

Abstract
Here we present evidence for previously unappreciated B-cell immune dysregulation during acute Epstein-Barr virus (EBV)-associated infectious mononucleosis (IM). Longitudinal analyses revealed that patients with acute IM have undetectable EBV-specific neutralizing antibodies and gp350-specific B-cell responses, which were associated with a significant reduction in memory B cells and no evidence of circulating antibody-secreting cells. These observations correlate with dysregulation of tumor necrosis factor family members BAFF and APRIL and increased expression of FAS on circulating B cells.
TEXT
Epstein-Barr virus (EBV) is a human herpesvirus that infects more than 90% of the world's population (1). While benign and asymptomatic for the most part, EBV infection has the potential to cause many nonmalignant and malignant diseases of lymphoid and epithelial origins (2). EBV infection during childhood is mostly asymptomatic; however, primary exposure during adolescence manifests itself as infectious mononucleosis (IM) in 30 to 70% of cases (3–7). It is not known why some individuals are more likely than others to develop clinical symptoms from delayed infection (8). While CD8+ T cells play a central protective role in the control of latent EBV infection, they are also likely to be the main mediators of disease during IM (9, 10). These observations suggest that other immune response mediators are likely important for the prevention of acute symptomatic EBV infection. Observations from a recent phase II clinical trial suggested that the induction of neutralizing antibodies can prevent symptomatic acute IM following primary infection (11). Despite these encouraging results, very little emphasis has been placed upon the investigation of humoral immunity during primary infection, although it was shown over 40 years ago that efficient EBV neutralization does not develop until well after convalescence (12), suggesting that defects in humoral immunity could contribute to the disease burden during acute IM. The goal of this study was therefore to investigate the role of humoral immunity during primary symptomatic EBV infection. We hypothesized that this increased viral replication during acute IM may be linked to impaired B-cell responses.
To test this hypothesis, we assessed EBV-specific neutralizing antibody responses at the time of diagnosis of acute IM and at least 6 months following recovery from clinical symptoms of acute viral infection. Neutralizing antibody levels were assessed with an EBV transformation assay as previously described (13, 14). As shown in Fig. 1A, none of the patients with acute IM had detectable anti-EBV neutralizing antibody responses during the acute stage of infection and the levels of neutralizing antibodies significantly increased as these patients recovered from acute viral infection. The levels of EBV-neutralizing antibodies in many patients remained well below the levels seen in asymptomatic virus carriers, even after recovery from acute IM (Fig. 1A).
FIG 1.

Delayed induction of gp350-specific neutralizing antibody response following acute EBV infection. (A) Serial dilutions of heat-inactivated plasma were incubated with EBV B95-8 and then with PBMC from an EBV-seronegative donor for 6 weeks. Data represent the effective dilution of plasma that inhibits B-cell transformation by 50%. (B) EBV gp350-specific Ig titers were evaluated by enzyme-linked immunosorbent assay. Data represent the inverse titer that induces 50% of the maximal optical density at 450 nm. (C) EBV gp350-specific IgG titers were evaluated by enzyme-linked immunosorbent assay. Data represent the inverse titer that induces 50% of the maximal optical density at 450 nm. (D) Frequency of IgG-secreting gp350-specific B cells determined by ELISPOT assay. PBMCs from IM patients and latent virus carriers were cultured in vitro for 6 days to stimulate antibody production from MBCs. Data represent the proportion of antigen-specific cells relative to the total IgG-producing B-cell population. Statistical analysis was performed with a Wilcoxon matched-pair signed-rank test to compare measurements at two time points for the same individual, and comparison of unpaired groups was performed by Mann-Whitney test. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Earlier studies have shown that EBV-encoded glycoprotein gp350 is one of the major immunodominant antigens in antiviral neutralizing antibody responses (15, 16). To determine whether lack of viral neutralization was associated with impaired induction of a gp350-specific response, gp350 antibody titers were assessed in the serum of IM patients. As shown in Fig. 1B and C, the levels of anti-gp350 Ig and total anti-gp350 IgG in patients with acute IM were significantly lower than the levels of gp350-specific Ig and IgG in patients who had recovered from clinical symptoms of acute viral infection and in asymptomatic virus carriers. To further confirm the impaired antiviral humoral responses during acute IM, we next quantitated the circulating EBV-specific memory B cells (MBCs) with enzyme-linked immunospot (ELISPOT) assays. Consistent with the data presented in Fig. 1A, most patients with acute infection had significantly lower numbers of gp350-specific MBCs than did age-matched healthy virus carriers (Fig. 1D). A significant increase in gp350-specific MBCs was observed following the resolution of acute IM symptoms.
