

ABSTRACT
Numerous studies have demonstrated that CD8+ T lymphocytes suppress virus replication during human immunodeficiency virus (HIV)/simian immunodeficiency virus (SIV) infection. However, the mechanisms underlying this activity of T cells remain incompletely understood. Here, we conducted CD8+ T lymphocyte depletion in 15 rhesus macaques (RMs) infected intravenously (i.v.) with SIVmac239. At day 70 postinfection, the animals (10 progressors with high viremia and 5 controllers with low viremia) were CD8 depleted by i.v. administration of the antibody M-T807R1. As expected, CD8 depletion resulted in increased virus replication, more prominently in controllers than progressors, which correlated inversely with predepletion viremia. Of note, the feature of CD8+ T lymphocyte predepletion that correlated best with the increase in viremia postdepletion was the level of CD8+ T-bet+ lymphocytes. We next found that CD8 depletion resulted in a homogenous increase of SIV RNA in superficial and mesenteric lymph nodes, spleen, and the gastrointestinal tract of both controllers and progressors. Interestingly, the level of SIV DNA increased postdepletion in both CD4+ central memory T lymphocytes (TCM) and CD4+ effector memory T lymphocytes (TEM) in progressor RMs but decreased in the CD4+ TCM of 4 out of 5 controllers. Finally, we found that CD8 depletion is associated with a greater increase in CD4+ T lymphocyte activation (measured by Ki-67 expression) in controllers than in progressors. Overall, these data reveal a differential impact of CD8+ T lymphocyte depletion between controller and progressor SIV-infected RMs, emphasizing the complexity of the in vivo antiviral role of CD8+ T lymphocytes.
IMPORTANCE In this study, we further dissect the impact of CD8+ T lymphocytes on HIV/SIV replication during SIV infection. CD8+ T lymphocyte depletion leads to a relatively homogenous increase in viral replication in peripheral blood and tissues. CD8+ T lymphocyte depletion resulted in a more prominent increase in viral loads and CD4+ T lymphocyte activation in controllers than in progressors. Interestingly, we found T-bet expression on CD8+ T lymphocytes to be the best predictor of viral load increase following depletion. The levels of SIV DNA increase postdepletion in both CD4+ TCM and TEM in progressor RMs but decrease in the CD4+ TCM of controllers. The findings described in this study provide key insights into the differential functions of CD8+ T lymphocytes in controller and progressor RMs.
INTRODUCTION
Several lines of evidence indicate that CD8+ T lymphocytes mediate control of virus replication during both human immunodeficiency virus (HIV) infection of humans and simian immunodeficiency virus (SIV) infection of rhesus macaques (RMs). First, the postpeak decline of viremia in acute HIV infection is coincident with the expansion of HIV-specific T cells (1, 2). Second, during both acute and chronic HIV/SIV infection, immune pressure mediated by HIV/SIV-specific CD8+ T lymphocytes is manifested by viral escape mutations (3). Third, there is a clear association between the presence of certain major histocompatibility complex (MHC) class I alleles and disease progression during both HIV infection of humans and SIV infection of RMs (4, 5). Fourth, HIV-1-infected individuals with polyfunctional HIV-1-specific T cells appear to progress less rapidly than those whose T lymphocytes have more limited functionality (6). While compelling, all of these studies are correlative in nature and fail to establish a direct cause-effect relationship. In this context, the most convincing evidence for a direct effect of CD8+ T lymphocytes in suppressing virus replication came from a series of studies in which these cells were depleted in vivo in SIV-infected RMs (7–10). These studies clearly demonstrated that antibody-mediated in vivo depletion of CD8+ T lymphocytes is consistently associated with increased virus replication and faster disease progression (11). Despite this strong evidence indicating that CD8+ T lymphocytes suppress virus replication during HIV/SIV infections, these cells ultimately fail to prevent disease progression in the vast majority of HIV-infected individuals and SIV-infected RMs.
The mechanisms by which CD8+ T lymphocytes exert an antiviral effect in vivo still are incompletely understood. Conceivably, these mechanisms can be summarized by two major functions: (i) CD8+ T lymphocytes may reduce production of virions on a per-cell basis either by direct killing of infected cells or by decreasing the rate of virus production via noncytolytic mechanisms, and/or (ii) CD8+ T lymphocytes may reduce the number of productively infected cells either by inhibiting the spread of infection (i.e., via production of β-chemokines or other cytokines) or by limiting the number of targets (i.e., activated CD4+ T lymphocytes) available for infection. While the exact in vivo contribution of these nonmutually exclusive antiviral effects by CD8+ T lymphocytes has not yet been defined, there is preliminary evidence that they all are involved (12–15). Further, there are several basic aspects of how depletion of CD8+ T lymphocytes affects SIV replication that have not yet been fully elucidated. These aspects include (i) the kinetics of virus replication post-CD8 depletion in progressor (i.e., high-viremia) versus controller (i.e., low-viremia) animals; (ii) the anatomic location of productively infected cells that support increased viremia post-CD8 depletion; (iii) how CD8 depletion impacts the frequency of infected cells within the main memory CD4+ T lymphocyte subsets; (iv) the features of the SIV-specific CD8+ T lymphocyte response prior to depletion that correlate best with the increase of virus replication postdepletion; and (v) the impact of CD8+ T lymphocyte depletion on CD4+ T lymphocyte activation.
In this study, we set out to investigate these aspects of the antiviral effect of CD8+ T lymphocytes in 15 SIV-infected RMs (10 progressors and 5 controllers) that underwent CD8+ T lymphocyte depletion at day 70 postinfection. The main results of this study are that (i) CD8+ T lymphocyte depletion is followed by an increase in virus replication that was more prominent in SIV-infected controllers than in progressors, and this increase in virus replication was relatively homogenous in various lymphoid organs and tissues; (ii) the levels of T-bet in CD8+ T lymphocytes prior to depletion predicted the increase in virus replication postdepletion; (iii) the levels of SIV DNA in CD4+ central memory T lymphocytes (TCM) increased postdepletion in progressor RMs but decreased in controllers; and (iv) CD8+ T lymphocyte depletion was associated with a greater increase in CD4+ T lymphocyte activation in controllers than in progressors. We concluded that these data reveal a complex role of CD8+ T lymphocytes in controlling virus replication in SIV-infected RMs that includes significant differences between controllers and progressors.
