

ABSTRACT
Herpesviruses are nuclear-replicating viruses that have successfully evolved to evade the immune system of humans, establishing lifelong infections. ICP27 from herpes simplex virus is a multifunctional regulatory protein that is functionally conserved in all known human herpesviruses. It has the potential to interact with an array of cellular proteins, as well as intronless viral RNAs. ICP27 plays an essential role in viral transcription, nuclear export of intronless RNAs, translation of viral transcripts, and virion host shutoff function. It has also been implicated in several signaling pathways and the prevention of apoptosis. Although much is known about its central role in viral replication and infection, very little is known about the structure and mechanistic properties of ICP27 and its homologs. We present the first crystal structure of ICP27 C-terminal domain at a resolution of 2.0 Å. The structure reveals the C-terminal half of ICP27 to have a novel fold consisting of α-helices and long loops, along with a unique CHCC-type of zinc-binding motif. The two termini of this domain extend from the central core and hint to possibilities of making interactions. ICP27 essential domain is capable of forming self-dimers as seen in the structure, which is confirmed by analytical ultracentrifugation study. Preliminary in vitro phosphorylation assays reveal that this domain may be regulated by cellular kinases.
IMPORTANCE ICP27 is a key regulatory protein of the herpes simplex virus and has functional homologs in all known human herpesviruses. Understanding the structure of this protein is a step ahead in deciphering the mechanism by which the virus thrives. In this study, we present the first structure of the C-terminal domain of ICP27 and describe its novel features. We critically analyze the structure and compare our results to the information available form earlier studies. This structure can act as a guide in future experimental designs and can add to a better understanding of mechanism of ICP27, as well as that of its homologs.
INTRODUCTION
Herpes simplex virus (HSV) and other human herpesviruses (HHVs; e.g., Epstein-Barr virus and varicella-zoster virus) have evolved a unique strategy of virion host shutoff, in which they efficiently suppress the host protein synthesis. At the onset of the lytic phase, viral immediate-early (IE) genes are expressed first. These IE proteins are responsible for inducing the expression of viral early and late genes. The key IE protein of HSV, infectious cell protein 27 (ICP27), helps in the host shutoff function (1, 2). This 63-kDa protein consists of 512 amino acids and is involved in multiple functions, such as stalling host pre-RNA processing, exporting intronless viral mRNAs out of the nucleus, shuttling between host nucleus and cytoplasm facilitated by its nuclear export signal (NES) and nuclear localization signal (NLS), and interacting with RNA via its RGG box (3, 4). Most of these functions are conserved across the Herpesviridae family, and ICP27 homologs are present in all known HHVs. Well-characterized homologs include IE4 in varicella-zoster virus, UL69 in human cytomegalovirus, EB2 (or SM) protein in Epstein-Barr virus, and open reading frame 57 (ORF57) in Kaposi's sarcoma-associated herpesvirus.
Carmody and Wente (5) have shown that, in eukaryotes, the process of pre-mRNA maturation and export from the nucleus is coupled with a splicing process. This involves the splicing factors (SRp20, 9G8, and ASF/SF2), the exon-junction complex, and the export proteins (TREX complex, TAP/NXF1, Aly/REF, and UAP56) that associate with the pre-mRNA and mRNA. Some of these proteins remain bound to the mRNA until they exit the nucleus through the nuclear pore complex. In an HSV-infected cell, ICP27 has been proposed to recruit SR protein kinase 1, resulting in unusual phosphorylation of splicing factors (SR proteins) (6, 7). Consequently, spliceosome assembly is stalled and host pre-mRNA cannot be processed (8, 9). It is reported that ICP27 interacts directly with export factors such as Aly/REF, making them unavailable to the host mRNAs (10, 11). HSV-1 mRNAs are predominantly intronless and hence not dependent on host splicing machinery for maturation. However, they do need an adaptor to help them translocate out of the nucleus by interacting with the host export factors. ICP27 appears to act as that multi-adaptor protein, binding to viral mRNAs and several host export proteins (e.g., Aly/REF, SRp20, 9G8, and TAP/NXF1) (11–13). In this process, it has been proposed to undergo reversible head-to-tail intramolecular association that may regulate its binding to TAP/NXF1 (14). Malik et al. (15) reported that ICP27 interacts with nucleoporin Nup62 of the nuclear pore complex to export the viral RNAs to the cytoplasm. Once in the cytoplasm, ICP27 is proposed to recruit cellular translation initiation factors such as eukaryotic translation initiation factor 3 (eIF3) and eIF4G, poly(A)-binding proteins, and polyribosomes, thus promoting translation of viral mRNAs (16, 17).
Based on several assays and predictions by protein sequence analysis, ICP27 is proposed to have multiple functional domains. ICP27 mainly binds to intronless viral mRNAs, mainly transcribed during the IE and late stages of the viral lytic cycle. The N-terminal RGG-box motif (residues 138 to 152) of ICP27 is reported to be responsible for this interaction (4). Unlike other known RGG-box-containing proteins that preferentially bind to G-quartet structures in mRNA, ICP27 seems to prefer GC-rich regions in mRNA that are flexible and without secondary structures (18). In addition, three KH-like domains have been proposed to be located in the C-terminal part of protein that too may bind to mRNA (19, 20). Other functional domains of ICP27 include the NES, NLS, and a putative zinc finger-like motif of CCHC-type (4, 21). Secondary structure predictions indicate that the N-terminal region up to ∼240 residues is disordered. Circular dichroism (CD) analysis and nuclear magnetic resonance (NMR) studies on the N-terminal half (residues 1 to 160) of the protein have in fact confirmed that this region is predominantly random-coils with no rigid three-dimensional structure (22). Another NMR study revealed that a short stretch of ICP27 (residues 103 to 110) was sufficient for binding to the RNA recognition motif of Aly/REF and was linear in the structure (23). For the C-terminal half of ICP27, structure predictions indicate multiple alpha helices and a few interspersed beta strands. This region has been proposed to interact with several proteins such as Nup62, SAP145, SRp20, and TAP/NXF-1 (8, 12–15). As mentioned above, this domain is predicted to encompass the three KH domains and a zinc finger-like motif of CCHC type (19, 21). The C-terminal half is also proposed to undergo self-association that assists in its import into the nucleus (24). This part of ICP27 is conserved across the human herpesvirus family and is essential for its functions (25).
The sequence similarity between the ICP27 homologs from human herpesviruses is low but significant in regions. They display similarities in the secondary structure predictions, with N-terminal half being disordered and the C-terminal primarily consisting of α-helices (25). The other well-studied ICP27 homologs, namely, ORF57 (Kaposi's sarcoma-associated virus [KSHV]) and EB2 protein (Epstein-Barr virus [EBV]), have been reported to bind to SR proteins such as SF2/ASF, U2AF, and SRp20 (26, 27), as in the case of ICP27. ORF57 has also been shown to interact with a TREX complex component, i.e., Aly/REF (28), and with PYM that helps recruit 40S ribosomal subunit (29). Similarities in the main functions of the homologs suggest that they may have related protein structure and mechanisms of action. However, thus far no experimental information exist regarding the structures of any of the homologs.
