

ABSTRACT
The assembly of influenza A virus at the plasma membrane of infected cells leads to release of enveloped virions that are typically round in tissue culture-adapted strains but filamentous in strains isolated from patients. The viral proteins hemagglutinin (HA), neuraminidase (NA), matrix protein 1 (M1), and M2 ion channel all contribute to virus assembly. When expressed individually or in combination in cells, they can all, under certain conditions, mediate release of membrane-enveloped particles, but their relative roles in virus assembly, release, and morphology remain unclear. To investigate these roles, we produced membrane-enveloped particles by plasmid-derived expression of combinations of HA, NA, and M proteins (M1 and M2) or by infection with influenza A virus. We monitored particle release, particle morphology, and plasma membrane morphology by using biochemical methods, electron microscopy, electron tomography, and cryo-electron tomography. Our data suggest that HA, NA, or HANA (HA plus NA) expression leads to particle release through nonspecific induction of membrane curvature. In contrast, coexpression with the M proteins clusters the glycoproteins into filamentous membrane protrusions, which can be released as particles by formation of a constricted neck at the base. HA and NA are preferentially distributed to differently curved membranes within these particles. Both the budding intermediates and the released particles are morphologically similar to those produced during infection with influenza A virus. Together, our data provide new insights into influenza virus assembly and show that the M segment together with either of the glycoproteins is the minimal requirement to assemble and release membrane-enveloped particles that are truly virus-like.
IMPORTANCE Influenza A virus is a major respiratory pathogen. It assembles membrane-enveloped virus particles whose shapes vary from spherical to filamentous. Here we examine the roles of individual viral proteins in mediating virus assembly and determining virus shape. To do this, we used a range of electron microscopy techniques to obtain and compare two- and three-dimensional images of virus particles and virus-like particles during and after assembly. The virus-like particles were produced using different combinations of viral proteins. Among our results, we found that coexpression of one or both of the viral surface proteins (hemagglutinin and neuraminidase) with the viral membrane-associated proteins encoded by the M segment results in assembly and release of filamentous virus-like particles in a manner very similar to that of the budding and release of influenza virions. These data provide novel insights into the roles played by individual viral proteins in influenza A virus assembly.
INTRODUCTION
Influenza A virus, which is a major human pathogen, assembles enveloped virions at the plasma membrane of the host cell. The viral envelope is studded with the viral glycoproteins hemagglutinin (HA) and neuraminidase (NA). The membrane further contains the viral ion channel M2 (1) and is coated with a helical array of M1 proteins on its internal surface (2). Influenza A virus has a segmented, negative-sense RNA genome, with one copy of each of its eight genomic RNAs packaged into the virion as a ribonucleoprotein particle (vRNP) (3). Virus assembly begins with HA- and NA-dependent recruitment of M1 to the plasma membrane. M1 in turn interacts with vRNPs (4). The M2 protein is thought to play a role in the final membrane scission step (5), and the sialidase activity of NA is required to prevent the newly produced virion from attachment to the producer cell via HA interacting with surface sialic acid residues (4). NA was reported to concentrate at the end of the virion opposite the vRNP, where it may be able to efficiently perform its enzymatic function of releasing the virus from the cell (2, 6). Released influenza A virions are pleiomorphic. Spherical forms of virions predominate in laboratory-adapted influenza A virus strains, such as PR8 or A/WSN/1933 (7, 8), whereas primary or low-passage-number isolated viruses are typically filamentous (9–13). The virion morphology also depends on the virus strain and host cell type (14). A recent study showed that PR8 becomes predominantly filamentous when passaged in guinea pigs, suggesting that the filamentous morphology is advantageous in vivo (8).
Expression of influenza virus proteins by use of a plasmid-derived expression system makes it possible to study the roles of individual proteins during assembly and release, as well as their influence on the morphology of the virion, in a systematic manner. It has been shown that expression of HA alone in 293T cells causes the release of spherical, HA-containing vesicles into the supernatant when exogenous NA enzymatic activity is supplemented (15). NA and M2 have also been reported to induce release of membrane-enveloped vesicles when expressed alone, though at much lower levels (15), and the level of NA release when this protein is expressed individually seems to be strain dependent (16). Furthermore, expression of NA alone from certain influenza A virus strains can produce particles with a filamentous morphology (16). M1 does not normally traffic to the plasma membrane when expressed alone, and consequently, it does not drive particle assembly and release unless it is artificially recruited to the plasma membrane by addition of a targeting peptide (17) or is overexpressed using a vaccinia virus expression system (18). M1 can be recruited to the plasma membrane by HA or NA via the respective cytoplasmic tail (19, 20), and also by the M2 protein (17).
The M segment (segment 7) of influenza A virus, which encodes the M1 and M2 proteins, appears to contain genetic determinants of filamentous morphology (2, 21–28). Surprisingly, however, plasmid-derived coexpression of all viral proteins from the filamentous A/Udorn/72 influenza virus strain resulted in the formation of spherical particles, and the omission of M1 did not alter their morphology as seen by negative-staining electron microscopy (EM) (15). These results suggested that plasmid-derived expression of influenza A virus proteins may not faithfully recapitulate the viral assembly and release pathway and indicated that HA or NA, but not M1, is required for particle assembly and release (4). Interestingly, M2, a key mediator of membrane fission in influenza A virus budding, was also dispensable for particle release upon plasmid-based expression of influenza A virus proteins (5). Thus, formation of differently shaped vesicles and virus-like particles (VLPs) following expression of influenza A virus proteins in the absence of viral infection may differ from assembly and release of the complete influenza A virus, and the structural roles of individual viral proteins in this process are currently incompletely understood.
For this report, we analyzed particles produced by individual or combined expression of influenza A virus HA, NA, and M segment and compared their morphology with that of influenza A virions. Particle release was characterized biochemically and by EM, electron tomography (ET), and cryo-ET to give a detailed view of membrane alterations, glycoprotein sorting, and particle morphology. These data reconcile some of the apparent contradictions present in the literature and provide novel insights into the roles of individual viral proteins in influenza A virus assembly.
MATERIALS AND METHODS
Plasmids.
DNA sequences corresponding to the coding regions of HA (A/Aichi/2/68), NA (A/Singapore/1/57), and the M segment (A/Hong Kong/1/68), cloned into the eukaryotic expression vector pCAGGS (29), were a kind gift from H. D. Klenk, Marburg, Germany. Transfection with the plasmid corresponding to the M segment (henceforth referred to as “M”) led to expression of both M1 and M2. Sequences of HA (A/Hong Kong/1/68) and NA (A/Hong Kong/1/68) from pHW2000_HA_Hong Kong 68 and pHW2000_NA_Hong Kong 68 (both gifts from J. Stech, Riems, Germany) were cloned into pCAGGS by using EcoRI and NotI restriction sites.
