

ABSTRACT
Infection of the lower respiratory tract by influenza A viruses results in increases in inflammation and immune cell infiltration in the lung. The dynamic relationships among the lung microenvironments, the lung, and systemic host responses during infection remain poorly understood. Here we used extensive systematic histological analysis coupled with live imaging to gain access to these relationships in ferrets infected with the 2009 H1N1 pandemic influenza A virus (H1N1pdm virus). Neutrophil levels rose in the lungs of H1N1pdm virus-infected ferrets 6 h postinfection and became concentrated at areas of the H1N1pdm virus-infected bronchiolar epithelium by 1 day postinfection (dpi). In addition, neutrophil levels were increased throughout the alveolar spaces during the first 3 dpi and returned to baseline by 6 dpi. Histochemical staining revealed that neutrophil infiltration in the lungs occurred in two waves, at 1 and 3 dpi, and gene expression within microenvironments suggested two types of neutrophils. Specifically, CCL3 levels, but not CXCL8/interleukin 8 (IL-8) levels, were higher within discrete lung microenvironments and coincided with increased infiltration of neutrophils into the lung. We used live imaging of ferrets to monitor host responses within the lung over time with [18F]fluorodeoxyglucose (FDG). Sites in the H1N1pdm virus-infected ferret lung with high FDG uptake had high levels of proliferative epithelium. In summary, neutrophils invaded the H1N1pdm virus-infected ferret lung globally and focally at sites of infection. Increased neutrophil levels in microenvironments did not correlate with increased FDG uptake; hence, FDG uptake may reflect prior infection and inflammation of lungs that have experienced damage, as evidenced by bronchial regeneration of tissues in the lungs at sites with high FDG levels.
IMPORTANCE Severe influenza disease is characterized by an acute infection of the lower airways that may progress rapidly to organ failure and death. Well-developed animal models that mimic human disease are essential to understanding the complex relationships of the microenvironment, organ, and system in controlling virus replication, inflammation, and disease progression. Employing the ferret model of H1N1pdm virus infection, we used live imaging and comprehensive histological analyses to address specific hypotheses regarding spatial and temporal relationships that occur during the progression of infection and inflammation. We show the general invasion of neutrophils at the organ level (lung) but also a distinct pattern of localized accumulation within the microenvironment at the site of infection. Moreover, we show that these responses were biphasic within the lung. Finally, live imaging revealed an early and sustained host metabolic response at sites of infection that may reflect damage and repair of tissues in the lungs.
INTRODUCTION
The annual impact of seasonal influenza A viruses (IAV) on public health is fairly predictable and is mostly preventable in normal, healthy populations with currently available vaccine strategies (1–3). However, novel IAV variants arising from antigenic shifts in zoonotic reservoirs are not predictable and may not be immediately preventable with contemporary vaccines (4–6). In the past 100 years, IAV with high transmissibility and virulence (1918 H1N1 IAV), high transmissibility and moderate virulence (2009 H1N1 pandemic IAV), and low transmissibility and high virulence (H5N1 and H7N9 IAV) have emerged (7–9). The virulence factors of IAV have been attributed to specific mutations in the viral proteins hemagglutinin (HA) and polymerase basic protein 2 (PB2) (10, 11). Moreover, the specific sialic acid-binding affinity of the HA determines the types of cells infected in the respiratory tract and hence the location of infection (i.e., upper versus lower respiratory tract) (12, 13). In general, IAV strains that are able to infect the lower respiratory tract cause more inflammation and have greater lethality (13–16).
Controlled inflammation is required for the resolution of IAV infection, yet increased inflammation is correlated with more-severe influenza disease, and a dysregulated immune response to infection with highly pathogenic avian influenza (HPAI) viruses (e.g., H5N1 IAV) may be fatal (17, 18). Seasonal H3N2 and H1N1 IAV typically infect only the upper respiratory tract and result in mild inflammation of the nasopharynx and trachea. Increased inflammation in the lung was observed in cases of infection with the 2009 pandemic H1N1 (H1N1pdm) IAV, which caused higher mortality than seasonal strains (19–24). Studies with humans and ferrets show that HPAI (e.g., H5N1 IAV) and H1N1pdm viruses infect and replicate well in the lower respiratory tract (15, 25), yet cases of H1Npdm virus infection showed much lower lethality than H5N1 IAV infection and did not cause severe immune dysregulation. Thus, the precise relationship between the location of infection and the location of inflammation is complex, and pathogenesis may depend on other specific immune responses to certain IAV isolates.
We know very little about how most viral respiratory infections and the resulting immune responses progress in real time within the local microenvironments of the upper and/or lower respiratory tract in humans or in animal models. Live, whole-body imaging of pathogens or host responses may offer a bridge to gain insight into these dynamics (26). During the 2009 H1N1pdm virus outbreak, molecular imaging of patients revealed that [18F]fluorodeoxyglucose (FDG) expression increases and becomes concentrated in sites of consolidation and ground-glass opacities within the lung (22, 23, 27, 28). This motivated our exploration of live FDG imaging in the ferret model of IAV infection (29). The ferret serves as a critical animal model with which to ask questions regarding IAV disease progression, given the similarity of its clinical signs and disease with those in humans (29–32). As observed for human cases of H1N1pdm virus disease, H1N1pdm virus-infected ferrets had a focal pattern of FDG uptake in the lungs that correlated with bronchiolitis and bronchitis histopathology scores (29).
We and others have shown that IAV infection initiates in discrete foci of the respiratory tract (29, 33, 34). Viral replication triggers inflammation in the form of cytokines, chemokines, and the infiltration of innate immune cells into the lungs (20, 35, 36). Cellular uptake of FDG in acute lung injury correlates with inflammatory cell infiltration (37, 38). We and others have hypothesized that cellular uptake of FDG in the lung following infection may be due to the infiltration of inflammatory cells, e.g., neutrophils and monocytes (29, 39, 40). Neutrophils are among the first cells to respond to many types of inflammation, particularly inflammation associated with bacterial infection (41–43). Neutrophils perform many functions during bacterial infection (e.g., phagocytosis, release of antibacterial proteases, and generation of reactive oxygen species), yet very little is known about their contributions during viral infections (44). In mild and severe human cases of influenza, levels of neutrophils and neutrophil chemoattractant signals are elevated in blood and nasal lavage fluid and are independent of bacterial coinfection (20, 22, 45–48). In mouse models of influenza, depletion of neutrophils results in increased virus replication and increased disease severity (49–52). However, it is known that neutrophil levels are increased in the lungs of mice during severe IAV infection (16, 17, 53–55).
Currently, we know very little about the timing and spatial distribution of neutrophils as they infiltrate the lungs following viral infection or, importantly, about how they function in disease resolution or exacerbation. Here we used an extensive systematic histological analysis coupled with live imaging to probe the temporal dynamics of virus distribution and inflammation within microenvironments of the lungs of ferrets infected with a clinical H1N1pdm IAV isolate. Given their importance early in infection, we focused on neutrophils and hypothesized that neutrophils would migrate preferentially to sites of IAV infection, creating an inflammatory microenvironment (40, 41, 44, 56). IAV distribution, neutrophil accumulation, and selected immune gene expression profiles were measured within discrete sections of a lung lobe. We show that the recruitment of neutrophils to the lung following infection occurs in two waves, coincides with chemokine gene expression, and may reflect two different states of neutrophil activation. Using extensive histological sampling, we show that neutrophilic inflammation correlates with the temporal and spatial distribution of IAV in the lungs of infected ferrets. Importantly, neutrophil recruitment and accumulation occur at the tissue level (i.e., the whole lung) and at sites of IAV infection. Finally, we show that FDG uptake in the infected lungs is not due to neutrophilic infiltrates but correlates with epithelial cell proliferation and regeneration following infection.
MATERIALS AND METHODS
Virus and viral titers.
Ferrets were infected with a clinical isolate of IAV H1N1pdm (A/Kentucky/180/2010), taken in a fatal case of H1N1pdm influenza (57), that has been described previously in ferret (29), DBA/2 mouse (57), and human primary cell culture (58) models. The virus was grown in 10-day-old embryonated hen eggs and was diluted in phosphate-buffered saline (PBS). The concentration of virus stock was determined using a 50% tissue culture infectious dose (TCID50) assay in Madin-Darby canine kidney cells as described previously (29).
Ferrets, ferret virus challenge, and ethics statement.
Four-month-old (16- to 20-week-old) female Fitch ferrets were obtained from Triple F Farms (Sayre, PA, USA). Ferrets were determined to be seronegative for the A/Uruguay/716/2007 (H3N2), seasonal A/Brisbane/59/2007 (H1N1), and A/Kentucky/180/2010 (H1N1pdm) subtypes of IAV by a hemagglutination inhibition assay (29). Animals were housed for an acclimation period of at least 1 week at the University of Louisville Regional Biocontainment Laboratory prior to virus challenge. (This research facility is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.) Animals were maintained on a 12-h light/dark cycle and were provided Teklad laboratory animal diet 2072 (Harlan Laboratories, Madison, WI) and water ad libitum. The research described here complied with federal statutes and regulations relating to animal experimentation, including the Animal Welfare Act, and adhered to the principles stated in the Guide for the Care and Use of Laboratory Animals (59). Experiments involving ferrets were approved by the University of Louisville Institutional Animal Care and Use Committee.
Ferrets were inoculated with 106 TCID50s of influenza A/Kentucky/180/2010 (H1N1pdm) virus as a 1-ml bolus. To prepare animals for infection with the virus, ferrets were sedated by intramuscular administration of 0.05 mg atropine, 5.0 mg ketamine, and 0.08 mg dexmedetomidine per kg of body weight. Sedated ferrets were held with their noses pointing upwards and necks extended while 0.5 ml of virus or PBS was given per naris. Following intranasal (i.n.) inoculation, animals were held in place for 1 min before anesthesia was reversed with 0.4 mg/kg atipamezole. Following infection, ferrets were assessed daily for temperature and weight and were monitored twice daily for clinical signs (dyspnea, nasal and ocular discharges, loss of appetite, neurological signs, sneezing, lethargy, anorexia, diarrhea, and other abnormalities) through the completion of the study. Body temperatures were measured via subcutaneous implanted microchip IPTT transponders (BioMedic Data Systems, Inc.). Right caudal lung tissues were snap-frozen in liquid nitrogen and were stored at −80°C until they were homogenized and stored in TRIzol reagent (Invitrogen). Blood was taken in sodium-EDTA tubes and was stored in TRIzol. Other lung tissues were taken for immunohistochemistry (IHC) as described below.
Analysis of gene expression levels by reverse transcription-PCR (RT-PCR).
Previously, the right caudal lung lobe of each ferret was divided into four sections, and each section was homogenized for the quantification of virus by determining the TCID50 (29). Following homogenization, a portion of the lung tissue was placed in TRIzol reagent and was stored at −80°C. Here, total RNA was extracted from these samples, and 1 μg RNA was used to make cDNA using SuperScript III reverse transcriptase (Life Technologies). Ferret mRNA gene-specific primers were generated by using published ferret sequences (Table 1). Primers were designed to span exons when possible by comparison to known human and canine splice sites. Primers were chosen to identify cell type-specific targets (e.g., granulocyte colony-stimulating factor receptor [G-CSFR; encoded by the Csf3r gene], myeloperoxidase [MPO; encoded by the Mpo gene], and neutrophil elastase [ELANE; encoded by the Ela2 gene] transcripts, which are specific to neutrophils; monocyte colony-stimulating factor receptor [M-CSFR; encoded by the Csf1r gene], which is used as a macrophage/monocyte marker; NCR1 [encoded by the Ncr1 gene], a natural killer cell receptor; and CD11b [encoded by the Itgam gene], an integrin expressed on inflammatory leukocytes), as well as soluble inflammatory signals (interleukin-6 [IL-6; encoded by the Il6 gene], CXCL8 [also known as interleukin-8; encoded by the Il8 gene], interferon beta [IFN-β; encoded by the Ifnb1 gene], CCL2 [also known as monocyte chemotactic protein 1; encoded by the Ccl2 gene], CCL3 [also known as monocyte-inducible protein 1α; encoded by the Ccl3 gene], and tumor necrosis factor alpha [TNF-α; encoded by the Tnf gene]). Real-time PCR was performed using SYBR green dye on a ViiA 7 real-time PCR machine (Applied Biosystems). Primers were validated using mitogen-stimulated whole blood from donor ferrets (Marshall Farms) (data not shown). Measurements of fold increases in transcript levels were compared to the transcript levels of housekeeping genes (glyceraldehyde-3-phosphate dehydrogenase [GAPDH; encoded by the Gapdh gene]) and average transcript levels in mock-infected animals by using the ΔΔCT method; the fold change is expressed here as the 2−ΔΔCT value.
TABLE 1.
Primers used to identify ferret leukocyte-specific and inflammatory genes
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| ELANE | GCTGCTGAATGACATCGTG | CACGTTCCTGTTGATGGTG |
| CCL2 | CAGAAGTGGGTCCGGGATTC | TTCATGGCTTTGCAGTTTGGG |
| CSF3R | AGAGCTGGAAGATGGAGCAT | TAGAGCTGAAAGGGCCTGAT |
| CCL3 | GGTCTTCTCTGCACCATTTG | CTTGCGTGTAATCTGCTTGG |
| MMP9 | AGCGAGAGACTCTTCACCCA | CAGTGGTGCAGGTGGAGTAA |
| CSF1R | ATGGAGACACCAAGCTCACA | CATCTTGGAAGCCTACGTGA |
| CD11B | CTGGAGCTGCCTGTGAAATA | ATAATGCGGCTGGTCTTCTC |
| TNFA | CCAGATGGCCTCCAACTAAT | CAGCTTCAGGGTTTGCTACA |
| IL-6 | ACCGGTCTTCAGGAGTTTCA | AAGAACTGGAGCAGGTGTTTG |
| IFNB | TCCCAGAAGTTCCAGAAGGA | TCGACAATGCTCTCATTCCA |
| IL-8 | GTGAAGCTGCAGTTCTGACG | GGGCCACTGTCAATCACTCT |
| SLC2A1 | CTGCTGAGCGTCATCTTCAT | TTCTTCAGCACACTCTTGGC |
| NCR1 | GCACCGAGAAGCAGAATCTT | GCTGGTACTTCACAGCCTCA |
| GZMA | AGCTCACTGTGTCCTGAACAA | TCAAAGCACGGATAAGGAAA |
| MPO | TCCCTGAGGAGGAAGAGATG | GCTGGTGTCAGCATGTCAGT |
| GAPDH | AGCTGAATGGGAAGCTCACT | CTGCTTCACCACCTTCTTGA |
| IAV HA | CACCAGTCCACGATTGCAATA | ATGGGAGGCTGGTGTTTATAGC |
Histology.