We present here the first structure of the C-terminal domain (residues 190 to 512) of ICP27 (ICP27-CTD) solved by X-ray crystallography to a resolution of 2.0 Å. The structure reveals a dimer, where N- and C-terminal regions form extensions surrounding and inserting into the neighboring subunit. We show that the zinc-binding motif of ICP27 is unique to other known zinc-binding domains. Further analysis by analytical ultracentrifugation also supports that ICP27-CTD exists as dimer in solution. The structural and mechanistic implications of this novel information are discussed.
MATERIALS AND METHODS
Subcloning, protein expression, and purification.
Vector containing the gene for ICP27 from HSV-1 (GenBank accession number ABI63515) was generously provided by Jurgen Haas, University of Edinburgh. ICP27 gene encoding residues 190 to 512 of the C-terminal domain, here referred to ICP27-CTD, was subcloned into vector pNIC-Bsa4 (GenBank accession number EF198106) with N-terminal 6×His tag and tobacco etch virus (TEV) protease cleavage site, using ligation-independent cloning. It was subsequently expressed in the BL21(DE3) Rosetta strain of Escherichia coli, grown at 37°C in Terrific broth, supplemented with 50 μg of kanamycin/ml and 34 μg of chloramphenicol/ml, to an optical density at 600 nm of 2.0. Expression was induced with 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) at 18°C in the LEX system (Harbinger Biotech, Canada). Cells were harvested after 16 h, resuspended in lysis buffer (100 mM HEPES [pH 8.0], 500 mM NaCl, 10 mM imidazole, 10% [vol/vol] glycerol) supplemented with 0.1 mg of lysozyme/ml, 1 μl of protease inhibitor cocktail III (Calbiochem)/ml, 1 mM MgCl2, 2 μl of Benzonase (Merck) per 750 ml of culture, and 1 mM tris(2-carboxyethyl)phosphine (TCEP), and stored at −80°C.
Cells were lysed on ice by sonication. The lysates were centrifuged at 47,000 × g for 30 min at 4°C and then filtered through a 1.2-μm-pore-size membrane. The filtrates were subjected to immobilized metal-ion affinity chromatography (IMAC) using a Ni-nitrilotriacetic acid Superflow column (Qiagen), pre-equilibrated with buffer (20 mM HEPES [pH 7.5], 500 mM NaCl, 10 mM imidazole, 10% [vol/vol] glycerol, 1 mM TCEP), and eluted with elution buffer (20 mM HEPES [pH 7.5], 500 mM NaCl, 500 mM imidazole, 10% [vol/vol] glycerol, 1 mM TCEP). The IMAC eluates were then subjected to second step of purification by size-exclusion chromatography using a HiLoad 16/60 Superdex 200 Prep-Grade column (GE Healthcare) pre-equilibrated with 20 mM HEPES (pH 7.5), 300 mM NaCl, 10% (vol/vol) glycerol, and 1 mM TCEP. Protein fractions were analyzed by SDS-PAGE and Western blotting. Fractions with the highest purity were collected and concentrated in a centrifugal concentrator with a molecular-weight cutoff of 10 kDa (Sartorius Stedim Biotech). Final protein concentrations were assessed by determining the UV absorbance at 280 nm. The ICP27-CTD protein was then stored at −80°C.
For SeMet-labeled ICP27-CTD, the cells were grown in the M9 selenomethionine (SeMet) high-yield growth medium (Shanghai Medicilon, Inc.). Cultivation and purification was performed as previously described for the native protein. Incorporation of the selenomethionine in the final protein was confirmed by mass spectrometry.
Crystallization, data collection, data processing, and structure refinement.
ICP27-CTD protein (37.87 kDa) was concentrated to ∼14 mg/ml (native) and ∼21 mg/ml (SeMet labeled) and was crystallized by the hanging-drop vapor diffusion method. Next, 2 μl of protein was mixed with 1 μl of reservoir solution, consisting of 0.2 M sodium thiocyanate and 4% benzamidine hydrochloride. The crystals grew overnight at room temperature. The protein crystallized in two different forms: long rods and rock-like crystals. Crystals were soaked in cryoprotectant consisting of mother liquor and 25% (vol/vol) glycerol and were then flash-frozen in liquid N2. X-ray diffraction experiments (at 100 K) and subsequent data collections for crystals of native and SeMet-labeled ICP27-CTD were performed at the National Synchrotron Radiation Research Center (NSRRC) in Taiwan, the Australian Synchrotron (AS) in Australia, and the Diamond Light Source (DLS) in the United Kingdom.
The rock-like crystals belonged to space group C2. For the SeMet-labeled crystals, a single-wavelength anomalous dispersion (SAD) data set at 2.0-Å resolution was collected at peak wavelength for selenium (0.97893 Å) at the NSRRC (13B1 beamline). Native data were collected at 2.73 Å at the DLS and at 3.0 Å at the AS. All data sets were integrated and scaled using HKL2000 (30). ICP27-CTD structure was solved with SAD method for the SeMet crystals and by molecular replacement for the native crystals using the SeMet crystal structure. Structure refinement was carried out using maximum-likelihood method of the phenix.refine program from the PHENIX suite (31). Manual model building was performed by COOT (32). The data statistics are summarized in Table 1. In order to detect zinc atoms, the crystal was irradiated with X-rays at the K-edge for zinc (1.283 Å). A sharp absorption peak was observed, indicating presence of zinc atoms in the crystal (data not shown).
TABLE 1.
Crystallographic data, phasing, and refinement statistics
| Parameter | Data seta |
|
|---|---|---|
| Selenomethionine | Native | |
| Data collection | ||
| Radiation source | NSRRC 13B1 | DLS MX I04 |
| Space group | C121 | C121 |
| Unit cell dimensions | ||
| a, b, c (Å) | 156.983, 120.335, 79.037 | 157.88, 121.4, 79.76 |
| α, β, γ (°) | 90.0, 100.46, 90.0 | 90, 100.46, 90 |
| Wavelength (Å) | 0.97893 | 0.97950 |
| Resolution range (Å) | 94.91–2.0 (2.07–1.997) | 39.16–2.73 (2.83–2.73) |
| Total no. of reflections | 234,835 (22,892) | 293,493 (21,627) |
| Unique no. of reflections | 89,418 (9,157) | 39,132 (2,847) |
| Rmergeb (%) | 9.8 (67.7) | 17.2 (78.4) |
| Mean I/σ<I> | 8.5 | 8.7 |
| Completeness (%) | 91.47 (93.91) | 99.9 (99.8) |
| Redundancy | 2.6 (2.5) | 7.5 (7.6) |
| Refinement | ||
| Rwork/Rfreec (%) | 21.68/24.62 | 22.7/27.66 |
| No. of atoms (non-H) | 6,383 | 6,375 |
| Protein | 6,090 | 6,264 |
| Ligand/ion | 3 Zn | 3 Zn |
| Water | 290 | 108 |
| Model quality | ||
| Estimated coordinate error (Å) | 0.24 | 0.39 |
| RMSD | ||
| Bond length (Å) | 0.022 | 0.01 |
| Bond angle (°) | 1.749 | 1.20 |
| Ramachandran plot | ||
| Favored region (%) | 98.0 | 96.0 |
| Allowed region (%) | 1.87 | 3.38 |
| Outliers (%) | 0.13 | 0.62 |
The highest-resolution values are indicated in parentheses.
