

ABSTRACT
To clarify the function(s) of the herpes simplex virus 1 (HSV-1) major virion structural protein UL47 (also designated VP13/14), we screened cells overexpressing UL47 for UL47-binding cellular proteins. Tandem affinity purification of transiently expressed UL47 coupled with mass spectrometry-based proteomics technology and subsequent analyses showed that UL47 interacted with cell protein p32 in HSV-1-infected cells. Unlike in mock-infected cells, p32 accumulated at the nuclear rim in HSV-1-infected cells, and this p32 recruitment to the nuclear rim required UL47. p32 formed a complex(es) with HSV-1 proteins UL31, UL34, Us3, UL47, and/or ICP22 in HSV-1-infected cells. All these HSV-1 proteins were previously reported to be important for HSV-1 nuclear egress, in which nucleocapsids bud through the inner nuclear membrane (primary envelopment) and the enveloped nucleocapsids then fuse with the outer nuclear membrane (de-envelopment). Like viral proteins UL31, UL34, Us3, and UL47, p32 was detected in primary enveloped virions. p32 knockdown reduced viral replication and induced membranous invaginations adjacent to the nuclear rim containing primary enveloped virions and aberrant localization of UL31 and UL34 in punctate structures at the nuclear rim. These effects of p32 knockdown were reduced in the absence of UL47. Therefore, the effects of p32 knockdown in HSV-1 nuclear egress were similar to those of the previously reported mutation(s) in HSV-1 regulatory proteins for HSV-1 de-envelopment during viral nuclear egress. Collectively, these results suggested that p32 regulated HSV-1 de-envelopment and replication in a UL47-dependent manner.
IMPORTANCE In this study, we have obtained data suggesting that (i) the HSV-1 major virion structural protein UL47 interacted with host cell protein p32 and mediated the recruitment of p32 to the nuclear rim in HSV-1-infected cells; (ii) p32 was a component of the HSV-1 nuclear egress complex (NEC), whose core components were UL31 and UL34; and (iii) p32 regulated HSV-1 de-envelopment during viral nuclear egress. It has been reported that p32 was a component of human cytomegalovirus NEC and was required for efficient disintegration of nuclear lamina, which has been thought to facilitate HSV-1 primary envelopment during viral nuclear egress. Thus, p32 appeared to be a core component of herpesvirus NECs, like UL31 and UL34 homologs in other herpesviruses, and to play multiple roles in herpesvirus nuclear egress.
INTRODUCTION
Herpesvirus nucleocapsids are too large to traverse the nuclear lamina or cross the inner (INM) and outer (ONM) nuclear membranes through nuclear pores. Therefore, herpesviruses appear to have evolved a unique nuclear egress mechanism in which progeny nucleocapsids assembled in the nucleus acquire primary envelopes by budding through the INM into the perinuclear space (primary envelopment), the space between the INM and ONM, and enveloped nucleocapsids then fuse with the ONM to release de-enveloped nucleocapsids into the cytoplasm (de-envelopment) (1, 2). A heterodimeric complex of herpes simplex virus 1 (HSV-1) proteins UL31 and UL34, which are conserved in all known herpesviruses, is critical for HSV-1 primary envelopment during viral nuclear egress and has been designated the nuclear egress complex (NEC) (1–6). Recently, the HSV-1 NEC has been reported to form a complex with the HSV-1 serine/threonine protein kinase Us3, major HSV-1 structural protein UL47 (also designated VP13/14), and HSV-1 regulatory protein ICP22 (7, 8). Among these recently identified components of the HSV-1 NEC, UL47 and ICP22 have been shown to be important for HSV-1 primary envelopment, based on the observations that a UL47-null or ICP22-null mutation significantly reduced the number of primary enveloped virions in the perinuclear space and induced accumulation of capsids in the nucleus (7, 8). In contrast, Us3 has been reported to play an important role in de-envelopment of HSV-1 nucleocapsids. In cells infected with recombinant Us3-null mutant viruses, recombinant viruses encoding enzymatically inactive Us3, a recombinant virus encoding UL31 with mutations in its Us3 phosphorylation sites, or a recombinant virus with mutations in gB and gH, which abolish Us3 phosphorylation of gB and gH expression, membranous structures are induced adjacent to the nuclear rim that are invaginations of the INM into the nucleoplasm and contain primary enveloped virions. There is also an aberrant accumulation of primary enveloped virions in the perinuclear space and in the induced invagination structures in these cells (9–12). It appears that Us3 is also involved in the primary envelopment of nucleocapsids, since Us3 was shown to phosphorylate lamins A and C: phosphorylation of these lamins leads to dissolution of the nuclear lamina, which is believed to facilitate HSV-1 nucleocapsid access to the INM (13–16).
