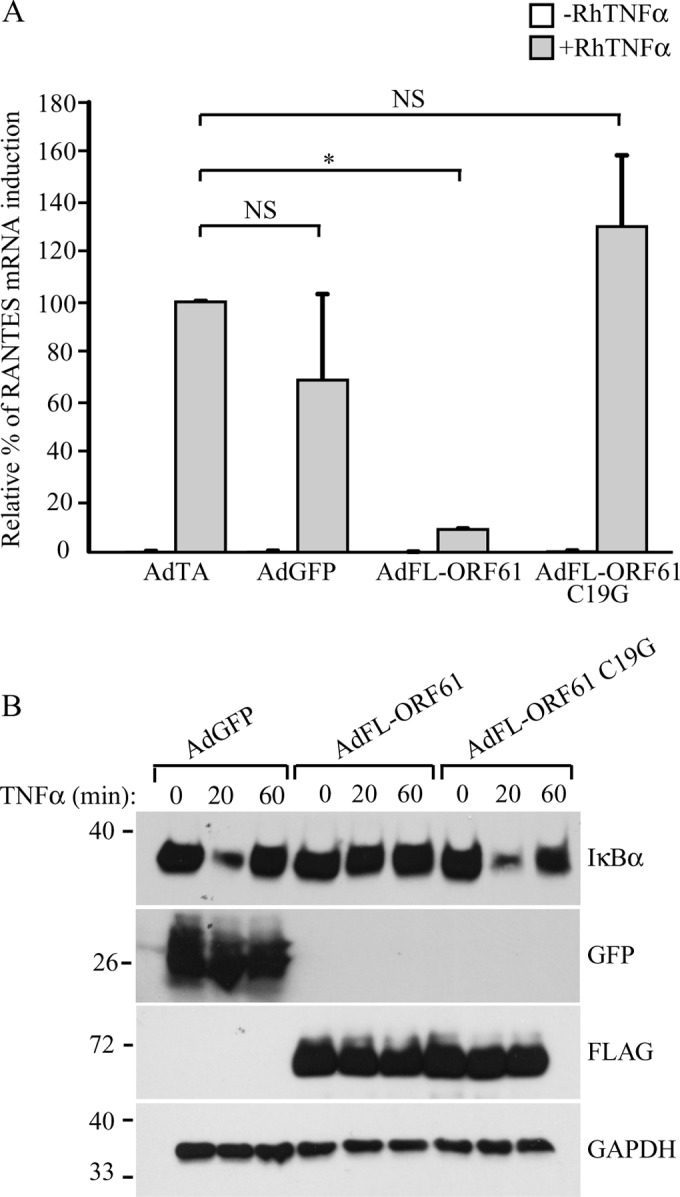
Inhibition of NF-κB signaling by SVV ORF61 is RING domain dependent. TRFs were infected with AdTA (MOI, 7) or coinfected with AdTA and AdGFP, AdFL-ORF61, or AdFL-ORF61 C19G (MOI, 15) for 48 h. (A) At 42 h p.i., the cells were stimulated with 100 ng/ml RhTNF-α for 6 h. RANTES mRNA expression was measured by reverse transcription-PCR and qPCR using specific primers. The data were normalized to the level of GAPDH mRNA expression measured in each sample, and the relative fold change of RANTES was determined. The graph shows relative fold changes normalized to the induction in the control cells (AdTA only). Shown are the means and standard deviations of the results of two independent experiments with three replicates per sample in each experiment. NS, not significant; *, P < 0.05. (B) At 48 h p.i., the TRFs were incubated with 100 ng/ml RhTNF-α for the indicated times. Whole lysates of the cells were analyzed for IκBα, ORF61, and GFP expression by SDS-PAGE and Western blotting using specific antibodies. GAPDH was used as a protein-loading control. The results from one out of three independent experiments are shown.
ORF61 deletion does not restore NF-κB signaling in SVV-infected cells.
To assess the contribution of ORF61 to the inhibition of NF-κB signaling by SVV, we deleted ORF61 by BAC mutagenesis, and SVVΔ61 was recovered. PCR analysis of DNA isolated from TRFs that were either mock infected or infected with wt SVV or SVVΔ61 confirmed deletion of the ORF61 gene (Fig. 6A). To address if phosphorylation and turnover of IκBα were differentially affected in the absence of ORF61, we stimulated the mock-, wt-, and SVVΔ61-infected cells with TNF-α for the indicated times and monitored IκBα and pIκBα levels by Western blotting. Mock-infected cells displayed diminished levels of IκBα after 20 min of TNF-α stimulation, and newly synthesized IκBα appeared after 60 min (Fig. 6B). In contrast, IκBα was not degraded in both wt- and SVVΔ61-infected cells, and phosphorylation of IκBα was observed only in mock-infected cells (Fig. 6B). Since phosphorylation of IκBα was not affected by ORF61, this result is consistent with a separate viral inhibitory mechanism acting upstream of ORF61 to prevent IκBα degradation by reducing phosphorylation. Thus, SVV seems to encode multiple mechanisms to inhibit NF-κB activation, and deletion of ORF61 alone does not restore IκBα degradation and release of NF-κB.
FIG 6.