Lungs were inflated with 10% neutral buffered formalin and were fixed for 48 h. Lungs were trimmed, placed in cassettes, dehydrated through ethanol to d-limonene, paraffin embedded, and sectioned at a thickness of 5 μm. For systematic sampling of the lungs, the right and left caudal lung lobes (RCa and LCa, respectively) were trimmed into four consecutive transverse slices approximately 0.5 cm thick, moving from the lung hilum to the posterior of the lobe, as illustrated in Fig. 1. Sections (numbered 1, 2, and 3 in Fig. 1) followed the main bronchus to approximately the lower middle of the lung (Fig. 1, left). The sections were paraffin embedded, and two serial 5-μm sections were mounted on each glass slide (Fig. 1, center). Slides were dewaxed in 3 xylene washes and were rehydrated in decreasing ethanol gradients to water for IHC, and regions of the slides were photographed and analyzed (e.g., Fig. 1, right, boxed areas).
FIG 1.
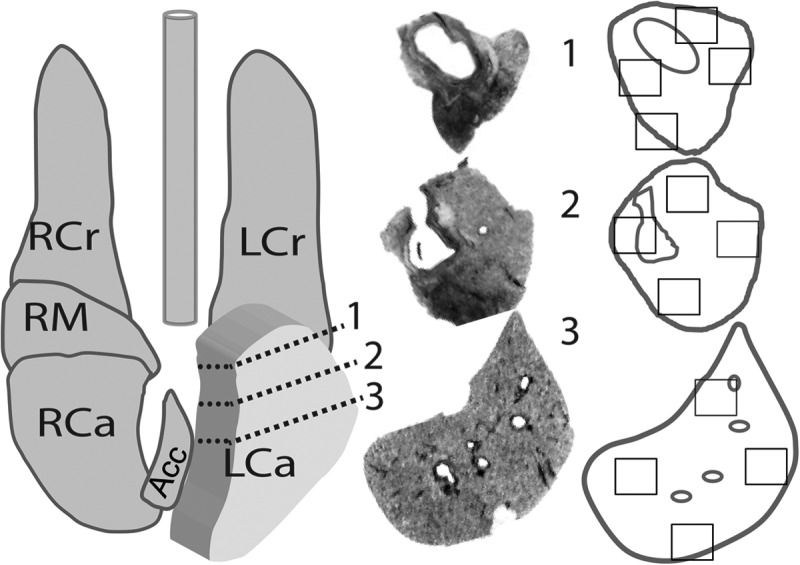
Sampling scheme for experiments on the temporal and spatial distribution of inflammation and infection in ferrets after H1N1pdm virus challenge. Comprehensive histological sampling of ferret lungs was performed at time points following intranasal infection with 106 TCID50s of H1N1pdm virus (A/Kentucky/180/10)/ferret. Samples were taken at 0.25, 1, and 3 dpi from 2 infected ferrets per time point, and from 2 mock-infected ferrets at 3 dpi, immediately following PET-CT imaging. (Left) Diagram of a ferret lung showing the method by which the left caudal lobe was divided (dashed lines on expanded lobe) for histology. The right caudal lobe was similarly divided into at least 4 cross sections along the main branching bronchus (cuts not shown). Ferret lung lobes are labeled as follows: RCr, right cranial lobe; LCr, left cranial lobe; RCa, right caudal lobe; LCa, left caudal lobe; RM, right middle lobe; Acc, accessory lobe. (Center) The resulting cross sections of tissues are shown as black-and-white photos of histological slides, numbered according to the locations on the lung diagram. (Right) Outlines of the tissues were given to a blinded observer for the selection of four regions for IHC analysis (boxed areas). This sampling scheme for IHC was used to analyze the pattern of neutrophil infiltration, as well as computed tomography and positron emission tomography images.
IHC and tissue staining.
Rehydrated tissue slides were incubated in 3% hydrogen peroxide and avidin-biotin blocking buffer (Vector Laboratories) for 15 min each at room temperature before being blocked in 5% fetal calf serum for 1 h at room temperature. For the detection of IAV nucleoprotein (NP), antigen was retrieved with 1% pronase in a CaCl2 solution and was detected with a primary monoclonal antibody (HB65 clone EVS238; EastCoast Bio, Inc.) (60), and avidin-biotin complex amplification (Vector Laboratories) was used to visualize the antibody with diaminobenzidine (DAB) peroxidase as the substrate. The slides were dehydrated through increasing ethanol gradients, cleared in xylenes, and mounted under a cover glass with a toluene solution. Neutrophils were stained pink by a 30-min incubation in naphthol AS-D chloroacetate esterase (NACE) at 37°C according to the manufacturer's instructions (Sigma-Aldrich), and one serial section was counterstained with hematoxylin to identify anatomical features. Slides were mounted with a cover glass under an aqueous mounting medium (VectaMount AQ; Vector Laboratories). For each NACE-stained slide, the next consecutive slide was similarly stained for IAV antigen. For both NACE and NP staining, one section per slide was counterstained with hematoxylin for anatomical reference, and a second, unstained section was used for image processing as described below. The Ki-67 and histone H3 antigens were retrieved by heating rehydrated slides in a 72°C water bath overnight in target retrieval buffer (Dako). A monoclonal antibody cross-reactive to ferret Ki-67 (clone MIB-1; Dako) was used as described above and stained with the DAB peroxidase substrate, and a rabbit polyclonal anti-human histone H3 antibody (Abcam) was stained red with the Vector Red alkaline phosphatase substrate (Vector Laboratories). Sirius Red staining was performed by incubating rehydrated tissue slides for 30 min in saturated picric acid containing 0.1% Sirius Red and 0.1% Fast Green and then dehydrating and mounting the slides under Permount mounting medium (Fisher Scientific).
IHC analyses.
Images from IHC slides were analyzed with the “Threshold Colour” plugin of the Fiji package of ImageJ software (61). Briefly, pixels of a certain hue and saturation (bright pink for NACE-stained neutrophils, red for Sirius Red-stained collagen, and brown for DAB-stained NP-positive or Ki-67-positive cells) were selected, and the percentage of total pixels was calculated for each image. The percentage of consolidation was determined by converting each image to a high-contrast 8-bit (black-and-white) image and calculating the percentage of black pixels (i.e., the number of pixels containing tissue, eliminating airspace). A Java computer language script (available on request) was generated to analyze all images in the same way using validated settings. Neutrophil density is reported here as the percentage of NACE-positive pixels divided by the percentage of consolidation in a single image. To validate this measurement, two independent observers counted the number of NACE-positive neutrophils in 50 images. There was a 95% correlation between the automated method and visual inspection (data not shown).
Comprehensive, systematic sampling of the caudal lung lobes was performed by a blinded observer choosing four regions per NACE-stained tissue section and photographing three images per region at ×20 magnification (Fig. 1, right, boxed areas). To correlate the number of neutrophils with foci of IAV infection, NP antigen-positive areas were photographed, and their anatomical locations (i.e., bronchial epithelium, bronchiolar epithelium, submucosal epithelium, alveolar region) were recorded. The same site was located on the NACE-stained slides and was photographed (i.e., a serial section of tissue no more than 15 μm away from the NP-positive site). IAV NP antigen-negative regions were anatomically site-matched to NP-positive foci located in the same general regions (e.g., a region of bronchiolar epithelium that was antigen negative was selected on a slide where bronchiolar epithelium was antigen positive, and therefore the antigen-negative region was always <0.5 cm from the original antigen-positive focus). NACE-positive neutrophils, IAV NP-positive cells, and areas of Sirius Red-stained collagen were quantified using automated image analysis as described above. The number of Ki-67-positive cells per slice was estimated by two observers using a scoring index (62, 63).
Ferret molecular imaging.
Ferrets were anesthetized by isoflurane induction, and 2 mCi of FDG was administered intraperitoneally as described previously (29, 64). The dose was confirmed before and after injection with an Atomlab 500 dose calibrator (Biodex Medical Systems Inc., Shirley, NY). Ferrets were kept warm under sedation with isoflurane for 1 h prior to intubation and during imaging using the Siemens trimodal platform as described previously (29, 64). Following imaging, ferrets were removed from isoflurane, given intramuscular ketamine (5 mg/kg) and dexmedetomidine (0.08 mg/kg), and immediately euthanized by cardiac exsanguination.
PET-CT image analysis.
After positron emission tomography (PET) and computed tomography (CT) images were acquired from the trimodal scanner, PET images were aligned to CT anatomical reference space in order to provide one-to-one voxel correspondence between anatomical and functional images. Since pathology on CT (e.g., consolidation) can confound true lung volume and boundary estimation, the lungs were segmented from CT images using a robust algorithm (i.e., interactive region growing) proposed in reference 65, by which users had a chance to correct any missegmented lung portion interactively if necessary. Then significant uptake regions were segmented using an affinity propagation-based PET delineation algorithm (66). Once all the metabolically active lesions were identified and delineated, maximum standard uptake values (SUVmax) and maximum intensity values were calculated as an evaluation metric for FDG uptake. These measurements were taken from regions coinciding with histopathology slices, which were manually located on the registered and segmented PET-CT scans. The alignment between histology and PET-CT images was ensured by processing 9-parameter affine registration and using prior knowledge about anatomy, particularly the airway structures and the lung lobes. For airway structure extraction, we used the hybrid algorithm for delineation of the airway tree as proposed in reference 67. Due to the 2-dimensional (2D) nature of the histopathology slices, registration operations were all carried out in 2D. After alignment of the histopathology slices into PET-CT scans, in vivo imaging correlation with histopathology slices was explored.
Statistical analyses.
All statistical analyses were performed using R, version 2.13.0, and GraphPad Prism, version 5. In general, nonparametric statistical tests were used as stated in Results; even when the sample size was large, nonindependent measurements for the same ferret made parametric analyses undesirable. Pairwise post hoc tests were adjusted for multiple comparisons using Bonferroni's method. For generalized linear modeling of neutrophil density, variables such as the time postinfection, the individual ferret, the lung lobe in the ferret, and the sample region within the lung lobe approximated a nested block design with repeated measures within ferrets, and within lobes within ferrets. The fully factorial model was reduced using stepwise regression to find the best-fitting model as determined by significant reduction in the Akaike information criterion. For all statistical tests, the type I error rate was set to an α value of 0.05, although P values of <0.1 are given consideration here due to sample size limitation and pseudoreplication as discussed below.
RESULTS
We have shown previously that infection of ferrets with the H1N1pdm virus (A/Kentucky/180/2009) causes moderate disease, evidenced by a brief febrile period, low weight loss, and clinical signs recorded during infection (i.e., lethargy, rhinorrhea) (29). Virus was detected in nasal swabs, throat swabs, nasal turbinates, and right caudal lung lobes on 1, 2, and 3 days postinfection (dpi) but was undetectable by 7 dpi (one animal had small amounts of virus in nasal and throat swabs on 7 dpi) (29). Histopathology revealed focal bronchitis and bronchiolitis characterized by infiltration of leukocytes (neutrophils and exudate macrophages) during H1N1pdm virus infection (29). Cellular uptake of FDG correlates with leukocytic infiltration in certain model systems (38–40). In this ferret model, we have observed progressive increases in FDG uptake in distinct foci of the H1N1pdm virus-infected ferret lung (29). Therefore, here, we tested the overarching hypothesis that FDG uptake during infection of ferrets with IAV was due to infiltrating neutrophils and macrophages. We first determined which leukocytes were likely to exhibit increased expression in these regions by measurement of immune gene expression profiles within discrete lung microenvironments.
Lung microenvironments of IAV infection have increased CCL2 and CCL3 gene expression.
First, the expression of signature immune genes was analyzed in the right caudal lobes of one mock-infected ferret and two H1N1pdm virus-infected ferrets on 1, 2, 3, and 7 dpi. As stated above, the right caudal lobe had the greatest levels of FDG uptake following infection (29). Primers for IAV HA and inflammatory cytokines and chemokines were used to confirm the presence of the virus and the host immune response to infection. Genes were chosen to detect cytokines and chemokines that are known to show increased expression during the acute phase of influenza virus infection and that are important for leukocyte activation and chemoattraction (e.g., IFN-β, TNF-α, IL-6, CCL2, CCL3, and CXCL8). First, IAV HA mRNA was detected at 1, 2 and 3 dpi, but not 7 dpi, and was not uniformly distributed within the right caudal lobe (data not shown). This matched the pattern of IAV infection as measured by the TCID50 assay and reported previously for the same ferret lung sections (29). Fold changes in the expression of inflammatory cytokine (IFN-β, TNF-α, IL-6) and chemokine (CCL2, CCL3, CXCL8) genes in sections were compared to expression in mock-infected controls. No significant change in inflammatory expression levels was detected in the blood (data not shown). In some lung sections from infected ferrets, but not in all, levels of the cytokines IL-6 and TNF-α (not shown) were elevated at 2 dpi. Other inflammatory signals (e.g., IFN-β, CXCL8) were not elevated above those in mock-infected animals (data not shown). The expression levels of the CCL2 and CCL3 genes were both slightly increased at 1 dpi and significantly increased at 3 dpi (P < 0.05) (Fig. 2).