MATERIALS AND METHODS
Animals.
Eighteen female adult rhesus macaques were infected intravenously (i.v.) with 3,000 50% tissue culture infectious doses (TCID50) of SIVmac239. All RMs were Mamu-B*08 and -B*17 negative; three Mamu-A*01-positive RMs were assigned to each experimental group, and two Mamu-A*01 RMs were assigned to the control group. Five animals each were sacrificed at day 3, day 7, and day 14 after CD8 depletion. Mock-depleted RMs were sacrificed 7 days postdepletion. Blood and tissues were collected throughout the study period, and plasma viral loads were monitored on a weekly basis. All animals were housed at the Yerkes National Primate Research Center and maintained in accordance with NIH guidelines. Studies were approved by the Emory University Institutional Animal Care and Use Committee.
CD8+ T lymphocyte depletion.
Ten weeks after SIV infection, 15 RMs were depleted of CD8+ T lymphocytes by i.v. treatment with 50 mg/kg of body weight of the rhesus recombinant monoclonal antibody M-T807R1 (National Institutes of Health Nonhuman Primate Reagent Resource). Three nondepleted control animals were mock depleted with a primatized control IgG1 OKT3 antibody reactive against the human CD3 molecule (NIH Nonhuman Primate Resource Reagent). The efficacy of the depletion in blood was determined by flow-cytometric analysis and complete blood cell counts at multiple time points after administration of the depleting reagent. Depletion efficiency in tissues was determined by flow-cytometric analysis as a fraction of CD8+ T lymphocytes from samples obtained predepletion.
Sample collection and processing.
Peripheral blood mononuclear cells (PBMC) were isolated from blood by gradient density centrifugation (Ficoll). Lymphocytes were isolated from freshly obtained lymph node and rectal biopsies by passing them through a 70-μm cell strainer and lysing red blood cells with ACK lysis buffer (Life Technologies).
Immunophenotyping, cytokine responses, and flow cytometry.
Multicolor flow-cytometric analysis was performed on lymphocytes isolated from peripheral blood and tissues according to standard procedures. The antibodies used were anti-CD4 allophycocyanin (APC)-Cy7 (clone OKT4; BioLegend), anti-CD4 Pacific blue (clone OKT4; BioLegend), anti-CD8 QDot 705 (clone 3B5; Invitrogen), anti-CD8 brilliant violet 711 (clone RPA-T8; BioLegend), Ki-67 fluorescein isothiocyanate (FITC) (clone B56; BD Biosciences), anti-CD3 Pacific blue (clone SP34-2; BD Biosciences), anti-CD3 APC-Cy7 (clone SP34-2; BD Biosciences), anti-CD95 phycoerythrin (PE)-Cy5 (clone DX2; BioLegend), anti-CCR7 (clone 3D12; BD Biosciences), anti-CCR5 (clone 3A9; BD Biosciences), anti-CD28 ECD (clone 28.2; Beckman Coulter), anti-CD28 PE-Cy7 (clone 28.2; eBioscience), anti-CD16 Alexa 700 (clone 3G8; BioLegend), anti-CD56 QDot 605 (clone MEM-188; Invitrogen), anti-CD20 eFluor 650 (clone 2H7; eBioscience), anti-CD62L PE (clone Sk11; BD Biosciences), anti-interleukin-2 (IL-2) (clone RAT; Life Technologies), anti-MIP-1α PE (clone D21; Fisher), anti-MIP-1β PE (clone 11A3; Fisher), gamma interferon (IFN-γ) APC (clone B27; BD Pharmingen), and tumor necrosis factor alpha (TNF-α) Alexa 700 (clone MAb 11; BD Pharmingen). Samples assessed for Ki-67 expression were surface stained first with the appropriate antibodies and then fixed and permeabilized using BD Perm 2 (BD Pharmingen) and stained intracellularly with Ki-67. Cytokine staining was performed on frozen PBMC after stimulation with pools of 15-mer peptides spanning the sequences of three major antigenic proteins of SIVmac239 (gag, pol, and env) as described previously (16). Flow cytometry data were acquired and samples were analyzed on an LSRII flow cytometer driven by the BD FACSDiva software package (version 6.1.3; BD Biosciences). Analyses of the acquired data were performed using FlowJo software (version 9.6.3; Tree Star). Further statistical analyses were performed using Prism 5.0 and Excel (Microsoft Office 2011) software.
Plasma viral loads.
The quantitative real-time reverse transcriptase PCR (RT-PCR) assay to determine SIVmac239 plasma viral load was performed as previously described (17).
Sorting of CD4+ T lymphocyte subsets.
Freshly isolated PBMC were stained with anti-CD4 Pacific blue (clone OKT4; BioLegend), anti-CD3 APC-Cy7 (clone SP34-2; BD Biosciences), anti-CD95 PE-Cy5 (clone DX2; BioLegend), anti-CD28 PE-Cy7 (clone 28.2; eBioscience), and anti-CD62L PE (clone Sk11; BD Biosciences). CD3+ CD4+ T lymphocytes were characterized as either naive (TN; CD28+ CD95− CD62L+), central memory (TCM; CD28+/− CD95+ CD62L+), or effector memory (TEM; CD28+/− CD95+ CD62L−) and sorted on a FACSAria III cell sorter (BD Biosciences).
Quantitative PCR for SIV gag DNA.
Quantification of SIVmac gag DNA from sorted naive, central memory, and effector memory CD4+ T lymphocytes was performed as previously described (18). Simultaneous PCR was performed for monkey albumin gene copy number and used to determine cell number quantifications. Samples with undetectable SIV DNA were assigned a level of half of the lower limit of detection for graphical purposes and statistical analysis.
SIV in situ hybridization and quantitative image analysis.
SIV in situ hybridization was performed on 5-μm tissue sections with SIV-digoxigenin-labeled antisense riboprobes as previously described (19). Quantification of SIV RNA-positive cells was performed by counting positive cells detected by nitrotetrazolium blue–5-bromo-4-chloro-3-indolylphosphate (purple-black color) in 10 randomly selected fields totaling a minimum of 120 mm2.