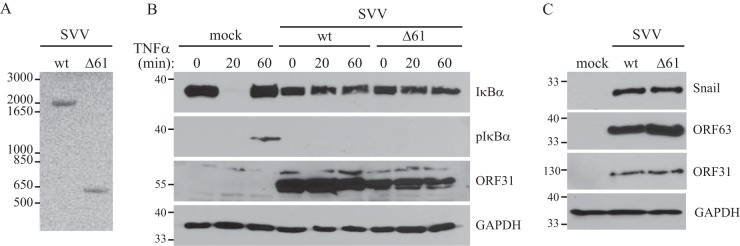
SVV ORF61 is not required for the inhibition of NF-κB signaling and Snail accumulation. TRFs were mock infected or infected with SVV wt or an ORF61 deletion mutant (Δ61) at a 5:1 ratio for 48 h. (A) PCR was performed on DNA extracted from the infected cells, and the presence of the ORF61 gene was studied using flanking primers. (B) At 48 h p.i., the cells were incubated with 100 ng/ml RhTNF-α for the indicated times. Lysates of the cells were analyzed for IκBα and phosphorylated IκBα by SDS-PAGE and Western blotting using specific antibodies. Samples were stained for ORF31 to confirm SVV infection, and GAPDH was used as a protein-loading control. (C) Lysates of the infected cells were analyzed for Snail expression by SDS-PAGE and Western blotting using a specific antibody. Infection was confirmed using antibodies for ORF63 and ORF31, and GAPDH was used as a protein-loading control. The results of one representative experiment out of three independent experiments are shown.
Since the targeting of β-TrCP by ORF61 results in the accumulation of Snail, we also compared Snail levels in cells infected with wt SVV or SVV with ORF61 deleted by Western blotting. However, compared to mock-infected cells, Snail accumulated in both wt- and SVVΔ61-infected cells (Fig. 6C). These data indicate that SVV interference with the degradation of Snail by the SCFβ-TrCP complex is not limited to ORF61.
VZV ORF61 blocks the ubiquitination of IκBα.
Next, we determined whether VZV blocks IκBα activation by the same mechanism as SVV. MRC5 cells were either mock or VZV.eGFP infected, and 48 h p.i., the cells were stimulated with TNF-α for the indicated times. IκBα was stabilized in VZV.eGFP-infected cells compared to mock-infected cells, but unlike SVV, we did not observe inhibition in IκBα phosphorylation (Fig. 7A). To determine whether VZV ORF61 was able to inhibit IκBα degradation, we stably expressed VZV ORF61 under the control of a tetracycline-inducible promoter in THF. These fibroblasts express the rtTA protein, and incubation with doxycycline (Dox) induced ORF61 expression (Fig. 7B and C). The THF rtTA ORF61VZV cells were incubated with 1 μg/ml Dox, and after 48 h, the cells were treated with TNF-α for the indicated times. In the absence of Dox, IκBα was degraded after 20 min of cytokine treatment, while Dox-dependent induction of ORF61 prevented IκBα degradation (Fig. 7B). To control for the effects of Dox-induced protein overexpression, we included a THF rtTA line that stably expressed the GAG protein of SIV (THF rtTA GAG). We did not observe increased IκBα stability in these cells, indicating that this was specifically induced by ORF61 (data not shown). Using VZV ORF61-expressing cells, we further examined whether ORF61 inhibited ubiquitination of IκBα. THF rtTA ORF61VZV were incubated with Dox for 48 h, and during the last 4 h, MG132 was added to the cultures to inhibit the proteasome. Upon stimulation of control cells with TNF-α for 1 h, polyubiquitinated IκBα was captured by TUBEs and visualized by Western blotting (Fig. 7C). In contrast, polyubiquitinated IκBα was not detected in ORF61-expressing cells. Together, these data indicate that, similar to SVV ORF61, VZV ORF61 inhibits ubiquitination of IκBα.
FIG 7.

VZV ORF61 inhibits the ubiquitination of IκBα. (A) MRC5 cells were mock or VZV infected for 48 h at a 5:1 ratio and stimulated with 100 ng/ml HuTNF-α at the indicated times. Lysates of the cells were analyzed for (phosphorylated) IκBα expression by SDS-PAGE and Western blotting using specific antibodies. The lysates were stained for ORF31 expression to control for infection, and GAPDH was used as a loading control. (B) THF rtTA ORF61VZV were incubated with 1 μg/ml Dox for 48 h, after which they were stimulated with 100 ng/ml HuTNF-α to activate NF-κB signaling. Lysates from the cells were stained for IκBα, ORF61, and GAPDH using specific antibodies in SDS-PAGE and Western blotting. (C) THF rtTA ORF61VZV were incubated with 1 μg/ml Dox for 44 h and incubated in 50 μM MG132 for 3 h, followed by stimulation with 100 ng/ml RhTNF-α for 1 h. Polyubiquitinated IκBα was immunoprecipitated from the lysates using TUBEs. Whole lysates and immunoprecipitated complexes were analyzed by SDS-PAGE and Western blotting using an IκBα-specific antibody. Input lysates were analyzed for ORF61 expression using a specific antibody, and GAPDH was used as a protein-loading control. Shown are the results of one representative experiment out of three independent experiments.
DISCUSSION
The results presented in this study suggest that SVV inhibits NF-κB activation by sequentially inhibiting phosphorylation and degradation of IκBα, thus retaining the NF-κB subunits p50 and p65 in the cytosol, preventing transcription of antiviral genes. Our data indicate that SVV ORF61 is responsible for the stabilization of IκBα downstream of phosphorylation. The viral protein interacts with β-TrCP, and this interaction likely prevents the ubiquitination and subsequent degradation of IκBα by the SCFβ-TrCP complex. β-TrCP facilitates the ubiquitination of several target genes, including Snail. We found increased levels of Snail in SVV ORF61-expressing cells, indicating that ORF61 broadly impedes protein degradation mediated by β-TrCP. We further showed that the RING domain of SVV ORF61 is critical for its ability to prevent IκBα degradation, suggesting that the ubiquitin ligase activity of ORF61 might be involved in inhibiting ubiquitination of IκBα. Similar to SVV ORF61, expression of VZV ORF61 inhibited ubiquitination of IκBα, indicating that the ORF61 proteins of the two viruses prevent NF-κB activation by similar mechanisms.
