

Abstract
Replicon particles of Rift Valley fever virus, referred to as nonspreading Rift Valley fever virus (NSR), are intrinsically safe and highly immunogenic. Here, we demonstrate that NSR-infected human dendritic cells can activate CD8+ T cells in vitro and that prophylactic and therapeutic vaccinations of mice with NSR encoding a tumor-associated CD8 peptide can control the outgrowth of lymphoma cells in vivo. These results suggest that the NSR system holds promise for cancer immunotherapy.
TEXT
Dendritic cells (DCs) are the most potent antigen-presenting cells in the body and are instrumental in directing adaptive immune responses against pathogens and tumors. DCs are naturally targeted by many arboviruses. Infection induces DC maturation and presentation of virus-associated antigens, explaining the interest in using these viruses as therapeutic vectors of tumor-associated antigens. Members of the positive-strand RNA virus families Togaviridae and Flaviviridae have been extensively evaluated for cancer immunotherapy (1–4), some of which have already entered clinical trials (2). Remarkably, arboviruses with segmented negative-strand RNA genomes have remained largely unexplored in this field. The genome segments of these viruses form double-stranded RNA panhandles that, in some viruses, contain a 5′ triphosphate. This structure is an optimal ligand for the cytoplasmic pattern recognition receptor RIG-I and thereby an excellent inducer of adjuvanting innate immune responses (5). We therefore propose that arboviruses with segmented negative-strand RNA genomes that activate RIG-I hold promise for cancer immunotherapy.
Rift Valley fever virus (RVFV), a member of the Bunyaviridae family, was recently shown to productively infect immature DCs with high efficiency (6). Whereas wild-type RVFV counteracts innate immune responses via the NSs protein (7), infection of DCs with RVFV variants lacking the NSs protein was shown to result in strong interferon (IFN) responses triggered by RIG-I signaling (8). We recently developed RVFV particles that lack not only the gene for NSs but also the glycoprotein-encoding genome segment (9). The resulting nonspreading RVFV (NSR) particles are unable to produce progeny virions, ensuring their safe application (9). Moreover, the absence of the gene for NSs provides a slot for the expression of a gene of interest. Vaccination with NSR particles was shown to be highly effective in protecting livestock from RVFV (10–12), and NSR particles expressing the influenza virus hemagglutinin gene protected mice from lethal influenza (13). The remarkable efficacy of NSR-based vaccines in both inbred and outbred animals, particularly its association with Th1-type immune responses (13), prompted the present study on the use of NSR for cancer immunotherapy.
Initially, we investigated whether NSR-infected human monocyte-derived DCs (MoDCs) can activate CD8+ T cells in vitro. MoDCs were derived from peripheral blood mononuclear cells of healthy donors by Ficoll-Isopaque density gradient centrifugation (GE Healthcare Bio-Sciences AB) and cultured as previously described (14). Infection of DCs was remarkably efficient, resulting in over 90% green fluorescent protein (GFP)-positive cells under optimal conditions (data not shown). To study the T-cell activation capacity of infected DCs, an NSR variant was constructed that encodes the immunodominant NLVPMVATV epitope of human cytomegalovirus pp65 (pp65495-503) fused to the C terminus of GFP (Fig. 1A). NSR particles encoding this fusion protein (NSRNLV) and particles encoding GFP only (NSRGFP) were used to infect DCs obtained from an HLA A2-positive donor. As a positive control, DCs were incubated with a synthetic NLVPMVATV peptide (1 μM), and culture medium was used as a negative control. After overnight incubation, HLA A*0201-restricted, NLVPMVATV-specific CD8+ T cells (14) were added to the DCs and cocultured for 4.5 h. T cells were harvested, stained for CD3, CD8, and CD107a (LAMP-1) surface markers, and subsequently intracellularly stained for IFN-γ and tumor necrosis factor (TNF). The results of this experiment demonstrate that NSR can be successfully used as a vector to deliver specific immunogenic epitopes to human DCs, which trigger an effector function in corresponding CD8+ T cells (Fig. 1B).
FIG 1.

(A) Schematic representation of the NSR S segment encoding GFP (top) or the C-terminally fused pp65495-503 epitope (bottom) in an antigenomic orientation. (B) NSR-infected human DCs activate antigen-specific CD8+ T cells. HLA A2+ DCs were infected with NSRGFP or NSRNLV and cultured overnight. As a negative control, DCs were left untreated; as positive control, DCs were loaded with synthetic peptide pp65495-503. An A2-restricted CD8+ T cell clone specific for the pp65495-503 epitope was added for 4.5 h of incubation in the presence of GolgiStop (Becton Dickinson) and analyzed for activation. The expression of IFN-γ, TNF, and LAMP-1 in the CD8/CD3-positive cell population is depicted. All three parameters are induced specifically in NSRNLV-infected cells and not in NSRGFP-infected cells. FS, forward scatter.