Cells.
293T human embryonic kidney cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS), l-glutamine, and penicillin-streptomycin at 37°C and 5% CO2.
Antibodies.
Polyclonal rabbit serum raised against a peptide homologous to a 25-amino-acid sequence at the C terminus of HA1 (A/X31 [H3N2]) was a kind gift from J. Zimmerberg (National Institutes of Health, Bethesda, MD). Polyclonal goat serum raised against NA (A/Singapore/1/57 [H2N2]) was obtained from BEI Resources (Manassas, VA). A monoclonal mouse anti-M1 antibody (30) was a gift from J. Yewdell (National Institutes of Health, Bethesda, MD). A monoclonal mouse anti-M2 antibody (14C2) was purchased from Abcam (Cambridge, MA). Antibodies against influenza virus proteins were diluted 1:2,000 or 1:1,000 (mouse anti-M2) for Western blotting and 1:150 for immunofluorescence microscopy. Mouse anti-β-actin (Sigma-Aldrich) was used at a 1:5,000 dilution for Western blotting. Alexa 647–donkey anti-goat, Alexa 405–goat anti-rabbit, and Alexa 488–goat anti-mouse (Invitrogen) were used as secondary antibodies for immunofluorescence microscopy, at a dilution of 1:1,000.
Particle preparation.
For Western blot analysis, 293T cells were seeded in a 6-cm-diameter tissue culture dish (1 × 106 cells/dish) and grown in medium without antibiotics. Twenty-four hours later, cells were transfected with the appropriate plasmids by using Trans-IT LT1 (Mirus) according to the manufacturer's instructions. Particles were produced using the following plasmid amounts: 0.5 μg pCAGGS-HA, 0.5 μg pCAGGS-NA, and 1 μg pCAGGS-M. At 5 h posttransfection, if required, exogenous bacterial neuraminidase from Clostridium perfringens (Sigma-Aldrich) was added at a concentration of 100 mU/ml. At 48 h posttransfection, the supernatant was collected and clarified by centrifugation at 2,000 × g for 10 min at 4°C. Subsequently, the supernatant was layered onto a 1-ml 30% (wt/vol) sucrose-NTE (100 mM NaCl, 10 mM Tris [pH 7.4], 1 mM EDTA) cushion and centrifuged at 200,000 × g for 2 h at 4°C in a Beckman Optima centrifuge using a TLA 100.4 rotor (Beckman Coulter, Fullerton, CA). The pellet was resuspended in 1× lithium dodecyl sulfate (LDS) loading buffer (NuPAGE) containing 100 mM dithiothreitol.
For EM, particles were prepared as described above, with the following modifications: cells were seeded in 6-well plates at 5 × 105 cells/well; transfection was carried out using FuGENE 6 (Roche); at 5 h posttransfection, the medium was exchanged for serum-free DMEM supplemented with l-glutamine and penicillin-streptomycin; 2 ml of supernatant was layered onto a 0.3-ml sucrose gradient and centrifuged at 165,000 × g in a TLA 120.1 rotor; and the pellet was resuspended in NTE.
To prepare cell lysates for Western blot analysis, transfected cells were lysed in 750 μl of lysis buffer (20 mM Tris-Cl [pH 7.4], 2% sodium dodecyl sulfate [SDS]) and homogenized by passage 3× through a 500-μl Hamilton syringe, and samples were prepared by adding 4× LDS loading buffer (NuPAGE) and dithiothreitol (final concentration, 100 mM). Cell lysates and released particles were analyzed in 4 to 12% SDS-polyacrylamide gels (NuPAGE) and then transferred to polyvinylidene difluoride membranes (Invitrogen) by using a Fastblot 44 semidry system (Biometra, Göttingen, Germany). Membranes with proteins were blocked in 5% low-fat milk in Tris-buffered saline (TBS) and then incubated with appropriate antibodies diluted in TBS containing 3% bovine serum albumin (Sigma) and 1% Tween 20 (Bio-Rad) for 1 h. Proteins were quantified with an Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE) and analyzed with the ImageJ-based image analysis program FIJI (31), using a gel macro.
Preparation of influenza virus A/Hong Kong/8/68.
Egg-propagated and sucrose gradient-purified influenza A virus (A/Hong Kong/8/68) was purchased from Charles River Laboratories (Wilmington, MA). 293T cells were seeded on a polylysine-coated 24-well plate (2.5 × 103 cells/well) 1 day prior to infection. Confluent cells were washed twice with serum-free SFM4 MegaVir medium (Thermo Fisher Scientific, Inc.) and inoculated with 100 μl of A/Hong Kong/8/68 (106.6 50% egg infective doses [EID50]). After 2 h, the virus inoculum was removed and cells were washed twice with SFM4 MegaVir medium. Infection was carried out in DMEM supplemented with l-glutamine, penicillin-streptomycin, 10% FCS, and 25 μg/ml of tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin (Sigma) at 37°C. At 5 days postinfection, the culture medium was clarified by centrifugation at 2,000 × g for 10 min at 4°C, and the supernatant was centrifuged at 130,000 × g for 30 min at 4°C in a TLA 100.3 rotor. The pellet was resuspended in NTE and used for cryo-EM. For thin-section EM, the infection was carried out with the following modifications: 293T cells were seeded on a polylysine-coated 6-well plate (3 × 106 cells/well), inoculated with 100 μl of A/Hong Kong/8/68 (3 × 106.6 EID50), fixed with 2.5% glutaraldehyde at 48 h postinfection, and processed as described for plasmid-derived particles.
Fluorescence microscopy.
293T cells (5 × 105 cells) were seeded on coverslips in 6-well plates. After 24 h, the cells were transfected with pCAGGS-HA, -NA, and -M as described above and fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) at 48 h posttransfection. Cells were permeabilized with 0.1% Triton X-100 in PBS, blocked in 5% bovine serum albumin (Sigma) in PBS, and incubated with antibodies against influenza virus proteins. After washing (3 times with PBS), the cells were incubated with Alexa 647–donkey anti-goat for 1 h, washed again, and then incubated with Alexa 405–goat anti-rabbit and Alexa 488–goat anti-mouse for 1 h. Confocal slices (4 μm thick) were acquired with a 100× objective, using a Zeiss 510 confocal microscope with random sampling, and transfected cells were quantified using FIJI.
Sample preparation for thin-section electron microscopy.