Deletion of ORF61 from SVV did not restore NF-κB signaling, consistent with SVV encoding at least one additional protein that inhibits NF-κB activation upstream of ORF61, most likely at the level of IκBα phosphorylation. Phosphorylation of IκBα by the IKK complex is required for its ubiquitination and degradation by the SCFβ-TrCP complex (17, 18). Infection with SVV resulted in low levels of phosphorylated IκBα, and treatment with TNF-α did not increase phosphorylation. Since SVV ORF61 alone did not block the phosphorylation of IκBα and phosphorylation was still observed in SVVΔ61-infected cells, we concluded that an unknown SVV protein interferes with phosphorylation, followed by ORF61 preventing ubiquitination of IκBα in a sequential process. The sequential inhibition of IκBα degradation thus underscores the importance of NF-κB-mediated innate immunity in controlling varicella viruses.
VZV infection also leads to the stabilization of IκBα (Fig. 7A), confirming previous observations in VZV-infected dendritic cells (21) and fibroblasts (20). Moreover, we demonstrate that isolated expression of VZV ORF61 prevents ubiquitination of IκBα, similar to SVV ORF61. However, unlike SVV, we observed that TNF-α-induced phosphorylation of IκBα was not inhibited in VZV-infected cells. This observation is consistent with a publication by Jones and Arvin that reported IκBα phosphorylation in VZV-infected cells (20). These results suggest both overlapping and divergent mechanisms by which SVV and VZV target the NF-κB pathway, a somewhat unexpected finding, given the high homology between SVV and VZV (55).
At present, the mechanism by which SVV blocks IκBα phosphorylation is unknown. We have not yet explored whether only TNF-α-induced IκBα phosphorylation is inhibited or whether SVV prevents IκBα phosphorylation regardless of the stimulus. One possibility is that SVV targets the IKK complex to inhibit NF-κB signaling, a strategy commonly used by viruses. For example, the VACV protein B14 and the hepatitis C virus protein NS5B inhibit IKK-mediated phosphorylation of IκBα by directly interacting with IKKβ or IKKα, respectively (56–58). In addition, the adenovirus protein E1A was shown to prevent UV-initiated IKKβ activation but did not affect the expression levels of the protein (59, 60). In contrast, the herpes simplex virus 1 protein ICP27 prevents IκBα phosphorylation by binding to the protein IκBα itself (61). The homolog of ICP27 in VZV is the ORF4 protein (62, 63). SVV has an ORF4 homolog (4) and, in addition, encodes ORFA, which is a 293-amino-acid-long truncated version of ORF4 that is not found in VZV (64, 65). It is therefore possible that ORFA is involved in the inhibition of IκBα phosphorylation that was uniquely observed in SVV-infected cells.
SVV ORF61 was shown to prevent the TNF-α-induced ubiquitination of IκBα. Typically, IκBα is phosphorylated by the activated IKK complex, creating a docking motif for β-TrCP to interact with IκBα and catalyze its ubiquitination (17, 18). We demonstrate that SVV ORF61 interacts with β-TrCP. In VZV-infected cells, ORF61 localizes predominantly to the nucleus (66). Fractionation studies revealed that SVV ORF61 in AdFL-ORF61-transduced cells is also mostly nuclear, though small amounts of protein were detected in the cytoplasm (data not shown). This raises the question of how a nuclear protein can affect a mostly cytosolic pathway. NF-κB–IκBα complexes have been shown to shuttle between the nucleus and the cytoplasm (15). The nuclear complexes can be activated by TNF-α, resulting in the degradation of IκBα by proteasomes residing in the nucleus (15, 16). Correspondingly, β-TrCP has been reported to localize to the nucleus through an interaction with heterogeneous nuclear ribonucleoprotein U (hnRNP-U) (67). We speculate that ORF61 interacts with nuclear β-TrCP, thereby both preventing nuclear IκBα degradation and preventing β-TrCP from translocating to the cytosol to act on cytoplasmic IκBα. Additionally, there is a small cytoplasmic fraction of ORF61 that could target cytoplasmic β-TrCP directly.
Amino acids 366 to 371 of ORF61 represent a motif (LSGPIKS) that is highly similar to the phosphodegron motifs found in β-TrCP substrates (DSGΦXS) (17, 18). Comparable phosphodegron-like (PDL) motifs were found in viral proteins that target the NF-κB pathway by interfering with the β-TrCP function. The Epstein-Barr virus (EBV) latent membrane protein 1 (LMP1) and the VACV A49 protein were shown to block the degradation of IκBα by binding to β-TrCP (40, 68). Mutagenesis analysis demonstrated that the PDL motifs of both proteins were required for the inhibition in NF-κB activation (40, 68). Additionally, the NSP1 protein of the porcine rotavirus (RV) strain OSU was demonstrated to stabilize activated IκBα by binding to and degrading β-TrCP (69). Morelli et al. showed that the N-terminal RING domain and the C-terminal PDL motif of OSU NSP1 were required for the inhibition of NF-κB signaling (70). We show that ORF61-mediated inhibition of NF-κB activation requires an intact RING domain (Fig. 5). However, SVV ORF61 did not reduce the levels of β-TrCP expressed in HEK 293T cells (Fig. 4C), suggesting that the ubiquitin ligase function of ORF61 is not required to mediate degradation of β-TrCP. Interestingly, a comparison of many different human and porcine RV strains with respect to their capacities to inhibit and degrade β-TrCP revealed that while all NSP1 homologs that include a PDL motif were able to inhibit NF-κB activation, not all affected the expression levels of β-TrCP (70). This indicates that β-TrCP degradation was not required for RV NSP1 to inhibit the SCF complex, suggesting that it is the interaction between NSP1 and β-TrCP that disrupts the protein's ability to ubiquitinate IκBα. Similarly, the RING domain of ORF61 might be required for a stable interaction. Alternatively, SVV ORF61 might mediate the degradation of other members of the SCFβ-TrCP complex.