Statistical analyses.
Statistical analyses were conducted using GraphPad Prism 5.0. Kruskal-Wallis one-way analysis of variance tests were performed to determine differences between groups in all of the tissues (see Fig. 2). Mann-Whitney U tests were used to perform analyses between pre- and postdepletion frequencies (see Fig. 2, 4, and 5). Spearman rank correlation tests were used to analyze all correlations (see Fig. 2, 3, and 4). All P values less than 0.05 were defined as significant.
FIG 2.

Plasma and tissue viral loads following CD8+ T lymphocyte depletion. Plasma viral load measured longitudinally for each animal. The dotted black line indicates the administration of depleting antibody. The shaded gray area indicates postdepletion time points. (B) Correlation between fold change in viral load (viral load postdepletion/viral load predepletion) with predepletion viral load for all experimentally depleted animals. (C) Number of SIV RNA+ cells within tissues of CD8-depleted controllers (n = 5), progressors (n = 10), and mock-depleted animals (n = 3). Statistical analyses were performed using Spearman rank correlation tests, Kruskal-Wallis one-way analysis of variance, and Mann-Whitney U tests.
FIG 4.

SIV DNA in CD4+ TN, TCM, and TEM increases postdepletion in progressor but not controller RMs. (A) Comparison of cell-associated SIV DNA levels in naive, central memory, and effector memory CD4+ T lymphocytes of controllers (n = 5) and progressors (n = 10) pre- and postdepletion. (B) Correlations between plasma viral load and cell-associated SIV DNA within TEM (B and C) and TCM (D and E) at predepletion and postdepletion time points. Statistical analyses were performed using Mann-Whitney U tests, and Spearman rank correlation tests were used to analyze all correlations. ns, not significant.
FIG 5.
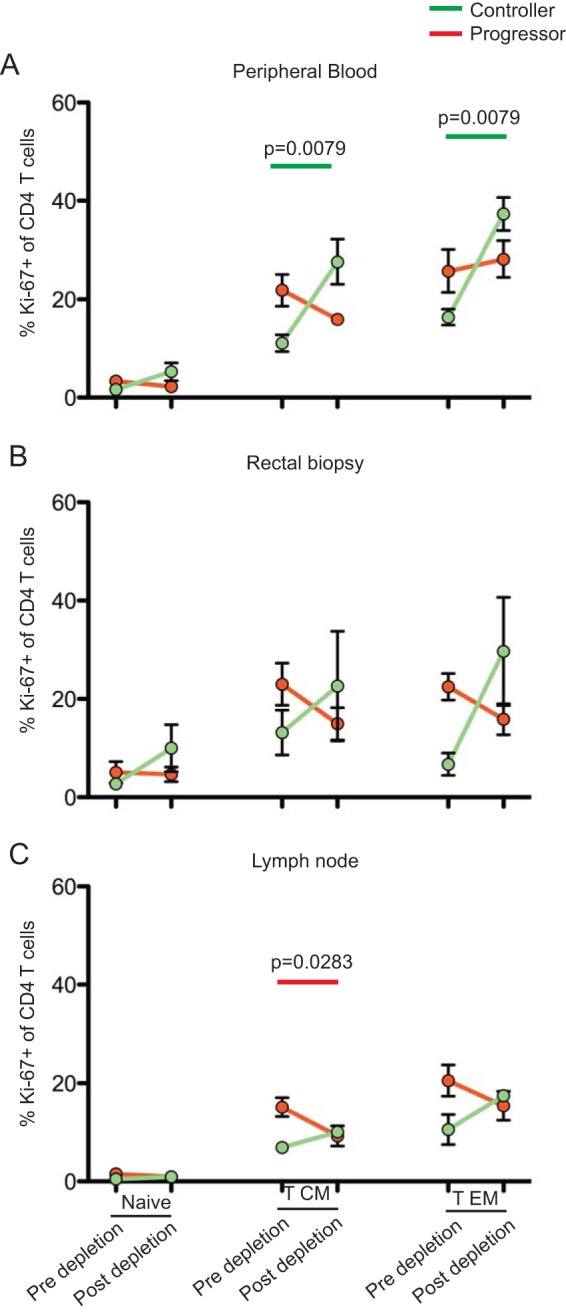
CD8+ T lymphocyte depletion is associated with greater increases in CD4+ T lymphocyte activation in controllers than in progressors. Comparative frequency of Ki-67+ CD4+ T lymphocytes within naive, central memory, and effector memory CD4+ T lymphocytes pre- and postdepletion isolated from the peripheral blood (A), rectal mucosa (B), and lymph nodes (C) of progressors (n = 10) and controllers (n = 5). Statistical analyses were performed using Mann-Whitney U tests.
FIG 3.

T-bet expression in CD8+ T lymphocytes is the best correlate of the level of viral load after CD8 depletion. Correlations between T-bet-expressing CD8+ T lymphocytes isolated predepletion from lymph nodes (A) and rectal biopsy specimens (B) with plasma viral load at necropsy. Correlations between T-bet expression in NK cells, defined as CD3− CD16+ CD8+ cells, from lymph nodes (C) and Ki-67 expression in NK cells from whole blood (D) with plasma viral load at necropsy. (E and F) Levels of expression of several markers on CD8+ T lymphocytes and NK cells were assessed within peripheral blood, lymph node, and rectal mucosa. None were found to correlate with plasma viral loads after CD8+ T lymphocyte depletion. Controllers are shown in green, and progressors are in red. Spearman rank correlation tests were used to analyze all correlations.
RESULTS
Experimental design.
