

ABSTRACT
Viruses are causally associated with a number of human malignancies. In this study, we sought to identify new virus-cancer associations by searching RNA sequencing data sets from >2,000 patients, encompassing 21 cancers from The Cancer Genome Atlas (TCGA), for the presence of viral sequences. In agreement with previous studies, we found human papillomavirus 16 (HPV16) and HPV18 in oropharyngeal cancer and hepatitis B and C viruses in liver cancer. Unexpectedly, however, we found HPV38, a cutaneous form of HPV associated with skin cancer, in 32 of 168 samples from endometrial cancer. In 12 of the HPV38-positive (HPV38+) samples, we observed at least one paired read that mapped to both human and HPV38 genomes, indicative of viral integration into the host DNA, something not previously demonstrated for HPV38. The expression levels of HPV38 transcripts were relatively low, and all 32 HPV38+ samples belonged to the same experimental batch of 40 samples, whereas none of the other 128 endometrial carcinoma samples were HPV38+, raising doubts about the significance of the HPV38 association. Moreover, the HPV38+ samples contained the same 10 novel single nucleotide variations (SNVs), leading us to hypothesize that one patient was infected with this new isolate of HPV38, which was integrated into his/her genome and may have cross-contaminated other TCGA samples within batch 228. Based on our analysis, we propose guidelines to examine the batch effect, virus expression level, and SNVs as part of next-generation sequencing (NGS) data analysis for evaluating the significance of viral/pathogen sequences in clinical samples.
IMPORTANCE High-throughput RNA sequencing (RNA-Seq), followed by computational analysis, has vastly accelerated the identification of viral and other pathogenic sequences in clinical samples, but cross-contamination during the processing of the samples remain a major problem that can lead to erroneous conclusions. We found HPV38 sequences specifically present in RNA-Seq samples from endometrial cancer patients from TCGA, a virus not previously associated with this type of cancer. However, multiple lines of evidence suggest possible cross-contamination in these samples, which were processed together in the same batch. Despite this potential cross-contamination, our data indicate that we have detected a new isolate of HPV38 that appears to be integrated into the human genome. We also provide general guidelines for computational detection and interpretation of pathogen-disease associations.
INTRODUCTION
Viruses are major causes of human morbidity and mortality, and some are known to be risk factors for tumorigenesis. Among the latter, human papillomavirus 16 (HPV16) and HPV18 are established causes of cervical and oropharyngeal carcinomas, HPV38 is associated with skin cancer (1, 2), and hepatitis B virus infection is associated with hepatocellular carcinoma (3). Cancer genome characterization initiatives, including The Cancer Genome Atlas (TCGA), have provided next-generation sequencing (NGS) data on thousands of tumors and various human cancers, and these data are valuable sources for understanding the transcriptional basis of tumorigenesis, as well as variations in tumors (4). Computational techniques have been used to quantify viral presence in many cancers, thereby accelerating the identification of virus-disease associations (5–7). However, the increasing sensitivity of these techniques may also reveal previously unappreciated viral contaminations that could be erroneously associated with the disease (8–10).
Here, we used computational techniques for identifying viral sequences in tumor samples. Unexpectedly, we found sequences matching HPV38 in endometrial cancer patients, with evidence of viral integration, a novel and potentially interesting finding. Upon further investigation, however, we noted that the HPV38 strain contained 10 novel mutations, and all the HPV38-positive (HPV38+) samples were from the same sequencing batch on a single plate, raising the possibility of cross-contamination. Our findings indicate the importance of being aware of batch- and plate-specific results for TCGA and other large-scale sequencing databases. Accordingly, we provide guidelines for the analysis of these types of data.
MATERIALS AND METHODS
Obtaining RNA-Seq data sets.
All high-throughput RNA sequencing (RNA-Seq) data sets for this study were obtained from TCGA research network (http://cancergenome.nih.gov/). Only RNA-Seq data from the Illumina platform with paired-end sequencing after May 2012 were used. The data were handled in accordance with the Data Use Certification Agreement for dbGaP study accession number phs000178. All data were downloaded using the GeneTorrent program from the Cancer Genome Hub (https://cghub.ucsc.edu/).
Filtering human, bacterial, and abundant sequencing reads.
All unmapped reads were initially extracted from TCGA BAM (binary sequence alignment map) files using samtools (11). The low-quality reads were removed using process_shortreads (12), with command line options “-c -q -s 17 -w 0.15 -filter_illumina -no_read_trimming.” Potential PCR clones were removed using the clone_filter program (12) with default settings. Reads were also discarded if they mapped to the human reference genome (hg19, GRCh37, including all alternative haplotypes), the human transcriptome, abundant sequences [including vector sequences (http://www.ncbi.nlm.nih.gov/tools/vecscreen/univec/), phage sequences (13), and poly(A/C) sequences], or bacterial rRNA and/or genomic sequences (13). The human transcriptome included GENECODE v19 comprehensive transcripts (14), RefSeq genes, all human mRNAs (15), UCSC genes (16), Ensemble genes (17), lincRNAs (18), and human rRNA sequences. The data for GENECODE, RefSeq, UCSC, and Ensemble transcripts were obtained using the UCSC Table Browser in March 2014. Mapping was performed using the Burrows-Wheeler Aligner (BWA) (19) with command line options “l = 28, k = 3, n = 0.1, and q = 20.”
Detecting viral sequences.