Our data demonstrate that VZV ORF61 also interferes with the ubiquitination of IκBα, thereby stabilizing the protein. In contrast to SVV ORF61, however, the VZV protein does not have an obvious PDL motif. Thus, it is possible that VZV ORF61 prevents IκBα ubiquitination and degradation via a different strategy than SVV ORF61. Alternatively, VZV ORF61 might associate with the SCFβ-TrCP complex via a different motif.
The SCFβ-TrCP complex is involved in the degradation of multiple host proteins, including β-catenin, Snail (18), and p105 (71). Since ectopic expression of SVV ORF61 resulted in the stabilization of Snail (Fig. 4B), we concluded that the inhibition of β-TrCP affects other SCFβ-TrCP substrates, as well. Human immunodeficiency virus type 1 (HIV-1) Vpu also interacts with β-TrCP via its PDL motif (72). This interaction prevents TNF-α- and virus-induced degradation of IκBα (73). Like SVV ORF61, Vpu was shown to affect other β-TrCP targets, as well (74). In addition, Vpu utilizes the E3 ubiquitin ligase activity of the SCFβ-TrCP complex to degrade the antiviral factor tetherin and CD4 (72, 75). Mutagenesis studies showed that tetherin degradation is dependent on the interaction between Vpu and β-TrCP (72). It would be interesting to study whether ORF61 redirects the SCFβ-TrCP complex and regulates the stability of other endogenous or viral proteins.
Unexpectedly, deletion of ORF61 from the viral genome did not result in restored degradation of Snail (Fig. 6C). SCFβ-TrCP complex-mediated degradation of Snail is dependent on phosphorylation of its phosphodegron motif by GSK-3β (76). Interestingly, GSK-3β is also involved in TNF-α-, LPS-, and IL-1β-induced NF-κB activation (77), and Takada et al. showed that treatment of GSK-3β−/− mouse fibroblasts with TNF-α did not result in IKK activation (77). Since we observed that SVV inhibits IκBα phosphorylation independently of ORF61, it is conceivable that SVV interferes with GSK-3β function, thereby preventing IKK activation and Snail degradation, even in the absence of ORF61, which would explain Snail accumulation in SVVΔ61-infected cells. Interestingly, Liu and Cohen showed that VZV ORF12 drives the activation of GSK-3β by the phosphatidylinositol 3-kinase/Akt pathway in infected cells (78). The authors speculated that this process is required for entry of VZV into the cells and for protection from apoptosis, since ORF12 is a tegument protein. It is thus possible that SVV activates GSK-3β via ORF12 immediately following infection to aid efficient replication and subsequently inhibits phosphorylation of GSK-3β through the unidentified protein to prevent NF-κB activation in the infected cells.
In conclusion, we have shown that SVV inhibits NF-κB-driven protein expression via at least two sequentially operating strategies. We have established that SVV ORF61 interferes with the ubiquitination of IκBα by binding to β-TrCP. This interaction is likely dependent on the PDL motif that is present in the SVV ORF61 sequence. SVV also prevents the phosphorylation of IκBα, although the mechanism of this inhibition is presently unknown. It is common for viruses to inhibit signaling pathways at multiple levels. For example, IRF3-driven cytokine expression is inhibited by at least three different VZV proteins, including ORF61 (22), ORF47 (79), and IE62 (80). Such sequential inhibitory mechanisms along a signal transduction pathway likely serve as fail-safe strategies to efficiently block an immune response. The viral resources devoted to inhibiting a given pathway are likely directly proportional to the antiviral impact of the respective immune response pathway. Thus, ORF61 might be responsible for eliminating residual phosphorylated IκBα that escaped the upstream inhibitory mechanism. Deletion of ORF61 alone thus does not restore NF-κB-mediated innate immunity, which might explain why SVV lacking ORF61 was still able to establish primary and latent infection in rhesus macaques, as reported by Meyer et al. (81). However, SVV with ORF61 deleted displayed reduced viral gene expression in vivo, which could either be related to the transactivator function of ORF61 or due to increased expression of antiviral genes that suppress viral gene expression. In addition, there was an increased frequency of plasmacytoid dendritic cells in the bronchoalveolar lavage (BAL) fluid and an increase in IFN-β gene expression in SVVΔORF61-infected animals (81). Enhanced recruitment of dendritic cells and enhanced cytokine expression could be the direct result of increased NF-κB activity by infected cells due to lack of ORF61 inhibition, or lack of ORF61 could indirectly affect innate immune responses by reduced transactivation of other viral genes that interfere with innate signaling pathways, such as IE62 (80) and ORF63 (82, 83). If deletion of ORF61, together with the yet to be identified protein that inhibits IκBα phosphorylation, restores NF-κB activation, it is to be expected that SVV infection will be severely attenuated by innate immunity while possibly maintaining or even improving the induction of SVV-specific adaptive immunity. Further delineation of NF-κB-inhibitory pathways by SVV and VZV might thus lead to improved vaccine design.
ACKNOWLEDGMENTS
This work was supported by the Training Program in Virology of the National Institutes of Health through grant NIH 5 T32 AI074494 to T.W., by the American Heart Association through grant 0730325N awarded to V.R.D., by the National Center for Research Resources and the Office of Research Infrastructure Programs (ORIP) through grant P51OD011092 to K.F., and by an EMBO Long Term Fellowship to M.C.V.