To investigate the in vivo role of CD8+ T lymphocytes as mediators of antiviral immune responses during SIVmac239 infection of RMs, we depleted these cells from a cohort of 15 adult Indian RMs. All animals were infected intravenously with 3,000 TCID50 of SIVmac239, and CD8+ T lymphocytes were depleted by i.v. administration of the primatized monoclonal antibody M-T807R1 at day 70 postinfection. Ten RMs were defined as progressors, with high viremia at the time of CD8 depletion, and 5 RMs were defined as controllers due to low viral loads predepletion. The study also included three control SIV-infected, mock-depleted animals. In all animals, several tissues, including blood, bone marrow (BM), lymph nodes (LN), and rectal mucosa via rectal biopsy specimen (RB), were sampled at multiple time points throughout the study (Fig. 1A). The SIV-infected CD8-depleted RMs then underwent selective necropsy at days 3 to 14 after depletion to extensively investigate the effect of this procedure in various lymphoid tissues and cell subsets. As shown in Fig. 1B and C for two representative RMs and for the whole group of animals, respectively, treatment with M-T807R1 resulted in depletion of >99% of circulating CD8+ T lymphocytes. As expected based on previous studies, the level of depletion was lower in tissues than in peripheral blood and ranged between 50 and 70% in lymph nodes and between 86 and 96% in the rectal mucosa (data not shown). Depletion of CD8+ lymphocytes is not complete in all tissues, possibly due to the differential rate of clearance of cells (20). However, since the CD8 molecule is critical for the function of MHC class I recognition, CD8+ lymphocytes are functionally impaired following binding of the depleting CD8 antibody (21). In addition to depleting CD8+ T lymphocytes, administration of CD8-depleting antibody also depletes NK and NKT cells that express high levels of the CD8 molecule (12).
FIG 1.

Experimental design and effective depletion of CD8+ T lymphocytes. (A) Study design depicting timeline of SIV infection, antibody-mediated CD8 depletion, and necropsy. Eighteen female rhesus macaques were intravenously infected with 3,000 TCID50 of SIVmac239. CD8-depleting antibody (M-T807R1) was administered at 70 days postinfection to 15 RMs. Five CD8-depleted animals each were necropsied 3, 7, and 14 days postdepletion. Three RMs were mock depleted using primatized control IgG1 antibody at 70 days postinfection and necropsied 7 days after mock depletion. (B) Representative flow cytometry plots of live CD3+ T lymphocytes isolated from peripheral blood depicting CD8+ T lymphocyte levels predepletion (5 days before depletion) and postdepletion (6 days after depletion). (C) Longitudinal assessment of CD8+ T lymphocyte frequencies (as a percentage of CD3+ T cells) in peripheral blood for all experimentally depleted (black; n = 15) and mock-depleted (purple; n = 3) animals at pre- and postdepletion (shaded gray) time points. The dotted black line indicates the administration of depleting antibody.
CD8+ T lymphocyte depletion results in a greater increase in SIV viremia in controllers than in progressors.
The 15 SIV-infected RMs that were part of this study included 10 normal progressors, with viral loads prior to CD8+ T lymphocyte depletion ranging between 22,200 and 17,200,000 SIV RNA copies/ml of plasma, and five controllers, with viral loads prior to CD8+ T lymphocyte depletion ranging between 50 and 1,300 SIV RNA copies/ml of plasma (Fig. 2A). Consistently with previous studies, treatment with the CD8-depleting antibody resulted in an increase in viral loads in both groups of animals, while, as expected, no change in the level of viremia was observed in the three animals that were mock depleted. Interestingly, the increase in viral load was more pronounced in the group of controllers than in the progressor RMs, with an average fold increase of 154 in progressors and 5,018 in controllers. At necropsy, however, the viral load of progressor RMs remained higher than that of controllers (P = 0.0013) (Fig. 2A). In addition, as shown in Fig. 2B, we found a significant inverse correlation between baseline viremia and the fold change in viremia after CD8 depletion (P = 0.0004). Overall, these data are consistent with the established hypothesis that CD8+ T lymphocytes exhibit better suppression of virus replication in controller SIV-infected RMs than in progressors.
CD8+ T lymphocyte depletion results in a homogenous increase in virus production within lymphoid tissues.
While several studies have shown that CD8+ T lymphocyte depletion in SIV-infected RMs results in increased virus replication (6–9), very few data are available with respect to the specific effect of this treatment in different anatomic sites. In this study, we examined the levels of SIV RNA in five different tissues at necropsy, including mesenteric LN, superficial LN, lamina propria of the rectum, lymphoid aggregates in the rectum, and the spleen. As these tissues could not be collected longitudinally in the animals, we used the three mock-depleted animals as controls. As shown in Fig. 2C, the levels of SIV RNA were consistently higher in CD8-depleted progressor RMs than in mock-depleted animals, and this effect was most pronounced in the spleen, mesenteric LN, and lymphoid aggregates of the rectum. Controller RMs also exhibited a nonsignificant trend toward higher levels of virus replication than mock-depleted animals. Interestingly, the levels of SIV RNA after CD8 depletion were similar between controller and progressor RMs in lamina propria of the rectum, while higher levels of virus replication were seen at all other sites examined in progressors compared to controllers. Of note, SIV RNA was found in both follicles and paracortex (i.e., T lymphocyte area) in the lymph nodes of CD8+ lymphocyte-depleted controller RMs, similar to the pattern observed in CD8+ lymphocyte-depleted progressors (data not shown). This observation is consistent with two recent studies linking the controller status with a compartmentalization of SIV in the lymph node follicles (22, 23). Overall, these histological data suggest that CD8+ T lymphocyte depletion is followed by a diffuse increase of SIV replication in several lymphoid organs and tissues.
T-bet expression in CD8+ T lymphocytes is the best correlate of the level of viral load postdepletion.
Relatively little is known regarding the specific feature(s) of total and/or SIV-specific CD8+ T lymphocyte responses that best correlate with the level of viral rebound after CD8 depletion. To address this issue, we examined a large number of CD8+ T lymphocyte features in our cohort of SIV-infected progressor and controller RMs and investigated any potential correlation with the observed changes in viremia postdepletion in blood and tissues Specifically, we measured the level of Gag-specific cells by tetramer staining in blood, lymph nodes, and rectum; levels of cells producing IFN-γ, TNF, IL-2, or MIP-1β after Gag-peptide stimulation; and the level of CD8+ T lymphocytes expressing Ki-67, T-bet, or Eomesodermin. As shown in Fig. 3, the only significant correlation that we observed was between the levels of T-bet expression in CD8+ T lymphocytes isolated from both lymph nodes and rectal biopsies before CD8+ T lymphocyte depletion and the level of plasma viremia after depletion (Fig. 3A and B). The list of CD8+ T lymphocyte features examined before CD8+ T lymphocyte depletion that did not correlate with the level of viremia postdepletion is shown in Fig. 3E. Taken together, these data suggest a role for the expression of the transcription factor T-bet in CD8+ T lymphocytes of SIV-infected RMs as a predictor of the increase in viremia that follows CD8+ T lymphocyte depletion in these animals. Since the CD8-depleting antibody also leads to depletion of NK cells, we assessed expression of various immunological markers on CD3− CD8+ NK cells. We found that both T-bet expression on NK cells in lymph nodes and Ki-67 expression on NK cells derived from whole blood correlated with postdepletion viral load (Fig. 3C and D). No other correlations between markers of NK cell activation/differentiation and postdepletion viral load were found (Fig. 3F).