For thin-section electron microscopy, transfected or infected 293T cells were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) for 1 h at room temperature and then fixed with OsO4 in 0.1 M cacodylate buffer (pH 7.2) for 30 min at 4°C. Specimens were subsequently dehydrated with ethanol, stained en bloc with 6% uranyl acetate in 70% ethanol for 2 h at 4°C, and further dehydrated with 80%, 90%, 95%, and, finally, 100% ethanol. To detach the cells, the dish was partially dissolved by use of propylene oxide. The cells were further embedded in epoxy resin (Glycid ether 100; Carl Roth, Karlsruhe, Germany) and polymerized for 24 h at 60°C. Fifty- and 300-nm sections were obtained with a Leica Ultracut UCT microtome and a diamond knife (Diatome).
Electron microscopy and tomography.
Thin sections were poststained with 2% lead citrate in water and examined in a Morgagni electron microscope (FEI). Three-hundred-nanometer sections were decorated with 10-nm protein A-gold particles on both sides of the section, and single- or dual-axis electron tomography was performed in an FEI Tecnai F30 electron microscope operated at 300 kV and equipped with an FEI Eagle charge-coupled device (CCD) camera. Tomographic tilt ranges were typically from +60° to −60°, with an angular increment of 1°. For cryo-electron tomography, isolated particles were resuspended in NTE buffer applied on holey-carbon grids and vitrified by plunging into liquid ethane using a Vitrobot machine (FEI). Vitrified samples were imaged for tomography in an FEI TF30 Polara instrument equipped with a Gatan GIF2002 energy filter and operated at 300 kV. Tomographic tilt series were acquired at a defocus of −5 μm at 0.494 nm/pixel, typically from +60° to −60° (angular increment, 3°), with a total dose of 6,000 to 10,000 e−/nm2. ET and cryo-EM of influenza virus-infected cells and virions, respectively, were performed on a Tecnai 20 microscope operated at 200 kV and equipped with a Gatan K2 base camera. Tomograms were reconstructed using the IMOD software suite (32).
RESULTS
Release of M1 is driven by HA or NA.
To assess the roles of influenza A virus proteins in virus particle assembly and structure, we transfected 293T cells with expression vectors containing DNA sequences corresponding to the coding regions of the HA (A/Aichi/2/68), NA (A/Singapore/1/57), and M (A/Hong Kong/1/68) viral RNA segments, either individually or in combinations. For combinations where NA was not expressed, exogenous NA was added to the medium to complement its enzymatic function. To verify the efficiency of cotransfection, cells transfected with the combination of all three plasmids were fixed at 48 h posttransfection and immunostained with antibodies against the HA, NA, and M1 proteins (Fig. 1A). Approximately 72% of the cells were productively transfected, among which 84% expressed all three proteins; 12% of transfected cells coexpressed two proteins, and 4% expressed only one protein (n = 70 cells).
FIG 1.

Expression and release of HA, NA, and M segment proteins. (A) Fluorescence micrographs of 293T cells cotransfected with expression plasmids for HA (A/Aichi/2/68), NA (A/Singapore/1/57), and the M segment (A/Hong Kong/1/68). Cells were fixed, permeabilized, and labeled with anti-HA (blue), anti-NA (red), and anti-M1 (green) antibodies at 48 h posttransfection. White arrows indicate filamentous protrusions. (B) Western blot analysis of lysates and of pellets after supernatant centrifugation through a 30% sucrose cushion. 293T cells were transfected with expression plasmids for HA (A/Aichi/2/68), NA (A/Singapore/1/57), and M (A/Hong Kong/1/68) alone or in combination, as indicated at the top of the figure, and harvested at 48 h posttransfection. M gives rise to expression of the M1 (28 kDa) and M2 (14 kDa) proteins. exoNA, treatment of transfected cells with exogenous NA to substitute for the enzymatic function of influenza virus NA. Respective antisera are indicated on the left; anti-actin was used to immunostain cell lysates as a loading control.
To assess the extent of particle release, cleared culture medium was centrifuged through a 30% sucrose cushion, and the pellet was collected. Influenza A virus protein levels in this fraction were compared to those in whole-cell lysates by Western blotting (Fig. 1B). When expressed alone, both HA and NA yielded release of particles carrying the respective protein. In contrast, transfection of DNA corresponding to the M segment (henceforth referred to as “M”) resulted in efficient expression of M1 and M2 (Fig. 1B) but did not lead to release of particles carrying M proteins unless HA or NA (or both) was coexpressed. When M was coexpressed with NA (NAM) or both HA and NA (HANAM), M1/M2-carrying particles were released much more efficiently than with coexpression with HA only (Fig. 1B).
Influenza A virus glycoproteins remodel the plasma membrane.
To assess morphological changes of the plasma membrane caused by expression of viral proteins, transfected cells were fixed at 48 h posttransfection and prepared for EM by being embedded in epoxy resin and thin sectioned according to standard protocols. Representative examples of thin sections for the different transfections as well as for untransfected cells are shown in Fig. 2. When M was transfected alone, the plasma membrane did not show any striking ruffling or formation of protrusions compared to untransfected cells, and no M1-specific electron density was observed. In all other cases, expression of the viral proteins strongly altered the morphology of the plasma membrane. When HA was expressed alone, the plasma membrane appeared to be ruffled and showed irregularly shaped protrusions of variable size (Fig. 2A). No regular filamentous protrusions were observed, however. In addition to plasma membrane alterations, HA expression also caused modification of some intracellular membrane compartments to form cisternae with tightly apposed membranes (Fig. 2B).
FIG 2.

EM analysis of the plasma membranes of cells expressing influenza A virus structural proteins. 293T cells either left untransfected or transfected with expression plasmids for HA (A/Aichi/2/68), NA (A/Singapore/1/57), and M (A/Hong Kong/1/68), alone or in combination, were processed for thin-section EM at 48 h posttransfection. (A) Images of representative sections of the cell surface. The (co)transfected construct(s) is indicated in the upper left region of each section. HA(+exoNA), treatment of HA-expressing cells with exogenous NA to substitute for the enzymatic NA activity. (B) HA expression alone induced formation of apposed membrane structures in the cytoplasm. The right panel shows an enlargement of the boxed region in the left panel.
When NA was expressed alone, the plasma membrane showed a mixture of filamentous and irregular protrusions (Fig. 2A). HA and NA expressed in combination (HANA) yielded a similar mixture of filamentous and irregular plasma membrane protrusions (Fig. 2A). In all cases where M was transfected in combination with HA, NA, or both, filamentous protrusions were seen at the plasma membrane that appeared more ordered and straighter than those in the experiments lacking M (Fig. 2A, compare upper and middle rows).
To assess the contribution of the enzymatic activity of NA to altered plasma membrane morphology, we also transfected 293T cells with HA, with (HAM) or without M, in the absence of added exogenous NA (Fig. 2A, third row). Membrane protrusions also formed in this case but did not point away from the cell surface as observed when exogenous NA was added. Instead, these protrusions appeared to be packed against the plasma membrane surface, most likely due to binding of HA to sialic acid residues in the absence of NA-mediated sialic acid removal. This effect was particularly striking for the filamentous protrusions formed in HA- and M-transfected cells (Fig. 2A).