UL47, a major structural protein in the HSV-1 virion tegument (17), is an RNA binding protein (18) and shuttles between the cytoplasm and nucleus in infected cells (19). It has been reported that UL47 plays an important role in viral replication and pathogenicity, based on studies showing that recombinant UL47 mutant viruses have reduced growth and reduced pathogenicity in cell cultures and/or a mouse model (20, 21). Although the precise mechanism(s) by which UL47 acts in viral replication and pathogenicity remains unknown at present, the functions of UL47 in HSV-1-infected cells have been gradually elucidated. UL47 functions include (i) regulation of subcellular localization of cellular and viral proteins, such as HSV-1 regulatory proteins ICP27 and Us3 kinase (20, 22) and cell polyadenylate-binding protein PABC1 (22); (ii) promotion of primary envelopment of nucleocapsids as described above (8); and (iii) regulation of the viral endoribonuclease responsible for virus host protein synthesis shutoff (23).
In this study, to further clarify the role(s) of UL47 in HSV-1-infected cells, we attempted to identify cellular proteins that interacted with UL47 by tandem affinity purification of transiently expressed UL47 coupled with mass spectrometry-based proteomics technology. We then focused on p32 (also designated C1qBP, TAP, and HABP), which was identified as one of the putative UL47-interacting cell proteins.
The p32 cell protein is primarily localized in the mitochondrial matrix, but it has also been reported to be present at the cell surface and in the nucleus and cytoplasm (24, 25). p32 is considered to be a multicompartmental protein capable of interacting with a wide range of cellular proteins in different subcellular compartments, such as the lamin B receptor, transcription factor TFIIB, high-molecular-weight kininogen, protein kinase C, hyaluronic acids, proapoptotic factor HRK, fibrinogen, and tumor suppressor ARF (26–33). In addition, p32 has been reported to interact with a wide variety of viral proteins, such as human immunodeficiency virus Rev (34) and Tat (35); an adenovirus core protein (36); rubella virus capsid and replicase proteins (37, 38); a hepatitis C virus core protein (39), Epstein-Barr virus (EBV) EBNA-1 (40); HSV-1 ICP27, open reading frame (ORF) P, and ICP34.5 (41–43); human cytomegalovirus (HCMV) UL97, UL50, and UL53 proteins (44, 45); herpesvirus saimiri (HVS) ORF73 protein (46); and murine gammaherpesvirus 68 (MHV-68) M2 (47). The ubiquity of p32 interactions with these viral proteins suggested its importance in the replication of diverse viruses. In agreement with this, it has recently been reported that p32 was required for efficient HSV-1 replication (41). HSV-1 ICP27 and ICP34.5 have been shown to cause redistribution of p32 to the nucleus and nuclear rim, respectively (41, 42). Furthermore, both ICP34.5 and p32 were shown to be required for efficient HSV-1 nuclear egress and for proper phosphorylation and redistribution of the nuclear lamina, thereby suggesting that p32 interacted with ICP34.5 and facilitated HSV-1 nuclear egress by regulating disintegration of the nuclear lamina (41).
Therefore, in the present study, after identification of p32 as a putative UL47-interacting cell protein, we investigated the effect(s) of the interaction between p32 and UL47 in HSV-1-infected cells.
MATERIALS AND METHODS
Cells and viruses.
293T, Vero, and HEp-2 cells have been described previously (48, 49). The viruses have been described previously (8, 20, 50) and included the following: HSV-1 wild-type strain HSV-1(F); recombinant virus YK511, encoding an enzymatically inactive Us3 mutant in which lysine at Us3 position 220 was replaced with methionine (Us3K220M); recombinant virus YK513, in which the Us3K220M mutation in YK511 was repaired (Us3K220M-repair); recombinant virus YK524, encoding UL47 fused to fluorescent protein mRFP1 (mRFP1-UL47); recombinant virus YK527, encoding mRFP1-UL47 and carrying the Us3K220M mutation (mRFP1-UL47/Us3K220M); recombinant virus YK528, in which Us3K220M in YK527 was repaired (mRFP1-UL47/Us3K220M-repair); recombinant virus YK545, a UL47-null mutant virus in which the UL47 gene was disrupted by insertion of a foreign gene cassette just downstream of the UL47 start codon (ΔUL47); recombinant virus YK546, in which the foreign gene cassette inserted into the UL47 locus of YK545 (ΔUL47) was excised (ΔUL47-repair); recombinant virus YK539, in which UL31 was fused to an MEF tag (MEF-UL31); recombinant virus YK538, in which UL34 was fused to an MEF tag (MEF-UL34); and recombinant virus YK536, in which UL47 was fused to an MEF tag (MEF-UL47) (Fig. 1).
FIG 1.