We created a novel comprehensive database of viral sequences by combining 3,931 known viral sequences from the European Nucleotide Archive (13) and the partially overlapping set of 28,038 viral sequences from the RINS (20) database. Viral sequences were combined using the nrdb program (21) to remove trivial redundancies. For each RNA-Seq library, all filtered reads were mapped to this virus database using the BWA (19), and the number of high-quality and unique reads that mapped to each virus was recorded. We used the “bwa aln” program with command line options “k = 1 and n = 1,” followed by “bwa sampe,” which provides 98% sequence homology for our typical read length of 50 bp (i.e., it allows one mismatch over the read length). For each library, only viruses with at least 10 mapped reads were reported.
Finding single nucleotide polymorphisms (SNPs).
Reads mapping to the HPV38 genome (accession number U31787) from all RNA-Seq libraries were piled up using samtools mpileup (11) with the command line options “−ugf U31787.fa,” followed by variant detection using bcftools (11) with the command line option “−vcg.” In total, 12 variants with raw read depths (DP) of >5 and quality (QUAL) of >10 were detected. Two of 12 mutations matched other known isolates (S418 and FA125) of HPV38 when BLASTed, whereas the remaining 10 were novel mutations.
Detecting viral integration in the human genome.
A BWA index database containing both human (hg19) and HPV38 (accession number U31787) genomes was created. Selected paired-end RNA-Seq libraries were aligned to this database using the “bwa mem” program (22) with default settings. For each library, chimeric read pairs (reads with one end mapping to the human genome and the mate (i.e. the other end) mapping to the HPV38 genome) were extracted. All chimeric reads were further examined using BLAST (23) to ensure the quality of alignment to both the human and HPV38 genomes.
Gene expression analysis.
The expression data were downloaded from TCGA data portal RNASeq V2 platform (https://tcga-data.nci.nih.gov/tcgafiles/ftp_auth/distro_ftpusers/anonymous/tumor/ucec/cgcc/unc.edu/illuminahiseq_rnaseqv2/rnaseqv2/unc.edu_UCEC.IlluminaHiSeq_RNASeqV2.Level_3.1.12.0/). Differentially expressed genes between samples grouped as HPV38+ and HPV38 negative (HPV38−) were obtained using the EBSeq program (24); we assumed that samples within a group were replicates. The number of differentially regulated genes was determined using a false discovery rate (FDR) value of <0.05 as a cutoff from the EBSeq output. The randomization between HPV38+ and HPV38− samples was done by randomly switching the group assignment for each sample in order to determine if there was a correlation between gene expression and whether a sample was HPV38+ or HPV38−.
RESULTS
Viral sequences in cancer RNA-Seq samples.
We developed a computational pipeline to search for viral sequences in NGS data (Fig. 1). In agreement with previous studies (5, 6), the most commonly observed viral sequences in our 2,102 compiled RNA-Seq data sets (see Table S1 in the supplemental material) comprising 21 cancers belonged to HPV, hepatitis virus, and herpesviruses, which collectively were present in ∼9.5% of cancer patients (Table 1). HPV sequences mainly corresponding to HPV16 were found in abundance (>10,000 reads, on average) in many patients (87%) with squamous cell cervical carcinoma (CESC), consistent with the known association of cervical carcinoma with HPV (25) (see Table S2 in the supplemental material). Nearly a quarter (24.3%) of patients with hepatocellular carcinoma had sequences corresponding to hepatitis virus, and ∼11% of patients with head and neck cancers (HNSC) had sequences from HPV16, consistent with prior reports of association of hepatitis virus (25) and HPV (26) with these tumors. Similar to another study (6), we also observed low abundance (<200 reads, on average) of Epstein-Barr virus (EBV), cytomegalovirus (CMV), and human herpesvirus 6B (HHV-6B) in several cancers. For example, 17 of 80 patients with adenocarcinoma of the colon (COAD) or rectum (READ) had EBV, CMV, or HHV-6B (Table 1). Notably, we did not detect any viral sequences in data sets of adrenocortical carcinoma (ACC), diffused large B-cell lymphoma (DLBL), glioblastoma multiforme (GBM), kidney chromophobe (KICH), kidney renal papillary cell carcinoma (KIRP), lung adenocarcinoma (LAUD), and thyroid carcinoma (THCA).
FIG 1.
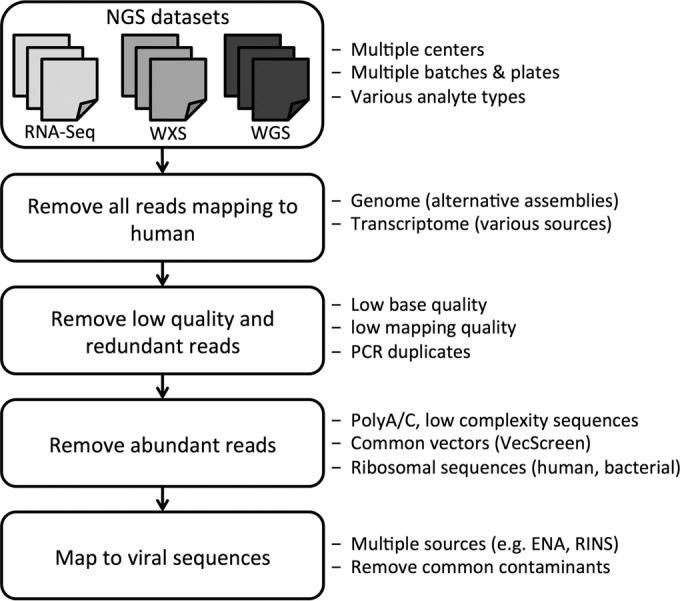
Computational pipeline for identifying viral sequences in NGS data. NGS data (RNA-Seq, WXS, and WGS) from cancer samples were obtained from the Cancer Genomics Hub. Sequencing reads that were mapped to the human genome and/or transcriptome were removed. The low-quality, redundant, low-complexity, and ribosomal reads and those that were mapped to common plasmid and vector sequences were also removed. The remaining reads were mapped to the viral database to quantify the viral presence (see Materials and Methods).