We thank Tihana Lenac Roviš, Stipan Jonjić, Susanne Bailer, and Jürgen G. Haas for development and production of the VZV- and SVV-specific monoclonal antibodies; Amruta Bhusari for excellent technical assistance; and the Virology Core at the Oregon National Primate Research Center for producing the ORF61 adenoviruses.
REFERENCES
- 1.Gilden D, Cohrs RJ, Mahalingam R, Nagel MA. 2009. Varicella zoster virus vasculopathies: diverse clinical manifestations, laboratory features, pathogenesis, and treatment. Lancet Neurol 8:731–740. doi: 10.1016/S1474-4422(09)70134-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kinchington PR, Goins WF. 2011. Varicella zoster virus-induced pain and post-herpetic neuralgia in the human host and in rodent animal models. J Neurovirol 17:590–599. doi: 10.1007/s13365-011-0069-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vafai A, Wellish M, Gilden DH. 1988. Expression of varicella-zoster virus in blood mononuclear cells of patients with postherpetic neuralgia. Proc Natl Acad Sci U S A 85:2767–2770. doi: 10.1073/pnas.85.8.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gray WL. 2004. Simian varicella: a model for human varicella-zoster virus infections. Rev Med Virol 14:363–381. doi: 10.1002/rmv.437. [DOI] [PubMed] [Google Scholar]
- 5.Messaoudi I, Barron A, Wellish M, Engelmann F, Legasse A, Planer S, Gilden D, Nikolich-Zugich J, Mahalingam R. 2009. Simian varicella virus infection of rhesus macaques recapitulates essential features of varicella zoster virus infection in humans. PLoS Pathog 5:e1000657. doi: 10.1371/journal.ppat.1000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balachandran S, Beg AA. 2011. Defining emerging roles for NF-kappaB in antivirus responses: revisiting the interferon-beta enhanceosome paradigm. PLoS Pathog 7:e1002165. doi: 10.1371/journal.ppat.1002165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dutta J, Fan Y, Gupta N, Fan G, Gelinas C. 2006. Current insights into the regulation of programmed cell death by NF-[kappa]B. Oncogene 25:6800–6816. doi: 10.1038/sj.onc.1209938. [DOI] [PubMed] [Google Scholar]
- 8.Ogawa H, Iimura M, Eckmann L, Kagnoff MF. 2004. Regulated production of the chemokine CCL28 in human colon epithelium. Am J Physiol Gastrointest Liver Physiol 287:G1062–G1069. doi: 10.1152/ajpgi.00162.2004. [DOI] [PubMed] [Google Scholar]
- 9.Hayden MS, West AP, Ghosh S. 2006. NF-[kappa]B and the immune response. Oncogene 25:6758–6780. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- 10.Hayden MS, Ghosh S. 2008. Shared principles in NF-kappaB signaling. Cell 132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 11.Stylianou E, O'Neill LA, Rawlinson L, Edbrooke MR, Woo P, Saklatvala J. 1992. Interleukin 1 induces NF-kappa B through its type I but not its type II receptor in lymphocytes. J Biol Chem 267:15836–15841. [PubMed] [Google Scholar]
- 12.Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. 1999. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem 274:10689–10692. doi: 10.1074/jbc.274.16.10689. [DOI] [PubMed] [Google Scholar]
- 13.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 14.Karin M. 1999. How NF-kB is activated: the role of the IkB kinase (IKK) complex. Oncogene 18:7. [DOI] [PubMed] [Google Scholar]
- 15.Johnson C, Van Antwerp D, Hope TJ. 1999. An N-terminal nuclear export signal is required for the nucleocytoplasmic shuttling of IkappaBalpha. EMBO J 18:6682–6693. doi: 10.1093/emboj/18.23.6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Renard P, Percherancier Y, Kroll M, Thomas D, Virelizier JL, Arenzana-Seisdedos F, Bachelerie F. 2000. Inducible NF-kappaB activation is permitted by simultaneous degradation of nuclear IkappaBalpha. J Biol Chem 275:15193–15199. doi: 10.1074/jbc.275.20.15193. [DOI] [PubMed] [Google Scholar]
- 17.Kanarek N, Ben-Neriah Y. 2012. Regulation of NF-kappaB by ubiquitination and degradation of the IkappaBs. Immunol Rev 246:77–94. doi: 10.1111/j.1600-065X.2012.01098.x. [DOI] [PubMed] [Google Scholar]
- 18.Lau AW, Fukushima H, Wei W. 2012. The Fbw7 and betaTRCP E3 ubiquitin ligases and their roles in tumorigenesis. Front Biosci (Landmark ed) 17:2197–2212. doi: 10.2741/4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le Negrate G. 2012. Viral interference with innate immunity by preventing NF-kappaB activity. Cell Microbiol 14:168–181. doi: 10.1111/j.1462-5822.2011.01720.x. [DOI] [PubMed] [Google Scholar]
- 20.Jones JO, Arvin AM. 2006. Inhibition of the NF-kappaB pathway by varicella-zoster virus in vitro and in human epidermal cells in vivo. J Virol 80:5113–5124. doi: 10.1128/JVI.01956-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sloan E, Henriquez R, Kinchington PR, Slobedman B, Abendroth A. 2012. Varicella-zoster virus inhibition of the NF-kappaB pathway during infection of human dendritic cells: role for open reading frame 61 as a modulator of NF-kappaB activity. J Virol 86:1193–1202. doi: 10.1128/JVI.06400-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu H, Zheng C, Xing J, Wang S, Li S, Lin R, Mossman KL. 2011. Varicella-zoster virus immediate-early protein ORF61 abrogates the IRF3-mediated innate immune response through degradation of activated IRF3. J Virol 85:11079–11089. doi: 10.1128/JVI.05098-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moriuchi H, Moriuchi M, Smith HA, Straus SE, Cohen JI. 1992. Varicella-zoster virus open reading frame 61 protein is functionally homologous to herpes simplex virus type 1 ICP0. J Virol 66:7303–7308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith MC, Boutell C, Davido DJ. 2011. HSV-1 ICP0: paving the way for viral replication. Future Virol 6:421–429. doi: 10.2217/fvl.11.