To investigate the feasibility of using NSR for cancer immunotherapy, we made use of C57BL/6 Kh (B6, H-2b) mice and E.G7-OVA cells, a chicken ovalbumin (OVA) gene-transfected clone of mouse lymphoma EL4 cells obtained from the American Type Culture Collection (Manassas, VA, USA). (The animal experiments described here were conducted in accordance with the Dutch Law on Animal Experiments [Wod, ID no. BWBR0003081] and approved by the Animal Ethics Committee of the Central Veterinary Institute [permit no. 2014101.c].) An NSR variant was constructed that encodes the CD8-restricted SIINFEKL epitope of OVA (OVA257-264) fused to GFP (NSROVA) (Fig. 2A). To determine whether NSROVA vaccination elicits a cellular immune response specific for this epitope, mice were vaccinated twice via the intramuscular route (thigh muscle), at a 1-week interval, with 108 50% tissue culture infective doses of NSROVA (Fig. 2B). Control mice were vaccinated with NSRGFP. One week after the second vaccination, spleen cells were collected and evaluated for the ability to produce IFN-γ after stimulation with a synthetic SIINFEKL peptide (10 μg/ml; Invitrogen) in an enzyme-linked immunospot assay (ELISPOT) assay (10). This experiment demonstrated that NSROVA vaccination elicits a SIINFEKL-specific cellular immune response (Fig. 2C).
FIG 2.
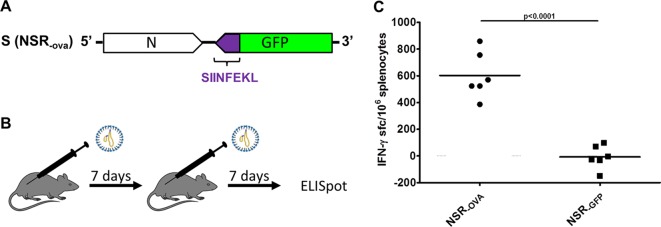
(A) Schematic representation of the NSR S segment encoding the OVA257-264 peptide fused to GFP in an antigenomic orientation. (B) Representation of the regimens used for vaccination with NSROVA or control NSRGFP vaccine. (C) ELISPOT assay showing IFN-γ responses of splenocytes collected from mice that were vaccinated with either NSRGFP or NSROVA (n = 6). Symbols represent individual counts of IFN-γ spot-forming cells (sfc). Statistical significance (Student's t test) is indicated.
We finally asked whether prophylactic or therapeutic vaccination with NSROVA can reduce the outgrowth of E.G7-OVA tumor cells (Fig. 3A). Mice were subcutaneously inoculated with 106 E.G7-OVA cells and euthanized by cervical dislocation when the tumor size reached 5,000 mm3. Both prophylactic and therapeutic vaccinations via the intramuscular route with NSROVA resulted in increased survival times compared to those of control mice that had received NSRGFP (Fig. 3B). Therapeutic vaccination resulted in complete tumor clearance in 2/10 mice, whereas prophylactic vaccination resulted in clearance of tumors in 6/10 mice. As expected, recultured tumor cells collected from NSRGFP-vaccinated mice upon necropsy were found to express OVA, as determined by a commercial OVA enzyme-linked immunosorbent assay (ELISA; Agro-Bio). Surprisingly, tumor cells collected from mice vaccinated with NSROVA did not express detectable levels of OVA (Fig. 3C). This finding suggests that small numbers of the inoculated cells did not express OVA or lost OVA expression in time and that tumor cells expressing OVA were efficiently cleared by either prophylactic or therapeutic vaccination.
FIG 3.

(A) Prophylactic and therapeutic regimens used for vaccination with NSROVA or control NSRGFP vaccine. The primary therapeutic vaccination was applied when more than 50% of the E.G7-OVA-inoculated mice had palpable tumors. (B) Survival curves of mice prophylactically (P, solid lines) or therapeutically (T, interrupted lines) vaccinated with NSRGFP or NSROVA. Statistical analyses were performed with GraphPad Prism. Log-rank (Mantel-Cox) tests revealed statistically significant differences (P < 0.0001) between NSRGFP and NSROVA vaccinations with either a prophylactic or a therapeutic regimen. (C) Detection of OVA in supernatants of cultured E.G7-OVA cells by ELISA. Cells were recultured for 7 days after being collected from mice (n = 3/group) that had reached the humane endpoint. Statistical significance (Student's t test with Welch's correction for unequal variances) is shown. E.G7-OVA cells that were used for the inoculation of mice were used to obtain a positive-control (+ Control) sample.
The present work demonstrates that NSR particles can successfully deliver an immunogenic peptide to human DCs and that these cells are capable of activating CD8+ T cells. In addition, vaccination of mice with NSR expressing a single CD8-restricted epitope resulted in the complete clearance of lymphoma cells expressing the targeted antigen. Future experiments will address the efficacy of NSR vaccination to control outgrowth of tumors expressing self-antigens. Considering that self-antigens are much less immunogenic, NSR particles will be designed to express not only CD8+ peptides but also CD4+ and B-cell epitopes.