Rmerge = Σ Σ |Ii − <I>|/Σ Σ Ii, where is Ii the intensity measurement of reflection h and <I> is the average intensity from multiple observations.
Rwork = Σ ||Fo| − |Fc||/Σ |Fo|, where Fo and Fc are the observed and calculated structure factors, respectively; Rfreeis equivalent to Rwork, but 5% of the measured reflections have been excluded from the refinement andset aside for cross-validation.
Sedimentation equilibrium by analytical ultracentrifugation.
A sedimentation equilibrium experiment was performed on ICP27-CTD at 20°C using a Beckman XL-I analytical ultracentrifuge. Next, 130 μl each of homogenized ICP27-CTD samples with optical densities at 280 nm of 0.3, 0.5, and 0.8 were prepared. The buffer system used was composed of 20 mM HEPES (pH 7.5), 300 mM NaCl, and 10% (vol/vol) glycerol. Centrifugation was carried out in a carbon-epoxy centerpiece with six quartz windows for references and samples. The samples were centrifuged until they reached equilibrium (15 h per speed) and run at 10,500, 12,700, and 15,500 rpm. The absorbance data were collected at 280 nm. Partial specific volumes and monomer molecular weights were calculated using the program SEDNTERP (33). The partial specific volume of ICP27-CTD was 0.732291 cm3/g. Scan data sets were analyzed by SEDFIT software (34). Data fitting was done to check the possibilities of the monomeric state and dimeric, trimeric, and tetrameric associations.
Phosphorylation of Ser/Thr residues.
For phosphorylation with protein kinase A (PKA), 100 μg of ICP27-CTD was incubated with 200 U of PKA type I catalytic subunit (New England BioLabs [NEB]) in 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, and 500 μM ATP. For phosphorylation with casein kinase II (CKII), 100 μg of ICP27-CTD was incubated with 250 U of CKII (NEB) in 20 mM Tris-HCl (pH 7.5), 50 mM KCl, 10 mM MgCl2, and 500 μM ATP. For phosphorylation with protein kinase C (PKC), 100 μg of ICP27-CTD was incubated with 160 mU of PKC (Sigma-Aldrich) in 20 mM HEPES (pH 7.5), 10 mM MgCl2, and 500 μM ATP. All reactions were carried out overnight at 30°C. To determine whether phosphorylation had occurred, the sample was incubated with PhosphoProtein purification resin (Qiagen), which binds specifically to phosphorylated proteins. The manufacturer's protocol was followed to elute the bound protein, and fractions were analyzed using SDS-PAGE. The protein was analyzed with liquid chromatography-tandem mass spectrometry to identify phosphorylated residues.
Protein structure accession code.
Atomic coordinates and structure factors for ICP27-CTD have been deposited in the RCSB Protein Data Bank (PDB) under accession code 5BQK.
RESULTS
Overall structure of ICP27-CTD.
Native and SeMet-labeled ICP27-CTD were purified to homogeneity and crystallized in isomorphous crystal forms belonging to the space group C2 with three molecules in the asymmetric unit (ASU). The SeMet-labeled protein crystals diffracted much better than the native ones in spite of similar crystal morphology and packing. Diffraction data were collected, and SeMet-derived phases allowed for initial automatic model building using AutoBuild of the Phenix suite. The structures were subsequently refined at resolutions of 2.73 Å (native) and 2.0 Å (SeMet labeled) with Rfree values of 27.66 and 24.72%, respectively (Table 1). The electron densities for the 6×His tag, the N-terminal residues 190 to 241, and the loop L2 residues 297 to 300 are not visible, suggesting that these regions are flexible in the crystal. Two of the three monomers in the ASU form a tight homodimer with approximate dimensions of 63.4 by 59.4 by 40.7 Å (Fig. 1). The third molecule in the ASU forms the same type of dimer with a crystallographically related molecule. The overall structure of the dimer reveals a coffee bean shaped molecule with the two subunits interacting in a yin-yang-like mode. Along the dimer interface, a prominent central groove (∼11 to 13.0 Å wide) is formed.
FIG 1.
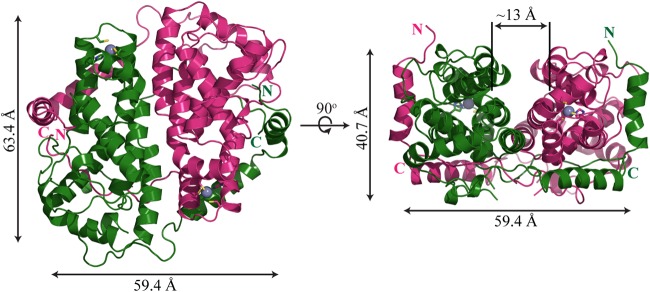
ICP27-CTD dimer. A diagram representation of ICP27-CTD is shown in its dimeric conformation. The two monomers (in magenta and green) are positioned in a yin-yang orientation. The dimensions of the homodimer were measured with PyMOL. The 90° rotated view shows a prominent groove running along the length of the dimer interface.
The structure of each monomer shows a compact core formed by 10 α-helices of various lengths, separated by loop regions (Fig. 2A and C). The N and C termini of the domain protrude out on the same side, although at different ends of the extended monomer. We termed them the N-terminal arm and C-tail, respectively. ICP27-CTD has been proposed to harbor a zinc-binding site, and this is found in the structure at the same end of the monomer from where the C terminus protrudes (Fig. 2C). A DALI search performed with the ICP27-CTD monomer reveals no overall structural resemblance to any known protein structure in the PDB, supporting the notion that ICP27-CTD has a novel fold.
FIG 2.

Overall structure of ICP27-CTD. (A) A schematic representation shows that the ICP27-CTD monomer consists of 10 α-helices (α1 to α10), separated by nine loops (L1 to L9) of varied lengths. The model starts at A242 and ends at F512 with the residues from 190 to 241 and His6-TEV tag missing. The first and last residues of each helix are indicated. The red dashed line indicates the four residues (G298 to G301) with missing electron densities. The orange circles indicate the positions of the zinc-binding residues. The schematic was prepared using TopDraw software in the CCP4 suite. (B) Diagram representation of ICP27-CTD monomer (magenta) in close contact with the second monomer, shown as a gray surface representation. The N-terminal arm of the first monomer is wrapped around the second monomer, whereas the C-terminal region is buried. The zinc ion is represented as a purple-gray sphere and is tetrahedrally coordinated by cysteine and histidine residues shown as sticks. (C) Diagram representation of ICP27-CTD monomer shown in two views rotated 90° along the x axis. The helical core is magenta, and the N and C termini are gray. The rotated figure shows the hollow region between the globular domain and the N-terminal arm.
A striking feature of the dimer structure is that both the N and C termini extend from the core domain to make extensive interactions with the other protomer of the dimer. The C terminus extends into to the helical core of the other monomer and is enclosed in the subunit (Fig. 2B). The N-terminal region, on the other hand, forms a long arm segment, which hugs the neighboring subunit. This involves primarily helices 1 and 2, where helix 1 interacts with the central part of the neighboring subunit, whereas helix 2 interacts with the region harboring the Zn binding site (Fig. 2B).
Biophysical characterization of the dimer.