FIG 2.
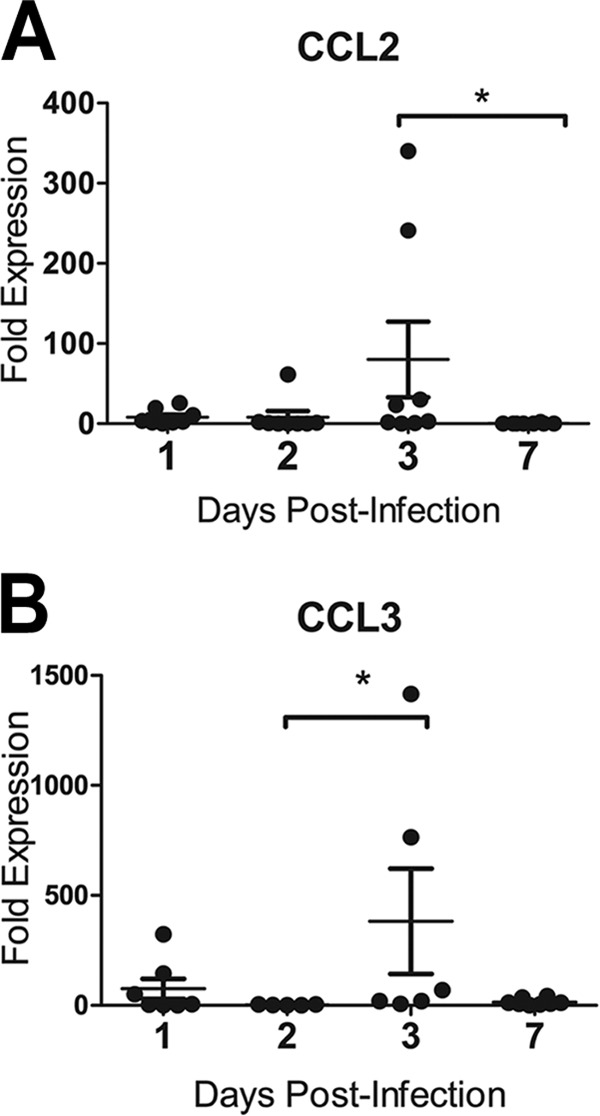
Inflammatory cytokine and chemokine gene expression in the ferret lung after infection with influenza A virus (H1N1pdm). Female ferrets (2 per time point) were infected with 106 TCID50s each of H1N1pdm virus (A/Kentucky/180/2010). The right caudal lung lobe from each H1N1pdm virus-infected and mock-infected (1 ferret per time point) ferret was taken upon euthanasia on days 1, 2, 3, and 7 postinfection and was divided into four sections. Each section was homogenized separately, and cDNA was synthesized for gene expression analysis. Ferret gene-specific primers were developed to cross exons, and RT-PCR was performed to identify the expression of the genes encoding leukocyte chemoattractant chemokines CCL2 (A) and CCL3 (B). Fold expression is compared to the expression of housekeeping controls (GAPDH) and to expression in mock-infected animals by using the ΔΔCT method. Asterisks indicate significant differences between days by nonparametric statistical tests, corrected for multiple comparisons.
Neutrophil-specific gene expression is detected in lung microenvironments.
Primers for leukocyte-specific genes that encode cell surface receptors and other cell-specific proteins were used to determine the presence of neutrophils (G-CSFR, MPO, ELANE, CD11b), macrophages (M-CSFR, CD11b), and natural killer cells (NCR1, granzyme A, CD11b) in the lung. Expression of the monocyte/macrophage-specific gene Csf1r (encoding M-CSFR) was increased slightly at 2 and 3 dpi, and increased expression of the natural killer cell receptor gene Ncr1 (encoding NCR1) was detected in some sections at 2 and 3 dpi (data not shown). Expression of the neutrophil growth factor receptor gene Csf3r (encoding G-CSFR) was increased in lung sections at 3 dpi (P < 0.05) (Fig. 3A). Neutrophil-specific granule genes (e.g., Mpo, Elane) were not significantly upregulated relative to expression in mock-infected animals (data not shown).
FIG 3.
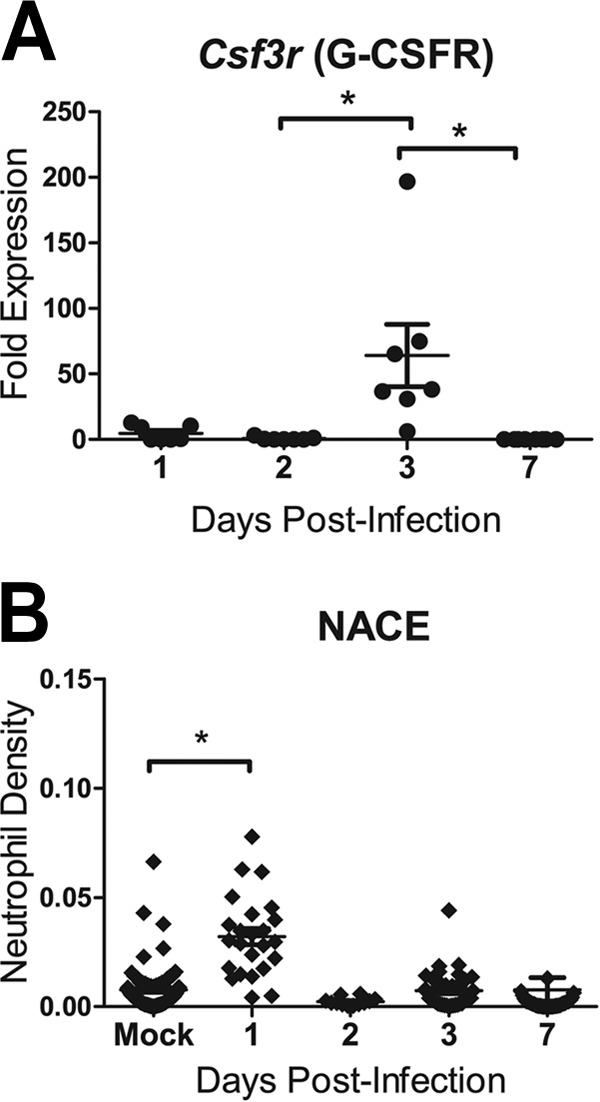
Neutrophils were detected in ferret lung sections after infection with influenza A virus (H1N1pdm). Female ferrets (2 per time point) were infected with 106 TCID50s each of the H1N1pdm virus (A/Kentucky/180/2010). (A) The right caudal lung lobe from H1N1pdm virus-infected and mock-infected (1 ferret per time point) ferrets was taken upon euthanasia on days 1, 2, 3, and 7 postinfection and was divided into four sections. Each section was homogenized separately, and cDNA was synthesized for gene expression analysis. Ferret gene-specific primers were developed to cross exons, and RT-PCR was performed to identify the expression of a neutrophil-expressed gene (Csf3r, encoding G-CSFR) relative to the expression of housekeeping controls and to expression in mock-infected animals. Asterisks indicate significant differences between days by nonparametric statistical tests, corrected for multiple comparisons. (B) Other lung lobes (left cranial, right cranial, left caudal, and middle) taken from each ferret were prepared for IHC, and one slice from each was stained for neutrophils (NACE). Neutrophils were quantified in images (taken with a 20× objective) from 3 regions on each of the NACE-stained slides by using ImageJ image analysis software. The number of NACE-positive pixels divided by the number of total tissue-containing pixels is presented as the neutrophil density. The asterisk indicates a significant difference in distribution from that in mock-infected ferrets by nonparametric statistical tests.
Neutrophils show biphasic infiltration into the lungs of IAV-infected ferrets.
The major inflammatory signals in the right caudal lung sections were the neutrophil-specific gene Csf3r and the genes encoding the leukocyte chemoattractants CCL2 and CCL3. To confirm the temporal pattern of neutrophils detected by gene expression in the lungs of these ferrets, NACE staining was used to label neutrophil-specific granules. One histological preparation per lung lobe (except for the right caudal lobe, which was used for gene expression, as described above) from H1N1pdm virus-infected and mock-infected ferrets was stained and inspected for neutrophils. To estimate the number of neutrophils within the stained histological sections, image analysis software was used to determine the number of NACE-positive pixels per unit of tissue in an image (taken with a 20× objective) of the NACE stained lung (expressed as the estimated neutrophil density), as described in Materials and Methods. Image analysis of sites chosen haphazardly within each tissue slice revealed that the overall density of neutrophils in the lung was at a maximum at 1 dpi (P < 0.05) (Fig. 3B). Thus, the major peak of neutrophils did not correspond to the major peak of neutrophil-specific gene expression in the lung. The lung lobes other than the right caudal lobe showed a wide range of neutrophil densities, similarly to the gene expression data, suggesting that there were foci of neutrophil infiltration throughout the lungs.
Neutrophils may hone to specific sites of IAV infection in the lungs.
To determine the precise distribution of IAV within the lungs, IHC was used to detect NP antigen in paraffin-embedded sections of the lung lobes. Infection was detected in bronchial, submucosal, and bronchiolar epithelial cells (Fig. 4A to D) but not in alveolar epithelium (not shown). IAV NP was detected in sections from all infected ferrets at 1 and 3 dpi and from one animal at 2 dpi but not in sections from infected animals at 7 dpi. H1N1pdm virus infection was highly focal in the lung and was primarily restricted to sites in bronchioles and bronchi (Fig. 4A to D). Similarly, the pattern of antigen positivity matched the patterns of IAV mRNA expression, chemokine- and neutrophil-specific gene expression, and NACE staining. Counterstaining of NP-labeled sections with hematoxylin allowed the identification of increased numbers of inflammatory macrophages and neutrophils, some of which were antigen positive (Fig. 4D), at foci of infection in the bronchioles. Therefore, neutrophils appeared to hone to the sites of infection in specific anatomic regions.
FIG 4.
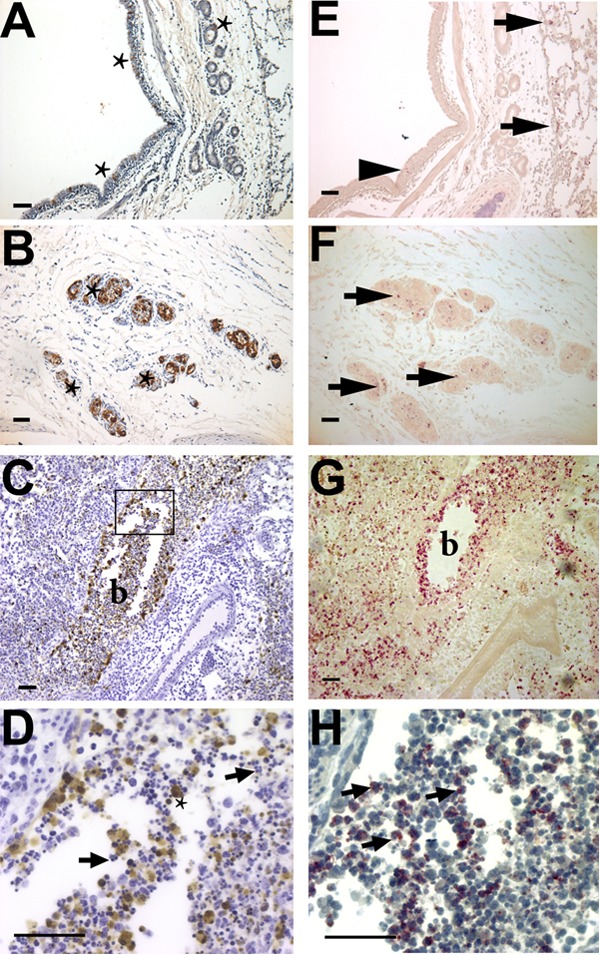
Immunohistochemical staining of influenza virus nucleoprotein and neutrophils in ferret lungs. Shown are histological preparations from the right caudal lobe of a female ferret 1 day after infection with 106 TCID50s of an H1N1pdm virus isolate (A/KY/180/2010). (A to D) The influenza antigen (NP) is stained brown, with a hematoxylin (blue) counterstain. (E to H) Neutrophils are stained pink with NACE in a serial section of the same region of the lung as the IHC NP stain (without a counterstain in panels E to G to show the bright pink NACE stain; with the blue hematoxylin counterstain in panel H). (A) Influenza virus-infected cells (stars) in the pseudostratified columnar epithelium of the bronchus of a ferret 2 days postinfection. (B) Infected seromucinous glands (three are marked with stars, but more are visible). (C) Infection of the bronchiolar epithelium (lumen marked “b”) was more common than infection of other sites, and the antigen can be seen within luminal cells. (D) Magnification of the region boxed in panel C. Many NP-positive cells (star) are macrophages, and the lumen of the bronchiole is filled with polymorphonuclear neutrophils (arrows indicate two, but many more are visible). (E) Few neutrophils are present at sites of influenza virus-infected epithelial cells within the epithelial layer of the bronchus (arrowhead), but more are present within the alveolar spaces adjacent to these sites (arrows). (F) Inflammatory cells within the infected submucosal glands are neutrophils (arrows indicate three areas, but more are present in each gland). (G) Extensive neutrophil infiltration in the bronchiolar lumen (b). (H) Magnification of a serial section of panel G that is stained for NACE and counterstained with hematoxylin to show the morphological features defining neutrophils (arrows show three, but many more are visible). Images were taken with a 10× (A to C and E to G) or 40× (D and H) objective. Bars, 50 μm.