TABLE 1.
Association of viruses with human cancer
| Cancer | No. of samples | No. of patientsa |
Other virusesb | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HBV | HCV | EBV | CMV | HHV-6B | HPV16 | HPV38 | |||
| ACC (adrenocortical carcinoma) | 79 | ||||||||
| BLCA (bladder urothelial carcinoma) | 119 | 1 | 3 | 1 | 1 | 3 (HPV6) | |||
| BRCA (breast invasive carcinoma) | 125 | 1 | |||||||
| CESC (cervical squamous cell carcinoma) | 91 | 1 | 1 | 59 | 11 (HPV18), 9 (HPV45), 3 (HPV59), 13 (HPVother) | ||||
| COAD (colon adenocarcinoma) | 44 | 2 | 4 | 2 | 2 (HPyV6) | ||||
| DLBC (diffuse large B-cell lymphoma) | 28 | ||||||||
| GBM (glioblastoma multiforme) | 95 | ||||||||
| HNSC (head and neck squamous cell carcinoma) | 123 | 2 | 1 | 14 | 2 (HPV33), 2 (HPV35), 1 (HPV56), 1 (HHV-1) | ||||
| KICH (kidney chromophobe) | 66 | ||||||||
| KIRC (kidney renal clear cell carcinoma) | 67 | 1 (HPV18), 1 (HPV94) | |||||||
| KIRP (kidney renal papillary cell carcinoma) | 120 | ||||||||
| LGG (brain lower grade glioma) | 100 | 1 | |||||||
| LIHC (liver hepatocellular carcinoma) | 115 | 22 | 6 | 1 | 1 | 1 (HPV18), 1 (AAV2) | |||
| LUAD (lung adenocarcinoma) | 125 | ||||||||
| LUSC (lung squamous cell carcinoma) | 125 | 3 | 1 | 1 (HPV30) | |||||
| PRAD (prostate adenocarcinoma) | 124 | 1 | 1 | ||||||
| READ (rectum adenocarcinoma) | 36 | 5 | 4 | ||||||
| SKCM (skin cutaneous melanoma) | 82 | 1 | 1 | ||||||
| THCA (thyroid carcinoma) | 123 | ||||||||
| UCEC (uterine corpus endometrioid carcinoma) | 168 | 30c | |||||||
| UCS (uterine carcinosarcoma) | 57 | 1 | 1 (HPV5), 1 (HPV38b), 1 (HPV133) | ||||||
| Total | 2,012 | 23 | 6 | 14 | 17 | 5 | 77 | 30 | 55 |
The number of patients with each cancer type with observed viral sequences in the RNA-Seq data. HHV, human herpesvirus; HPV, human papillomavirus; HCV, hepatitis C virus; HBV, hepatitis B virus; CMV, cytomegalovirus. The numbers after the virus abbreviations represent subtypes.
The number of other viruses that were found in each cancer type, with the virus name in parentheses. AAV, adeno-associated virus; HPyV, human polyomavirus.
See Discussion in the text.
HPV38 sequences were present in endometrial cancer TCGA RNA-Seq samples.
Unexpectedly, we found that 32 out of 168 RNA-Seq samples with uterine corpus endometrioid carcinoma had sequencing reads mapping uniquely to HPV38 (see Materials and Methods), with the number of reads corresponding to HPV38 ranging from 14 to 554 (mean = 72 reads per sample) (Table 2). This is ∼200-fold less than the number of HPV16 reads in cervical or head and neck carcinomas or hepatitis B virus (HBV) reads in liver cancer but within a 3-fold range of the number of reads observed for EBV, CMV, and HHV-6B in other cancers. We next examined whole-exome sequencing (WXS) data from other sequencing centers available for selected samples to potentially verify the HPV38 association with endometrial carcinoma, but we did not detect any samples containing HPV38 sequences. This is in contrast to HPV16 or HBV, where viral presence in RNA-Seq data correlated with identifying the viral sequences in the WXS data, as well (9). Similarly, we detected EBV and HHV-6B reads in the WXS data for most of the samples where these viruses were detected by RNA-Seq (Table 3). Surprisingly, however, the majority of samples with CMV sequences by RNA-Seq did not have CMV sequences in the WXS data (Table 4). Similar patterns were observed when examining the WXS data for samples where EBV, HHV-6B, and CMV were detected by RNA-Seq (6) (see Table S3 in the supplemental material). The absence of viral sequences in the WXS data could reflect contamination of the RNA-Seq but not the WXS libraries, or it could represent some intrinsic limitations related to the genome coverage of the WXS libraries. Indeed, in one of the two available whole-genome sequencing (WGS) libraries, we detected significantly more reads matching CMV than in the WXS data, even after normalizing for library size (P < 1E−15) (Table 4).
TABLE 2.