ICP27 has been reported to form dimers that help in its import from cytoplasm into the nucleus (24). We found ICP27-CTD to form dimers in a yin-yang orientation, as seen in the crystal structure. To confirm that the dimeric state of ICP27-CTD exists even in solution, we performed an analytic ultracentrifugation sedimentation equilibrium experiment. The data could be best fit to a monomer-dimer self-association model (Fig. 3). The association constant (Ka) for this model is 1.8113 × 106 M−1 with an root mean square deviation (RMSD) of 0.005 and chi-square value of 0.76828. The values for the other models are condensed in Table 2.
FIG 3.

Analytical ultracentrifugation profile of ICP27-CTD. The sedimentation equilibrium analysis of ICP27-CTD is shown with experimental data in the top graph and the residuals of fit in the lower graph. The data were collected for three different concentrations of protein (0.8, 0.5, and 0.3 absorbance units) at 10,500, 12,700, and 15,500 rpm. The data are fit to a monomer-dimer model, showing that ICP27-CTD is capable of forming self-associating dimers even at low concentrations.
TABLE 2.
Analytical centrifugation data of ICP27-CTD after fitting in various monomer/n-mer self-association models
| ICP27-CTD association | n | Log Ka | Ka (M−1) | RMSD | Chi-square value |
|---|---|---|---|---|---|
| Monomer-monomer | 1 | 0.0271 | 1.6313E+01 | ||
| Monomer-dimer | 2 | 6.258 | 1.8113E+06 | 0.0050 | 7.6828E–01 |
| Monomer-trimer | 3 | 9.092 | 1.2359E+09 | 0.0070 | 1.5265E+00 |
| Monomer-tetramer | 4 | 13.021 | 1.0495E+13 | 0.0081 | 3.1062E+00 |
Zinc-binding site of ICP27.
The structure reveals a zinc-binding site coordinated by residues C400, H479, C483, and C488 (Fig. 4). Residues C483, C488, H502, and C508 have previously been proposed to be Zn ligands in ICP27 (21), whereas C400 and H479 are unanticipated Zn ligands, although they are highly conserved in all ICP27 homologs (Fig. 5 and Fig. 6, lower inset). Residues H479, C483, and C488 are located in a compact helix-loop-helix motif (helices 9 and 10), where H479 and C488 are found in the C- and N-terminal loops of the helices, respectively, whereas C483 is found in the interconnecting loop. C400, on the other hand, is located in a distant preceding helix 6 that is packing onto the helix-loop-helix motif of the other zinc ligands using extensive hydrophobic and polar interactions.
FIG 4.
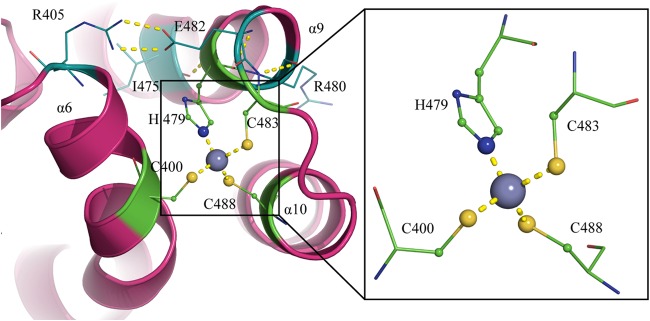
Zinc-binding motif of ICP27-CTD. A diagram representation of the zinc-binding motif shows the protein in magenta and the zinc ion as a gray sphere. The zinc ion is tetrahedrally coordinated by C400, H479, C483, and C488, shown by yellow dashed lines. H479, C483, and C488 form a helix-loop-helix structure. H479 is back-coordinated by E482, which in turn is stabilized by I475 and R405. R405 is in close proximity to the zinc-binding residue C400, providing more stability between helix and the helix-loop-helix. C483 is coordinated by R480.
FIG 5.

Comparison of ICP27 with its homologs. ICP27-CTD (HHV1) sequence is aligned with its various homologs from the alphaherpesvirus family (IE4 from HHV3, UL54 from bovine herpesvirus 5 [BoHV5], UL54 from suid herpesvirus 1 [SuHV1], and UL54 from equine herpesvirus 9 [EHV9]), the betaherpesvirus family (UL69 from HHV5), and the gammaherpesvirus family (ORF57 from HHV8). Secondary structural elements of ICP27-CTD are drawn above the sequences as green α-helices with black-lined loops, with a red dashed line showing the position of the four missing residues. Residues conserved in all sequences are highlighted in red boxes with a white font; residues with similar sequences are highlighted in a red font. Residues involved in the zinc-binding motif of ICP27-CTD are marked with a red star. A black box is used to indicate the residues forming the C-tail, a yellow box indicates the hydrophobic pocket region, and a purple box indicates the flexible cap region as observed in ICP27-CTD structure. Residues involved in key interactions between the hydrophobic pocket and the C-tail are marked with a blue star. Orange triangles indicate the positions of residues phosphorylated in the in vitro assay and analyzed by mass spectrometry. Multiple sequence alignment was performed using CLUSTAL Omega, and sequence similarities were rendered using ESPript3. The sequence of ICP27 homolog from HHV4, i.e., the EB2 protein, is manually aligned to the above multiple sequence alignment. The conserved residues are boxed in red.
FIG 6.

Mapping of amino acid sequence conservation on ICP27-CTD structure. Based on multiple sequence alignment of ICP27 and its homologs, the conserved amino acid residues are mapped on the ICP27-CTD structure. Depending on the extent of conservation, the residues are colored from red, for highest conservation, to blue, for nonconserved residues. The upper inset shows the hydrophobic pocket, lined with conserved residues, along with the C-tail inserted in it. The lower inset displays the highly conserved zinc-binding residues. UCSF Chimera was used for rendering by sequence conservation. The final images were prepared in PyMOL.
The occupancy of the Zn atoms in the different subunits (in both native and SeMet-derivative structures) appears to be close to 100%. The coordination sphere of the Zn is tetrahedral with coordination distances for the cysteine sulfur of 1.9 to 2.5 Å and with histidine nitrogen of ∼2.0 Å (Fig. 4, inset). H479 is stabilized by back coordination to E482, which in turn makes a salt bridge to R405, which sits in the same helix as the distant C400 ligand (Fig. 4). C483 is also stabilized by back coordination, in this case by hydrogen bonds to the main chain amino group of R480. The partial positive charge provide by this interaction indicate that C483 might be in its deprotonated form. C400 and C488, however, have no direct interactions contributed by protein residues, but there are several well-ordered water molecules available for stabilizing interactions with these groups. The compactness of the Zn-binding motif suggests that it is not a catalytic zinc site but rather has a structural zinc finger-like function. Zinc fingers are compact structural motifs, where the zinc serves as a central core to allow the stabilization of small domains. They typically serve as versatile domains for interactions with nucleic acids (35, 36) and other proteins (37). A comparison with previously reported structures of zinc finger domains (38) reveals that ICP27 is structurally distinct and therefore provides a novel fold for a zinc finger domain. The motif is also unique in that it spans a large distance of 78 residues between the first and second zinc-binding residues.
Trans-subunit interaction of the N- and C-terminal arms of ICP27-CTD.