SIV DNA in CD4+ TN, TCM, and TEM increases postdepletion in progressor RMs but not in controllers.
HIV/SIV replication occurs at different levels in specific CD4+ T lymphocyte subsets (24, 25). Relatively little is known, however, about the role played by CD8+ T lymphocytes in controlling virus replication in the subsets of naive, central memory, and effector memory CD4+ T lymphocytes (TN, TCM, and TEM, respectively). To address this question, we sorted CD4+ TN, TCM, and TEM, as determined based on the expression of CD28, CD95, and CD62L, before and after CD8+ T lymphocyte depletion in the 15 SIV-infected RMs included in this study, and measured the level of cell-associated SIV DNA by RT-PCR. As shown in Fig. 4A, we found that the levels of SIV DNA increased almost uniformly in CD4+ TN (9/10 animals), TCM (10/10), and TEM (10/10) sorted from progressors, while in controllers the levels of SIV DNA increased in CD4+ TEM of 4/5 RMs but in only 2/5 and 1/5 RMs for the TN and TCM subsets, respectively. Of note, we found a significant correlation between SIV plasma viremia and the levels of SIV DNA in CD4+ TEM before and after CD8+ T lymphocyte depletion (Fig. 4B and C) and between plasma viremia and SIV DNA in CD4+ TCM after CD8+ depletion (Fig. 4E). However, no correlation was found between SIV viremia and SIV DNA levels in CD4+ TCM before CD8+ depletion (Fig. 4D). Overall, these data are consistent with the possibility that CD8+ T lymphocyte-mediated control of infection operates through different mechanisms in specific CD4+ T lymphocyte subsets.
CD8+ lymphocyte depletion is associated with a greater increase in CD4+ T cell activation in controllers than in progressors.
The increase in virus replication that follows CD8+ lymphocyte depletion in SIV-infected RMs may be, at least in part, the result of increased levels of CD4+ T cell activation that can be caused by factors such as the homeostatic response to CD8+ T cell depletion, increased availability of proliferative cytokines, and reactivation of latent viruses, such as cytomegalovirus (CMV). To address the potential role of CD4+ T cell activation in determining the levels of virus replication before and after CD8+ lymphocyte depletion, we measured the expression of the proliferation marker Ki-67 in CD4+ TN, TCM, and TEM isolated from the blood, lymph nodes, and recta (biopsy samples) of our cohort of 15 SIV-infected RMs (Fig. 5). As shown in Fig. 5, we found that Ki-67 expression remained stable overall in CD4+ T cells of progressors (and, in fact, even decreased in LN-derived TCM), consistent with the fact that, in these animals, the levels of CD4+ T cell activation was already very high prior to CD8 depletion. In contrast, the level of Ki-67 expression increased in controllers, with a statistically significant difference for both TCM and TEM in peripheral blood and a similar, albeit nonsignificant, trend in lymph node and rectal biopsy specimens. Overall, these data indicate that CD8+ T lymphocyte depletion is followed by a more pronounced increase in CD4+ T cell activation in controller SIV-infected RMs than in progressors.
DISCUSSION
There are many lines of scientific evidence supporting the role of CD8+ T lymphocytes in controlling virus replication in the setting of HIV or SIV infection. Perhaps the most convincing observation is that in vivo experimental depletion of CD8+ T cells in SIV-infected macaques is consistently followed by a significant increase in the level of virus replication. While this observation has been confirmed in numerous studies (8, 12), there are several aspects of this phenomenon that remain relatively poorly understood. For example, there is a lack of evidence regarding (i) the cellular and anatomic origin of the increased virus replication, (ii) the contribution of cytolytic versus noncytolytic mechanisms of virus control (13, 15), (iii) the role played by CD8+ T lymphocytes versus CD8+ NK cells, which also are depleted by this treatment, and (iv) the contribution of the enhanced activation of CD4+ T lymphocytes that follows CD8+ T lymphocyte depletion in promoting higher levels of virus replication (11, 26). The current study was aimed at providing some insights into these poorly understood aspects of the antiviral role of CD8+ T lymphocytes during HIV/SIV infections.
The main findings of this study are that (i) CD8+ T lymphocyte depletion was followed by an increase in virus replication that was more prominent in SIV-infected controller than progressor RMs and was relatively homogenous in various lymphoid organs and tissues, (ii) the levels of T-bet expression in CD8+ T lymphocytes prior to depletion was the best predictor of the magnitude of the increase of virus replication postdepletion, (iii) the levels of SIV DNA after CD8+ T lymphocyte depletion increased in all CD4+ T lymphocyte subsets (i.e., TN, TCM, and TEM) in progressors but only in TEM in controllers, with a decline of SIV DNA in TCM of 4 out of 5 animals, and (iv) CD8+ T lymphocyte depletion was associated with a greater increase in CD4+ T lymphocyte activation in controllers than in progressors. While some of these results overall are confirmatory of previously published work by us and others, several of the above-described findings are novel and deserve particular attention, especially regarding the differential role of CD8+ T lymphocytes in controller versus progressor SIV-infected RMs.