RNA-Seq samples with HPV38 sequences
| Sample no. | TCGA barcode | No. of readsa |
|---|---|---|
| S01 | TCGA-AJ-A3OK-01A-12R-A22K-07 | 554 |
| S02 | TCGA-B5-A3F9-01A-21R-A22K-07 | 214 |
| S03 | TCGA-AX-A3FX-01A-11R-A22K-07 | 137 |
| S04 | TCGA-AJ-A3NC-01A-11R-A22K-07 | 129 |
| S05 | TCGA-AX-A3GB-01A-11R-A22K-07 | 124 |
| S06 | TCGA-B5-A3FC-01A-11R-A22K-07 | 108 |
| S07 | TCGA-EY-A3QX-01A-11R-A22K-07 | 85 |
| S08 | TCGA-K6-A3WQ-01A-11R-A22K-07 | 79 |
| S09 | TCGA-AJ-A3OJ-01A-11R-A22K-07 | 77 |
| S10 | TCGA-AJ-A3NF-01A-11R-A22K-07 | 48 |
| S11 | TCGA-AX-A3FV-01A-11R-A22K-07 | 48 |
| S12 | TCGA-BG-A3PP-01A-11R-A22K-07 | 43 |
| S13 | TCGA-AJ-A3I9-01A-11R-A22K-07 | 42 |
| S14 | TCGA-AX-A3FW-01A-11R-A22K-07 | 42 |
| S15 | TCGA-BK-A13B-01A-51R-A22K-07 | 42 |
| S16 | TCGA-FI-A3PV-01A-11R-A22K-07 | 42 |
| S17 | TCGA-EO-A3L0-01A-11R-A22K-07 | 36 |
| S18 | TCGA-AJ-A3QS-01A-11R-A22K-07 | 32 |
| S19 | TCGA-A5-A3LO-01A-11R-A22K-07 | 30 |
| S20 | TCGA-AJ-A3TW-01A-11R-A22K-07 | 30 |
| S21 | TCGA-D1-A3JQ-01A-11R-A22K-07 | 26 |
| S22 | TCGA-KP-A3W0-01A-21R-A22K-07 | 26 |
| S23 | TCGA-A5-A1OH-01A-21R-A22K-07 | 24 |
| S24 | TCGA-B5-A1MS-01B-11R-A22K-07 | 24 |
| S25 | TCGA-D1-A3JP-01A-31R-A22K-07 | 24 |
| S26 | TCGA-AJ-A3NH-01A-11R-A22K-07 | 22 |
| S27 | TCGA-KP-A3VZ-01A-11R-A22K-07 | 22 |
| S28 | TCGA-A5-A3LP-01A-11R-A22K-07 | 20 |
| S29 | TCGA-AJ-A3NE-01A-11R-A22K-07 | 14 |
| S30 | TCGA-AJ-A3NG-01A-11R-A22K-07 | 14 |
| S31 | TCGA-AX-A3FZ-01A-11R-A22K-07 | 14 |
| S32 | TCGA-KJ-A3U4-01A-11R-A22K-07 | 14 |
The number of reads that uniquely mapped to HPV38 from RNA-Seq samples from endometrial cancer patients.
TABLE 3.
RNA-Seq and corresponding WXS samples with EBV and HHV-6B sequences
| Sequence | TCGA barcode |
Cancer | No. of readsa |
||
|---|---|---|---|---|---|
| WXS | RNA-Seq | RNA-Seq | WXS | ||
| EBV | TCGA-CK-6747-01A-11D-1835-10 | TCGA-CK-6747-01A-11R-1839-07 | COAD (colon) | 18 | 8 |
| TCGA-D5-6920-01A-11D-1924-10 | TCGA-D5-6920-01A-11R-1928-07 | COAD (colon) | 26 | 8 | |
| TCGA-AG-3725-01A-11D-1733-10 | TCGA-AG-3725-01A-11R-1736-07 | READ (rectum) | 66 | 42 | |
| TCGA-CI-6624-01C-11D-1826-10 | TCGA-CI-6624-01C-11R-1830-07 | READ (rectum) | 34 | 2 | |
| TCGA-DC-6160-01A-11D-1657-10 | TCGA-DC-6160-01A-11R-1660-07 | READ (rectum) | 34 | 96 | |
| TCGA-EI-6511-01A-11D-1733-10 | TCGA-EI-6511-01A-11R-1736-07 | READ (rectum) | 348 | 206 | |
| TCGA-EI-6882-01A-11D-1924-10 | TCGA-EI-6882-01A-11R-1928-07 | READ (rectum) | 14 | 8 | |
| TCGA-FC-A4JI-01A-11D-A257-08 | TCGA-FC-A4JI-01A-11R-A250-07 | PRAD (prostate) | 48 | 6 | |
| TCGA-DK-A2I4-01A-11D-A21A-08 | TCGA-DK-A2I4-01A-11R-A21D-07 | BLCA (bladder) | 24 | 0 | |
| TCGA-CR-7392-01A-11D-2012-08 | TCGA-CR-7392-01A-11R-2016-07 | HNSC (head and neck) | 12 | 0 | |
| TCGA-DQ-7593-01A-11D-2229-08 | TCGA-DQ-7593-01A-11R-2232-07 | HNSC (head and neck) | 40 | 10 | |
| TCGA-18-3407-01A-01D-0983-08 | TCGA-18-3407-01A-01R-0980-07 | LUSC (lung [squamous cell]) | 18 | 114 | |
| TCGA-66-2769-01A-02D-1522-08 | TCGA-66-2769-01A-02R-0851-07 | LUSC (lung [squamous cell]) | 44 | 0 | |
| TCGA-98-7454-01A-11D-2042-08 | TCGA-98-7454-01A-11R-2045-07 | LUSC (lung [squamous cell]) | 60 | 0 | |
| HHV-6B | TCGA-A6-5665-01B-03D-2298-08 | TCGA-A6-5665-01A-01R-1653-07 | COAD (colon) | 30 | 236 |
| TCGA-AD-6899-01A-11D-1924-10 | TCGA-AD-6899-01A-11R-1928-07 | COAD (colon) | 12 | 212 | |
| TCGA-DK-A2I2-01A-11D-A17V-08 | TCGA-DK-A2I2-01A-11R-A180-07 | BLCA (bladder) | 46 | 2 | |
| TCGA-EA-A50E-01A-21D-A26G-09 | TCGA-EA-A50E-01A-21R-A26T-07 | CESC (cervix) | 24 | 18 | |
| TCGA-S3-A6ZG-01A-22D-A32I-09 | TCGA-S3-A6ZG-01A-22R-A32P-07 | BRCA (breast) | 40 | 42 | |
Number of reads matching the indicated virus in RNA-Seq and corresponding WXS data.