The C-terminal end of ICP27-CTD consists of the highly hydrophobic stretch formed by residues 502 to 512. This stretch makes an intriguing insertion into the core of the neighboring subunit (Fig. 7A). Also, the N-terminal residues 242 to 278 form an extended arm-like structure, constituted by helices 1 and 2 that wrap around the neighboring subunit. Together with other interactions at the subunit interface, these trans-subunit interactions of the N- and C-terminal arms are likely to assist in making ICP27-CTD a stable dimer, as confirmed by the biophysical study discussed above.
FIG 7.

Interactions of the C-tail residues with the conserved hydrophobic pocket region. (A) ICP27-CTD C-tail of one molecule (magenta diagram representation) is lodged in the hydrophobic pocket of second molecule (surface representation colored by B-factor values). The lower wall of the pocket forms a flexible cap (as seen by a high B-factor value shown in orange-red colors). (B) Conserved C-tail residues (magenta diagram and stick representation) Y505, F506, and Y507 are sandwiched via hydrophobic interactions with conserved residues L315 and E358 from the pocket region. (C) Several buried charges are conserved around the C-tail interaction region, making polar interactions (orange dashes). E358 forms a salt bridge with Y505 and F506 of the C-tail. R340 from the pocket and K504 from the C-tail make stabilizing interactions with E358. E347 has its carboxylate group only 4.3 Å away from K504. (D) C-tail residues S510 and L511 make hydrophobic interactions with the conserved YXPG motif. The motif is, in turn, stabilized by the conserved D357.
The C-terminal motif composed of residues HGKYFYCNSLF in HSV1 and HSV2, has a high degree of conservation in herpesviruses (Fig. 5; Fig. 6, upper inset) and is likely to be functionally important. We therefore label this motif the C-tail. In the ICP27-CTD structure, the C-tail is buried in the neighboring subunit, where it makes extensive interactions with helices 3, 4, and 5 and loop regions preceding helix 3 (residues 292 to 312). F506 and Y507 are the key interacting residues in the C-tail that are highly conserved. They are localized in an extended hydrophobic pocket formed by residues from helices 3, 4, and 5 (Fig. 7B). A number of highly or partly conserved hydrophobic residues constitute this pocket, including the fully conserved L315 that makes an interaction with the F506 side chain (Fig. 5). The extended backbone structure of Y505 and F506 is stabilized by the buried E358, which makes hydrogen bonds to the amino groups of Y505 and F506. E358 is buried and fully conserved in herpes sequences, and the importance of this residue is supported by a further salt-link-like interaction with the buried and highly conserved R340 (Fig. 5 and 7C). K504 is also buried in the same region and makes polar interactions with E358. E347 is buried with its carboxylate group 4.3 Å from K504 (Fig. 7C). Buried charges are unusual, and it is likely that this conserved and buried network of charged residues is important for the function of ICP27.
The C-tail extends through the subunit, exiting the hydrophobic pocket and emerging close to the N-terminal arm. The side chains of the C-tail L511 and F512 make hydrophobic interactions on the other side of the subunit to the N-terminal arm motif in the region between helices 1 and 2. The C-tail residues S510 and L511 also interact with a conserved YXPG motif (residues 463 to 466) preceding helix 9 (Fig. 7D). The extended conformation of this motif is stabilized by the conserved and buried E357, which forms a charge-compensating side chain to the N termini of helix 10. Finally, the extended loop region between residues 302 to 312 forms a cap on the C-tail. The side chain of the conserved F303 interacts with the main chain of F506 and the aliphatic region of K504. The aliphatic region of R309 lines the aromatic Y507 of the C-tail, both of which are frequent residues in ICP27 homologs.
Although the high conservation of motifs involved in binding of the C-tail in the subunit structure is very clear and indicative of a key role in the function of ICP27, conservation is less evident in the helical N-terminal arm. Both helices of this arm are amphiphilic in nature, and the hydrophobic face is interacting with the core subunit, while the polar residues are predominantly exposed to the solvent. The residual sequence conservation in other herpes homologs, however, is sufficient to support that this motif is present on most, if not all ICP27 homologs.
Effect of in vitro phosphorylation of ICP27-CTD.
ICP27 is involved in a series of processes in the nucleus and the cytoplasm, where it is attributed to a number of different protein interactions. Host protein-protein interactions are often regulated by phosphorylation, and phosphorylation events are reported to regulate a number of herpesvirus proteins (39–43). ICP27 is observed to have both stable and transient phosphorylations (44). Due to the multitude of functions executed by ICP27-CTD, it is possible that it harbors regulatory phosphorylation sites. These could change the structural properties to, for example, activate the protein for specific protein-protein interactions during particular steps of its functional cycle. It has been shown that ICP27 is phosphorylated in vivo at S114, probably by the cellular kinases PKA and CKII (45). To investigate which additional potential serine/threonine sites are accessible for phosphorylation, and thereby identify potential functional regions, we performed an in vitro phosphorylation of purified ICP27-CTD using three different kinases—PKA, CKII, and PKC—and subsequently analyzed the phosphorylation patterns by mass spectrometry, the results of which are shown in the Table 3. Interestingly, a number of sites are found in the region surrounding the C-tail interaction site, including S290, S311, T314, and S321 in the loop region, which caps the C-tail (Fig. 8A). This region capping the C-tail has a high B-factor value (Fig. 7A), indicating higher flexibility and therefore potential availability as the substrate for kinases. Similarly, multiple phosphorylation sites are found in close proximity of the zinc-binding motif, including S266, S268, S272, T478, S484, S485, and S493 (Fig. 7B). These may be involved in phosphorylation-induced local conformational changes. A detailed study of the effects of phosphorylation in this region may shed some light on possible regulatory mechanisms of ICP27.
TABLE 3.
ICP27-CTD residues phosphorylated in vitro using different cellular kinases and identified by mass spectrometry
| Kinase | Phosphorylated residues |
|---|---|
| PKA | T244, S266, S268, S290, S311, T314, S493 |
| CKII | T244, T249, S266, S272, S290, T478, S484, S493 |
| PKC | T220, T244, T248, S255, S268, S272, S290, T314, S321, T335, S484, S485, S493 |
FIG 8.

Phosphorylation sites of ICP27-CTD. In vitro phosphorylation assays using PKA, CKII, and PKC kinases, followed by analysis using mass spectrometry revealed several phosphorylated sites in ICP27-CTD (shown in orange). Molecule 1 is depicted in gray, and molecule 2 is depicted in light pink. (A) Many of the observed sites were concentrated in the flexible loop capping the C-tail, mainly S290, S311, T314, and S321. Considering the high flexibility of the capping loop, there exists a possibility of phosphorylation-induced local conformation changes. (B) Most other sites are concentrated in the region in close to the zinc-binding motif, mainly S266, S268, S272, T478, S484, S485, and S493.
DISCUSSION
The functions of a large number of herpes proteins are still poorly understood at the structural level. This can partly be attributed to the challenges of purifying and determining structures from viral proteins, which are, in general, more flexible and unstable (46–48). We particularly faced these challenges when overexpressing and purifying the flexible N-terminal domain of ICP27, between residues 1 to 210. Most of the constructs that expressed suffered from heavy degradation and were very difficult to purify. The few protein constructs of the N-terminal domain that we managed to purify did not crystallize in any crystallization screens (data not shown). We have managed to produce a stable construct of the essential C-terminal domain of ICP27. This has helped us to determine a high-resolution crystal structure, providing the first structural information on this important protein.