To delineate the potential reason for the lack of EBV-specific neutralizing antibodies, we performed a longitudinal analysis of the frequency of MBCs (CD3− CD19+ CD20+ CD27hi) and plasmablasts (CD3− CD19+ CD20lo CD27hi CD38hi) in the peripheral blood of IM patients. Representative gating analyses of these B-cell subsets are shown in Fig. 2A. These analyses revealed a significant reduction in the frequency of MBCs during acute infection (Fig. 2B). The frequency of MBCs remained low even after recovery compared with that in healthy virus carriers (Fig. 2B). Interestingly, despite ongoing viral replication, there was no significant increase in the frequency of plasmablasts in acute IM patients compared with that in asymptomatic healthy virus carriers (Fig. 2C). This finding is in contrast to previous studies of other viral infections, including those caused by influenza, dengue and respiratory syncytial viruses, which showed that increased frequencies of plasmablasts in the peripheral blood are readily detectable during the acute phase of infection (17–19).
FIG 2.

Impact of acute EBV infection on the peripheral B-cell compartment. PBMCs from IM patients and latent virus carriers were incubated with fluorescently labeled antibodies specific for CD19, CD20, CD27, and CD38, and then the frequencies of MBCs and plasmablasts were assessed by flow cytometry. (A) Representative analysis of the gating strategy for MBCs and plasmablasts from a single donor is shown. SSC, side scatter; FSC, side scatter; ASC, antibody-secreting cells. (B, C) Box-and-whisker plots represent the mean and range of either MBCs (B) or plasmablasts (C) in PBMCs. Dots represent data from individual donors. Statistical analysis was performed by one-way analysis of variance. *, P < 0.05; ****, P < 0.0001.
We next explored the possibility that the low frequency of plasmablasts in acute IM is due to the inability of B cells to mature into plasmablasts. Peripheral blood mononuclear cells (PBMCs) from acute IM patients and latent EBV carriers were stimulated with polyclonal B-cell stimulators and assessed for the frequencies of plasmablasts and total IgG-producing B cells. Consistent with the data presented above, conversion of B cells to plasmablasts was significantly lower in acute IM patients than in latent EBV carriers (Fig. 3A and B). The efficacy of B-cell conversion also appeared to inversely correlate with the severity of clinical symptoms during acute EBV infection. Although the difference was not statistically significant, patients diagnosed with severe clinical symptoms typically showed fewer IgG-producing cells (Fig. 3C). In vitro infection of healthy PBMCs with EBV did not prevent the conversion of B cells into IgG-producing cells (data not shown), so direct infection of B cells by EBV is unlikely to lead to a defect in the maturation of MBCs into plasmablasts.
FIG 3.