Comprehensive sampling of the lung lobes for neutrophil infiltration.
The preliminary data suggested that neutrophils infiltrate the lungs and hone to sites of IAV infection, driven by chemokine gradients (e.g., CCL3). However, the data were correlative because the sampling was not comprehensive and data were obtained from different lobes (i.e., gene expression data from the right caudal lobe; neutrophil and NP staining data from the four remaining lung lobes). Therefore, we performed a second ferret study to confirm these observations using systematic histological sampling of the lung. Six female ferrets were infected with 106 TCID50s each of the H1N1pdm virus (and 2 ferrets were mock infected), and 2 animals each were euthanized at 0.25, 1, and 3 dpi. Left and right caudal lung lobes were formalin fixed, sectioned into four 0.5-cm cross sections from the hilum moving posteriorly, embedded in paraffin, and prepared for histology (three cross sections are shown in Fig. 1). Tissue slices were stained with NACE and were photographed to quantify neutrophils. Methodical sampling provided data for a generalized linear model to establish factors important for neutrophil distribution in the lung. Four regions from each slice were chosen blindly, and three nonoverlapping images were taken from each region to quantify the number of NACE-positive pixels as a function of total tissue (excluding airways) per image using ImageJ (Fig. 1). This established a 3D grid of sampling locations within the caudal lung lobes, wherein three images were taken from locations within each caudal lung lobe (48 images per lung lobe).
Neutrophils infiltrate the right caudal lobe at early times after infection.
Stepwise model testing of the generalized linear model revealed that the best-fitting model included the time postinfection, the lung lobe (left versus right), and the distance from the lung hilum. Post hoc analyses confirmed that neutrophil levels were significantly increased at 1 dpi and 3 dpi (Fig. 5A) (P < 0.001 in the left and right caudal lobes), as in the previous study (Fig. 3B); however, greater neutrophil density in the lungs of infected animals than in those of mock-infected animals was found as early as 0.25 dpi (Fig. 5A) (P <0.05 in the left caudal lobe; the difference in the right caudal lobe was nonsignificant). The distribution of neutrophils was higher in the right caudal lobes than in the left caudal lobes at 0.25 and 1 dpi (Fig. 5A) (P < 0.001) but not at other time points. Additionally, it is noteworthy that a range of neutrophil densities was observed, yet, in general, infected animals had higher baseline levels of neutrophils in either lobe at 0.25, 1, and 3 dpi. Thus, neutrophils appeared to be focally distributed throughout the lung, and these observations (e.g., higher levels in the right caudal lobe) suggest that they are driven by IAV infection.
FIG 5.

Neutrophils in the lungs of ferrets after infection with influenza A virus (H1N1pdm). Ferrets were infected with 106 TCID50s of an H1N1pdm virus isolate (A/KY/180/2010), and lung tissues were taken upon euthanasia and were processed for histology. Caudal lung lobes, taken from two infected ferrets per day (0.25, 1, and 3 dpi), were systematically divided and were prepared for histology. Neutrophils in sections histochemically stained with NACE were quantified by using image analysis software, and data are expressed as the neutrophil density (the number of neutrophil-positive pixels per total tissue pixels on each image). (A) Neutrophil density was quantified over time. Data are divided by lobe. Large asterisks indicate significant differences from mock-infected animals, and significant differences between lobes are indicated by bars with small asterisks above them (P < 0.05 by nonparametric statistical tests). (B) Influenza virus nucleoprotein (NP) was identified on histological sections by IHC. The neutrophil density was calculated from serial NACE-stained sections; data were grouped according to distance from known NP-positive cells; and sites of IAV-positive cells were matched to distant sites within the same ferret (red, pink, and light-pink dots indicate NP-positive sites and sites <1 cm and >1 cm from NP-positive sites, respectively) and to sites in mock-infected animals (black dots). Data are plotted over time, and means at a given time point are indicated by lines colored according to distance (red, medium-gray, and light-gray lines for NP-positive sites and sites <1 cm and >1 cm from NP-positive sites, respectively). The asterisk indicates a significant difference between sites at a given time point. (C) Neutrophil density was calculated from pictures taken at sites of known influenza virus infection (NP positive), and data were grouped by anatomic region (alveoli, bronchioles, and bronchi with submucosal [SM] glands). These images were site-matched within the same ferret (<1 cm from known IAV-positive sites) to anatomic regions that had no IAV-positive cells (NP negative). Note that NP-positive cells at alveolar sites were leukocytes and that data for mock-infected animals are not shown on this panel but are shown in panels A and B. Asterisks indicate significant differences between NP-positive and NP-negative sites.
Neutrophil levels are increased at sites of IAV infection.
To test the hypothesis that neutrophil levels were increased at foci of IAV-infected epithelial cells, IHC staining for IAV NP was performed in serial sections of NACE-stained tissues taken from the right and left caudal lobes as described above (Fig. 4). Using NP-stained serial sections as a reference, images were taken from three areas of NACE-stained tissue slices defined by their distances from NP-positive sites. First, three nonoverlapping images were taken from each antigen-positive site. For each antigen-positive site, two other sites were chosen, and three nonoverlapping images were analyzed from each site: the antigen-positive site, a site <1 cm from the antigen-positive site (within the same histology slice), and a site >1 cm from the antigen-positive site (from another histology slice within the same lobe). These three groups of images were also compared to images taken of NACE-stained mock-infected controls, and image analysis software was used to calculate the estimated neutrophil density as described above. There was no linear relationship between the distance from the focus of an infected cell and the density of neutrophils, i.e., neutrophil levels at all sites were higher than those for mock-infected control animals (Fig. 5B) (P < 0.01). However, a pattern was revealed by resolving the spatial distribution over time: neutrophil density was significantly greater at NP-positive sites and at sites <1 cm from known NP-positive sites at 1 dpi (Fig. 4 and 5B) (P < 0.01). There was no difference in spatial distribution at 3 dpi, and neutrophil density remained higher than that for mock-infected controls (Fig. 5B) (P < 0.01).
Neutrophil levels are increased at specific locations of IAV infection.
To test the hypothesis that anatomical location explained the distribution of neutrophils after infection, sites of IAV-positive bronchi (as seen, for example, in Fig. 4A), seromucinous glands (Fig. 4B), and bronchioles (Fig. 4C and D) were identified (no antigen-positive alveolar epithelial cells were detected). These sites and antigen-negative sites within the same ferret (in similar anatomic regions) were located on NACE-stained serial sections, imaged (Fig. 4E to G), and analyzed using image analysis software to calculate the estimated neutrophil density as described above. Neutrophil density was significantly higher in IAV-positive bronchioles than in controls (Fig. 4C, D, G, and H and 5C) (P < 0.01). Neutrophil levels in infected bronchi or seromucinous glands were not higher than those in antigen-negative regions (Fig. 4E and F and 5C). Furthermore, neutrophil levels were increased in bronchioles and alveoli where no IAV-positive antigen was detected by IHC (Fig. 5C). Interestingly, certain areas containing neutrophils in the lumens of bronchioles near IAV-positive epithelial cells had extracellular histone H3 staining (data not shown), an indicator of neutrophil extracellular trap formation (NETosis). However, there were insufficient sites of NETosis, and a lack of available reagents for the ferret model, to allow us to investigate this phenomenon further at this time.
Analysis of PET-FDG images to achieve direct spatial correspondence with histology.
We have reported previously that molecular imaging of ferrets using FDG-PET showed a progressive increase in inflammation following infection with the H1N1pdm virus (correlated with bronchiolitis and bronchitis pathology scores) that was greatest in the right caudal lobe but also focally distributed throughout the other lung lobes (29). We hypothesized that this might be due to increased inflammatory cell infiltration at sites after infection (29, 38–40). Therefore, we injected infected ferrets with FDG and imaged them by PET-CT prior to euthanasia at 0.25, 1, and 3 dpi (Fig. 6). Using the same ferrets in the second study, we analyzed PET-CT data similarly to histological data (i.e., with a 3D grid) in order to achieve direct spatial correspondence between the histology and the imaging. Automated airway segmentation of CT images was first performed to extract the lungs and airways from the images (Fig. 6). Then stacked 3D image constructions were used to locate the exact positions of tissues taken for IHC by using anatomical features as references (such as the branching of bronchi, the location of the heart, and bone structure), and gross anatomical photographs taken during necropsy were also used to ensure accurate correspondence between CT and IHC. Next, automated registration of PET-CT images was performed to ensure that the images were aligned, which then allowed us to measure the uptake of FDG at sites taken for histology (Fig. 6).
FIG 6.

Rendering of PET-CT images of ferret lungs at different time points after infection with the H1N1pdm virus. Stacked whole-lung images give 3-dimensional views at three time points following infection with 106 TCID50s of the H1N1pdm virus (A/KY/180/2010). Lungs from only one ferret per time point (including a mock-infected ferret at 3 dpi) are shown. Ferrets were injected with an intraperitoneal dose of radiolabeled FDG, and lungs were imaged using PET-CT 1 h later. For each image, the light pink surface corresponds to the border of the lung that was segmented from CT images by an automated CT analysis. PET-CT fusion images were used to define the uptake of FDG within the segmented lung space, and the intensity of FDG uptake is represented by small colored volumes within the lung borders. Regions of FDG uptake are colored according to voxel intensity within the FDG-PET image, as shown on the color scale on the right (from <0.05 [blue] to >0.5 [dark red]).
FDG uptake did not match the pattern of neutrophil infiltration.
We tested whether FDG uptake was associated with sites of increased neutrophil infiltration. The stacked CT images were used to locate the regions on which NACE image analysis had been performed, and FDG uptake (expressed as SUVmax) was determined on PET images from the same regions. Globally, there was an early increase in FDG SUVmax at 0.25 dpi, which immediately decreased below the levels for mock-infected animals (Fig. 6 and 7A). For both the left and right caudal lobes, uptake in mock-infected animals was significantly greater than uptake in infected animals at 1 dpi (P < 0.05) (Fig. 7A). Temporally, this did not align with the pattern of neutrophil infiltration (Fig. 5A). Due to differences in resolution between the two modalities (PET-CT and IHC), we averaged the neutrophil density from three nonoverlapping images from each region measured by IHC and compared this to a single SUVmax from that region. Over all time points, there was no association between FDG uptake and neutrophil density (data not shown). However, there was a significant linear association between SUVmax and neutrophil density at 1 dpi, although the association was comparatively weak (R2 = 0.26) and was likely due to outliers (nonzero slope; P < 0.001) (data not shown). This finding was very surprising; therefore, we attempted to determine what was responsible for the FDG uptake, using this data set, for which we had confidence in our correspondence between PET-CT and histology.
FIG 7.
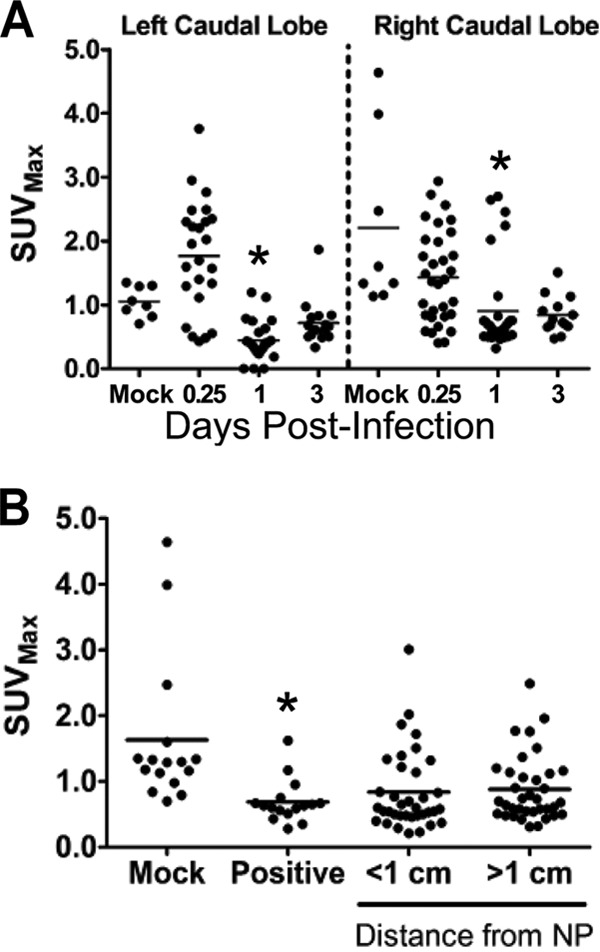
Distribution of cellular uptake of radiolabeled FDG during influenza virus infection in ferrets. Female ferrets were infected with 106 TCID50s of the H1N1pdm virus (A/KY/180/2010). A radiolabeled glucose analog (FDG) was injected at 0.25, 1, and 3 days postinfection, and ferrets (2 per time point) were imaged 1 h later. Ferrets were euthanized immediately after imaging, and lung tissues were prepared for histology. (A) The temporal distribution of FDG uptake (maximum standard uptake value [SUVmax] within a defined volume) was measured in images taken by positron emission tomography after locating histology slices by using anatomical references in registered computed tomography images. These areas matched areas analyzed for neutrophil infiltration and approximate a 3D grid of each ferret caudal lung lobe. (B) Spatial distribution of FDG uptake at sites in the ferret lung grouped by distance from IAV nucleoprotein (NP)-positive cells within the lungs. The presence of the H1N1pdm virus was measured by immunohistochemical staining for viral nucleoprotein. Asterisks indicate statistically significant differences from mock-infected animals.
FDG uptake did not match sites of IAV infection.