TABLE 4.
RNA-Seq and corresponding WXS/WGS samples with CMV sequences
| Cancer | TCGA barcode |
No. of readsa |
||
|---|---|---|---|---|
| WXS | RNA-Seq | RNA-Seq | WXS | |
| COAD (colon) | TCGA-A6-5657-01A-01D-1650-10 | TCGA-A6-5657-01A-01R-A32Z-07 | 42 | 4 |
| COAD (colon) | TCGA-D5-6529-01A-11D-1771-10 | TCGA-D5-6529-01A-11R-1774-07 | 98 | 8 |
| COAD (colon) | TCGA-D5-6536-01A-11D-1719-10 | TCGA-D5-6536-01A-11R-1723-07 | 58 | 0 |
| READ (rectum) | TCGA-CI-6619-01B-11D-1826-10 | TCGA-CI-6619-01B-11R-1830-07 | 1,860 | 66 |
| READ (rectum) | TCGA-CL-4957-01A-01D-1733-10 | TCGA-CL-4957-01A-01R-1736-07 | 156 | 38 |
| READ (rectum) | TCGA-EI-6884-01A-11D-1924-10 | TCGA-EI-6884-01A-11R-1928-07 | 32 | 0 |
| READ (rectum) | TCGA-AG-3731-01A-11D-1733-10 | TCGA-AG-3731-01A-11R-1736-07 | 20 | 0 |
| PRAD (prostate) | TCGA-FC-A4JI-01A-11D-A257-08 | TCGA-FC-A4JI-01A-11R-A250-07 | 44 | 0 |
| LIHC (liver) | TCGA-CC-5263-01A-01D-A12Z-10 | TCGA-CC-5263-01A-01R-A131-07 | 56 | 0 |
| HNSC (head and neck) | TCGA-D6-6826-01A-11D-1912-08 | TCGA-D6-6826-01A-11R-1915-07 | 166 | 0 (8) |
| BLCA (bladder) | TCGA-FD-A3SN-01A-12D-A22Z-08 | TCGA-FD-A3SN-01A-12R-A22U-07 | 638 | 4 (176) |
| BLCA (bladder) | TCGA-PQ-A6FN-01A-11D-A31L-08 | TCGA-A6-6649-01A-11R-1774-07 | 320 | 4 |
| BLCA (bladder) | TCGA-DK-A1AF-01A-11D-A13W-08 | TCGA-DK-A1AF-01A-11R-A13Y-07 | 2,120 | 10 |
| CESC (cervix) | TCGA-EA-A50E-01A-21D-A26G-09 | TCGA-EA-A50E-01A-21R-A26T-07 | 20 | 0 |
| SKCM (skin) | TCGA-EB-A3Y7-01A-11D-A23B-08 | TCGA-EB-A3Y7-01A-11R-A239-07 | 12 | 0 |
| UCS (uterine) | TCGA-N5-A4RM-01A-11D-A28R-08 | TCGA-NA-A4QV-01A-11R-A28V-07 | 188 | 0 |
| UCS (uterine) | TCGA-NA-A4QV-01A-11D-A28R-08 | TCGA-PQ-A6FN-01A-11R-A31N-07 | 412 | 0 |
The number of reads matching the indicated virus in RNA-Seq and the corresponding WXS data. The two numbers in parentheses are from corresponding WGS data with TCGA barcodes of TCGA-D6-6826-01A-11D-1911-02 and TCGA-FD-A3SN-01A-12D-A233-26, respectively.
Identification of an HPV38 strain with 10 new SNPs.
Because many variants of HPV38 have been discovered (27), we examined our HPV38+ samples for novel variants. We pooled the reads mapping to HPV38 from all HPV38+ samples and used samtools (11) to detect high-quality variants (see Materials and Methods). We detected 12 mutations compared to the reference HPV38 (accession number U31787), 10 of which were novel mutations while 2 matched mutations in the S418 and FA125 subtypes (Table 5). Eight mutations were in coding regions (E1, E2, L1, and L2), and 4 were in noncoding regions. We then examined whether HPV38+ samples contained different variants of HPV38. Whenever the mutation region was covered (>1 read) in a sample, the sample indeed contained that mutation (Fig. 2), i.e., none of the HPV38+ samples had a single nucleotide variation (SNV) that was absent in other samples, suggesting that the reads mapped to HPV38 in HPV38+ samples are from the same source, consistent with cross-contamination.
TABLE 5.