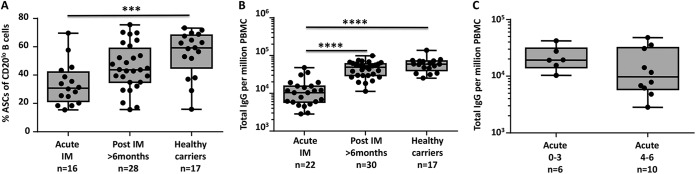
Acute EBV infection is associated with a global defect in conversion into antibody-secreting cells (ASC). PBMCs from IM patients and latent virus carriers were cultured in vitro for 6 days with a B-cell stimulation cocktail (pokeweed mitogen extract, Staphylococcus aureus protein A, CpG ODN 2006, and interleukin-10) to stimulate conversion into plasmablasts. Cultured cells were then assessed for the frequency of plasmablasts by flow cytometry or for the capacity to produce IgG by ELISPOT assay. Box-and-whisker plots represent the mean and range of either plasmablasts (A) or IgG-producing cells (B, C). Dots represent data from individual donors. Statistical analysis was performed by one-way analysis of variance. ***, P < 0.001; ****, P < 0.0001.
Finally, the impairment of B-cell-mediated immune regulation during acute IM was further evident by dysregulated expression of tumor necrosis factor (TNF) superfamily ligands and receptors BAFF (B-cell-activating factor) and APRIL (B-cell proliferation-inducing ligand), which play a critical role in B-cell survival, maturation, and activation (20, 21). These ligands mediate their activity through interaction with BAFF-R (BAFF receptor) and BCMA (B-cell maturation antigen) (22). Earlier studies have shown that a defect in signaling through these receptors can cause impairment of T-cell-dependent humoral immunity and in the generation of MBCs (21). Acute IM patients showed increased levels of BAFF in plasma and downregulated expression of BAFF-R on circulating B cells (Fig. 4A and B). In contrast, the levels of APRIL in plasma were significantly lower, whereas BCMA expression on circulating B cells was significantly increased (Fig. 4C and D). Furthermore, we also observed significantly higher levels of FAS on B cells and FAS ligand (FAS-L) in plasma during acute IM, which decreased as the disease resolved and eventually reached the levels seen in healthy virus carriers (Fig. 4F). This increased expression of FAS on B cells may indicate increased susceptibility to FAS-mediated death.
FIG 4.

Dysregulation of TNF family members BAFF and APRIL during acute IM. Concentrations of BAFF (A) and APRIL (C) in IM patients and latent virus carriers were determined by enzyme-linked immunosorbent assay. Data represent the mean ± the standard error of the mean of nine IM patients during and after IM and of 10 latent virus carriers. PBMCs from IM patients and healthy virus carriers were assessed for the expression of BAFF-R (B) and BCMA (D) by flow cytometry. Data represent the mean fluorescence intensity (MFI) of BAFF-R and BCMA expression on CD19+ B cells ± the standard error of the mean. (E) The expression of FAS on B cells from IM patients and latent virus carriers was assessed by flow cytometry. Dot points represent the mean fluorescent intensities of FAS expression on CD19+ B cells from individual samples. (F) FAS-L expression in the plasma of IM patients and latent virus carriers was determined with a BD cytometric bead array kit. Dot points represent the concentrations of FAS-L in individual samples. Statistical analysis was performed with a Wilcoxon matched-pair signed-rank test to compare measurements at two time points for the same individual, and comparison of unpaired groups was performed by Mann-Whitney test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
In this study, we have shown that during primary symptomatic EBV infection, there is delayed induction of EBV-specific neutralizing antibodies and gp350-specific MBCs and that this is coincident with homeostatic dysregulation in the global B-cell compartment. Interestingly, while other EBV-specific antibody responses, including those targeting EBV nuclear antigen 1, are not induced until well after convalescence, B-cell responses to the viral capsid antigens (VCA) and early antigens (EA) are readily detected during acute EBV infection. While the precise mechanism of the differential kinetics of B-cell responses to VCA/EA and gp350 remains to be determined, we hypothesize that while early priming of B-cell responses to VCA/EA can be induced rapidly after infection because of the abundance of these proteins or their ease of processing by the immune system, B-cell responses to other less abundant or less efficiently processed antigens are not induced as easily. Subsequently, the onset of B-cell immune dysregulation restricts the induction of an efficient neutralizing antibody response, which likely contributes to the increased viral load, leading to uncontrolled activation of EBV-specific CD8+ T cells. Further studies on the mechanism of this impaired B-cell immunity may explain how the immune dysregulation observed during acute IM leads to the poor induction of EBV-specific neutralizing antibody responses and help us to design better therapeutic and/or prophylactic strategies to prevent acute IM symptoms and perhaps reduce the risk of developing more serious EBV-associated diseases (23).