We tested the hypothesis that FDG uptake occurs in sites of H1N1pdm virus infection in ferret lungs. Using an approach similar to that described above for NACE-stained sections, we measured FDG-PET uptake (expressed as SUVmax) at sites of IAV-positive cells determined by IHC. We also measured SUVmax at two other sites based on distance from IAV-positive cells: <1 cm and >1 cm from any known IAV-positive area; both were measured in regions within the same ferret lobe. There was significantly less FDG uptake at sites of IAV-positive cells than in mock-infected animals (P < 0.01 for all regions compared to mock-infected animals) (Fig. 7B). Because this approach was also potentially confounded by the disparity in resolution between PET-CT and microscopic imaging modalities, we infected immortalized cell lines with several IAV isolates in order to test whether IAV infection may increase the uptake of glucose in vitro. We experimentally infected several human cells and cell lines (e.g., A549 cells and the well-differentiated human bronchial epithelial cell line 16HBEo−), and we observed slightly increased uptake of FDG with some but not all treatments (data not shown). Thus, we may infer that increased FDG uptake in infected ferrets was not directly associated with IAV infection.
FDG uptake measured proliferation in the lungs following IAV infection.
We then tested the hypothesis that proliferating cells were responsible for the increased FDG uptake in lung tissues by detecting Ki-67 protein with IHC. Ki-67 is a nuclear protein that is present in large amounts during cellular proliferation. In order to estimate the level of proliferating cells, we used a Ki-67 proliferation index (Fig. 8; images show examples for scores 0 to 3), which is an established method for grading tumors clinically (62). To sample the lungs, 3 images were taken for each anatomical region (e.g., bronchus, bronchiole, alveolus) from 4 stained tissue slices per ferret from both the right and left caudal lobes (2 ferrets per time point[0.25, 1, and 3 dpi]), and at least 1 slice each from the left caudal, right cranial, left cranial, and right middle lobes from each ferret (2 animals per time point [1, 2, 3, and 6 dpi]). This sampling resulted in 18 to 50 images per anatomic region per time point. Two independent observers blindly scored the images, and the scores were averaged. No significant regions of proliferating epithelium were detected in ferrets imaged at 0.25 dpi, 1 dpi, or 3 dpi; therefore, we included histological samples from the previous study described above (i.e., samples from two infected ferrets and one mock ferret were taken at each of 1, 2, 3, and 7 dpi; the right caudal lobe is excluded from these sections) (29). Both the alveolar and bronchiolar regions had baseline levels of proliferating cells, and there was no difference between lung tissues taken from mock-infected and infected animals in these anatomical regions (not shown). However, there was an increase over time in the level of proliferating cells in the bronchi, and the Ki-67 proliferation index value at 7 dpi was significantly higher than that for mock-infected animals (P < 0.01) (Fig. 8).
FIG 8.
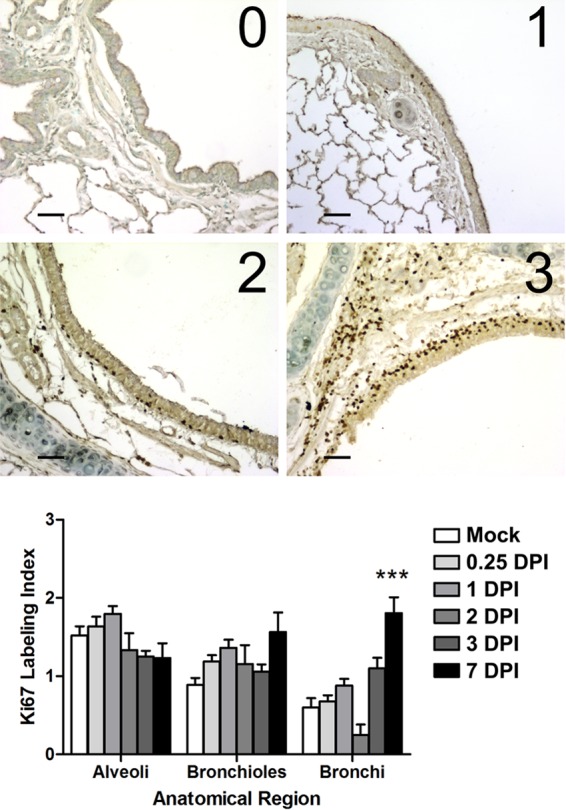
Proliferation of lung epithelial cells measured by the Ki-67 labeling index. (Top) Images representative of the scoring system (labeling index scores 0 to 3) used to grade the extent of lung cellular proliferation (Ki-67 antigen positivity) in immunohistochemical sections taken from ferrets infected with 106 TCID50s of the H1N1pdm virus (A/KY/180/2010). Bar, 50 μm. (Bottom) The left and right cranial, left caudal, and middle lobes were taken at necropsy (at 0.25, 1, 2, 3, and 7 dpi) and were prepared for histology. Bronchi, bronchioles, and alveolar spaces were analyzed in the lobes of each ferret per time point (18 to 50 images [not shown] per anatomic region per time point). The images were scored by two independent observers for the levels of Ki-67 positive cells. Shown is the mean proliferation index per anatomic region over time (error bars represent the standard errors of the means). Asterisks indicate a significant difference (***, P < 0.01) from the score for mock-infected animals by a nonparametric statistical test.
DISCUSSION
The pathology of human infection with IAV is often known only from lethal endpoints, taken at autopsy (18, 22, 45, 46). In humans, severe IAV infection affects the lower respiratory tract and results in massive infiltration of neutrophils (22, 23). Similarly, severe IAV disease in ferrets is characterized by early infiltration of neutrophils (17, 68). With respect to neutrophils in the lungs during IAV infection, pathology reports from lethal human influenza pneumonia typically note the presence of suppurative necrosis, mixed cellular infiltrates, or neutrophilic infiltration (20, 22, 24, 30, 45, 46). However, few studies have directly addressed simple questions about the relationship between neutrophils and IAV infection. For example, when do neutrophils arrive in the lungs following IAV infection? What is the pattern of neutrophil infiltration, i.e., is it specific to particular anatomical locations? Is it specific to sites of IAV? Or is it specific to sites of damage and necrosis? How important are the various inflammatory signals released in viral microenvironments to the activation and chemoattraction of neutrophils during IAV infection? Answers to these questions will provide a basis for answering more-important questions, such as how the severity of disease affects neutrophil infiltration (or vice versa). Severe IAV infection often results in a form of acute lung injury and may lead to the formation of acute respiratory distress syndrome (ARDS). Neutrophils are implicated both in the development of ARDS and in recovery from ARDS (41–43). Based on mouse studies, neutrophils play an important role in protection from lethal disease during IAV infection; however, much remains unknown about the relationship between neutrophils and IAV infection in the ferret model (49–51, 53, 54).
Although the ferret model is important for the study of IAV pathogenesis and has been used for this purpose for nearly 80 years, this model is challenging. For example, no ferret-specific reagents are commercially available, and the cross-reactive antibodies are unverified (69, 70). To overcome the limitations in the availability of immunological reagents, we have explored the use of molecular imaging as a tool for studying the pathogenesis of IAV in the ferret lung (29). Molecular imaging modalities, such as PET, rely on in vivo accumulation of injectable non-species-specific radioisotope-labeled probes that can be imaged in 3 dimensions, and images can be fused with CT images to provide anatomical references (65, 71). The use of such modalities in infectious disease imaging is quickly developing (39, 65), and preliminary data provide the foundation for the use of FDG-PET imaging in the diagnosis or prognosis of clinical cases of IAV (23, 27, 29). To further develop this tool, and to expand the tools available for the use of the ferret model, we established a model of FDG-PET during the infection of ferrets with the H1N1pdm virus (29, 64). To complement the comprehensive 3D nature of the images provided by PET-CT fusion, we chose a systematic sampling of the lung to measure infiltrating neutrophils in ferrets. Our sampling approach differed from methods used previously to determine the spatial and temporal dynamics of IAV infection in the ferret (21, 68, 72), which typically focused measurements to the site of infection and/or lung lesions. Additionally, we relied on gene expression in discrete sections of tissue to test changes in the viral microenvironment, because antibody-based staining of tissues (e.g., IHC) or cells (e.g., flow cytometry) is currently unavailable for nearly all desirable ferret antigens. In doing so, we were able to test many hypotheses about neutrophil infiltration following H1N1pdm virus infection.
Neutrophils migrate into the lungs early and accumulate in sites of infected bronchioles. The increase in neutrophil density in the lungs until 1 dpi may be due to an increase in chemokine transcription within specific lung microenvironments (73, 74). Leukocyte chemoattractant chemokines, such as CXCL8, CCL2, and TNF-α, are known to be present at sites of infection following experimental human IAV infection (47), and levels are higher in patients with the H1N1pdm virus than in those with seasonal IAV (48, 75). Previously, we and others have shown increased secretion of leukocyte chemoattractant chemokines (TNF-α, CCL2, CCL3, and CXCL8) following in vitro infection of a well-differentiated normal human bronchial epithelial cell culture with the H1N1pdm virus relative to that with seasonal IAV infection (58, 76). In IAV-infected ferrets, we and others have shown increased expression of other leukocyte chemoattractant genes (CCL2, CCL3, and CXCL10), as well as leukocyte-specific genes (G-CSFR, M-CSFR, CD11b, and cathepsin B [CTSB]), in the blood (77–79). Therefore, it is likely that neutrophils infiltrate the lungs after H1N1pdm virus infection by following chemokine gradients to the site of infection. Moreover, we observed that the distribution of neutrophils was higher in the right caudal lobes at 1 dpi (Fig. 5A), which matched our observations and those of others that i.n. and aerosol challenges of ferrets with IAV show a preference for infection of the right caudal lobe (29, 30, 33).
The specific signal that drives neutrophil chemoattraction in ferrets during IAV infection remains unknown. Here we demonstrate that CCL3 was upregulated in discrete locations within the lung that were correlated with the presence of IAV HA mRNA, neutrophils, and a neutrophil-specific gene (G-CSFR) (Fig. 2 and 3). The significant increase in gene expression for CCL3 occurred at 3 dpi, which indicates that another chemoattractant may be more important for early recruitment at 1 dpi (Fig. 2). Similarly, depletion of neutrophils from a mouse model of influenza virus infection decreased the amount of CCL3, suggesting that neutrophils may also be a source of this chemokine (50). In this study, we found that ferrets did not upregulate CXCL8 following infection with the H1N1pdm virus. CXCL8 is the major chemoattractant chemokine for neutrophils during many human infections (80). It is known that neutrophil chemoattraction is somewhat species specific and that some mammals do not possess CXCL8. For example, in mice, the chief neutrophil chemoattractant chemokines are CXCL1 and CXCL2 (81). In humans, neutrophils display the chemokine receptors CCR1 and CCR5, which are important for their chemotaxis and oxidative burst in response to CCL3. The ferret genome contains orthologs for these gene sequences (NCBI Gene IDs, 101693329 and 101692743, respectively) (73, 82); however, it is not known if these are similarly expressed on ferret neutrophils. Additionally, neutrophil chemoattractants not commonly associated with viral infection, such as eicosanoid molecules (leukotriene) and complement, were not measured (44). In general, sectioning of the lung lobe revealed the highly focal nature of inflammatory gene expression in the lungs, which matched the focal nature of viral distribution in this ferret model. Although not shown, the increased expression of IL-6 and TNF in certain sections but not others indicates a complex relationship between infection and immune response.
We measured the focal nature of infection in two ways: (i) sampling of discrete sections of a single lung lobe for gene expression and virus titration and (ii) an unbiased, semicomprehensive histological analysis of two lung lobes. By use of NACE-stained tissues, the latter approach showed a range of neutrophil densities that was greater in infected animals than in mock-infected animals. By resolving the distance from NP-positive cells over time, we showed that neutrophils become greatly enhanced at specific viral foci by 1 dpi (Fig. 5B). We observed occasional NETosis at sites of IAV-infected cells, but we were unable to make any further statistical correlation to spatial or temporal aspects of disease without further sampling. Together, these observations indicate that neutrophils migrate to sites of IAV infection and perform their functions there. Thus, we conclude that inflammation caused by IAV was focal and was driven by the presence of IAV-infected cells, yet the entire lung experienced increased infiltration as a result of infection. This may be due to the specific migratory patterns of neutrophils through the lung to sites of inflammation (83). The increased “background” neutrophil infiltration in infected animals (i.e., before and after 1 dpi, and away from foci at 1 dpi) was not obvious by standard hematoxylin-and-eosin IHC: neutrophils were uniformly distributed throughout the lung at a density that was quantifiably greater than that in mock-infected controls.
By using the histochemical stain NACE, we detected two waves of neutrophils following H1N1pdm virus infection. As discussed above, the peak of the first wave occurred at 1 dpi and was concentrated at sites of influenza virus infection. A second wave of neutrophils was detected at 3 dpi, by both NACE staining and gene expression signatures. This biphasic pattern of neutrophils following H1N1pdm virus infection has been reported elsewhere (68). Neutrophil-specific transcriptional markers were detected in lung microenvironments only at 3 dpi (e.g., Csf3r [encoding the G-CSF receptor], and Itgam, encoding the alpha M subunit of the Mac-1 integrin leukocyte activation marker CD11b) (Fig. 3). Thus, there appear to be two different pools of neutrophils invading the lung following infection, which differ in their gene expression status, and the second may be responding to or producing CCL3. Currently, it is thought that steady-state neutrophils are mature, terminally differentiated cells with limited cell-specific gene regulation; e.g., mature neutrophils have preformed granules, but neutrophils themselves are capable of gene transcription in response to certain stimuli (84, 85). This may explain why gene expression of neutrophil granule contents was not detected (MPO, ELANE, CTSB). We hypothesize that the first wave, at 1 dpi, consists of mature neutrophils and that the second, at 3 dpi, represents a more immature population, perhaps as a result of emergency granulopoiesis, that has been recruited to the lung from the bone marrow as a direct result of IAV infection and systemic inflammation and thus contains mRNA for the neutrophil growth factor receptor, G-CSFR (44, 86). Additionally, these gene signatures provide support for the biphasic infiltration of neutrophils observed in other lung lobes, showing a relationship between systemic activation and recruitment of neutrophils and their subsequent concentration at specific viral microenvironments. These observations should be studied further, since these findings may indicate distinct functions of these two pools of neutrophils.