Schematic diagrams of the genome structures of wild-type HSV-1(F) and the relevant domains of the recombinant viruses used in this study. Line 1, wild-type HSV-1(F) genome; line 2, domains with the UL46 to UL48 and Us2 to Us5 genes; line 3, domains with the UL47 and Us3 genes; lines 4 to 11, recombinant viruses with mutations in the UL47 and/or Us3 genes; line 12, domains with the UL12 to UL14 genes; line 13, domains with the UL13 gene and a part of the UL14 gene; line 14, recombinant virus with a null mutation in the UL13 gene; line 15, domains with the UL30 to UL35 genes; line 16, domains of the UL34 and UL31 genes; line 17, recombinant virus encoding MEF-tagged UL31; line 18, recombinant virus encoding MEF-tagged UL34.
Plasmids.
Plasmid pSSCH-Luc encoding shRNA against firefly luciferase (Luc) mRNA was described previously (51). Plasmids were constructed for this study as follows. (i) To construct pcDNA-MEF-UL47 (Fig. 2A), an expression plasmid for UL47 fused to an MEF tag (MEF-UL47), the UL47 ORF without a start codon, was amplified by PCR from pBC1007 (52) and cloned into pcDNA-MEF (53). (ii) pcDNA-MEF-gB, an expression plasmid for gB fused to an MEF tag (MEF-gB), was constructed by cloning a DNA fragment encoding MEF-gB amplified by PCR from recombinant virus DNA expressing MEF-gB (53) into pcDNA4/HisMax C (Invitrogen). (iii) pGEX-p32, for generating a fusion protein of glutathione S-transferase (GST) and p32, was constructed by amplifying the entire p32 coding sequence by PCR from an EBV-transformed human peripheral blood lymphocyte MATCHMAKER cDNA library (Clontech) in frame with GST. (iv) pCMV-p32(F), an expression plasmid for p32 fused to three Flag tag repeats at its C terminus [p32(F)], was constructed by cloning the entire p32 coding sequence without a stop codon amplified by PCR from pGEX-p32 into p3xFlag-CMV-14 (Sigma). (v) pSSCH-p32, for generating a stable cell line expressing shRNA against the 3′ untranslated region (UTR) of p32 mRNA, was constructed as follows. Oligonucleotides 5′-TTTGATTATCATCCTAATATCATGGCTTCCTGTCACCATGATATTAGGATGATAATCTTTTTTG-3′ and 5′-AATTCAAAAAAGATTATCATCCTAATATCATGGTGACAGGAAGCCATGATATTAGGATGATAAT-3′ were annealed and cloned into the BbsI and EcoRI sites of pmU6 (53). The BamHI-SalI fragment of the resultant plasmid, containing the U6 promoter and the sequence encoding short hairpin RNA (shRNA) against the 3′ UTR of p32, was cloned into the BamHI and SalI sites of pSSCH, which is a derivative of retrovirus vector pMX containing a hygromycin B resistance gene, to produce pSSCH-p32. (vi) pMXs-p32, a retrovirus vector expressing p32, was constructed by cloning the entire p32 coding sequence amplified by PCR from pGEX-p32 into pMXs-puro (53).
FIG 2.
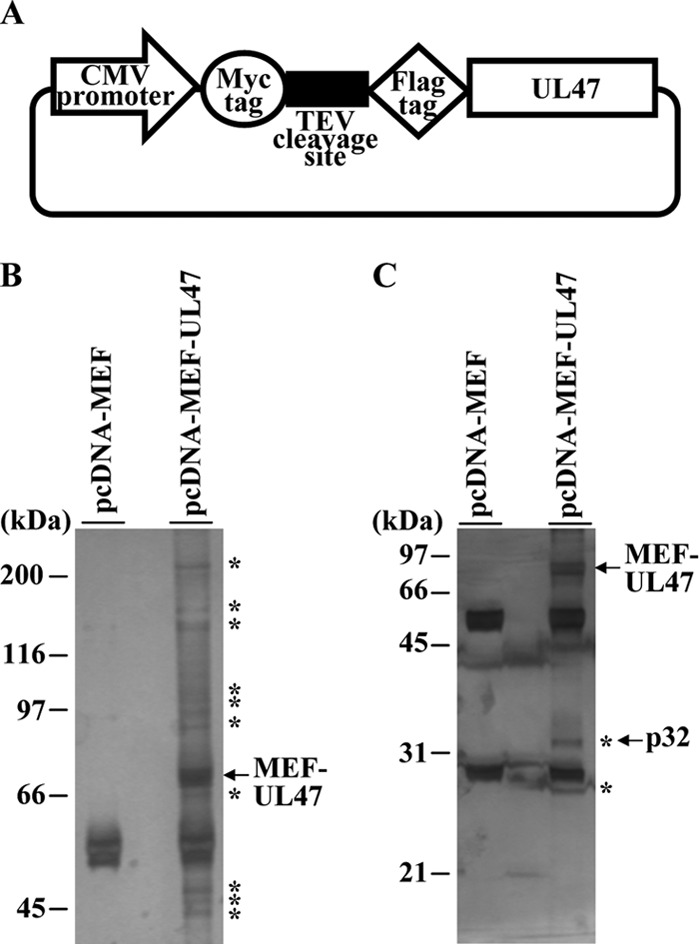
Identification of cellular proteins interacting with HSV-1 UL47. (A) Schematic diagram of expression plasmid pcDNA-MEF-UL47 encoding UL47 fused to an MEF tag. (B and C) 293T cells were transfected with the empty vector pcDNA-MEF or plasmid pcDNA-MEF-UL47, harvested, and immunoprecipitated with anti-Myc antibody and anti-Flag antibody. Immunoprecipitates were separated in 7.5% (B) or 12% (C) denaturing gels and silver stained. Bands marked with asterisks were excised, digested, and analyzed by mass spectrometry. The arrow marks MEF-UL47 and p32. Molecular mass markers are indicated on the left.