ET reveals different budding structures and glycoprotein distributions.
To obtain a more detailed view of the structures induced by expression of the different influenza A virus proteins at the plasma membrane, we cut 300-nm-thick sections of transfected, chemically fixed, and epoxy resin-embedded 293T cells and imaged them by ET. Computational reconstruction methods were used to achieve a 3D view of the cell structure within the section. This gave a clearer view of the ultrastructure than conventional thin-section EM and allowed the 3D topology of the cell surface to be visualized directly.
As suggested by thin-section EM, HA expression alone induced pleomorphic protrusions and ruffles in the membrane, rather than single, morphologically well-defined budding events (Fig. 3A). Expression of NA, alone or together with HA, induced a mixture of pleomorphic and quasi-filamentous protrusions. Coexpression of M with either HA or NA led to formation of much straighter filamentous protrusions, which were uniform in diameter along their length (Fig. 3A to C). Similar results were obtained when M was coexpressed with both NA and HA, suggesting that the filamentous phenotype does not depend on the type of glycoprotein (HA or NA) but is caused by the recruitment of M proteins to the budding site.
FIG 3.
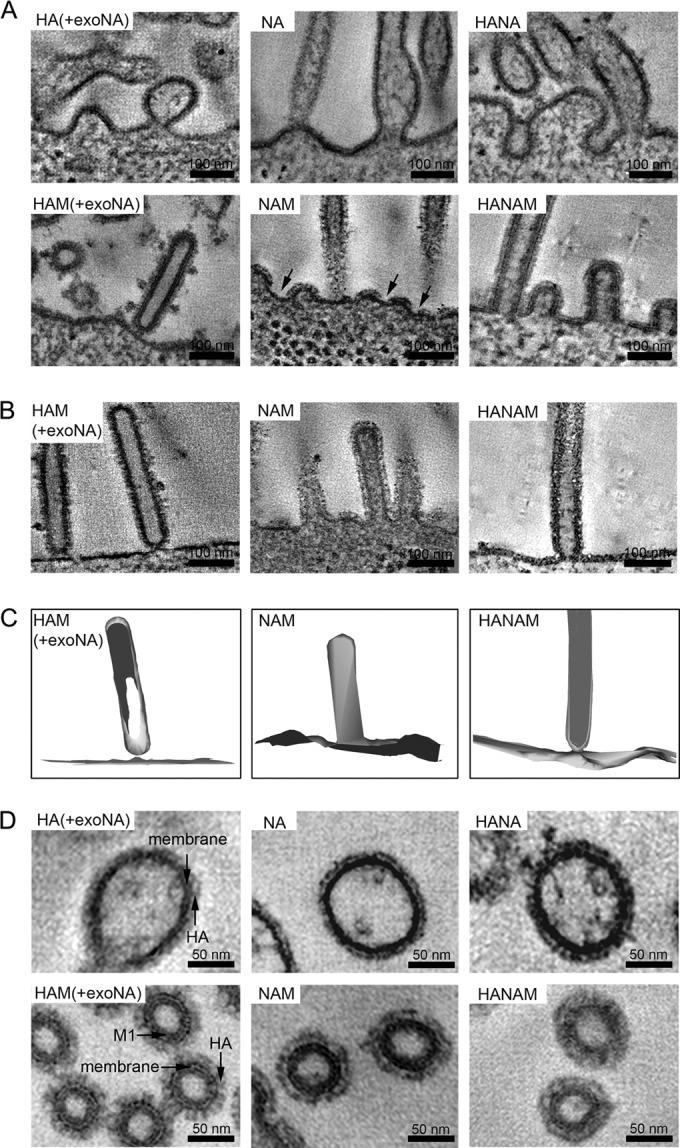
ET of the plasma membrane shows different budding intermediates and glycoprotein distributions. (A) Computational slices from electron tomograms of 293T cells transfected with expression plasmids for HA (A/Aichi/2/68), NA (A/Singapore/1/57), and M (A/Hong Kong/1/68), alone or in combination. Transfected cells were chemically fixed and embedded in epoxy resin at 48 h posttransfection. Arrows mark regions of the plasma membrane that appear to be free of glycoproteins. (B) Computational slices through budding intermediates corresponding to the 3D renderings shown in panel C. (C) 3D renderings of the different budding intermediates shown in panel B. (D) Transverse slices through budding particles. When M was coexpressed, a thickening of the membrane was observed.
Different stages of straight filament formation were observed in these studies. In the case of coexpression of the HA and M segments, elongated filaments predominated. These filaments often exhibited a narrow neck where they connected with the plasma membrane, suggestive of being in a late step of particle release (Fig. 3B); in other cases, they appeared to have completed budding but remained attached to the cell surface via a glycoprotein interaction (Fig. 3A and C). When NA and M were coexpressed, the cell surface exhibited a mixture of elongated filaments and small nascent budding structures (Fig. 3A). The elongated NAM filamentous protrusions were mostly open toward the cytoplasm, without a constricted neck (Fig. 3A and B). When HA, NA, and M were coexpressed in combination, a mixture of all these phenotypes was observed (often in the same cell), including small buds and extended filaments, with or without a constricted neck (Fig. 3A and B).
The ET images, which were clearer than the EM images, allowed an analysis of the distribution of viral glycoproteins on filamentous membrane protrusions. When M was coexpressed with HA, NA, or both, the membrane of the protrusions appeared thicker than that in pleomorphic protrusions lacking M. This difference was presumably caused by the presence of the M1 layer underneath the membrane (Fig. 3D). When the cells were transfected with HA, NA, or both, but without M, the membrane was covered by a uniform layer of glycoproteins, both along the protrusions and on the membrane between protrusions. In HAM-transfected cells, the membranes of the protrusions and of the budding structures were always densely coated with glycoproteins, while the plasma membrane between protrusions was heterogeneous, with some areas appearing more densely (Fig. 3A) and some more sparsely coated (Fig. 3B). A more prominent effect on the plasma membrane between protrusions was seen in NAM-transfected cells: the protrusions remained densely coated with glycoproteins in this case as well, while the membrane between protrusions appeared to lack glycoproteins (Fig. 3A, arrows). In cells where all three influenza A virus structural segments were coexpressed, a mixed phenotype was seen, with some cells showing glycoproteins only on protrusions and others also showing glycoproteins on the membrane between protrusions. These observations suggest that the M1 protein forms a layer underneath the membrane which induces clustering of both HA and NA into protrusions but that NA is clustered to a greater extent than HA.