The observation that the increase in viral load following CD8+ T lymphocyte depletion is more pronounced when virus replication is lower prior to depletion is consistent with what was observed in two previous studies (8, 10). In this study, however, we report for the first time a strong inverse correlation between predepletion viremia and fold change in viremia postdepletion. Overall, these data are quite consistent with the hypothesis that CD8+ T lymphocytes are key contributors to the suppression of virus replication that is observed in HIV/SIV-infected elite controllers (both humans and rhesus macaques) and is associated with specific MHC class I alleles (4, 27). CD8+ T lymphocytes contribute less to control in progressors and we see relatively little change in viral load in progressors following CD8+ T lymphocyte depletion, as has been described before (28). Interestingly, a detailed histological analysis of several lymphoid tissues and organs revealed that the frequency of virus-producing cells (i.e., SIV RNA positive by in situ hybridization) increases after CD8+ T lymphocyte depletion in a relatively uniform fashion in all examined tissues, with virus found in both B and T cell areas of lymph nodes in the CD8-depleted controllers. The latter observation is consistent with two recent studies showing a specific virus compartmentalization within lymph nodes of controller versus progressor SIV-infected RMs (22, 23).
While numerous studies have linked CD8+ T lymphocyte depletion with increased virus replication (7–12), it is not clear what particular function or phenotypic marker of CD8+ T lymphocytes is best correlated with this effect. In this study, we investigated potential correlations between a number of specific aspects of the CD8+ T lymphocytes, both SIV specific and as bulk populations, prior to CD8+ T lymphocyte depletion and the observed changes in viral load following depletion. We found that expression of the transcription factor T-bet in bulk CD8+ T cells was the only predictor of the levels of viral load after CD8+ T lymphocyte depletion. We did not find any correlation between the level of T-bet expression on SIV-specific CD8+ T lymphocytes and plasma viral load increase. This is a novel finding that is consistent with a possible role of T-bet as a transcription factor that promotes effector CD8+ T lymphocyte functions, including cytotoxic activity and production of multiple cytokines (29, 30), and that represses the expression of the inhibitory receptor PD-1, which suppresses many CD8+ T lymphocyte functions (31), and it has been associated with in vivo control of virus replication in HIV-infected humans (32, 33). The high predepletion levels of T-bet expression in SIV-infected progressors may reflect the high levels of activation and antigenic load which resulted from the poor control of virus occurring in progressors. Further studies involving in vivo blockade of T-bet-expressing CD8+ T lymphocytes would be needed to further delineate the role of T-bet in controlling virus replication in SIV-infected RMs, particularly with respect to whether the levels of T-bet expression in CD8+ T lymphocytes represent a cause or a consequence of prevailing levels of virus replication.
An intriguing novel finding of this study is that the impacts of CD8+ T lymphocyte depletion on the relative proportion of SIV DNA-positive cells within the memory subsets of CD4+ T lymphocytes (TCM and TEM) appear to be different in SIV-infected progressor and controller RMs. As expected, CD8 depletion resulted in increased levels of viral DNA in all CD4+ T lymphocyte compartments of progressors. However, in CD8-depleted controllers, the level of SIV DNA increased only in TEM; in fact, it decreased in TCM of 4 out of 5 animals. Remarkably, this decline of the frequency of SIV-infected CD4+ TCM occurred concomitantly with an increase in plasma viremia. Importantly, this decline in SIV DNA in CD4+ TCM does not appear to be due to trafficking of this cell subset into lymph nodes or the gastrointestinal tract, as the same decline was seen at these sites as well as in peripheral blood. At this time we have not identified a mechanism for this surprising finding, but it is tempting to speculate that, in CD8-depleted SIV-infected controllers, a substantial proportion of SIV-infected TCM differentiated to become TEM, perhaps as a result of the increased CD4+ T lymphocyte activation, or was killed by the reactivated virus in the absence of CD8+ T lymphocytes. The latter possibility would be consistent with the proposed hypothesis that CD8+ T lymphocytes suppress HIV/SIV replication through noncytolytic mechanisms that act at the level of provirus transcription (34).
Several studies have shown that CD8+ T lymphocyte depletion is followed by an increased fraction of activated/proliferating CD4+ T lymphocytes (11, 26). While the relative contribution of this phenomenon to the observed increase in viremia after CD8+ depletion remains unclear, two findings suggest that its role is in fact minor. First, the increased level of CD4+ T lymphocyte activation is observed for the most part at a time when the increase in virus replication has already occurred (12). Second, when the increase of CD4+ T lymphocyte activation following CD8+ T lymphocyte depletion is abrogated through blockade of IL-15 signaling, the increase in SIV replication still is present (11). In the current study, we found that the increase of CD4+ T lymphocyte activation, as measured by the proliferation marker Ki-67, was more pronounced in CD8-depleted controllers than progressors, indicating that, in SIV-infected RMs with high virus replication prior to CD8+ T lymphocyte depletion, a maximal level of CD4+ T lymphocyte activation was present already in the immune system of these animals.
Overall, the current set of results indicates that experimental depletion of CD8+ T lymphocytes during SIV infection of macaques remains a valuable research tool to investigate the mechanisms by which these cells suppress virus replication in vivo. Specifically, we have identified a series of differences between SIV-infected progressor and controller RMs with respect to the impact of CD8 depletion on the virology and immunology of SIV infection that provides novel insights into the in vivo function of these cells. Ultimately, a better understanding of the mechanisms responsible for the CD8+ T lymphocyte-mediated protection from HIV/SIV replication may result in interventions that most effectively harness these antiviral functions in the setting of prophylactic and therapeutic vaccines for HIV/AIDS.
ACKNOWLEDGMENTS
This work was supported primarily by grant R01-AI090797 to G.S. In addition, it was supported by the NIH/NCRR (grant P51RR000165) and currently is supported by the Office of Research Infrastructure Programs/OD (grant P51OD011132) to the Yerkes National Primate Research Center. A.C. gratefully acknowledges partial support by the American Foundation for AIDS Research (grant 108905-56-RGRL) and by an NICHD Child Health Research Career Development Award (K12 HD072245). This project has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract no. HHSN261200800001E.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
We thank the Emory Center for AIDS Research (CFAR) Virology Core for their technical support.