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moriuchi H, Moriuchi M, Straus SE, Cohen JI. 1993. Varicella-zoster virus (VZV) open reading frame 61 protein transactivates VZV gene promoters and enhances the infectivity of VZV DNA. J Virol 67:4290–4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Sommer M, Rajamani J, Arvin AM. 2009. Regulation of the ORF61 promoter and ORF61 functions in varicella-zoster virus replication and pathogenesis. J Virol 83:7560–7572. doi: 10.1128/JVI.00118-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagpal S, Ostrove JM. 1991. Characterization of a potent varicella-zoster virus-encoded trans-repressor. J Virol 65:5289–5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moriuchi H, Moriuchi M, Cohen JI. 1994. The RING finger domain of the varicella-zoster virus open reading frame 61 protein is required for its transregulatory functions. Virology 205:238–246. doi: 10.1006/viro.1994.1639. [DOI] [PubMed] [Google Scholar]
- 29.Perera LP, Mosca JD, Ruyechan WT, Hay J. 1992. Regulation of varicella-zoster virus gene expression in human T lymphocytes. J Virol 66:5298–5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Everett RD, Boutell C, McNair C, Grant L, Orr A. 2010. Comparison of the biological and biochemical activities of several members of the alphaherpesvirus ICP0 family of proteins. J Virol 84:3476–3487. doi: 10.1128/JVI.02544-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walters MS, Kyratsous CA, Silverstein SJ. 2010. The RING finger domain of Varicella-Zoster virus ORF61p has E3 ubiquitin ligase activity that is essential for efficient autoubiquitination and dispersion of Sp100-containing nuclear bodies. J Virol 84:6861–6865. doi: 10.1128/JVI.00335-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gray WL, Davis K, Ou Y, Ashburn C, Ward TM. 2007. Simian varicella virus gene 61 encodes a viral transactivator but is non-essential for in vitro replication. Arch Virol 152:553–563. doi: 10.1007/s00705-006-0866-0. [DOI] [PubMed] [Google Scholar]
- 33.Counter CM, Hahn WC, Wei W, Caddle SD, Beijersbergen RL, Lansdorp PM, Sedivy JM, Weinberg RA. 1998. Dissociation among in vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc Natl Acad Sci U S A 95:14723–14728. doi: 10.1073/pnas.95.25.14723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuss AW, Knodel M, Berberich-Siebelt F, Lindemann D, Schimpl A, Berberich I. 1999. A1 expression is stimulated by CD40 in B cells and rescues WEHI 231 cells from anti-IgM-induced cell death. Eur J Immunol 29:3077–3088. doi:. [DOI] [PubMed] [Google Scholar]
- 35.Mahalingam R, Wellish M, White T, Soike K, Cohrs R, Kleinschmidt-DeMasters BK, Gilden DH. 1998. Infectious simian varicella virus expressing the green fluorescent protein. J Neurovirol 4:438–444. doi: 10.3109/13550289809114543. [DOI] [PubMed] [Google Scholar]
- 36.Eisfeld AJ, Yee MB, Erazo A, Abendroth A, Kinchington PR. 2007. Downregulation of class I major histocompatibility complex surface expression by varicella-zoster virus involves open reading frame 66 protein kinase-dependent and -independent mechanisms. J Virol 81:9034–9049. doi: 10.1128/JVI.00711-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaffray E, Wood KM, Hay RT. 1995. Domain organization of I kappa B alpha and sites of interaction with NF-kappa B p65. Mol Cell Biol 15:2166–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lenac Rovis T, Bailer SM, Pothineni VR, Ouwendijk WJ, Simic H, Babic M, Miklic K, Malic S, Verweij MC, Baiker A, Gonzalez O, von Brunn A, Zimmer R, Fruh K, Verjans GM, Jonjic S, Haas J. 2013. Comprehensive analysis of varicella-zoster virus proteins using a new monoclonal antibody collection. J Virol 87:6943–6954. doi: 10.1128/JVI.00407-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ng TI, Keenan L, Kinchington PR, Grose C. 1994. Phosphorylation of varicella-zoster virus open reading frame (ORF) 62 regulatory product by viral ORF 47-associated protein kinase. J Virol 68:1350–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mansur DS, Maluquer de Motes C, Unterholzner L, Sumner RP, Ferguson BJ, Ren H, Strnadova P, Bowie AG, Smith GL. 2013. Poxvirus targeting of E3 ligase beta-TrCP by molecular mimicry: a mechanism to inhibit NF-kappaB activation and promote immune evasion and virulence. PLoS Pathog 9:e1003183. doi: 10.1371/journal.ppat.1003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hansen SG, Powers CJ, Richards R, Ventura AB, Ford JC, Siess D, Axthelm MK, Nelson JA, Jarvis MA, Picker LJ, Fruh K. 2010. Evasion of CD8+ T cells is critical for superinfection by cytomegalovirus. Science 328:102–106. doi: 10.1126/science.1185350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 43.Henkel JR, Apodaca G, Altschuler Y, Hardy S, Weisz OA. 1998. Selective perturbation of apical membrane traffic by expression of influenza M2, an acid-activated ion channel, in polarized Madin-Darby canine kidney cells. Mol Biol Cell 9:2477–2490. doi: 10.1091/mbc.9.9.