The lung is a known reservoir of steady-state neutrophils, perhaps due to the large blood volume of the lung capillaries (44, 87, 88). An early increase in airway and circulating blood neutrophil levels is a feature of IAV infection in humans (45, 46) and is common in experimental IAV infection in mice (17, 55, 57) and ferrets (32, 68, 89). We show that this happens as early as 6 h postinfection (Fig. 5A and B). However, increased numbers of neutrophils in the lower airways have been correlated with increased influenza disease severity (17, 21, 53, 68, 77), and fewer neutrophils are found in the lungs of ferrets infected with virus isolates causing less-severe influenza disease (68, 89). Excessive neutrophil infiltration may contribute to acute lung injury by causing alveolar-capillary damage (17, 50, 54). Conversely, neutrophils are essential to recovery from IAV infection as part of the innate immune response as well as the adaptive immune response (49, 51, 52, 90). For example, depleting mice of neutrophils prior to IAV infection leads to increased viral replication and more severe clinical disease (53). This suggests that the balance between damage from excessive neutrophil infiltration and the generation of controlled immune responses during IAV infection is a critical determinant of disease outcome. This study shows a unique pattern of neutrophils and suggests that they are honing to specific sites of infection and inflammation during H1N1pdm virus infection. It is not known how these patterns of infiltration will change in response to a more severe IAV infection or to infection with an HPAI virus that causes immune dysregulation.
As it pertains to our findings, a majority of research on neutrophils has been centered on their relationship to acute injury (particularly acute lung injury) and bacterial infection (reviewed in references 41 to 43). Indeed, pulmonary bacterial coinfections are clinically common during and following IAV infection and are thought to represent the major risk factor for mortality from an IAV infection. Bacterial coinfection was not evident in any of the ferrets examined by histopathology. No further effort was made to detect bacteria (e.g., by PCR, IHC, or direct culture), since commensal bacteria are known to exist throughout the respiratory tract, and any pathogenic bacterial infection would be distinctive on histopathology. Further, our methodology for ferret husbandry and i.n. infection is in line with contemporary standards, and we would not expect bacterial coinfection to occur in this controlled laboratory model. Previous studies have shown that neutrophils are capable of recognizing IAV and IAV-infected cells (91–94), although discrepancies in data suggest that neutrophil responses to IAV are dependent on the viral strain (92–95). Interest in the role of neutrophils in viral disease is quickly growing (56, 96–98), and the topic deserves further research.
We have hypothesized that increases in FDG uptake following infection are due to increases in inflammatory cell infiltration into the lungs (29, 38–40). Large FDG lesions (SUVmax, >3) begin to be detected in the lungs of IAV-infected ferrets at 3 dpi and increase (SUVmax, ≥5) until 7 dpi (29). However, the density of neutrophils in the lung did not match the temporal or spatial pattern of FDG uptake in the lung. We previously reported greater uptake of FDG in the right caudal lung compared to that in other lobes, where we and others have found increased viral replication and neutrophil infiltration (29, 30, 33). Another hypothesis to explain increased FDG uptake is that IAV increases cellular glucose metabolism (99). We found that infection with IAV isolates caused an increase in FDG uptake by immortalized human epithelial cell lines (A549) in vitro but not by differentiated human bronchial epithelial cells (16HBEo−) (29, 58; also data not shown). Increased glucose levels promote the acidification of cellular endosomal compartments, a step required for IAV infection of cells (99, 100). In hospital clinics, FDG is used to detect metabolically active tumors throughout the body, including the lungs, as it is taken into cells through glucose transporters and accumulates (71, 101, 102). In this study, we did not detect an increase in the gene expression of GLUT1 (Slc2a1) or GLUT4 (Slc2a4) in lung tissues (data not shown). Thus, we could not detect IAV-dependent increased glycolysis in vivo via uptake of FDG at the times tested in this study (Fig. 7). We have reported gradually increasing FDG uptake until 7 dpi (SUVmax, >5) (29), a time point after IAV is cleared from the lungs. Therefore, we cannot support the hypothesis that FDG uptake is directly related to neutrophil infiltration or inflammation within the IAV-infected microenvironment.
We tested the hypothesis that FDG uptake was related to lung regeneration and repair after IAV-induced injury and inflammation. IHC staining for the nuclear antigen Ki-67 detects cells undergoing proliferation (i.e., cells in the S, G2, and M phases of the cell cycle) (103) and is used on biopsy tissue as a diagnostic tool to confirm a tumor diagnosis or the presence of neoplastic tissue (104). In lung cancer, Ki-67 staining is known to be correlated with FDG uptake measured by PET imaging (105, 106). We found a low background of cell proliferation in the lungs, especially in bronchioles, but a higher level in alveolar spaces. There was a marked increase in the proliferation of cells within the bronchial epithelium up to 7 days after infection with IAV (Fig. 8). This matches the timing of FDG uptake in ferrets seen previously (29) and corresponds to the increase in bronchiolitis and the presence of bronchiolar cytonecrosis, which was replaced by metaplastic bronchiolar epithelium beginning as early as 3 dpi (detected fully at 7 dpi). It is still not known whether proliferating cells contribute to the early FDG uptake (<3 dpi) in infected ferrets. Small foci of FDG are often seen in locations within the thorax soon after infection. This may indicate the initiation of an adaptive immune response (e.g., the FDG uptake is due to activated migratory dendritic cells within mediastinal lymph nodes).
The often extensive cytonecrosis of the airway epithelium, followed by metaplasia of pneumocytes and large airway epithelium (depending on the severity of disease), is a common feature of infection by many IAV subtypes, including the H1N1pdm virus (22, 46). Normally, lung cells are quiescent and enter the cell cycle only after infection and injury, as part of the regeneration process (reviewed in reference 107). We have reported a significant enrichment of genes associated with cellular proliferation in primary human bronchial epithelial cell culture after IAV infection and found lower virus replication in undifferentiated than in well-differentiated bronchial epithelial cells (58). Others have shown that ferrets infected with mild seasonal viruses, but not HPAI viruses, also show significant enrichment of genes involved in cellular growth and proliferation (108). Recently, IAV has been used to elucidate the method by which the distal airways rebuild from basal stem cell-like cells, a process that may continue for as long as 1 month postinfection (109). Our studies suggest that FDG could be used to probe the recovery of the lungs after infection. Such data could be valuable for screening compounds that assist in the repair and regeneration of the lungs during this sensitive time.
Conclusions.
The microenvironmental dynamics of IAV infection are important for controlling virus replication, inflammation, and disease progression (13, 14, 25, 78, 91); however, processes of inflammation extend beyond the site of infection (17, 18, 35, 53). Whereas in vitro experimental approaches are important for defining cellular and subcellular effects of virus infection, well-defined relevant animal models are essential for understanding how these cellular interactions define organ and organismal responses to infection (14, 15, 31, 77). These data support the importance of the whole-organ response to infection and of immunological changes that occur at viral foci. Neutrophils migrate into the lungs early and accumulate in sites of infected bronchioles, potentially responding to chemokines released by IAV-infected cells. A second wave of neutrophils, which have altered gene expression profiles suggesting differences in maturation/phenotype, is detected at 3 dpi. Live imaging of infected ferrets suggested increased FDG uptake at sites of cellular proliferation, and not necessarily at sites of leukocytic infiltration. Future studies should address several questions raised by the data presented here, such as the site-specific actions of neutrophils, the mechanism behind the biphasic temporal distribution, and the phenomenon of epithelial regeneration in the context of the entire lung or the entire organism during infection.
ACKNOWLEDGMENTS
We thank the University of Louisville Regional Biocontainment Laboratory staff, in particular Marlene Steffen, and the Research Resource Facility's vivarium staff for assistance with ferret imaging studies. We also thank the Gavin Arteel and Juliane Ingeborg-Arteel laboratories, in particular Veronica Massey, for assistance with IHC.
Financial support was provided in part by the Commonwealth of Kentucky as a Clinical and Translational Science Pilot Project Program at the University of Louisville (to C.B.J.), the Clinical Research Training Program (a public-private partnership supported jointly by the U.S. National Institutes of Health [NIH] and Pfizer Inc. via a grant to the Foundation for NIH from Pfizer Inc.), and the Center for Infectious Disease Imaging in the Intramural Research Program of the NIH.
The funders had no role in the study design, data collection and analysis, the decision to publish, or the preparation of the manuscript.
REFERENCES
- 1.Preaud E, Durand L, Macabeo B, Farkas N, Sloesen B, Palache A, Shupo F, Samson SI, Vaccines Europe influenza working group. 2014. Annual public health and economic benefits of seasonal influenza vaccination: a European estimate. BMC Public Health 14:813. doi: 10.1186/1471-2458-14-813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu PJ, Santibanez TA, Williams WW, Zhang J, Ding H, Bryan L, O'Halloran A, Greby SM, Bridges CB, Graitcer SB, Kennedy ED, Lindley MC, Ahluwalia IB, LaVail K, Pabst LJ, Harris L, Vogt T, Town M, Singleton JA, Centers for Disease Control and Prevention. 2013. Surveillance of influenza vaccination coverage—United States, 2007–08 through 2011–12 influenza seasons. MMWR Surveill Summ 62(SS-04):1–28. [PubMed] [Google Scholar]
- 3.Nichol KL, Treanor JJ. 2006. Vaccines for seasonal and pandemic influenza. J Infect Dis 194(Suppl 2):S111–S118. doi: 10.1086/507544. [DOI] [PubMed] [Google Scholar]
- 4.Pearce MB, Belser JA, Houser KV, Katz JM, Tumpey TM. 2011. Efficacy of seasonal live attenuated influenza vaccine against virus replication and transmission of a pandemic 2009 H1N1 virus in ferrets. Vaccine 29:2887–2894. doi: 10.1016/j.vaccine.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 5.Hancock K, Veguilla V, Lu X, Zhong W, Butler EN, Sun H, Liu F, Dong L, DeVos JR, Gargiullo PM, Brammer TL, Cox NJ, Tumpey TM, Katz JM. 2009. Cross-reactive antibody responses to the 2009 pandemic H1N1 influenza virus. N Engl J Med 361:1945–1952. doi: 10.1056/NEJMoa0906453. [DOI] [PubMed] [Google Scholar]
- 6.Ellebedy AH, Ducatez MF, Duan S, Stigger-Rosser E, Rubrum AM, Govorkova EA, Webster RG, Webby RJ. 2011. Impact of prior seasonal influenza vaccination and infection on pandemic A (H1N1) influenza virus replication in ferrets. Vaccine 29:3335–3339. doi: 10.1016/j.vaccine.2010.08.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shinya K, Makino A, Kawaoka Y. 2010. Emerging and reemerging influenza virus infections. Vet Pathol 47:53–57. doi: 10.1177/0300985809354464. [DOI] [PubMed] [Google Scholar]
- 8.Palese P. 2004. Influenza: old and new threats. Nat Med 10:S82–S87. doi: 10.1038/nm1141. [DOI] [PubMed] [Google Scholar]
- 9.Kilbourne ED. 2006. Influenza pandemics of the 20th century. Emerg Infect Dis 12:9–14. doi: 10.3201/eid1201.051254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neumann G, Kawaoka Y. 2006. Host range restriction and pathogenicity in the context of influenza pandemic. Emerg Infect Dis 12:881–886. doi: 10.3201/eid1206.051336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tscherne DM, Garcia-Sastre A. 2011. Virulence determinants of pandemic influenza viruses. J Clin Invest 121:6–13. doi: 10.1172/JCI44947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stevens J, Blixt O, Glaser L, Taubenberger JK, Palese P, Paulson JC, Wilson IA. 2006. Glycan microarray analysis of the hemagglutinins from modern and pandemic influenza viruses reveals different receptor specificities. J Mol Biol 355:1143–1155. doi: 10.1016/j.jmb.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 13.Matrosovich MN, Matrosovich TY, Gray T, Roberts NA, Klenk HD. 2004. Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc Natl Acad Sci U S A 101:4620–4624. doi: 10.1073/pnas.0308001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Riel D, Munster VJ, de Wit E, Rimmelzwaan GF, Fouchier RA, Osterhaus AD, Kuiken T. 2007. Human and avian influenza viruses target different cells in the lower respiratory tract of humans and other mammals. Am J Pathol 171:1215–1223. doi: 10.2353/ajpath.2007.070248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Riel D, Munster VJ, de Wit E, Rimmelzwaan GF, Fouchier RA, Osterhaus AD, Kuiken T. 2006. H5N1 virus attachment to lower respiratory tract. Science 312:399. doi: 10.1126/science.1125548. [DOI] [PubMed] [Google Scholar]