ACKNOWLEDGMENTS
We thank Linda Jones for technical assistance in the studies described in this report.
This work was supported by National Health and Medical Research Council (NHMRC), Australia. R.K. is supported by an NHMRC senior principal research fellowship.
R.K. and C.S. designed this study and wrote the manuscript. A.P., N.T., V.D., M.W., D.J.M., and A.H. conducted various experimental studies. A.P., D.J.M., A.R., H.H.B., K.A.H., and M.W. provided critical intellectual input and also contributed to the writing of this report.
We have no conflict of interest to declare.
REFERENCES
- 1.Rickinson AB, Kieff E. 1996. Epstein-Barr virus, p 2397–2446. In Fields BN, Knipe DM, Howley PM (ed), Fields virology, vol 3 Lippincott-Raven Publishers, Philadelphia, PA. [Google Scholar]
- 2.Hislop AD, Ressing ME, van Leeuwen D, Pudney VA, Horst D, Koppers-Lalic D, Croft NP, Neefjes JJ, Rickinson AB, Wiertz EJ. 2007. A CD8+ T cell immune evasion protein specific to Epstein-Barr virus and its close relatives in Old World primates. J Exp Med 204:1863–1873. doi: 10.1084/jem.20070256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chervenick PA. 1989. Infectious mononucleosis: the classic clinical syndrome, p 29–34. In D. Schlossberg (ed), Infectious mononucleosis, vol 2 Springer Verlag, New York, NY. [Google Scholar]
- 4.Evans AS, Niederman JC, McCollum RW. 1969. Infectious mononucleosis—role of EB virus. N Engl J Med 280:112. [DOI] [PubMed] [Google Scholar]
- 5.Ginsburg CM, Henle W, Henle G, Horwitz CA. 1977. Infectious mononucleosis in children. Evaluation of Epstein-Barr virus-specific serological data. JAMA 237:781–785. [DOI] [PubMed] [Google Scholar]
- 6.Niederman JC, McCollum RW, Henle G, Henle W. 1968. Infectious mononucleosis. JAMA 203:139–143. [DOI] [PubMed] [Google Scholar]
- 7.Straus SE, Fleisher GR. 1989. Infectious mononucleosis: epidemiology and pathogenesis, p 8–28. In D. Schlossberg (ed), Infectious mononucleosis, vol 2 Springer Verlag, New York, NY. [Google Scholar]
- 8.Balfour HH Jr, Dunmire SK, Hogquist KA. 2015. Infectious mononucleosis. Clin Transl Immunology 4:e33. doi: 10.1038/cti.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balfour HH Jr, Odumade OA, Schmeling DO, Mullan BD, Ed JA, Knight JA, Vezina HE, Thomas W, Hogquist KA. 2013. Behavioral, virologic, and immunologic factors associated with acquisition and severity of primary Epstein-Barr virus infection in university students. J Infect Dis 207:80–88. doi: 10.1093/infdis/jis646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baumgarten E, Herbst H, Schmitt M, Seeger KH, Schulte Overberg U, Henze G. 1994. Life-threatening infectious mononucleosis: is it correlated with virus-induced T cell proliferation? Clin Infect Dis 19:152–156. doi: 10.1093/clinids/19.1.152. [DOI] [PubMed] [Google Scholar]
- 11.Sokal EM, Hoppenbrouwers K, Vandermeulen C, Moutschen M, Leonard P, Moreels A, Haumont M, Bollen A, Smets F, Denis M. 2007. Recombinant gp350 vaccine for infectious mononucleosis: a phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J Infect Dis 196:1749–1753. doi: 10.1086/523813. [DOI] [PubMed] [Google Scholar]
- 12.Hewetson JF, Rocchi G, Henle W, Henle G. 1973. Neutralizing antibodies to Epstein-Barr virus in healthy populations and patients with infectious mononucleosis. J Infect Dis 128:283–289. doi: 10.1093/infdis/128.3.283. [DOI] [PubMed] [Google Scholar]