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hardy S, Kitamura M, Harris-Stansil T, Dai Y, Phipps ML. 1997. Construction of adenovirus vectors through Cre-lox recombination. J Virol 71:1842–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gossen M, Bujard H. 1992. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A 89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gray WL, Zhou F, Noffke J, Tischer BK. 2011. Cloning the simian varicella virus genome in E. coli as an infectious bacterial artificial chromosome. Arch Virol 156:739–746. doi: 10.1007/s00705-010-0889-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. doi: 10.1016/0378-1119(95)00193-A. [DOI] [PubMed] [Google Scholar]
- 48.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hayden MS, Ghosh S. 2012. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 26:203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M Jr, Akira S, Yonehara S, Kato A, Fujita T. 2005. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol 175:2851–2858. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- 51.Li X, Fang Y, Zhao X, Jiang X, Duong T, Kain SR. 1999. Characterization of NFkappaB activation by detection of green fluorescent protein-tagged IkappaB degradation in living cells. J Biol Chem 274:21244–21250. doi: 10.1074/jbc.274.30.21244. [DOI] [PubMed] [Google Scholar]
- 52.Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, Akira S. 2002. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol 169:6668–6672. doi: 10.4049/jimmunol.169.12.6668. [DOI] [PubMed] [Google Scholar]
- 53.Hjerpe R, Aillet F, Lopitz-Otsoa F, Lang V, England P, Rodriguez MS. 2009. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep 10:1250–1258. doi: 10.1038/embor.2009.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schlessinger K, Hall A. 2004. GSK-3beta sets Snail's pace. Nat Cell Biol 6:913–915. doi: 10.1038/ncb1004-913. [DOI] [PubMed] [Google Scholar]
- 55.Gray WL. 2010. Simian varicella virus: molecular virology. Curr Top Microbiol Immunol 342:291–308. doi: 10.1007/82_2010_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Benfield CT, Mansur DS, McCoy LE, Ferguson BJ, Bahar MW, Oldring AP, Grimes JM, Stuart DI, Graham SC, Smith GL. 2011. Mapping the IkappaB kinase beta (IKKbeta)-binding interface of the B14 protein, a vaccinia virus inhibitor of IKKbeta-mediated activation of nuclear factor kappaB. J Biol Chem 286:20727–20735. doi: 10.1074/jbc.M111.231381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen RA, Ryzhakov G, Cooray S, Randow F, Smith GL. 2008. Inhibition of IkappaB kinase by vaccinia virus virulence factor B14. PLoS Pathog 4:e22. doi: 10.1371/journal.ppat.0040022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi SH, Park KJ, Ahn BY, Jung G, Lai MM, Hwang SB. 2006. Hepatitis C virus nonstructural 5B protein regulates tumor necrosis factor alpha signaling through effects on cellular IkappaB kinase. Mol Cell Biol 26:3048–3059. doi: 10.1128/MCB.26.8.3048-3059.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shao R, Tsai EM, Wei K, von Lindern R, Chen YH, Makino K, Hung MC. 2001. E1A inhibition of radiation-induced NF-kappaB activity through suppression of IKK activity and IkappaB degradation, independent of Akt activation. Cancer Res 61:7413–7416. [PubMed] [Google Scholar]
- 60.Shao R, Hu MC, Zhou BP, Lin SY, Chiao PJ, von Lindern RH, Spohn B, Hung MC. 1999. E1A sensitizes cells to tumor necrosis factor-induced apoptosis through inhibition of IkappaB kinases and nuclear factor kappaB activities. J Biol Chem 274:21495–21498. doi: 10.1074/jbc.274.31.21495. [DOI] [PubMed] [Google Scholar]
- 61.Kim JC, Lee SY, Kim SY, Kim JK, Kim HJ, Lee HM, Choi MS, Min JS, Kim MJ, Choi HS, Ahn JK. 2008. HSV-1 ICP27 suppresses NF-kappaB activity by stabilizing IkappaBalpha. FEBS Lett 582:2371–2376. doi: 10.1016/j.febslet.2008.05.044. [DOI] [PubMed] [Google Scholar]
- 62.Perera LP, Kaushal S, Kinchington PR, Mosca JD, Hayward GS, Straus SE. 1994. Varicella-zoster virus open reading frame 4 encodes a transcriptional activator that is functionally distinct from that of herpes simplex virus homology ICP27. J Virol 68:2468–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moriuchi H, Moriuchi M, Smith HA, Cohen JI. 1994. Varicella-zoster virus open reading frame 4 protein is functionally distinct from and does not complement its herpes simplex virus type 1 homolog, ICP27. J Virol 68:1987–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gray WL. 2012. The simian varicella virus ORF A is expressed in infected cells but is non-essential for replication in cell culture. Arch Virol 157:1803–1806. doi: 10.1007/s00705-012-1367-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mahalingam R, White T, Wellish M, Gilden DH, Soike K, Gray WL. 2000. Sequence analysis of the leftward end of simian varicella virus (EcoRI-I fragment) reveals the presence of an 8-bp repeat flanking the unique long segment and an 881-bp open-reading frame that is absent in the varicella zoster virus genome. Virology 274:420–428. doi: 10.1006/viro.2000.0465. [DOI] [PubMed] [Google Scholar]