- 16.Watanabe T, Tisoncik-Go J, Tchitchek N, Watanabe S, Benecke AG, Katze MG, Kawaoka Y. 2013. 1918 Influenza virus hemagglutinin (HA) and the viral RNA polymerase complex enhance viral pathogenicity, but only HA induces aberrant host responses in mice. J Virol 87:5239–5254. doi: 10.1128/JVI.02753-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perrone LA, Plowden JK, Garcia-Sastre A, Katz JM, Tumpey TM. 2008. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog 4:e1000115. doi: 10.1371/journal.ppat.1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, Hoang DM, Chau NV, Khanh TH, Dong VC, Qui PT, Cam BV, Ha DQ, Guan Y, Peiris JS, Chinh NT, Hien TT, Farrar J. 2006. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med 12:1203–1207. doi: 10.1038/nm1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maines TR, Jayaraman A, Belser JA, Wadford DA, Pappas C, Zeng H, Gustin KM, Pearce MB, Viswanathan K, Shriver ZH, Raman R, Cox NJ, Sasisekharan R, Katz JM, Tumpey TM. 2009. Transmission and pathogenesis of swine-origin 2009 A(H1N1) influenza viruses in ferrets and mice. Science 325:484–487. doi: 10.1126/science.1177238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shieh WJ, Blau DM, Denison AM, Deleon-Carnes M, Adem P, Bhatnagar J, Sumner J, Liu L, Patel M, Batten B, Greer P, Jones T, Smith C, Bartlett J, Montague J, White E, Rollin D, Gao R, Seales C, Jost H, Metcalfe M, Goldsmith CS, Humphrey C, Schmitz A, Drew C, Paddock C, Uyeki TM, Zaki SR. 2010. 2009 pandemic influenza A (H1N1): pathology and pathogenesis of 100 fatal cases in the United States. Am J Pathol 177:166–175. doi: 10.2353/ajpath.2010.100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Itoh Y, Shinya K, Kiso M, Watanabe T, Sakoda Y, Hatta M, Muramoto Y, Tamura D, Sakai-Tagawa Y, Noda T, Sakabe S, Imai M, Hatta Y, Watanabe S, Li C, Yamada S, Fujii K, Murakami S, Imai H, Kakugawa S, Ito M, Takano R, Iwatsuki-Horimoto K, Shimojima M, Horimoto T, Goto H, Takahashi K, Makino A, Ishigaki H, Nakayama M, Okamatsu M, Takahashi K, Warshauer D, Shult PA, Saito R, Suzuki H, Furuta Y, Yamashita M, Mitamura K, Nakano K, Nakamura M, Brockman-Schneider R, Mitamura H, Yamazaki M, Sugaya N, Suresh M, Ozawa M, Neumann G, Gern J, Kida H, Ogasawara K, Kawaoka Y. 2009. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature 460:1021–1025. doi: 10.1038/nature08260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gill JR, Sheng ZM, Ely SF, Guinee DG, Beasley MB, Suh J, Deshpande C, Mollura DJ, Morens DM, Bray M, Travis WD, Taubenberger JK. 2010. Pulmonary pathologic findings of fatal 2009 pandemic influenza A/H1N1 viral infections. Arch Pathol Lab Med 134:235–243. doi: 10.1043/1543-2165-134.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mollura DJ, Asnis DS, Crupi RS, Conetta R, Feigin DS, Bray M, Taubenberger JK, Bluemke DA. 2009. Imaging findings in a fatal case of pandemic swine-origin influenza A (H1N1). AJR Am J Roentgenol 193:1500–1503. doi: 10.2214/AJR.09.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munster VJ, de Wit E, van den Brand JM, Herfst S, Schrauwen EJ, Bestebroer TM, van de Vijver D, Boucher CA, Koopmans M, Rimmelzwaan GF, Kuiken T, Osterhaus AD, Fouchier RA. 2009. Pathogenesis and transmission of swine-origin 2009 A(H1N1) influenza virus in ferrets. Science 325:481–483. doi: 10.1126/science.1177127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicholls JM, Chan MC, Chan WY, Wong HK, Cheung CY, Kwong DL, Wong MP, Chui WH, Poon LL, Tsao SW, Guan Y, Peiris JS. 2007. Tropism of avian influenza A (H5N1) in the upper and lower respiratory tract. Nat Med 13:147–149. doi: 10.1038/nm1529. [DOI] [PubMed] [Google Scholar]
- 26.Bray M, Lawler J, Paragas J, Jahrling PB, Mollura DJ. 2011. Molecular imaging of influenza and other emerging respiratory viral infections. J Infect Dis 203:1348–1359. doi: 10.1093/infdis/jir038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bellani G, Guerra L, Pesenti A, Messa C. 2010. Imaging of lung inflammation during severe influenza A: H1N1. Intensive Care Med 36:717–718. doi: 10.1007/s00134-010-1756-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mollura DJ, Morens DM, Taubenberger JK, Bray M. 2010. The role of radiology in influenza: novel H1N1 and lessons learned from the 1918 pandemic. J Am Coll Radiol 7:690–697. doi: 10.1016/j.jacr.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jonsson CB, Camp JV, Wu A, Zheng H, Kraenzle JL, Biller AE, Vanover CD, Chu YK, Ng CK, Proctor M, Sherwood L, Steffen MC, Mollura DJ. 2012. Molecular imaging reveals a progressive pulmonary inflammation in lower airways in ferrets infected with 2009 H1N1 pandemic influenza virus. PLoS One 7:e40094. doi: 10.1371/journal.pone.0040094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moore IN, Lamirande EW, Paskel M, Donahue D, Qin J, Subbarao K. 2014. Severity of clinical disease and pathology in ferrets experimentally infected with influenza viruses is influenced by inoculum volume. J Virol 88:13879–13891. doi: 10.1128/JVI.02341-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belser JA, Katz JM, Tumpey TM. 2011. The ferret as a model organism to study influenza A virus infection. Dis Model Mech 4:575–579. doi: 10.1242/dmm.007823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stark GV, Long JP, Ortiz DI, Gainey M, Carper BA, Feng J, Miller SM, Bigger JE, Vela EM. 2013. Clinical profiles associated with influenza disease in the ferret model. PLoS One 8:e58337. doi: 10.1371/journal.pone.0058337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gustin KM, Belser JA, Wadford DA, Pearce MB, Katz JM, Tumpey TM, Maines TR. 2011. Influenza virus aerosol exposure and analytical system for ferrets. Proc Natl Acad Sci U S A 108:8432–8437. doi: 10.1073/pnas.1100768108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tran V, Moser LA, Poole DS, Mehle A. 2013. Highly sensitive real-time in vivo imaging of an influenza reporter virus reveals dynamics of replication and spread. J Virol 87:13321–13329. doi: 10.1128/JVI.02381-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, Martinborough E, Peach R, Oldstone MB, Rosen H. 2011. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 146:980–991. doi: 10.1016/j.cell.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meunier I, Embury-Hyatt C, Stebner S, Gray M, Bastien N, Li Y, Plummer F, Kobinger GP, von Messling V. 2012. Virulence differences of closely related pandemic 2009 H1N1 isolates correlate with increased inflammatory responses in ferrets. Virology 422:125–131. doi: 10.1016/j.virol.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 37.Chen DL, Mintun MA, Schuster DP. 2004. Comparison of methods to quantitate 18F-FDG uptake with PET during experimental acute lung injury. J Nucl Med 45:1583–1590. [PubMed] [Google Scholar]
- 38.Chen DL, Schuster DP. 2004. Positron emission tomography with [18F]fluorodeoxyglucose to evaluate neutrophil kinetics during acute lung injury. Am J Physiol Lung Cell Mol Physiol 286:L834–L840. doi: 10.1152/ajplung.00339.2003. [DOI] [PubMed] [Google Scholar]
- 39.Goldsmith SJ, Vallabhajosula S. 2009. Clinically proven radiopharmaceuticals for infection imaging: mechanisms and applications. Semin Nucl Med 39:2–10. doi: 10.1053/j.semnuclmed.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 40.Jones HA, Clark RJ, Rhodes CG, Schofield JB, Krausz T, Haslett C. 1994. In vivo measurement of neutrophil activity in experimental lung inflammation. Am J Respir Crit Care Med 149:1635–1639. doi: 10.1164/ajrccm.149.6.7516252. [DOI] [PubMed] [Google Scholar]
- 41.Abraham E. 2003. Neutrophils and acute lung injury. Crit Care Med 31:S195–S199. doi: 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- 42.Craig A, Mai J, Cai S, Jeyaseelan S. 2009. Neutrophil recruitment to the lungs during bacterial pneumonia. Infect Immun 77:568–575. doi: 10.1128/IAI.00832-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grommes J, Soehnlein O. 2011. Contribution of neutrophils to acute lung injury. Mol Med 17:293–307. doi: 10.2119/molmed.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kolaczkowska E, Kubes P. 2013. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 45.Kuiken T, Taubenberger JK. 2008. Pathology of human influenza revisited. Vaccine 26(Suppl 4):D59–D66. doi: 10.1016/j.vaccine.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taubenberger JK, Morens DM. 2008. The pathology of influenza virus infections. Annu Rev Pathol 3:499–522. doi: 10.1146/annurev.pathmechdis.3.121806.154316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hayden FG, Fritz R, Lobo MC, Alvord W, Strober W, Straus SE. 1998. Local and systemic cytokine responses during experimental human influenza A virus infection. Relation to symptom formation and host defense. J Clin Invest 101:643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee N, Wong CK, Chan PK, Chan MC, Wong RY, Lun SW, Ngai KL, Lui GC, Wong BC, Lee SK, Choi KW, Hui DS. 2011. Cytokine response patterns in severe pandemic 2009 H1N1 and seasonal influenza among hospitalized adults. PLoS One 6:e26050. doi: 10.1371/journal.pone.0026050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tate MD, Deng YM, Jones JE, Anderson GP, Brooks AG, Reading PC. 2009. Neutrophils ameliorate lung injury and the development of severe disease during influenza infection. J Immunol 183:7441–7450. doi: 10.4049/jimmunol.0902497. [DOI] [PubMed] [Google Scholar]
- 50.Tumpey TM, Garcia-Sastre A, Taubenberger JK, Palese P, Swayne DE, Pantin-Jackwood MJ, Schultz-Cherry S, Solorzano A, Van Rooijen N, Katz JM, Basler CF. 2005. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J Virol 79:14933–14944. doi: 10.1128/JVI.79.23.14933-14944.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fujisawa H. 2001. Inhibitory role of neutrophils on influenza virus multiplication in the lungs of mice. Microbiol Immunol 45:679–688. doi: 10.1111/j.1348-0421.2001.tb01302.x. [DOI] [PubMed] [Google Scholar]
- 52.Fujisawa H. 2008. Neutrophils play an essential role in cooperation with antibody in both protection against and recovery from pulmonary infection with influenza virus in mice. J Virol 82:2772–2783. doi: 10.1128/JVI.01210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tate MD, Brooks AG, Reading PC. 2008. The role of neutrophils in the upper and lower respiratory tract during influenza virus infection of mice. Respir Res 9:57. doi: 10.1186/1465-9921-9-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, Chow VT. 2011. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol 179:199–210. doi: 10.1016/j.ajpath.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Long JP, Kotur MS, Stark GV, Warren RL, Kasoji M, Craft JL, Albrecht RA, Garcia-Sastre A, Katze MG, Waters KM, Vasconcelos D, Sabourin PJ, Bresler HS, Sabourin CL. 2013. Accumulation of CD11b+ Gr-1+ cells in the lung, blood and bone marrow of mice infected with highly pathogenic H5N1 and H1N1 influenza viruses. Arch Virol 158:1305–1322. doi: 10.1007/s00705-012-1593-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jenne CN, Wong CH, Zemp FJ, McDonald B, Rahman MM, Forsyth PA, McFadden G, Kubes P. 2013. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe 13:169–180. doi: 10.1016/j.chom.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 57.Camp JV, Chu YK, Chung DH, McAllister RC, Adcock RS, Gerlach RL, Wiemken TL, Peyrani P, Ramirez JA, Summersgill JT, Jonsson CB. 2013. Phenotypic differences in virulence and immune response in closely related clinical isolates of influenza A 2009 H1N1 pandemic viruses in mice. PLoS One 8:e56602. doi: 10.1371/journal.pone.0056602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gerlach RL, Camp JV, Chu YK, Jonsson CB. 2013. Early host responses of seasonal and pandemic influenza A viruses in primary well-differentiated human lung epithelial cells. PLoS One 8:e78912. doi: 10.1371/journal.pone.0078912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.National Research Council. 1996. Guide for the care and use of laboratory animals, 7th ed National Academies Press, Washington, DC. [Google Scholar]
- 60.Nicholls JM, Wong LP, Chan RW, Poon LL, So LK, Yen HL, Fung K, van Poucke S, Peiris JS. 2012. Detection of highly pathogenic influenza and pandemic influenza virus in formalin fixed tissues by immunohistochemical methods. J Virol Methods 179:409–413. doi: 10.1016/j.jviromet.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 61.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ogino J, Asanuma H, Hatanaka Y, Matsuno Y, Gotoda H, Muraoka S, Tsuji T, Fukazawa Y, Yamashiro K, Kondo N, Iwaki H, Miyokawa N, Hasegawa T. 2013. Validity and reproducibility of Ki-67 assessment in gastrointestinal stromal tumors and leiomyosarcomas. Pathol Int 63:102–107. doi: 10.1111/pin.12038. [DOI] [PubMed] [Google Scholar]