All the experiments described above made use of HA from A/Aichi/2/68, NA from A/Singapore/1/57, and M from A/Hong Kong/1/68. To verify that the morphologies observed were not peculiar to the use of the corresponding strains and to rule out incompatibility effects of coexpressing structural proteins from different influenza A virus strains, we performed ET of the plasma membranes of cells expressing combinations of HA from A/Hong Kong/1/68, NA from A/Hong Kong/1/68, and M from A/Hong Kong/1/68 (Fig. 4). These experiments revealed results very similar to those reported above: coexpression of M with HA or NA (or both) again led to the production of long filamentous protrusions, which were straighter and better defined than those with NA alone, while the degree of membrane ruffling for expression of NA from A/Hong Kong/1/68 alone was lower than that seen with NA from A/Singapore/1/57.
FIG 4.

ET of plasma membrane alterations for expression of structural proteins from the influenza virus A/Hong Kong/1/68. Computational slices from electron tomograms show 293T cells transfected with plasmids containing NA, NAM, HAM, and HANAM, all from A/Hong Kong/1/68. Transfected cells were chemically fixed and embedded in epoxy resin at 48 h posttransfection. (A) Low-magnification overviews of the plasma membrane alterations. (B) Magnified views of panel A or of other tomograms of the same sample.
Cryo-ET of released particles.
To compare the respective alterations at the plasma membrane induced by the different influenza A virus membrane-associated proteins to the morphologies of extracellular particles released from transfected cells, we performed cryo-ET of purified particles. Overview images and higher-magnification views are shown in Fig. 5A. Particles formed by HA (A/Aichi/2/68), by NA (A/Singapore/1/57), or by coexpression of HA and NA were highly pleomorphic, with NA particles showing occasional extended filamentous regions (Fig. 5A, upper panels). In contrast, particles produced following coexpression of M with either HA, NA, or both HA and NA formed long filaments of up to several micrometers long (Fig. 5A, lower panels). Thus, expression of M promotes the formation of long filamentous particles.
FIG 5.
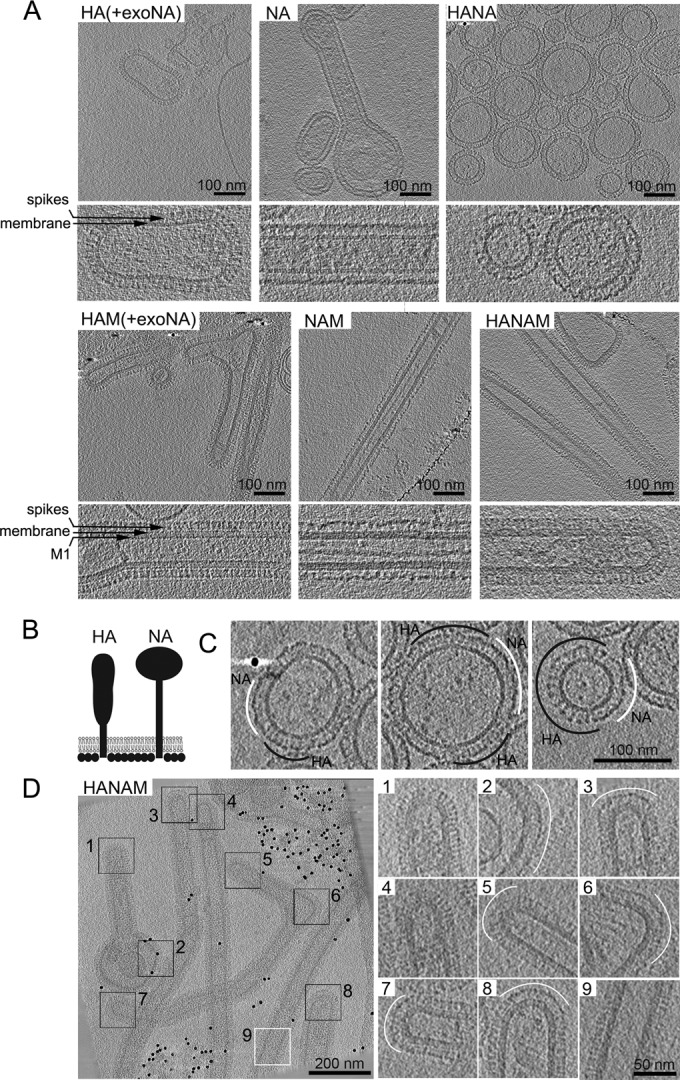
Cryo-ET of isolated released particles. The images show computational slices of cryo-electron tomograms of isolated particles produced from 293T cells expressing HA (A/Aichi/2/68), NA (A/Singapore/1/57), and M (A/Hong Kong/1/68), alone or in combination. (A) Pairs of panels show overview images (upper) and higher-magnification views (lower) for the different combinations (as indicated in each panel). The M1, membrane, and glycoprotein spike layers are annotated in the higher-magnification views. (B) Schematic representation of HA and NA shapes as seen by cryo-ET. (C) HA and NA formed separate clusters in membrane-enveloped particles carrying both HA and NA. HA and NA clusters are marked with black and white arcs, respectively. (D) HA and NA formed separate clusters in filamentous particles resulting from coexpression of HA, NA, and M. Membranes that curve in two dimensions (spherically curved), including tips and bends in filaments, are marked with black boxes. Panels 1 to 8 show tomogram slices of these areas at a higher magnification. NA clusters (white arcs) are preferentially found on the spherically curved areas. Note that one particle has NA clusters on both tips (boxes 5 and 7) and in a bend (box 6). HA is preferentially found along the cylindrically curved sides of the filamentous particle (white box 9).
Cryo-electron tomograms of particles produced from cells coexpressing M with one or both viral glycoproteins always exhibited a thickening of the plasma membrane that can be attributed to the M1 layer (Fig. 5A, compare larger-magnification images of the upper and lower panels). This layer covered the entire inside of the membrane, including both the sides and the tips of filamentous particles. HA and NA in released particles could be distinguished according to their characteristic shapes, which are represented schematically in Fig. 5B. NA showed less electron density in the stem region than HA (Fig. 5A, compare HA and HAM with NA and NAM). When HA and NA were coexpressed, they separated into clusters of variable size (Fig. 5C), which appeared to segregate into different regions of the filamentous particles when the M1 layer was present (Fig. 5D). HA mainly covered the sides of these filaments, whereas NA clusters mainly localized to the filament tips and were also found in bends or spherical bulges connected to filamentous particles. In some cases, multiple NA clusters were observed (NA clusters 5 to 7 in Fig. 5D are all on the same particle).
Particle formation by coexpression of M with one of the viral glycoproteins resembles assembly of influenza A virions.