ACKNOWLEDGMENTS
We thank Tuna Mutis and Anton Martens (VU University Medical Center, Department of Hematology) for useful discussions.
REFERENCES
- 1.Moran TP, Collier M, McKinnon KP, Davis NL, Johnston RE, Serody JS. 2005. A novel viral system for generating antigen-specific T cells. J Immunol 175:3431–3438. doi: 10.4049/jimmunol.175.5.3431. [DOI] [PubMed] [Google Scholar]
- 2.Morse MA, Hobeika AC, Osada T, Berglund P, Hubby B, Negri S, Niedzwiecki D, Devi GR, Burnett BK, Clay TM, Smith J, Lyerly HK. 2010. An alphavirus vector overcomes the presence of neutralizing antibodies and elevated numbers of Tregs to induce immune responses in humans with advanced cancer. J Clin Invest 120:3234–3241. doi: 10.1172/JCI42672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anraku I, Harvey TJ, Linedale R, Gardner J, Harrich D, Suhrbier A, Khromykh AA. 2002. Kunjin virus replicon vaccine vectors induce protective CD8+ T-cell immunity. J Virol 76:3791–3799. doi: 10.1128/JVI.76.8.3791-3799.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herd KA, Harvey T, Khromykh AA, Tindle RW. 2004. Recombinant Kunjin virus replicon vaccines induce protective T-cell immunity against human papillomavirus 16 E7-expressing tumour. Virology 319:237–248. doi: 10.1016/j.virol.2003.10.032. [DOI] [PubMed] [Google Scholar]
- 5.Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, Barchet W, Coch C, Janke M, Mihailovic A, Wardle G, Juranek S, Kato H, Kawai T, Poeck H, Fitzgerald KA, Takeuchi O, Akira S, Tuschl T, Latz E, Ludwig J, Hartmann G. 2009. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 31:25–34. doi: 10.1016/j.immuni.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lozach PY, Kühbacher A, Meier R, Mancini R, Bitto D, Bouloy M, Helenius A. 2011. DC-SIGN as a receptor for phleboviruses. Cell Host Microbe 10:75–88. doi: 10.1016/j.chom.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 7.Le May N, Mansuroglu Z, Leger P, Josse T, Blot G, Billecocq A, Flick R, Jacob Y, Bonnefoy E, Bouloy M. 2008. A SAP30 complex inhibits IFN-beta expression in Rift Valley fever virus infected cells. PLoS Pathog 4:e13. doi: 10.1371/journal.ppat.0040013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ermler ME, Yerukhim E, Schriewer J, Schattgen S, Traylor Z, Wespiser AR, Caffrey DR, Chen ZJ, King CH, Gale M Jr, Colonna M, Fitzgerald KA, Buller RM, Hise AG. 2013. RNA helicase signaling is critical for type I interferon production and protection against Rift Valley fever virus during mucosal challenge. J Virol 87:4846–4860. doi: 10.1128/JVI.01997-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kortekaas J, Oreshkova N, Cobos-Jimenez V, Vloet RP, Potgieter CA, Moormann RJ. 2011. Creation of a nonspreading Rift Valley fever virus. J Virol 85:12622–12630. doi: 10.1128/JVI.00841-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oreshkova N, van Keulen L, Kant J, Moormann RJ, Kortekaas J. 2013. A single vaccination with an improved nonspreading Rift Valley fever virus vaccine provides sterile immunity in lambs. PLoS One 8:e77461. doi: 10.1371/journal.pone.0077461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kortekaas J, Antonis AF, Kant J, Vloet RP, Vogel A, Oreshkova N, de Boer SM, Bosch BJ, Moormann RJ. 2012. Efficacy of three candidate Rift Valley fever vaccines in sheep. Vaccine 30:3423–3429. doi: 10.1016/j.vaccine.2012.03.027. [DOI] [PubMed] [Google Scholar]
- 12.Kortekaas J, Oreshkova N, van Keulen L, Kant J, Bosch BJ, Bouloy M, Moulin V, Goovaerts D, Moormann RJ. 2014. Comparative efficacy of two next-generation Rift Valley fever vaccines. Vaccine 32:4901–4908. doi: 10.1016/j.vaccine.2014.07.037. [DOI] [PubMed] [Google Scholar]
- 13.Oreshkova N, Cornelissen LA, de Haan CA, Moormann RJ, Kortekaas J. 2014. Evaluation of nonspreading Rift Valley fever virus as a vaccine vector using influenza virus hemagglutinin as a model antigen. Vaccine 32:5323–5329. doi: 10.1016/j.vaccine.2014.07.051. [DOI] [PubMed] [Google Scholar]
- 14.Flinsenberg TW, Compeer EB, Koning D, Klein M, Amelung FJ, van Baarle D, Boelens JJ, Boes M. 2012. Fcγ receptor antigen targeting potentiates cross-presentation by human blood and lymphoid tissue BDCA-3+ dendritic cells. Blood 120:5163–5172. doi: 10.1182/blood-2012-06-434498. [DOI] [PubMed] [Google Scholar]