The fold of ICP27-CTD is novel, but the weak yet significant sequence similarity supports that the overall fold is likely to be conserved in members of all herpes families (Fig. 5). ICP27-CTD is sufficient to form dimers in solution as observed from multiangle light scattering analysis (data not shown) and analytical ultracentrifugation data (Fig. 3). The core structure of ICP27 is formed by a bundle of 10 α-helices (Fig. 2A and C); this is in contrast to the structure predictions suggesting that the core fold of the C-terminal region of ICP27 should have three KH domains (19, 20). KH domains contain essential β-sheets and overall have different folds than seen in our structure (49). A fold for the KSHV ORF57 was also generated using computational methods, which is very different from the fold value observed in our structure (50). Many herpes proteins lack significant homology to known structures, but this warrants caution when using weak fold prediction methods for generating working models for their structural information.
The Zn-binding domain of ICP27-CTD also has a novel fold and is unique in the sense that the first metal ligand C400 is distant in sequence to the other three ligands (Fig. 4). Still, the fold of this domain is compact with the Zn in the center of the domain, and the domain has properties similar to those of other zinc finger-like domains. When the Zn domain is positioned on the surface of the protein, it is likely involved in interacting with other proteins. The exposed charge distribution of the domain does not support that it would be involved in binding RNA.
A striking feature of the structure of ICP27-CTD is the N- and C-terminal extensions inserting and wrapping around the partner monomer in the dimer (Fig. 1). Extended arms covering the surfaces of neighboring subunits, as the N-terminal arm of ICP27-CTD, is not unusual in protein complexes. In many cases, they probably have major roles in constitutively stabilizing the complex, but they might also have regulatory roles where they, for example, are released during the functional cycle or needed in dimeric form to perform function (51, 52). The N-terminal helices of ICP27-CTD appear to be largely aliphatic, interacting with their hydrophobic face onto the core domain. Since these may be relatively strong interactions, their role is more likely to constitutively stabilize the complex, rather than to serve as regulatory elements, although the later cannot be excluded. The C-terminal C-tail, on the other hand, forms an unusual structure by inserting itself into the core domain of neighboring ICP27-CTD (Fig. 7A). The high conservation of, and around, the C-tail in the structure suggests that this is important and potentially also plays a role in ICP27 function (Fig. 5). The structure around the C-tail indicates that the bundle of helices 3, 4, and 5 and loop L2 could be seen as a subdomain when they reside on one side of the C-tail. The loop shows significantly higher B values than the remaining core domain (Fig. 7A). A slight rotation of this subdomain could allow the C-tail to be released and exposed to the surface, where it could be involved in specific interactions. Alternatively, during the formation of the dimer, the flexibility of this subdomain could allow the C-tail to bind into the core domain. We have made extensive efforts to express ICP27 and its homologs with alterations in the C-tail region, but none of these proteins have been stable enough for biochemical or structural studies. We have instead phosphorylated ICP27-CTD with three different protein kinases to investigate potential regulatory sites and whether phosphorylation can change properties of the protein, including oligomerization and the C-tail behavior. Wilcox et al. reported that ICP27 harbors both stable and transient phosphorylations that may be regulating its activity within the cell (44). Phosphopeptide mapping of ICP27, performed using in vivo and in vitro techniques, revealed some probable phosphorylation sites, mapped to the N-terminal region of the protein (45). Based on these findings, it was shown that mutations in N-terminal phosphorylation sites may lead to altered interactions between ICP27 and Aly/REF and may result in defective viral replication (53, 54). In our study, several phosphorylation sites where mapped in the region of the C-tail binding pocket and also around the zinc-binding motif (Fig. 7A and B). We used a limited proteolysis approach to assess whether the overall structure was destabilized and whether the C-tail was released, i.e., being more exposed upon phosphorylation; however, no increase in proteolytic sensitivity was seen (data not shown). Further probing, including in vivo studies may be needed in order to determine conclusively the effects of phosphorylation in these key regions. It would be interesting to explore whether these phosphorylations in conserved regions of the protein could lead to or disrupt protein-protein interactions, disrupt the dimeric conformation, or act as some sort of a signal in switching between one or more of the various functions of ICP27.
ICP27 plays a role in multiple processes during its functional cycle and has been, in addition to mRNA binding, implicated in interactions with a number of different proteins (25). In most studies, however, it is not clear which of the ICP27 domains makes these interactions. Because only cellular interaction studies have been performed, it is debatable whether the interactions are direct or mediated through other protein components. Based on our structural information, the Zn-binding motif stands out as a likely region for interactions with other proteins, although other regions are also likely to be involved. It has been proposed that the ICP27-CTD makes direct interactions with mRNA, in addition to the well-characterized interactions made by the RGG motif in the N-terminal of ICP27 (17, 18). ORF57 contains two proposed RNA binding sites: N-terminal RGG1 (residues 138 to 140) and C-terminal RGG2 (residues 372 to 374) (55). This RGG2 corresponds to the RRR motif (residues 416 to 418) in ICP27. Although this motif is exposed on the surface of ICP27 protein, this surface region appears to lack the appropriate pockets and charge distribution for interactions with RNA.
In summary, we provide the first structural information of ICP27, which reveals a novel and interesting fold and a novel type of Zn-binding domain. The structural and biophysical data support that ICP27 is a constitutive dimer with an intriguing arm exchange structure. Weak yet significant sequence similarity support that these features are common for homologs in alpha-, beta-, and gammaherpesviruses. We also observed that the secondary structural CD profile of the ICP27 homolog, EB2 from EBV, is strikingly similar to that of ICP27-CTD (data not shown). The ICP27-CTD structure now provides a framework for re-evaluating previous structure predictions based on primary sequence, as well as predictions of interaction modes with potential protein partners. In particular, it should be valuable in guiding site-directed mutagenesis studies in vivo of the potential functional regions of ICP27 and their specific roles in making the multitude of interactions proposed for ICP27 function.
ACKNOWLEDGMENTS
We thank Jurgen Haas at the University of Edinburgh, Edinburgh, United Kingdom, for providing the full-length clone of ICP27 and the Protein Production Platform (Nanyang Technological University [NTU], Singapore) for the initial cloning and small-scale expression screening of the protein construct used in this study. Portions of this research were carried out at the National Synchrotron Radiation Research Center (NSRRC), a national user facility supported by the National Science Council of Taiwan, Republic of China. The Synchrotron Radiation Protein Crystallography Core Facility is supported by the National Core Facility Program for Proteomics and Structural Biology. This research was also undertaken on the MX1 and MX2 beamlines at the Australian Synchrotron (AS), Australia, and at the Diamond Light Source (DLS), United Kingdom. We are also grateful to the technical staff at the NSRRC (Taiwan), AS (Australia), and DLS (United Kingdom) synchrotrons for their assistance during diffraction data collection. We thank Wahyu Surya (NTU) for technical assistance in the AUC experiment, the NTU Proteomics and Mass spectrometry service facility, and the Experimental Therapeutics Centre mass spectrometry facility, Singapore.