REFERENCES
- 1.Koup RA, Safrit JT, Cao Y, Andrews CA, McLeod G, Borkowsky W, Farthing C, Ho DD. 1994. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol 68:4650–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borrow P, Lewicki H, Hahn BH, Shaw GM, Oldstone MB. 1994. Virus-specific CD8+ cytotoxic T lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J Virol 68:6103–6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goulder PJ, Watkins DI. 2004. HIV and SIV CTL escape: implications for vaccine design. Nat Rev Immunol 4:630–640. doi: 10.1038/nri1417. [DOI] [PubMed] [Google Scholar]
- 4.Carrington M, O'Brien SJ. 2003. The influence of HLA genotype on AIDS. Annu Rev Med 54:535–551. doi: 10.1146/annurev.med.54.101601.152346. [DOI] [PubMed] [Google Scholar]
- 5.Goulder PJ, Watkins DI. 2008. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat Rev Immunol 8:619–630. doi: 10.1038/nri2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, Lederman MM, Benito JM, Goepfert PA, Connors M, Roederer M, Koup RA. 2006. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 107:4781–4789. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, Lifton MA, Racz P, Tenner-Racz K, Dalesandro M, Scallon BJ, Ghrayeb J, Forman MA, Montefiori DC, Rieber EP, Letvin NL, Reimann KA. 1999. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283:857–860. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- 8.Jin X, Bauer DE, Tuttleton SE, Lewin S, Gettie A, Blanchard J, Irwin CE, Safrit JT, Mittler J, Weinberger L, Kostrikis LG, Zhang L, Perelson AS, Ho DD. 1999. Dramatic rise in plasma viremia after CD8(+) T cell depletion in simian immunodeficiency virus-infected macaques. J Exp Med 189:991–998. doi: 10.1084/jem.189.6.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matano T, Shibata R, Siemon C, Connors M, Lane HC, Martin MA. 1998. Administration of an anti-CD8 monoclonal antibody interferes with the clearance of chimeric simian/human immunodeficiency virus during primary infections of rhesus macaques. J Virol 72:164–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lifson JD, Rossio JL, Piatak M Jr, Parks T, Li L, Kiser R, Coalter V, Fisher B, Flynn BM, Czajak S, Hirsch VM, Reimann KA, Schmitz JE, Ghrayeb J, Bischofberger N, Nowak MA, Desrosiers RC, Wodarz D. 2001. Role of CD8(+) lymphocytes in control of simian immunodeficiency virus infection and resistance to rechallenge after transient early antiretroviral treatment. J Virol 75:10187–10199. doi: 10.1128/JVI.75.21.10187-10199.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okoye A, Park H, Rohankhedkar M, Coyne-Johnson L, Lum R, Walker JM, Planer SL, Legasse AW, Sylwester AW, Piatak M Jr, Lifson JD, Sodora DL, Villinger F, Axthelm MK, Schmitz JE, Picker LJ. 2009. Profound CD4+/CCR5+ T cell expansion is induced by CD8+ lymphocyte depletion but does not account for accelerated SIV pathogenesis. J Exp Med 206:1575–1588. doi: 10.1084/jem.20090356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klatt NR, Shudo E, Ortiz AM, Engram JC, Paiardini M, Lawson B, Miller MD, Else J, Pandrea I, Estes JD, Apetrei C, Schmitz JE, Ribeiro RM, Perelson AS, Silvestri G. 2010. CD8+ lymphocytes control viral replication in SIVmac239-infected rhesus macaques without decreasing the lifespan of productively infected cells. PLoS Pathog 6:e1000747. doi: 10.1371/journal.ppat.1000747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong JK, Strain MC, Porrata R, Reay E, Sankaran-Walters S, Ignacio CC, Russell T, Pillai SK, Looney DJ, Dandekar S. 2010. In vivo CD8+ T-cell suppression of SIV viremia is not mediated by CTL clearance of productively infected cells. PLoS Pathog 6:e1000748. doi: 10.1371/journal.ppat.1000748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elemans M, Seich Al Basatena NK, Klatt NR, Gkekas C, Silvestri G, Asquith B. 2011. Why don't CD8+ T cells reduce the lifespan of SIV-infected cells in vivo? PLoS Comput Biol 7:e1002200. doi: 10.1371/journal.pcbi.1002200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seich Al Basatena NK, Chatzimichalis K, Graw F, Frost SD, Regoes RR, Asquith B. 2013. Can non-lytic CD8+ T cells drive HIV-1 escape? PLoS Pathog 9:e1003656. doi: 10.1371/journal.ppat.1003656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunham R, Pagliardini P, Gordon S, Sumpter B, Engram J, Moanna A, Paiardini M, Mandl JN, Lawson B, Garg S, McClure HM, Xu YX, Ibegbu C, Easley K, Katz N, Pandrea I, Apetrei C, Sodora DL, Staprans SI, Feinberg MB, Silvestri G. 2006. The AIDS resistance of naturally SIV-infected sooty mangabeys is independent of cellular immunity to the virus. Blood 108:209–217. doi: 10.1182/blood-2005-12-4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garber DA, Silvestri G, Barry AP, Fedanov A, Kozyr N, McClure H, Montefiori DC, Larsen CP, Altman JD, Staprans SI, Feinberg MB. 2004. Blockade of T cell costimulation reveals interrelated actions of CD4+ and CD8+ T cells in control of SIV replication. J Clin Investig 113:836–845. doi: 10.1172/JCI19442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chahroudi A, Cartwright E, Lee ST, Mavigner M, Carnathan DG, Lawson B, Carnathan PM, Hashempoor T, Murphy MK, Meeker T, Ehnert S, Souder C, Else JG, Cohen J, Collman RG, Vanderford TH, Permar SR, Derdeyn CA, Villinger F, Silvestri G. 2014. Target cell availability, rather than breast milk factors, dictates mother-to-infant transmission of SIV in sooty mangabeys and rhesus macaques. PLoS Pathog 10:e1003958. doi: 10.1371/journal.ppat.1003958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Micci L, Alvarez X, Iriele RI, Ortiz AM, Ryan ES, McGary CS, Deleage C, McAtee BB, He T, Apetrei C, Easley K, Pahwa S, Collman RG, Derdeyn CA, Davenport MP, Estes