HPV38 mutations
| SNV no. | Gene | Positiona | SNV | Statusb |
|---|---|---|---|---|
| SNV01 | Noncoding | 45 | C→T | S418 |
| SNV02 | E1 | 1014 | C→A | Novel |
| SNV03 | 2113 | T→C | Novel | |
| SNV04 | E2 | 2993 | T→C | Novel |
| SNV05 | 3325 | C→G | FA125 | |
| SNV06 | 3767 | C→G | Novel | |
| SNV07 | 3920 | A→G | Novel | |
| SNV08 | Noncoding | 4030 | C→G | Novel |
| SNV09 | L2 | 5625 | G→C | Novel |
| SNV10 | L1 | 6523 | A→C | Novel |
| SNV11 | Noncoding | 7230 | G→C | Novel |
| SNV12 | 7378 | G→A | Novel |
Reference accession no. U31787.
Status of the mutation (novel or the strain of the reported mutation).
FIG 2.

HPV38 SNVs in endometrial cancer samples. For each sample (rows), the SNVs that were found in its sequencing reads mapping to HPV38 are shown. Black indicates that there was a read covering the SNV region and the read contained the SNV; gray marks the cases where there was no read covering the SNV region. We did not observe any read that covered an SNV region but did not contain the SNV.
HPV38 might have been integrated into the human host genome.
As other HPVs are known to integrate into the host genome (28), we asked whether HPV38 was also integrated into the human genome. We examined all chimeric paired-end reads where one end mapped to HPV38 and the mate mapped to the human genome (see Materials and Methods). Overall, we detected 19 such high-quality chimeric reads (Table 6). The integration sites seem to be random; however, the number of chimeric reads in our samples is small, so repeated patterns of integration analogous to those observed for HPV16 in cervical cancer (29) could be missed. Moreover, despite the high quality of the detected chimeric paired-end reads, some of them might represent artifacts that were generated by NGS during amplifications and annealing.
TABLE 6.
HPV38 integration sitesa
| Viral gene | Host gene | Host positionb | Sample no. |
|---|---|---|---|
| E6 | ST3GAL3c | chr1:44287568 | S26 |
| VRK2 | chr2:58036979 | S24 | |
| PTCHD4 | chr6:48280112 | S05 | |
| DCUN1D1c | chr3:182671521 | S17 | |
| MSX2 | chr5:174243847 | S14 | |
| C1orf100 | chr1:244498605 | S05 | |
| E7 | SMCR9 | chr17:17339681 | S12 |
| E1 | TSLP | chr5:110405535 | S24 |
| C3orf79 | chr3:153116968 | S21 | |
| RBM15Bc | chr3:51430732 | S28 | |
| SUCOc | chr1:172579894 | S28 | |
| NFKBID | chr19:36378125 | S03 | |
| LOC389602 | chr7:155895707 | S21 | |
| E2 | CCDC12c | chr3:47001968 | S22 |
| LOC389033 | chr2:130567052 | S24 | |
| Y_RNA | chr12:17858721 | S08 | |
| SCAMP1d | chr5:77651331 | S01 | |
| L1 | GPATCH2L | chr14:76778745 | S05 |
| LRP1Bc | chr2:141049612 | S01 |
The integration sites obtained from 19 high-quality chimeric reads (see Materials and Methods). Virus reference accession no. U31787; host reference genome, hg19.
Positions are within ±500-bp (the typical insert size) range of actual integration.
Integration is within the gene body (for the others, integration is upstream or downstream).
The corresponding chimeric read contains the novel A→G mutation at U31787 3920.
Differential expression between samples may represent a batch effect.
We next determined whether HPV38+ samples had different expression profiles than HPV38− samples. Indeed, we observed 61 genes that were significantly down- or upregulated in the 32 HPV38+ samples compared to the 136 HPV− samples, whereas when 32 samples were selected at random and compared to the remaining samples, >98% of the time, only 0 or 1 gene was differentially expressed. However, although the HPV38+ samples were from various sources, including the International Genomics Consortium, Gynecologic Oncology Group, and University of Pittsburgh, we realized that during the sequencing process at the sequencing center at the University of North Carolina, they were placed on the same plate (A22K) and grouped into the same batch (batch 228), whereas HPV38− samples were distributed among other plates and batches. This raises the possibility that the difference in expression profiles between HPV38+ and HPV38− samples was due to a batch effect, as noted generally for NGS (30) data and TCGA (31), rather than necessarily indicating an association between HPV38 and endometrial cancer.
DISCUSSION
We have identified sequencing reads that map uniquely to HPV38 in RNA-Seq data from endometrial cancer patients in the TCGA database. The HPV38 strain from these samples contained 10 novel mutations and was integrated into the genome, something not previously shown for HPV38. However, there were several indications of possible cross-contamination within these samples. First, all the HPV38+ samples were from the same batch (batch 228), and no other batch with endometrial cancer samples contained HPV38+ samples, a statistically highly unlikely situation if the association of HPV38 with endometrial carcinoma were real (Fisher exact test; P < 3E−27). Second, the same viral mutations, when detectable, were shared by all HPV38+ samples, suggesting a single source of infection/contamination. Third, the number of reads matching HPV38 in these samples was generally low (mean = 72 reads), and the corresponding WXS data did not contain any HPV38 sequence.