This research was funded by a startup grant at Nanyang Technological University as well as grants from the Swedish Research Council to Pär Nordlund.
REFERENCES
- 1.Hardwicke MA, Sandri-Goldin RM. 1994. The herpes simplex virus regulatory protein ICP27 contributes to the decrease in cellular mRNA levels during infection. J Virol 68:4797–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hardy WR, Sandri-Goldin RM. 1994. Herpes simplex virus inhibits host cell splicing, and regulatory protein ICP27 is required for this effect. J Virol 68:7790–7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mears WE, Rice SA. 1996. The RGG box motif of the herpes simplex virus ICP27 protein mediates an RNA-binding activity and determines in vivo methylation. J Virol 70:7445–7453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mears WE, Lam V, Rice SA. 1995. Identification of nuclear and nucleolar localization signals in the herpes simplex virus regulatory protein ICP27. J Virol 69:935–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carmody SR, Wente SR. 2009. mRNA nuclear export at a glance. J Cell Sci 122:1933–1937. doi: 10.1242/jcs.041236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sciabica KS, Dai QJ, Sandri-Goldin RM. 2003. ICP27 interacts with SRPK1 to mediate HSV splicing inhibition by altering SR protein phosphorylation. EMBO J 22:1608–1619. doi: 10.1093/emboj/cdg166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Souki SK, Sandri-Goldin RM. 2009. Arginine methylation of the ICP27 RGG box regulates the functional interactions of ICP27 with SRPK1 and Aly/REF during herpes simplex virus 1 infection. J Virol 83:8970–8975. doi: 10.1128/JVI.00801-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bryant HE, Wadd SE, Lamond AI, Silverstein SJ. 2001. Herpes simplex virus IE63 (ICP27) protein interacts with spliceosome-associated protein 145 and inhibits splicing prior to the first catalytic step. J Virol 75:4376–4385. doi: 10.1128/JVI.75.9.4376-4385.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindberg A, Kreivi JP. 2002. Splicing inhibition at the level of spliceosome assembly in the presence of herpes simplex virus protein ICP27. Virology 294:189–198. doi: 10.1006/viro.2001.1301. [DOI] [PubMed] [Google Scholar]
- 10.Chen IB, Sciabica KS, Sandri-Goldin RM. 2002. ICP27 interacts with the RNA export factor Aly/REF to direct herpes simplex virus type 1 intronless mRNAs to the TAP export pathway. J Virol 76:12877–12889. doi: 10.1128/JVI.76.24.12877-12889.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koffa MD, Clements JB, Izaurralde E, Wadd S, Wilson SA, Mattaj IW, Kuersten S. 2001. Herpes simplex virus ICP27 protein provides viral mRNAs with access to the cellular mRNA export pathway. EMBO J 20:5769–5778. doi: 10.1093/emboj/20.20.5769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen IB, Li L, Silva L, Sandri-Goldin RM. 2005. ICP27 recruits Aly/REF but not TAP / NXF1 to herpes simplex virus type 1 transcription sites although TAP/NXF1 is required for ICP27 export. J Virol 79:3949–3961. doi: 10.1128/JVI.79.7.3949-3961.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Escudero-Paunetto L, Li L, Hernandez FP, Sandri-Goldin RM. 2010. SR proteins SRp20 and 9G8 contribute to efficient export of herpes simplex virus 1 mRNAs. Virology 401:155–164. doi: 10.1016/j.virol.2010.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hernandez FP, Sandri-Goldin RM. 2010. Head-to-Tail intramolecular interaction of herpes simplex virus type 1 regulatory protein ICP27 is important for its interaction with cellular mRNA export receptor TAP/NXF1. mBio 1:e00268-10. doi: 10.1128/mBio.00268-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malik P, Tabarraei A, Kehlenbach RH, Korfali N, Iwasawa R, Graham SV, Schirmer EC. 2012. Herpes simplex virus ICP27 protein directly interacts with the nuclear pore complex through Nup62, inhibiting host nucleocytoplasmic transport pathways. J Biol Chem 287:12277–12292. doi: 10.1074/jbc.M111.331777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fontaine-Rodriguez EC, Taylor TJ, Olesky M, Knipe DM. 2004. Proteomics of herpes simplex virus-infected cell protein 27: association with translation initiation factors. Virology 330:487–492. doi: 10.1016/j.virol.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 17.Larralde O, Smith RWP, Wilkie GS, Malik P, Gray NK, Clements JB. 2006. Direct stimulation of translation by the multifunctional herpesvirus ICP27 protein. J Virol 80:1588–1591. doi: 10.1128/JVI.80.3.1588-1591.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corbin-Lickfett KA, Chen IB, Cocco MJ, Sandri-Goldin RM. 2009. The HSV-1 ICP27 RGG box specifically binds flexible, GC-rich sequences but not G-quartet structures. Nucleic Acids Res 37:7290–7301. doi: 10.1093/nar/gkp793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soliman TM, Silverstein SJ. 2000. Herpesvirus mRNAs are sorted for export via Crm1-dependent and -independent pathways. J Virol 74:2814–2825. doi: 10.1128/JVI.74.6.2814-2825.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soliman TM, Silverstein SJ. 2000. Identification of an export control sequence and a requirement for the KH domains in ICP27 from herpes simplex virus type 1. J Virol 74:7600–7609. doi: 10.1128/JVI.74.16.7600-7609.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vaughan PJ, Thibault KJ, Hardwicke MA, Sandri-Goldin RM. 1992. The herpes simplex virus immediate-early protein ICP27 encodes a potential metal binding domain and binds zinc in vitro. Virology 384:377–384. [DOI] [PubMed] [Google Scholar]
- 22.Corbin-Lickfett KA, Souki SK, Cocco MJ, Sandri-Goldin RM. 2010. Three arginine residues within the RGG box are crucial for ICP27 binding to herpes simplex virus 1 GC-rich sequences and for efficient viral RNA export. J Virol 84:6367–6376. doi: 10.1128/JVI.00509-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tunnicliffe RB, Hautbergue GM, Kalra P, Jackson BR, Whitehouse A, Wilson SA, Golovanov AP. 2011. Structural basis for the recognition of cellular mRNA export factor REF by herpes viral proteins HSV-1 ICP27 and HVS ORF57. PLoS Pathog 7:e1001244. doi: 10.1371/journal.ppat.1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhi Y, Sciabica K, Sandri-Goldin RM. 1999. Self-interaction of the herpes simplex type 1 regulatory protein ICP27. Virology 257:341–351. doi: 10.1006/viro.1999.9698. [DOI] [PubMed] [Google Scholar]
- 25.Sandri-Goldin RM. 2011. The many roles of the highly interactive HSV protein ICP27, a key regulator of infection. Future Microbiol 6:1261–1277. doi: 10.2217/fmb.11.119. [DOI] [PubMed] [Google Scholar]