- 13.Moss DJ, Pope JH. 1972. Assay of the infectivity of Epstein-Barr virus by transformation of human leucocytes in vitro. J Gen Virol 17:233–236. doi: 10.1099/0022-1317-17-2-233. [DOI] [PubMed] [Google Scholar]
- 14.Sashihara J, Burbelo PD, Savoldo B, Pierson TC, Cohen JI. 2009. Human antibody titers to Epstein-Barr Virus (EBV) gp350 correlate with neutralization of infectivity better than antibody titers to EBV gp42 using a rapid flow cytometry-based EBV neutralization assay. Virology 391:249–256. doi: 10.1016/j.virol.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffman GJ, Lazarowitz SG, Hayward SD. 1980. Monoclonal antibody against a 250,000-dalton glycoprotein of Epstein-Barr virus identifies a membrane antigen and a neutralizing antigen. Proc Natl Acad Sci U S A 77:2979–2983. doi: 10.1073/pnas.77.5.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thorley-Lawson DA, Geilinger K. 1980. Monoclonal antibodies against the major glycoprotein (gp350/220) of Epstein-Barr virus neutralize infectivity. Proc Natl Acad Sci U S A 77:5307–5311. doi: 10.1073/pnas.77.9.5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wrammert J, Koutsonanos D, Li GM, Edupuganti S, Sui J, Morrissey M, McCausland M, Skountzou I, Hornig M, Lipkin WI, Mehta A, Razavi B, Del Rio C, Zheng NY, Lee JH, Huang M, Ali Z, Kaur K, Andrews S, Amara RR, Wang Y, Das SR, O'Donnell CD, Yewdell JW, Subbarao K, Marasco WA, Mulligan MJ, Compans R, Ahmed R, Wilson PC. 2011. Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J Exp Med 208:181–193. doi: 10.1084/jem.20101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wrammert J, Onlamoon N, Akondy RS, Perng GC, Polsrila K, Chandele A, Kwissa M, Pulendran B, Wilson PC, Wittawatmongkol O, Yoksan S, Angkasekwinai N, Pattanapanyasat K, Chokephaibulkit K, Ahmed R. 2012. Rapid and massive virus-specific plasmablast responses during acute dengue virus infection in humans. J Virol 86:2911–2918. doi: 10.1128/JVI.06075-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee FE, Falsey AR, Halliley JL, Sanz I, Walsh EE. 2010. Circulating antibody-secreting cells during acute respiratory syncytial virus infection in adults. J Infect Dis 202:1659–1666. doi: 10.1086/657158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Darce JR, Arendt BK, Wu X, Jelinek DF. 2007. Regulated expression of BAFF-binding receptors during human B cell differentiation. J Immunol 179:7276–7286. doi: 10.4049/jimmunol.179.11.7276. [DOI] [PubMed] [Google Scholar]
- 21.Rauch M, Tussiwand R, Bosco N, Rolink AG. 2009. Crucial role for BAFF-BAFF-R signaling in the survival and maintenance of mature B cells. PLoS One 4:e5456. doi: 10.1371/journal.pone.0005456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneider P, MacKay F, Steiner V, Hofmann K, Bodmer JL, Holler N, Ambrose C, Lawton P, Bixler S, Acha-Orbea H, Valmori D, Romero P, Werner-Favre C, Zubler RH, Browning JL, Tschopp J. 1999. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med 189:1747–1756. doi: 10.1084/jem.189.11.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen JI. 2015. Epstein-Barr virus vaccines. Clin Transl Immunology 4:e32. doi: 10.1038/cti.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]