- 63.Hasegawa T, Yamamoto S, Nojima T, Hirose T, Nikaido T, Yamashiro K, Matsuno Y. 2002. Validity and reproducibility of histologic diagnosis and grading for adult soft-tissue sarcomas. Hum Pathol 33:111–115. doi: 10.1053/hupa.2002.30184. [DOI] [PubMed] [Google Scholar]
- 64.Wu A, Zheng H, Kraenzle J, Biller A, Vanover CD, Proctor M, Sherwood L, Steffen M, Ng C, Mollura DJ, Jonsson CB. 2012. Ferret thoracic anatomy by 2-deoxy-2-(18F)fluoro-d-glucose (18F-FDG) positron emission tomography/computed tomography (18F-FDG PET/CT) imaging. ILAR J 53:E9–E21. doi: 10.1093/ilar.53.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bagci U, Foster B, Miller-Jaster K, Luna B, Dey B, Bishai WR, Jonsson CB, Jain S, Mollura DJ. 2013. A computational pipeline for quantification of pulmonary infections in small animal models using serial PET-CT imaging. EJNMMI Res 3:55. doi: 10.1186/2191-219X-3-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Foster B, Bagci U, Ziyue X, Dey B, Luna B, Bishai W, Jain S, Mollura DJ. 2014. Segmentation of PET images for computer-aided functional quantification of tuberculosis in small animal models. IEEE Trans Biomed Eng 61:711–724. doi: 10.1109/TBME.2013.2288258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu Z, Bagci U, Foster B, Mansoor A, Mollura DJ. 2013. Spatially constrained random walk approach for accurate estimation of airway wall surfaces. Med Image Comput Comput Assist Interv 16(Part 2):559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van den Brand JM, Stittelaar KJ, van Amerongen G, Reperant L, de Waal L, Osterhaus AD, Kuiken T. 2012. Comparison of temporal and spatial dynamics of seasonal H3N2, pandemic H1N1 and highly pathogenic avian influenza H5N1 virus infections in ferrets. PLoS One 7:e42343. doi: 10.1371/journal.pone.0042343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martel CJ, Aasted B. 2009. Characterization of antibodies against ferret immunoglobulins, cytokines and CD markers. Vet Immunol Immunopathol 132:109–115. doi: 10.1016/j.vetimm.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 70.Rutigliano JA, Doherty PC, Franks J, Morris MY, Reynolds C, Thomas PG. 2008. Screening monoclonal antibodies for cross-reactivity in the ferret model of influenza infection. J Immunol Methods 336:71–77. doi: 10.1016/j.jim.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Czernin J, Benz MR, Allen-Auerbach MS. 2010. PET/CT imaging: the incremental value of assessing the glucose metabolic phenotype and the structure of cancers in a single examination. Eur J Radiol 73:470–480. doi: 10.1016/j.ejrad.2009.12.023. [DOI] [PubMed] [Google Scholar]
- 72.van den Brand JM, Stittelaar KJ, Leijten LM, van Amerongen G, Simon JH, Osterhaus AD, Kuiken T. 2012. Modification of the ferret model for pneumonia from seasonal human influenza A virus infection. Vet Pathol 49:562–568. doi: 10.1177/0300985811429812. [DOI] [PubMed] [Google Scholar]
- 73.Ramos CD, Canetti C, Souto JT, Silva JS, Hogaboam CM, Ferreira SH, Cunha FQ. 2005. MIP-1α[CCL3] acting on the CCR1 receptor mediates neutrophil migration in immune inflammation via sequential release of TNF-α and LTB4. J Leukoc Biol 78:167–177. doi: 10.1189/jlb.0404237. [DOI] [PubMed] [Google Scholar]
- 74.Reichel CA, Puhr-Westerheide D, Zuchtriegel G, Uhl B, Berberich N, Zahler S, Wymann MP, Luckow B, Krombach F. 2012. C-C motif chemokine CCL3 and canonical neutrophil attractants promote neutrophil extravasation through common and distinct mechanisms. Blood 120:880–890. doi: 10.1182/blood-2012-01-402164. [DOI] [PubMed] [Google Scholar]
- 75.Hagau N, Slavcovici A, Gonganau DN, Oltean S, Dirzu DS, Brezoszki ES, Maxim M, Ciuce C, Mlesnite M, Gavrus RL, Laslo C, Hagau R, Petrescu M, Studnicska DM. 2010. Clinical aspects and cytokine response in severe H1N1 influenza A virus infection. Crit Care 14:R203. doi: 10.1186/cc9324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zeng H, Pappas C, Katz JM, Tumpey TM. 2011. The 2009 pandemic H1N1 and triple-reassortant swine H1N1 influenza viruses replicate efficiently but elicit an attenuated inflammatory response in polarized human bronchial epithelial cells. J Virol 85:686–696. doi: 10.1128/JVI.01568-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rowe T, Leon AJ, Crevar CJ, Carter DM, Xu L, Ran L, Fang Y, Cameron CM, Cameron MJ, Banner D, Ng DC, Ran R, Weirback HK, Wiley CA, Kelvin DJ, Ross TM. 2010. Modeling host responses in ferrets during A/California/07/2009 influenza infection. Virology 401:257–265. doi: 10.1016/j.virol.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maines TR, Belser JA, Gustin KM, van Hoeven N, Zeng H, Svitek N, von Messling V, Katz JM, Tumpey TM. 2012. Local innate immune responses and influenza virus transmission and virulence in ferrets. J Infect Dis 205:474–485. doi: 10.1093/infdis/jir768. [DOI] [PubMed] [Google Scholar]
- 79.Ljungberg K, McBrayer A, Camp JV, Chu YK, Tapp R, Noah DL, Grimes S, Proctor ML, Liljestrom P, Jonsson CB, Bruder CE. 2012. Host gene expression signatures discriminate between ferrets infected with genetically similar H1N1 strains. PLoS One 7:e40743. doi: 10.1371/journal.pone.0040743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mukaida N. 2003. Pathophysiological roles of interleukin-8/CXCL8 in pulmonary diseases. Am J Physiol Lung Cell Mol Physiol 284:L566–L577. doi: 10.1152/ajplung.00233.2002. [DOI] [PubMed] [Google Scholar]
- 81.De Filippo K, Henderson RB, Laschinger M, Hogg N. 2008. Neutrophil chemokines KC and macrophage-inflammatory protein-2 are newly synthesized by tissue macrophages using distinct TLR signaling pathways. J Immunol 180:4308–4315. doi: 10.4049/jimmunol.180.6.4308. [DOI] [PubMed] [Google Scholar]
- 82.Wolpe SD, Davatelis G, Sherry B, Beutler B, Hesse DG, Nguyen HT, Moldawer LL, Nathan CF, Lowry SF, Cerami A. 1988. Macrophages secrete a novel heparin-binding protein with inflammatory and neutrophil chemokinetic properties. J Exp Med 167:570–581. doi: 10.1084/jem.167.2.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wagner JG, Roth RA. 2000. Neutrophil migration mechanisms, with an emphasis on the pulmonary vasculature. Pharmacol Rev 52:349–374. [PubMed] [Google Scholar]
- 84.Zhang X, Kluger Y, Nakayama Y, Poddar R, Whitney C, DeTora A, Weissman SM, Newburger PE. 2004. Gene expression in mature neutrophils: early responses to inflammatory stimuli. J Leukoc Biol 75:358–372. doi: 10.1189/jlb.0903412. [DOI] [PubMed] [Google Scholar]
- 85.Newburger PE, Subrahmanyam YV, Weissman SM. 2000. Global analysis of neutrophil gene expression. Curr Opin Hematol 7:16–20. doi: 10.1097/00062752-200001000-00004. [DOI] [PubMed] [Google Scholar]
- 86.Manz MG, Boettcher S. 2014. Emergency granulopoiesis. Nat Rev Immunol 14:302–314. doi: 10.1038/nri3660. [DOI] [PubMed] [Google Scholar]
- 87.Kreisel D, Nava RG, Li W, Zinselmeyer BH, Wang B, Lai J, Pless R, Gelman AE, Krupnick AS, Miller MJ. 2010. In vivo two-photon imaging reveals monocyte-dependent neutrophil extravasation during pulmonary inflammation. Proc Natl Acad Sci U S A 107:18073–18078. doi: 10.1073/pnas.1008737107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lien DC, Wagner WW Jr, Capen RL, Haslett C, Hanson WL, Hofmeister SE, Henson PM, Worthen GS. 1987. Physiological neutrophil sequestration in the lung: visual evidence for localization in capillaries. J Appl Physiol (1985) 62:1236–1243. [DOI] [PubMed] [Google Scholar]
- 89.McBrayer A, Camp JV, Tapp R, Yamshchikov V, Grimes S, Noah DL, Jonsson CB, Bruder CE. 2010. Course of seasonal influenza A/Brisbane/59/07 H1N1 infection in the ferret. Virol J 7:149. doi: 10.1186/1743-422X-7-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fujisawa H, Tsuru S, Taniguchi M, Zinnaka Y, Nomoto K. 1987. Protective mechanisms against pulmonary infection with influenza virus. I. Relative contribution of polymorphonuclear leukocytes and of alveolar macrophages to protection during the early phase of intranasal infection. J Gen Virol 68(Part 2):425–432. [DOI] [PubMed] [Google Scholar]
- 91.Ratcliffe DR, Nolin SL, Cramer EB. 1988. Neutrophil interaction with influenza-infected epithelial cells. Blood 72:142–149. [PubMed] [Google Scholar]
- 92.Daigneault DE, Hartshorn KL, Liou LS, Abbruzzi GM, White MR, Oh SK, Tauber AI. 1992. Influenza A virus binding to human neutrophils and cross-linking requirements for activation. Blood 80:3227–3234. [PubMed] [Google Scholar]
- 93.Hartshorn KL, Daigneault DE, White MR, Tauber AI. 1992. Anomalous features of human neutrophil activation by influenza A virus are shared by related viruses and sialic acid-binding lectins. J Leukoc Biol 51:230–236. [DOI] [PubMed] [Google Scholar]
- 94.Hartshorn KL, Daigneault DE, White MR, Tuvin M, Tauber JL, Tauber AI. 1992. Comparison of influenza A virus and formyl-methionyl-leucyl-phenylalanine activation of the human neutrophil. Blood 79:1049–1057. [PubMed] [Google Scholar]
- 95.Cassidy LF, Lyles DS, Abramson JS. 1989. Depression of polymorphonuclear leukocyte functions by purified influenza virus hemagglutinin and sialic acid-binding lectins. J Immunol 142:4401–4406. [PubMed] [Google Scholar]
- 96.Bataki EL, Evans GS, Everard ML. 2005. Respiratory syncytial virus and neutrophil activation. Clin Exp Immunol 140:470–477. doi: 10.1111/j.1365-2249.2005.02780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Koma T, Yoshimatsu K, Nagata N, Sato Y, Shimizu K, Yasuda SP, Amada T, Nishio S, Hasegawa H, Arikawa J. 2014. Neutrophil depletion suppresses pulmonary vascular hyperpermeability and occurrence of pulmonary edema caused by hantavirus infection in C.B-17 SCID mice. J Virol 88:7178–7188. doi: 10.1128/JVI.00254-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Raftery MJ, Lalwani P, Krautkrämer E, Peters T, Scharffetter-Kochanek K, Kruger R, Hofmann J, Seeger K, Krüger DH, Schönrich G. 2014. β2 Integrin mediates hantavirus-induced release of neutrophil extracellular traps. J Exp Med 211:1485–1497. doi: 10.1084/jem.20131092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kohio HP, Adamson AL. 2013. Glycolytic control of vacuolar-type ATPase activity: a mechanism to regulate influenza viral infection. Virology 444:301–309. doi: 10.1016/j.virol.2013.06.026. [DOI] [PubMed] [Google Scholar]
- 100.Marjuki H, Gornitzky A, Marathe BM, Ilyushina NA, Aldridge JR, Desai G, Webby RJ, Webster RG. 2011. Influenza A virus-induced early activation of ERK and PI3K mediates V-ATPase-dependent intracellular pH change required for fusion. Cell Microbiol 13:587–601. doi: 10.1111/j.1462-5822.2010.01556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kurata T, Oguri T, Isobe T, Ishioka S, Yamakido M. 1999. Differential expression of facilitative glucose transporter (GLUT) genes in primary lung cancers and their liver metastases. Jpn J Cancer Res 90:1238–1243. doi: 10.1111/j.1349-7006.1999.tb00702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.de Geus-Oei LF, van Krieken JH, Aliredjo RP, Krabbe PF, Frielink C, Verhagen AF, Boerman OC, Oyen WJ. 2007. Biological correlates of FDG uptake in non-small cell lung cancer. Lung Cancer 55:79–87. doi: 10.1016/j.lungcan.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 103.Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H. 1984. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol 133:1710–1715. [PubMed] [Google Scholar]
- 104.Brown DC, Gatter KC. 1990. Monoclonal antibody Ki-67: its use in histopathology. Histopathology 17:489–503. doi: 10.1111/j.1365-2559.1990.tb00788.x. [DOI] [PubMed] [Google Scholar]
- 105.Yamamoto Y, Nishiyama Y, Ishikawa S, Nakano J, Chang SS, Bandoh S, Kanaji N, Haba R, Kushida Y, Ohkawa M. 2007. Correlation of 18F-FLT and 18F-FDG uptake on PET with Ki-67 immunohistochemistry in non-small cell lung cancer. Eur J Nucl Med Mol Imaging 34:1610–1616. doi: 10.1007/s00259-007-0449-7. [DOI] [PubMed] [Google Scholar]
- 106.Buck AK, Halter G, Schirrmeister H, Kotzerke J, Wurziger I, Glatting G, Mattfeldt T, Neumaier B, Reske SN, Hetzel M. 2003. Imaging proliferation in lung tumors with PET: 18F-FLT versus 18F-FDG. J Nucl Med 44:1426–1431. [PubMed] [Google Scholar]
- 107.Kotton DN, Morrisey EE. 2014. Lung regeneration: mechanisms, applications and emerging stem cell populations. Nat Med 20:822–832. doi: 10.1038/nm.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cameron CM, Cameron MJ, Bermejo-Martin JF, Ran L, Xu L, Turner PV, Ran R, Danesh A, Fang Y, Chan PK, Mytle N, Sullivan TJ, Collins TL, Johnson MG, Medina JC, Rowe T, Kelvin DJ. 2008. Gene expression analysis of host innate immune responses during lethal H5N1 infection in ferrets. J Virol 82:11308–11317. doi: 10.1128/JVI.00691-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kumar PA, Hu Y, Yamamoto Y, Hoe NB, Wei TS, Mu D, Sun Y, Joo LS, Dagher R, Zielonka EM, Wang DY, Lim B, Chow VT, Crum CP, Xian W, McKeon F. 2011. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell 147:525–538. doi: 10.1016/j.cell.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]