To assess whether particle production by coexpression of influenza A virus structural proteins mimicked bona fide virion assembly, we infected 293T cells with influenza A virus (Hong Kong/8/68) and performed EM and ET on infected cells. Large numbers of filamentous protrusions were seen on the surfaces of infected cells (Fig. 6A) by thin-section EM. ET revealed the presence of small budding structures as well as filamentous protrusions that appeared to be attached to the plasma membrane either via a constricted neck or by glycoproteins (Fig. 6B). We did not observe any filamentous protrusions connected to the cell without a constricted neck. Both small budding structures and the filamentous protrusions contained electron-dense material, which may represent the vRNPs and was not observed in particles formed by coexpression of viral structural proteins. We also assessed the morphology of virions released from 293T cells by using cryo-EM. The virions assembled long filaments with a diameter and appearance similar to those of particles formed upon cotransfection of M with HA, NA, or both (compare Fig. 5 with Fig. 6C). Although the cryo-EM projection images are suggestive of the presence of NA at the tips of the filamentous virus particles, it is not possible to reliably distinguish NA and HA in these images.
FIG 6.
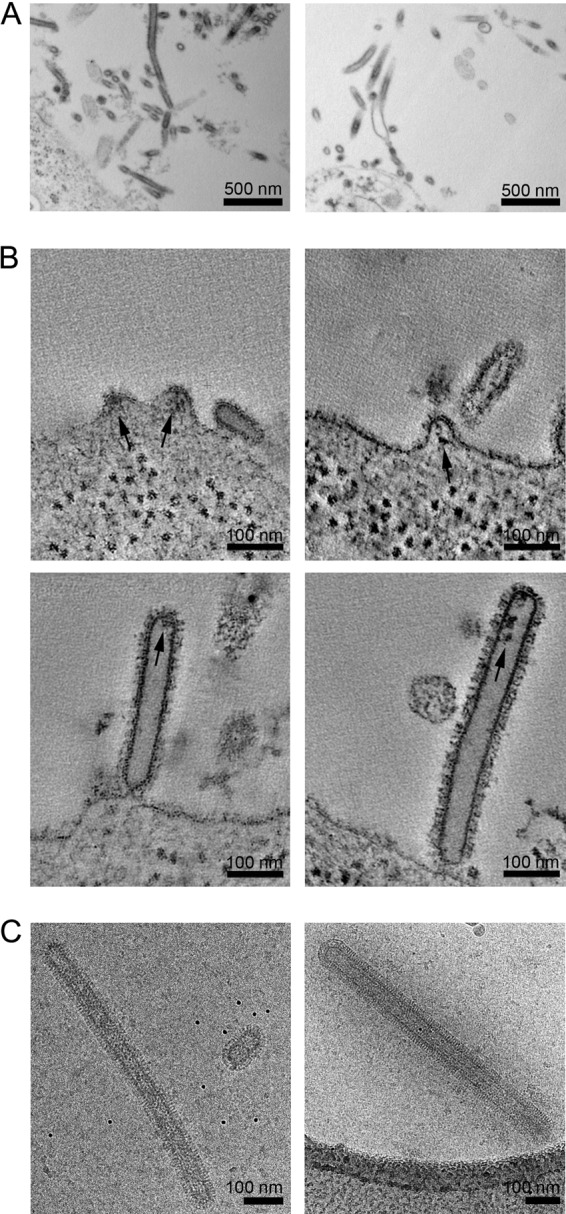
Morphologies of budding and released influenza virus A/Hong Kong/8/68. (A) Micrographs of thin-sectioned infected 293T cells at 2 days postinfection. Viruses budding from the plasma membrane of the 293T cell show a filamentous morphology. (B) Computational slices through electron tomograms showing the plasma membrane morphologies of infected cells, including small buds containing density likely corresponding to vRNP (arrows), virions attached to the cell via constricted necks, and released virions. (C) Cryo-electron micrographs of released virions.
DISCUSSION
In this study, we used a plasmid-derived expression system to produce influenza A virus proteins corresponding to the HA, NA, and M segments in 293T cells and analyzed their roles in particle assembly and morphology by using EM, ET, and cryo-ET, including a comparison with bona fide influenza A virus assembly. M encodes the M1 protein and can produce two other mRNAs by splicing: mRNA3 encodes a small peptide of nine amino acids, whose absence does not affect infection in tissue culture (33), and mRNA2 gives rise to the M2 ion channel, which appears to be critical for release of the viral bud (5). Splicing of the M segment mRNA has been reported to require the synthesis of other viral proteins (34), in addition to the host splicing factor SF2/ASF (35), and was therefore not expected to take place when M was transfected alone. Nevertheless, transfection of a plasmid carrying only M yielded production of M1 and M2 in the absence of any other influenza A virus protein, suggesting that influenza A virus M RNA splicing is independent of other viral proteins, at least in transfected 293T cells.
Expression of HA alone in transfected 293T cells led to the release of a particulate antigen that could be recovered following centrifugation through a sucrose cushion. Release of HA-derived particles into the culture medium was dependent on addition of exogenous NA activity to avoid immediate binding to the producer cells. Similar particles were formed in the absence of NA activity but remained stuck to the cell surface. The M1 protein did not lead to particle release when expressed alone and required one of the two glycoproteins (preferentially NA) to yield extracellular particles, consistent with previous reports (15, 17). In contrast to these reports, we did not observe M1 release when both M1 and M2 were expressed in the absence of influenza virus glycoproteins (15, 17). The increased release of M1 and M2 when these were coexpressed with NA compared to that on coexpression with HA may reflect different contributions of the glycoproteins during assembly or more efficient enzymatic cleavage by membrane-associated NA than by exogenously added NA. Expression of NA (either alone or in combination with other viral structural proteins) always led to release of extracellular particles, which was more efficient for NA from A/Singapore/1/57 than for NA from A/Hong Kong/1/68. These differences may be due to differences in the respective cellular expression levels.
Biochemical release assays do not provide information on the mode of antigen release or the form of the released particles, and we therefore characterized the morphologies of the plasma membrane and the released particles in the viral protein-expressing cells. We found that these morphologies changed according to which combination of proteins was expressed. Individual expression of HA induced polymorphic, bleb-like plasma membrane protrusions, while expression of NA from A/Singapore/1/57 alone yielded a mixture of polymorphic and quasi-filamentous protrusions (as also reported in reference 36). In both cases, the plasma membrane of the viral protein-expressing cells appeared to be completely coated with the viral proteins, and they did not form a particular budding structure. Expression of NA from A/Hong Kong/1/68 resulted in a smaller number of polymorphic protrusions. The morphology of the released particles reflected the plasma membrane morphology: HA expression led to the release of pleomorphic, roughly round particles, while NA expression led to the release of pleomorphic particles, some of which exhibited a quasi-filamentous morphology. Such heterogeneous, pleomorphic particles do not reflect the filamentous morphology of the influenza virus A/Hong Kong/68 propagated in 293T cells (Fig. 6), and we hypothesize that these particles are released in a nonspecific manner by the massive membrane distortion caused by the presence of large numbers of curvature-inducing proteins at the plasma membrane.