- 26.Majerciak V, Yamanegi K, Allemand E, Kruhlak M, Krainer AR, Zheng ZM. 2008. Kaposi's sarcoma-associated herpesvirus ORF57 functions as a viral splicing factor and promotes expression of intron-containing viral lytic genes in spliceosome-mediated RNA splicing. J Virol 82:2792–2801. doi: 10.1128/JVI.01856-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verma D, Bais S, Gaillard M, Swaminathan S. 2010. Epstein-Barr Virus SM protein utilizes cellular splicing factor SRp20 to mediate alternative splicing. J Virol 84:11781–11789. doi: 10.1128/JVI.01359-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boyne JR, Colgan KJ, Whitehouse A. 2008. Recruitment of the complete hTREX complex is required for Kaposi's sarcoma-associated herpesvirus intronless mRNA nuclear export and virus replication. PLoS Pathog 4:e1000194. doi: 10.1371/journal.ppat.1000194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyne JR, Jackson BR, Taylor A, Macnab SA, Whitehouse A. 2010. Kaposi's sarcoma-associated herpesvirus ORF57 protein interacts with PYM to enhance translation of viral intronless mRNAs. EMBO J 29:1851–1864. doi: 10.1038/emboj.2010.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. [DOI] [PubMed] [Google Scholar]
- 31.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of COOT. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laue T, Stafford W. 1999. Modern applications of analytical ultracentrifugation. Annu Rev Biophys Biomol Struct 28:75–100. doi: 10.1146/annurev.biophys.28.1.75. [DOI] [PubMed] [Google Scholar]
- 34.Schuck P. 2003. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal Biochem 320:104–124. doi: 10.1016/S0003-2697(03)00289-6. [DOI] [PubMed] [Google Scholar]
- 35.Klug A. 2010. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu Rev Biochem 79:213–231. doi: 10.1146/annurev-biochem-010909-095056. [DOI] [PubMed] [Google Scholar]
- 36.Miller J, McLachlan AD, Klug A. 1985. Repetitive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes. EMBO J 4:1609–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brayer KJ, Segal DJ. 2008. Keep your fingers off my DNA: protein-protein interactions mediated by C2H2 zinc finger domains. Cell Biochem Biophys 50:111–131. doi: 10.1007/s12013-008-9008-5. [DOI] [PubMed] [Google Scholar]
- 38.Krishna SS. 2003. Structural classification of zinc fingers: SURVEY AND SUMMARY. Nucleic Acids Res 31:532–550. doi: 10.1093/nar/gkg161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fujii H, Kato A, Mugitani M, Kashima Y, Oyama M, Kozuka-Hata H, Arii J, Kawaguchi Y. 2014. The UL12 protein of herpes simplex virus 1 is regulated by tyrosine phosphorylation. J Virol 88:10624–10634. doi: 10.1128/JVI.01634-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johannsen E, Luftig M, Chase MR, Weicksel S, Cahir-Mcfarland E, Illanes D, Sarracino D, Kieff E. 2004. Proteins of purified Epstein-Barr virus. Proc Natl Acad Sci U S A 101:16286–16291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malik P, Clements JB. 2004. Protein kinase CK2 phosphorylation regulates the interaction of Kaposi's sarcoma-associated herpesvirus regulatory protein ORF57 with its multifunctional partner hnRNP K. Nucleic Acids Res 32:5553–5569. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Medina-Palazon C, Gruffat H, Mure F, Filhol O, Vingtdeux-Didier V, Drobecq H, Cochet C, Sergeant N, Sergeant A, Manet E. 2007. Protein kinase CK2 phosphorylation of EB2 regulates its function in the production of Epstein-Barr virus infectious viral particles. J Virol 81:11850–11860. doi: 10.1128/JVI.01421-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tait AR, Straus SK. 2008. Phosphorylation of U24 from human herpes virus type 6 (HHV-6) and its potential role in mimicking myelin basic protein (MBP) in multiple sclerosis. FEBS Lett 582:2685–2688. doi: 10.1016/j.febslet.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 44.Wilcox KW, Kohn A, Sklyanskaya E, Roizman B. 1980. Herpes simplex virus phosphoproteins: phosphate cycles on and off some viral polypeptides and can alter their affinity for DNA. J Virol 33:167–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhi Y, Sandri-Goldin RM. 1999. Analysis of the phosphorylation sites of herpes simplex virus type 1 regulatory protein ICP27. J Virol 73:3246–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fogg MJ, Alzari P, Bahar M, Bertini I, Betton JM, Burmeister WP, Cambillau C, Canard B, Carrondo MA, Coll M, Daenke S, Dym O, Egloff MP, Enguita FJ, Geerlof A, Haouz A, Jones TA, Ma Q, Manicka SN, Migliardi M, Nordlund P, Owens RJ, Peleg Y, Schneider G, Schnell R, Stuart DI, Tarbouriech N, Unge T, Wilkinson AJ, Wilmanns M, Wilson KS, Zimhony O, Grimes JM. 2006. Application of the use of high-throughput technologies to the determination of protein structures of bacterial and viral pathogens. Acta Crystallogr D Biol Crystallogr 62:1196–1207. doi: 10.1107/S0907444906030915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tait AR, Straus SK. 2011. Overexpression and purification of U24 from human herpesvirus type-6 in Escherichia coli: unconventional use of oxidizing environments with a maltose binding protein-hexahistine dual tag to enhance membrane protein yield. Microb Cell Fact 10:51–62. doi: 10.1186/1475-2859-10-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tarbouriech N, Daenke S, Cusack S. 2006. Structural genomics of the Epstein-Barr virus. Acta Crystallogr D Biol Crystallogr 62(Pt 10):1276–1285. [DOI] [PubMed] [Google Scholar]
- 49.Valverde R, Edwards L, Regan L. 2008. Structure and function of KH domains. FEBS J 275:2712–2726. doi: 10.1111/j.1742-4658.2008.06411.x. [DOI] [PubMed] [Google Scholar]
- 50.Taylor A, Jackson BR, Noerenberg M, Hughes DJ, Boyne JR, Verow M, Harris M, Whitehouse A. 2011. Mutation of a C-terminal motif affects Kaposi's sarcoma-associated herpesvirus ORF57 RNA binding, nuclear trafficking, and multimerization. J Virol 85:7881–7891. doi: 10.1128/JVI.00138-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Austin J, Schreiber S. 1994. Proximity versus allostery: the role of regulated protein dimerization in biology. Chem Biol 1:131–136. doi: 10.1016/1074-5521(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 52.Marianayagam NJ, Sunde M, Matthews JM. 2004. The power of two: protein dimerization in biology. Trends Biochem Sci 29:618–625. doi: 10.1016/j.tibs.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 53.Corbin-Lickfett KA, Rojas S, Li L, Cocco MJ, Sandri-Goldin RM. 2010. ICP27 phosphorylation site mutants display altered functional interactions with cellular export factors Aly/REF and TAP/NXF1 but are able to bind herpes simplex virus 1 RNA. J Virol 84:2212–2222. doi: 10.1128/JVI.01388-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rojas S, Corbin-Lickfett KA, Escudero-Paunetto L, Sandri-Goldin RM. 2010. ICP27 phosphorylation site mutants are defective in herpes simplex virus 1 replication and gene expression. J Virol 84:2200–2211. doi: 10.1128/JVI.00917-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jackson BR, Noerenberg M, Whitehouse A. 2012. The Kaposi's sarcoma-associated herpesvirus ORF57 protein and its multiple roles in mRNA biogenesis. Front Microbiol 3:59. doi: 10.3389/fmicb.2012.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]