Given that the sensitivity of NGS techniques has markedly increased, many previously undetected details, including handling and bench contamination, are now evident. This presents a new challenge for computational techniques to detect and evaluate contamination sources. For example, HeLa cell contamination led to misidentification of HPV18 in noncervical cancers in TCGA (8, 9), and 293T contamination led to misidentification of viruses in ovarian cancer cell lines (10). To begin addressing this challenge, we propose guidelines (a flowchart) for finding viral sequences from NGS data (Fig. 3). These guidelines take into consideration the viral copy number, nucleotide variations in the virus, the viral integration sites, differential gene expression in individuals harboring the virus versus those lacking it, the sample source (batch and plate numbers), the availability of independent replicates (e.g., RNA-Seq, WXS, and WGS), and known common contaminations (e.g., plasmids and cell lines) as indicators of potential cross-contamination. These considerations/guidelines for the detection of viral sequences should be readily applicable to other pathogenic sequences, as well.
FIG 3.
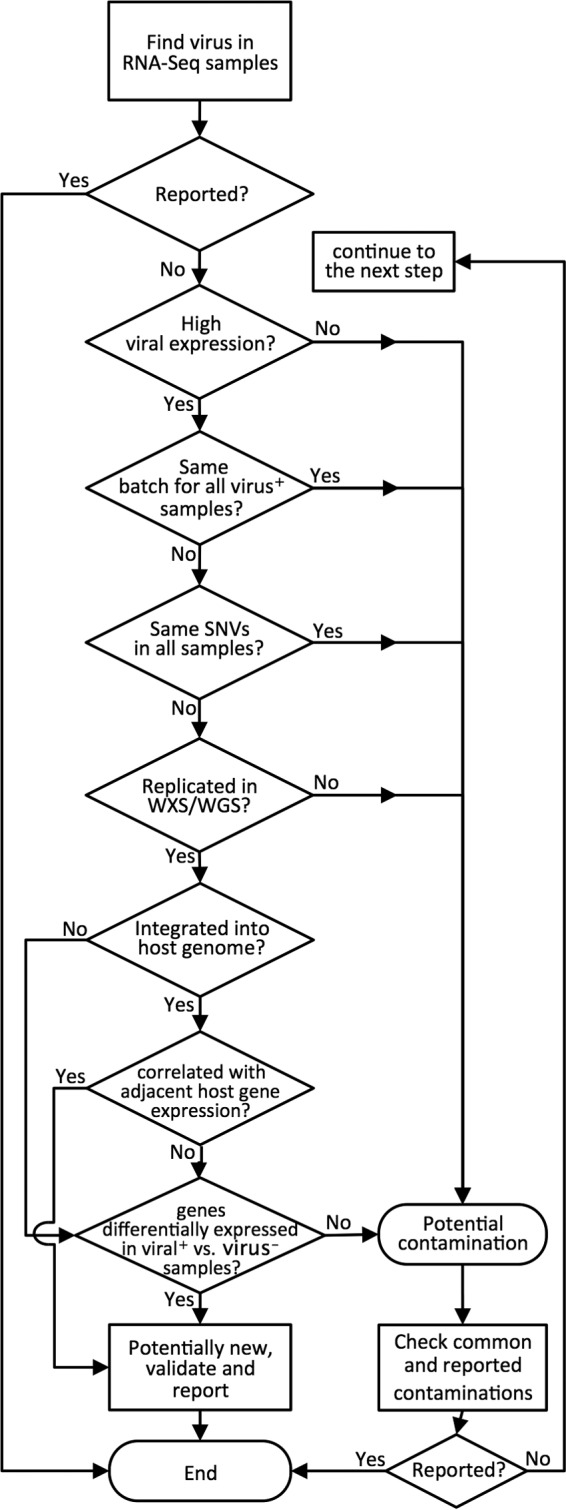
Proposed guidelines for using NGS data to identify viruses and/or other pathogens and distinguishing possible contamination. The order of some of the steps in the flowchart can be changed as needed.
Supplementary Material
ACKNOWLEDGMENTS
The results shown here are in part based upon data generated by the TCGA Research Network (http://cancergenome.nih.gov/) program. This study utilized the high-performance computing facility, helix/biowulf (http://biowulf.nih.gov/), at the National Institutes of Health.
This work was supported by the Division of Intramural Research, National Heart, Lung, and Blood Institute, National Institutes of Health, and a grant to M.K. from the NIH (K22-KHL125593A).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00822-15.