- 66.Reichelt M, Brady J, Arvin AM. 2009. The replication cycle of varicella-zoster virus: analysis of the kinetics of viral protein expression, genome synthesis, and virion assembly at the single-cell level. J Virol 83:3904–3918. doi: 10.1128/JVI.02137-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Davis M, Hatzubai A, Andersen JS, Ben-Shushan E, Fisher GZ, Yaron A, Bauskin A, Mercurio F, Mann M, Ben-Neriah Y. 2002. Pseudosubstrate regulation of the SCF(beta-TrCP) ubiquitin ligase by hnRNP-U. Genes Dev 16:439–451. doi: 10.1101/gad.218702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tang W, Pavlish OA, Spiegelman VS, Parkhitko AA, Fuchs SY. 2003. Interaction of Epstein-Barr virus latent membrane protein 1 with SCFHOS/beta-TrCP E3 ubiquitin ligase regulates extent of NF-kappaB activation. J Biol Chem 278:48942–48949. doi: 10.1074/jbc.M307962200. [DOI] [PubMed] [Google Scholar]
- 69.Graff JW, Ettayebi K, Hardy ME. 2009. Rotavirus NSP1 inhibits NFkappaB activation by inducing proteasome-dependent degradation of beta-TrCP: a novel mechanism of IFN antagonism. PLoS Pathog 5:e1000280. doi: 10.1371/journal.ppat.1000280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morelli M, Dennis AF, Patton JT. 2015. Putative E3 ubiquitin ligase of human rotavirus inhibits NF-kappaB activation by using molecular mimicry to target beta-TrCP. mBio 6:e02490-14. doi: 10.1128/mBio.02490-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Orian A, Gonen H, Bercovich B, Fajerman I, Eytan E, Israel A, Mercurio F, Iwai K, Schwartz AL, Ciechanover A. 2000. SCF(beta)(-TrCP) ubiquitin ligase-mediated processing of NF-kappaB p105 requires phosphorylation of its C-terminus by IkappaB kinase. EMBO J 19:2580–2591. doi: 10.1093/emboj/19.11.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mangeat B, Gers-Huber G, Lehmann M, Zufferey M, Luban J, Piguet V. 2009. HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its beta-TrCP2-dependent degradation. PLoS Pathog 5:e1000574. doi: 10.1371/journal.ppat.1000574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bour S, Perrin C, Akari H, Strebel K. 2001. The human immunodeficiency virus type 1 Vpu protein inhibits NF-kappa B activation by interfering with beta TrCP-mediated degradation of Ikappa B. J Biol Chem 276:15920–15928. doi: 10.1074/jbc.M010533200. [DOI] [PubMed] [Google Scholar]
- 74.Besnard-Guerin C, Belaidouni N, Lassot I, Segeral E, Jobart A, Marchal C, Benarous R. 2004. HIV-1 Vpu sequesters beta-transducin repeat-containing protein (betaTrCP) in the cytoplasm and provokes the accumulation of beta-catenin and other SCFbetaTrCP substrates. J Biol Chem 279:788–795. doi: 10.1074/jbc.M308068200. [DOI] [PubMed] [Google Scholar]
- 75.Margottin F, Bour SP, Durand H, Selig L, Benichou S, Richard V, Thomas D, Strebel K, Benarous R. 1998. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol Cell 1:565–574. doi: 10.1016/S1097-2765(00)80056-8. [DOI] [PubMed] [Google Scholar]
- 76.Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. 2004. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol 6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 77.Takada Y, Fang X, Jamaluddin MS, Boyd DD, Aggarwal BB. 2004. Genetic deletion of glycogen synthase kinase-3beta abrogates activation of IkappaBalpha kinase, JNK, Akt, and p44/p42 MAPK but potentiates apoptosis induced by tumor necrosis factor. J Biol Chem 279:39541–39554. doi: 10.1074/jbc.M403449200. [DOI] [PubMed] [Google Scholar]
- 78.Liu X, Cohen JI. 2013. Varicella-zoster virus ORF12 protein activates the phosphatidylinositol 3-kinase/Akt pathway to regulate cell cycle progression. J Virol 87:1842–1848. doi: 10.1128/JVI.02395-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vandevenne P, Lebrun M, El Mjiyad N, Ote I, Di Valentin E, Habraken Y, Dortu E, Piette J, Sadzot-Delvaux C. 2011. The varicella-zoster virus ORF47 kinase interferes with host innate immune response by inhibiting the activation of IRF3. PLoS One 6:e16870. doi: 10.1371/journal.pone.0016870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sen N, Sommer M, Che X, White K, Ruyechan WT, Arvin AM. 2010. Varicella-zoster virus immediate-early protein 62 blocks interferon regulatory factor 3 (IRF3) phosphorylation at key serine residues: a novel mechanism of IRF3 inhibition among herpesviruses. J Virol 84:9240–9253. doi: 10.1128/JVI.01147-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Meyer C, Kerns A, Haberthur K, Dewane J, Walker J, Gray W, Messaoudi I. 2013. Attenuation of the adaptive immune response in rhesus macaques infected with simian varicella virus lacking open reading frame 61. J Virol 87:2151–2163. doi: 10.1128/JVI.02369-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ambagala AP, Cohen JI. 2007. Varicella-zoster virus IE63, a major viral latency protein, is required to inhibit the alpha interferon-induced antiviral response. J Virol 81:7844–7851. doi: 10.1128/JVI.00325-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Verweij MC, Wellish M, Whitmer T, Malouli D, Lapel M, Jonjic S, Haas JG, DeFilippis VR, Mahalingam R, Fruh K. 2015. Varicella viruses inhibit interferon-stimulated JAK-STAT signaling through multiple mechanisms. PLoS Pathog 11:e1004901. doi: 10.1371/journal.ppat.1004901. [DOI] [PMC free article] [PubMed] [Google Scholar]