JD, Silvestri G, Lackner AA, Paiardini M. 2014. CD4 depletion in SIV-infected macaques results in macrophage and microglia infection with rapid turnover of infected cells. PLoS Pathog 10:e1004467. doi: 10.1371/journal.ppat.1004467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Veazey RS, Acierno PM, McEvers KJ, Baumeister SH, Foster GJ, Rett MD, Newberg MH, Kuroda MJ, Williams K, Kim EY, Wolinsky SM, Rieber EP, Piatak M Jr, Lifson JD, Montefiori DC, Brown CR, Hirsch VM, Schmitz JE. 2008. Increased loss of CCR5+ CD45RA− CD4+ T cells in CD8+ lymphocyte-depleted simian immunodeficiency virus-infected rhesus monkeys. J Virol 82:5618–5630. doi: 10.1128/JVI.02748-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmitz JE, Simon MA, Kuroda MJ, Lifton MA, Ollert MW, Vogel CW, Racz P, Tenner-Racz K, Scallon BJ, Dalesandro M, Ghrayeb J, Rieber EP, Sasseville VG, Reimann KA. 1999. A nonhuman primate model for the selective elimination of CD8+ lymphocytes using a mouse-human chimeric monoclonal antibody. Am J Pathol 154:1923–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Connick E, Folkvord JM, Lind KT, Rakasz EG, Miles B, Wilson NA, Santiago ML, Schmitt K, Stephens EB, Kim HO, Wagstaff R, Li S, Abdelaal HM, Kemp N, Watkins DI, MaWhinney S, Skinner PJ. 2014. Compartmentalization of simian immunodeficiency virus replication within secondary lymphoid tissues of rhesus macaques is linked to disease stage and inversely related to localization of virus-specific CTL. J Immunol 193:5613–5625. doi: 10.4049/jimmunol.1401161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukazawa Y, Lum R, Okoye AA, Park H, Matsuda K, Bae JY, Hagen SI, Shoemaker R, Deleage C, Lucero C, Morcock D, Swanson T, Legasse AW, Axthelm MK, Hesselgesser J, Geleziunas R, Hirsch VM, Edlefsen PT, Piatak M Jr, Estes JD, Lifson JD, Picker LJ. 2015. B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat Med 21:132–139. doi: 10.1038/nm.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paiardini M, Cervasi B, Reyes-Aviles E, Micci L, Ortiz AM, Chahroudi A, Vinton C, Gordon SN, Bosinger SE, Francella N, Hallberg PL, Cramer E, Schlub T, Chan ML, Riddick NE, Collman RG, Apetrei C, Pandrea I, Else J, Munch J, Kirchhoff F, Davenport MP, Brenchley JM, Silvestri G. 2011. Low levels of SIV infection in sooty mangabey central memory CD(4)(+) T cells are associated with limited CCR5 expression. Nat Med 17:830–836. doi: 10.1038/nm.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brenchley JM, Vinton C, Tabb B, Hao XP, Connick E, Paiardini M, Lifson JD, Silvestri G, Estes JD. 2012. Differential infection patterns of CD4+ T cells and lymphoid tissue viral burden distinguish progressive and nonprogressive lentiviral infections. Blood 120:4172–4181. doi: 10.1182/blood-2012-06-437608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mueller YM, Do DH, Boyer JD, Kader M, Mattapallil JJ, Lewis MG, Weiner DB, Katsikis PD. 2009. CD8+ cell depletion of SHIV89.6P-infected macaques induces CD4+ T cell proliferation that contributes to increased viral loads. J Immunol 183:5006–5012. doi: 10.4049/jimmunol.0900141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teixeira SL, de Sa NB, Campos DP, Coelho AB, Guimaraes ML, Leite TC, Veloso VG, Morgado MG. 2014. Association of the HLA-B*52 allele with non-progression to AIDS in Brazilian HIV-1-infected individuals. Genes Immun 15:256–262. doi: 10.1038/gene.2014.14. [DOI] [PubMed] [Google Scholar]
- 28.Kim EY, Veazey RS, Zahn R, McEvers KJ, Baumeister SH, Foster GJ, Rett MD, Newberg MH, Kuroda MJ, Rieber EP, Piatak M Jr, Lifson JD, Letvin NL, Wolinsky SM, Schmitz JE. 2008. Contribution of CD8+ T cells to containment of viral replication and emergence of mutations in Mamu-A*01-restricted epitopes in simian immunodeficiency virus-infected rhesus monkeys. J Virol 82:5631–5635. doi: 10.1128/JVI.02749-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sullivan BM, Juedes A, Szabo SJ, von Herrath M, Glimcher LH. 2003. Antigen-driven effector CD8 T cell function regulated by T-bet. Proc Natl Acad Sci U S A 100:15818–15823. doi: 10.1073/pnas.2636938100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jenner RG, Townsend MJ, Jackson I, Sun K, Bouwman RD, Young RA, Glimcher LH, Lord GM. 2009. The transcription factors T-bet and GATA-3 control alternative pathways of T-cell differentiation through a shared set of target genes. Proc Natl Acad Sci U S A 106:17876–17881. doi: 10.1073/pnas.0909357106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali MA, Intlekofer AM, Boss JM, Reiner SL, Weinmann AS, Wherry EJ. 2011. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat Immunol 12:663–671. doi: 10.1038/ni.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buggert M, Tauriainen J, Yamamoto T, Frederiksen J, Ivarsson MA, Michaelsson J, Lund O, Hejdeman B, Jansson M, Sonnerborg A, Koup RA, Betts MR, Karlsson AC. 2014. T-bet and Eomes are differentially linked to the exhausted phenotype of CD8+ T cells in HIV infection. PLoS Pathog 10:e1004251. doi: 10.1371/journal.ppat.1004251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hersperger AR, Martin JN, Shin LY, Sheth PM, Kovacs CM, Cosma GL, Makedonas G, Pereyra F, Walker BD, Kaul R, Deeks SG, Betts MR. 2011. Increased HIV-specific CD8+ T-cell cytotoxic potential in HIV elite controllers is associated with T-bet expression. Blood 117:3799–3808. doi: 10.1182/blood-2010-12-322727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mackewicz CE, Blackbourn DJ, Levy JA. 1995. CD8+ T cells suppress human immunodeficiency virus replication by inhibiting viral transcription. Proc Natl Acad Sci U S A 92:2308–2312. doi: 10.1073/pnas.92.6.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]