REFERENCES
- 1.Bzhalava D, Guan P, Franceschi S, Dillner J, Clifford G. 2013. A systematic review of the prevalence of mucosal and cutaneous human papillomavirus types. Virology 445:224–231. doi: 10.1016/j.virol.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 2.Thomas M, Narayan N, Pim D, Tomaic V, Massimi P, Nagasaka K, Kranjec C, Gammoh N, Banks L. 2008. Human papillomaviruses, cervical cancer and cell polarity. Oncogene 27:7018–7030. doi: 10.1038/onc.2008.351. [DOI] [PubMed] [Google Scholar]
- 3.Williams R. 2006. Global challenges in liver disease. Hepatology 44:521–526. doi: 10.1002/hep.21347. [DOI] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. 2013. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khoury JD, Tannir NM, Williams MD, Chen Y, Yao H, Zhang J, Thompson EJ, TCGA Network, Meric-Bernstam F, Medeiros LJ, Weinstein JN, Su X. 2013. Landscape of DNA virus associations across human malignant cancers: analysis of 3,775 cases using RNA-Seq. J Virol 87:8916–8926. doi: 10.1128/JVI.00340-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang KW, Alaei-Mahabadi B, Samuelsson T, Lindh M, Larsson E. 2013. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat Commun 4:2513. doi: 10.1038/ncomms3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strong MJ, O'Grady T, Lin Z, Xu G, Baddoo M, Parsons C, Zhang K, Taylor CM, Flemington EK. 2013. Epstein-Barr virus and human herpesvirus 6 detection in a non-Hodgkin's diffuse large B-cell lymphoma cohort by using RNA sequencing. J Virol 87:13059–13062. doi: 10.1128/JVI.02380-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salyakina D, Tsinoremas NF. 2013. Viral expression associated with gastrointestinal adenocarcinomas in TCGA high-throughput sequencing data. Hum Genomics 7:23. doi: 10.1186/1479-7364-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cantalupo PG, Katz JP, Pipas JM. 2015. HeLa nucleic acid contamination in The Cancer Genome Atlas leads to the misidentification of HPV18. J Virol 89:4051–4057. doi: 10.1128/JVI.03365-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borozan I, Wilson S, Blanchette P, Laflamme P, Watt SN, Krzyzanowski PM, Sircoulomb F, Rottapel R, Branton PE, Ferretti V. 2012. CaPSID: a bioinformatics platform for computational pathogen sequence identification in human genomes and transcriptomes. BMC Bioinformatics 13:206. doi: 10.1186/1471-2105-13-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. 2013. Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140. doi: 10.1111/mec.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leinonen R, Akhtar R, Birney E, Bower L, Cerdeno-Tarraga A, Cheng Y, Cleland I, Faruque N, Goodgame N, Gibson R, Hoad G, Jang M, Pakseresht N, Plaister S, Radhakrishnan R, Reddy K, Sobhany S, Ten Hoopen P, Vaughan R, Zalunin V, Cochrane G. 2011. The European Nucleotide Archive. Nucleic Acids Res 39:D28–D31. doi: 10.1093/nar/gkq967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, Barnes I, Bignell A, Boychenko V, Hunt T, Kay M, Mukherjee G, Rajan J, Despacio-Reyes G, Saunders G, Steward C, Harte R, Lin M, Howald C, Tanzer A, Derrien T, Chrast J, Walters N, Balasubramanian S, Pei B, Tress M, Rodriguez JM, Ezkurdia I, van Baren J, Brent M, Haussler D, Kellis M, Valencia A, Reymond A, Gerstein M, Guigo R, Hubbard TJ. 2012. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pruitt KD, Tatusova T, Maglott DR. 2007. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D. 2006. The UCSC known genes. Bioinformatics 22:1036–1046. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- 17.Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T, Durbin R, Eyras E, Gilbert J, Hammond M, Huminiecki L, Kasprzyk A, Lehvaslaiho H, Lijnzaad P, Melsopp C, Mongin E, Pettett R, Pocock M, Potter S, Rust A, Schmidt E, Searle S, Slater G, Smith J, Spooner W, Stabenau A, Stalker J, Stupka E, Ureta-Vidal A, Vastrik I, Clamp M. 2002. The Ensembl genome database project. Nucleic Acids Res 30:38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhaduri A, Qu K, Lee CS, Ungewickell A, Khavari PA. 2012. Rapid identification of non-human sequences in high-throughput sequencing datasets. Bioinformatics 28:1174–1175. doi: 10.1093/bioinformatics/bts100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gish W. 1992. The nrdb program. National Center for Biotechnology Information, Bethesda, MD: ftp://ncbi.nlm.nih.gov/pub/nrdb/README. [Google Scholar]
- 22.Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 24.Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BM, Haag JD, Gould MN, Stewart RM, Kendziorski C. 2013. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 29:1035–1043. doi: 10.1093/bioinformatics/btt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N. 1999. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 189:12–19. doi:. [DOI] [PubMed] [Google Scholar]
- 26.Ringstrom E, Peters E, Hasegawa M, Posner M, Liu M, Kelsey KT. 2002. Human papillomavirus type 16 and squamous cell carcinoma of the head and neck. Clin Cancer Res 8:3187–3192. [PubMed] [Google Scholar]
- 27.Kocjan BJ, Seme K, Cimerman M, Kovanda A, Potocnik M, Poljak M. 2009. Genomic diversity of human papillomavirus (HPV) genotype 38. J Med Virol 81:288–295. doi: 10.1002/jmv.21392. [DOI] [PubMed] [Google Scholar]
- 28.Hu Z, Zhu D, Wang W, Li W, Jia W, Zeng X, Ding W, Yu L, Wang X, Wang L, Shen H, Zhang C, Liu H, Liu X, Zhao Y, Fang X, Li S, Chen W, Tang T, Fu A, Wang Z, Chen G, Gao Q, Li S, Xi L, Wang C, Liao S, Ma X, Wu P, Li K, Wang S, Zhou J, Wang J, Xu X, Wang H, Ma D. 2015. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat Genet 47:158–163. doi: 10.1038/ng.3178. [DOI] [PubMed] [Google Scholar]
- 29.Schmitz M, Driesch C, Jansen L, Runnebaum IB, Durst M. 2012. Non-random integration of the HPV genome in cervical cancer. PLoS One 7:e39632. doi: 10.1371/journal.pone.0039632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leek JT, Scharpf RB, Bravo HC, Simcha D, Langmead B, Johnson WE, Geman D, Baggerly K, Irizarry RA. 2010. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet 11:733–739. doi: 10.1038/nrg2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lauss M, Visne I, Kriegner A, Ringner M, Jonsson G, Hoglund M. 2013. Monitoring of technical variation in quantitative high-throughput datasets. Cancer Inform 12:193–201. doi: 10.4137/CIN.S12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.