In contrast, coexpression of M with either HA or NA (or both) led to the formation of long, well-defined, and largely straight filamentous protrusions on the cell surface. This observation is consistent with previous studies showing that M can determine the filamentous morphology of extracellular particles upon plasmid-derived expression of influenza A virus structural proteins and with the mapping of genetic determinants of filamentous morphology to M1 and M2 (14, 21, 28). These results differ from those reported by Chen et al. (15), who observed no difference in the spherical morphology of extracellular particles when HA or NA was coexpressed with or without M1. This discrepancy might reflect the use of a different VLP purification protocol and the more advanced EM technology applied in the present study. Since 293T cells are not polarized, it is clear that the filamentous morphology does not require cell polarization (14). The filamentous protrusions observed in this study had an electron-dense layer on the inner site of the membrane, presumably formed by M1. NA from A/Singapore/1/57 and, to a lesser extent, HA became clustered in these filamentous protrusions. The protrusions appeared to be similar to those observed upon assembly of the complete influenza virus A/Hong Kong/8/68 in 293T cells. Furthermore, the released M-containing particles closely resembled the morphology of filamentous influenza viruses A/Hong Kong/8/68 (this study) and A/Udorn/72 (2, 37). Accordingly, we conclude that coexpression of the M segment with one or both of the influenza A virus glycoproteins gives rise to virus-like assembly sites and extracellular virus-like particles, while expression of only the glycoproteins mostly causes nonspecific membrane vesiculation.
We observed that the base of the HAM VLPs was often constricted to form a well-defined neck similar to the one found at the base of influenza A virions. In the case of the NAM construct, the filaments mostly connected to the plasma membrane without a neck and were thus open to the cytoplasm. Since the EM images represent a snapshot of the steady state, these data indicate either that the NAM construct is less efficient in neck constriction than the HAM construct and the complete virus or that particle scission after neck constriction is more efficient for the NAM construct than for the HAM construct, with more particles captured in the stage of neck constriction in the latter case. In either case, these data indicate that formation of straight filamentous particles occurs via well-defined budding intermediates and suggest that membrane scission and particle release occur via an intermediate with a constricted neck.
NA and HA at the plasma membrane in particle-producing cells and in the particle membrane could be distinguished based on their shape, as reported previously (38). When either HA or NA was coexpressed with M, the respective glycoprotein covered the entire surface of released filamentous particles. When both HA and NA were expressed together with M, they separated into distinct clusters. NA preferentially localized to regions where the membranes of filamentous particles curved in two dimensions (as in the surface of a sphere), such as tips or bends. HA preferentially localized to regions which curved in only one dimension (as in the side of a cylinder). This suggests that the preferential localization of NA and HA within filamentous particles may be governed by preferential sorting into regions of different membrane curvature. Within an assembling virion, this would localize NA toward the tips of the filament, as has been reported for extracellular virions: Calder et al. observed an NA cluster at the tip of filamentous influenza virus A/Udorn/72, which was opposite the vRNP-containing tip (2).
Taken together, (i) the formation of quasi-filamentous particles when NA was expressed alone, (ii) the efficient particle release when M1 was coexpressed with NA, (iii) the preference of NA for the tips of filamentous particles, and (iv) the clustering of NA into small nascent buds in NAM-expressing cells all point to NA playing an important role in influenza virus assembly and release, in addition to its well-known enzymatic function. Membrane curvature-mediated localization of NA to one or both tips of filaments would position NA ideally to play a role in the initiation of budding, in neck formation, in scission, and, as originally proposed by Murti and Webster (6), in enzymatic release of the extracellular particle from the cell surface. We suggest that it may modulate all of these steps and may thus play important structural roles in influenza virus assembly and release.
ACKNOWLEDGMENTS
We are grateful to Hans-Dieter Klenk for the pCAGGS-M, -NA, and -HA plasmids and to Juergen Stech for the pHW2000_HA_HK68, and pHW2000_NA_HK68 plasmids. We thank Elena Mekhedov, Hang Waters, and Joshua Zimmerberg for providing the A/Hong Kong/8/68 influenza virus and for technical support. We thank Jonathan W. Yewdell for the mouse anti-M1 antibody and BEI Resources for the goat anti-N2 and goat anti-HA3 antibodies.
This study was supported in part by Deutsche Forschungsgemeinschaft SFB1129 (projects 9 and 10 to H.-G. Kräusslich and J. A. G. Briggs, respectively). Electron microscopy was performed in the EMBL electron microscopy core facility, except that of influenza virions, which was performed at the NIH/NICHD, in the laboratory of Joshua Zimmerberg.
REFERENCES
- 1.Lamb RA, Zebedee SL, Richardson CD. 1985. Influenza virus M2 protein is an integral membrane protein expressed on the infected-cell surface. Cell 40:627–633. doi: 10.1016/0092-8674(85)90211-9. [DOI] [PubMed] [Google Scholar]
- 2.Calder LJ, Wasilewski S, Berriman JA, Rosenthal PB. 2010. Structural organization of a filamentous influenza A virus. Proc Natl Acad Sci U S A 107:10685–10690. doi: 10.1073/pnas.1002123107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palese P, Shaw ML. 2007. Orthomyxoviridae: the viruses and their replication, p 1647–1689. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Rossman JS, Lamb RA. 2011. Influenza virus assembly and budding. Virology 411:229–236. doi: 10.1016/j.virol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rossman JS, Jing X, Leser GP, Lamb RA. 2010. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 142:902–913. doi: 10.1016/j.cell.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murti KG, Webster RG. 1986. Distribution of hemagglutinin and neuraminidase on influenza virions as revealed by immunoelectron microscopy. Virology 149:36–43. doi: 10.1016/0042-6822(86)90084-X. [DOI] [PubMed] [Google Scholar]
- 7.Mosley VM, Wyckoff RW. 1946. Election micrography of the virus of influenza. Nature 157:263. [DOI] [PubMed] [Google Scholar]
- 8.Seladi-Schulman J, Steel J, Lowen AC. 2013. Spherical influenza viruses have a fitness advantage in embryonated eggs, while filament-producing strains are selected in vivo. J Virol 87:13343–13353. doi: 10.1128/JVI.02004-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Booy FP, Ruigrok RW, van Bruggen EF. 1985. Electron microscopy of influenza virus. A comparison of negatively stained and ice-embedded particles. J Mol Biol 184:667–676. [DOI] [PubMed] [Google Scholar]
- 10.Burnet FM, Lind PE. 1957. Studies on filamentary forms of influenza virus with special reference to the use of dark-ground-microscopy. Arch Gesamte Virusforsch 7:413–428. doi: 10.1007/BF01241959. [DOI] [PubMed] [Google Scholar]
- 11.Chu CM, Dawson IM, Elford WJ. 1949. Filamentous forms associated with newly isolated influenza virus. Lancet i:602. [DOI] [PubMed] [Google Scholar]
- 12.Donald HB, Isaacs A. 1954. Some properties of influenza virus filaments shown by electron microscopic particle counts. J Gen Microbiol 11:325–331. doi: 10.1099/00221287-11-2-325. [DOI] [PubMed] [Google Scholar]
- 13.Neumann G, Noda T, Kawaoka Y. 2009. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459:931–939. doi: 10.1038/nature08157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts PC, Compans RW. 1998. Host cell dependence of viral morphology. Proc Natl Acad Sci U S A 95:5746–5751. doi: 10.1073/pnas.95.10.5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen BJ, Leser GP, Morita E, Lamb RA. 2007. Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J Virol 81:7111–7123. doi: 10.1128/JVI.00361-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yondola MA, Fernandes F, Belicha-Villanueva A, Uccelini M, Gao Q, Carter C, Palese P. 2011. Budding capability of the influenza virus neuraminidase can be modulated by tetherin. J Virol 85:2480–2491. doi: 10.1128/JVI.02188-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang D, Harmon A, Jin J, Francis DH, Christopher-Hennings J, Nelson E, Montelaro RC, Li F. 2010. The lack of an inherent membrane targeting signal is responsible for the failure of the matrix (M1) protein of influenza A virus to bud into virus-like particles. J Virol 84:4673–4681. doi: 10.1128/JVI.02306-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gomez-Puertas P, Albo C, Perez-Pastrana E, Vivo A, Portela A. 2000. Influenza virus matrix protein is the major driving force in virus budding. J Virol 74:11538–11547. doi: 10.1128/JVI.74.24.11538-11547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ali A, Avalos RT, Ponimaskin E, Nayak DP. 2000. Influenza virus assembly: effect of influenza virus glycoproteins on the membrane association of M1 protein. J Virol 74:8709–8719. doi: 10.1128/JVI.74.18.8709-8719.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Enami M, Enami K. 1996. Influenza virus hemagglutinin and neuraminidase glycoproteins stimulate the membrane association of the matrix protein. J Virol 70:6653–6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bourmakina SV, Garcia-Sastre A. 2003. Reverse genetics studies on the filamentous morphology of influenza A virus. J Gen Virol 84:517–527. doi: 10.1099/vir.0.18803-0. [DOI] [PubMed] [Google Scholar]
- 22.Burleigh LM, Calder LJ, Skehel JJ, Steinhauer DA. 2005. Influenza A viruses with mutations in the M1 helix six domain display a wide variety of morphological phenotypes. J Virol 79:1262–1270. doi: 10.1128/JVI.79.2.1262-1270.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elleman CJ, Barclay WS. 2004. The M1 matrix protein controls the filamentous phenotype of influenza A virus. Virology 321:144–153. doi: 10.1016/j.virol.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 24.Iwatsuki-Horimoto K, Horimoto T, Noda T, Kiso M, Maeda J, Watanabe S, Muramoto Y, Fujii K, Kawaoka Y. 2006. The cytoplasmic tail of the influenza A virus M2 protein plays a role in viral assembly. J Virol 80:5233–5240. doi: 10.1128/JVI.00049-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCown MF, Pekosz A. 2006. Distinct domains of the influenza A virus M2 protein cytoplasmic tail mediate binding to the M1 protein and facilitate infectious virus production. J Virol 80:8178–8189. doi: 10.1128/JVI.00627-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roberts KL, Leser GP, Ma C, Lamb RA. 2013. The amphipathic helix of influenza A virus M2 protein is required for filamentous bud formation and scission of filamentous and spherical particles. J Virol 87:9973–9982. doi: 10.1128/JVI.01363-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roberts PC, Lamb RA, Compans RW. 1998. The M1 and M2 proteins of influenza A virus are important determinants in filamentous particle formation. Virology 240:127–137. doi: 10.1006/viro.1997.8916. [DOI] [PubMed] [Google Scholar]
- 28.Rossman JS, Jing X, Leser GP, Balannik V, Pinto LH, Lamb RA. 2010. Influenza virus M2 ion channel protein is necessary for filamentous virion formation. J Virol 84:5078–5088. doi: 10.1128/JVI.00119-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199. doi: 10.1016/0378-1119(91)90434-D. [DOI] [PubMed] [Google Scholar]
- 30.Yewdell JW, Frank E, Gerhard W. 1981. Expression of influenza A virus internal antigens on the surface of infected P815 cells. J Immunol 126:1814–1819. [PubMed] [Google Scholar]
- 31.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kremer JR, Mastronarde DN, McIntosh JR. 1996. Computer visualization of three-dimensional image data using IMOD. J Struct Biol 116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 33.Jackson D, Lamb RA. 2008. The influenza A virus spliced messenger RNA M mRNA3 is not required for viral replication in tissue culture. J Gen Virol 89:3097–3101. doi: 10.1099/vir.0.2008/004739-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inglis SC, Brown CM. 1981. Spliced and unspliced RNAs encoded by virion RNA segment 7 of influenza virus. Nucleic Acids Res 9:2727–2740. doi: 10.1093/nar/9.12.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shih SR, Nemeroff ME, Krug RM. 1995. The choice of alternative 5′ splice sites in influenza virus M1 mRNA is regulated by the viral polymerase complex. Proc Natl Acad Sci U S A 92:6324–6328. doi: 10.1073/pnas.92.14.6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lai JC, Chan WW, Kien F, Nicholls JM, Peiris JS, Garcia JM. 2010. Formation of virus-like particles from human cell lines exclusively expressing influenza neuraminidase. J Gen Virol 91:2322–2330. doi: 10.1099/vir.0.019935-0. [DOI] [PubMed] [Google Scholar]
- 37.Vijayakrishnan S, Loney C, Jackson D, Suphamungmee W, Rixon FJ, Bhella D. 2013. Cryotomography of budding influenza A virus reveals filaments with diverse morphologies that mostly do not bear a genome at their distal end. PLoS Pathog 9:e1003413. doi: 10.1371/journal.ppat.1003413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harris A, Cardone G, Winkler DC, Heymann JB, Brecher M, White JM, Steven AC. 2006. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc Natl Acad Sci U S A 103:19123–19127. doi: 10.1073/pnas.0607614103. [DOI] [PMC free article] [PubMed] [Google Scholar]