Identification of proteins that interact with UL47.
293T cells were transfected with pcDNA-MEF or pcDNA-MEF-UL47 using polyethylenimine as described previously (54), harvested at 48 h posttransfection, and lysed in 0.1% NP-40 buffer (50 mM Tris-HCl [pH 8.0], 120 mM NaCl, 50 mM NaF, 0.1% NP-40) containing a protease inhibitor cocktail (Nacalai Tesque). After centrifugation, the supernatants were immunoprecipitated with an anti-Myc monoclonal antibody and the immunoprecipitates were incubated with AcTEV protease (Invitrogen). After another centrifugation, the supernatants were immunoprecipitated with an anti-Flag monoclonal antibody and the immunoprecipitates were washed three times with wash buffer (50 mM Tris-HCl [pH 8.0], 120 mM NaCl, 50 mM NaF). The immunoprecipitates were analyzed by electrophoresis in 7.5% and 12% denaturing polyacrylamide gels and visualized by silver staining (Daiichikagaku) according to the manufacturer's instructions. Protein bands from cells transfected with pcDNA-MEF-UL47, but not with pcDNA-MEF (Fig. 2), were excised from the denaturing gels, digested in the gel with trypsin, and analyzed by nano-liquid chromatography tandem mass spectrometry (nanoLC-MS/MS) as described previously (53). For this analysis, we used Q-STAR Elite (AB SCIEX) coupled with Dina (KYA Technologies). The MS/MS signals were then analyzed against the human proteins in the RefSeq database (National Center for Biotechnology Information; 35,853 sequences as of 4 February 2013) using the Mascot algorithm (version 2.4.1; Matrix Science) with the following parameters: variable modifications, oxidation (Met), protein N-terminal acetylation, pyroglutamination (Gln); maximum missed cleavages, 2; peptide mass tolerance, 200 ppm; and MS/MS tolerance, 0.5 Da. Protein identification was based on the criterion of having at least one MS/MS data signal with a Mascot score greater than the threshold (P < 0.05).
Production and purification of GST fusion proteins in Escherichia coli.
GST fusion protein GST-p32 was expressed in E. coli that had been transformed with pGEX-p32 and was purified as described previously (52, 55).
Antibodies.
To generate rabbit polyclonal antibody to p32, rabbits were immunized with purified GST-p32 as described previously (48). Serum from the immunized rabbits was used as anti-p32 rabbit polyclonal antibody. Commercial rabbit polyclonal antibody against VP23 (CAC-CT-HSV-UL18; CosmoBio) and commercial mouse monoclonal antibodies against Flag (M2; Sigma), Myc (PL14; MBL), ICP8 (10A3; Chemicon), VP5 (3B6; Virusys), and β-actin (AC15; Sigma) were used in this study. Rabbit polyclonal antibody to UL34, UL31, ICP22, Us3 and UL47, mouse polyclonal antibody to UL31, and chicken polyclonal antibody to UL34 (a generous gift from R. Roller) were described previously (3, 7, 8, 50, 56). Rabbit polyclonal antibody to UL34 was used for immunoblotting, and chicken polyclonal antibody to UL34 was used for immunofluorescence.
Ethics statement.
All animal experiments were carried out in accordance with the Guidelines for Proper Conduct of Animal Experiments, Science Council of Japan. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the Institute of Medical Science, The University of Tokyo (IACUC protocol approval number 19-26).
Antibody analyses.
Immunoprecipitation, immunoblotting, and immunofluorescence were performed as described previously (48, 52).
Purification of virions.
Vero cells were infected with wild-type HSV-1(F) at a multiplicity of infection (MOI) of 0.01 for 48 h. For purification of extracellular virions, cell culture supernatants were harvested by low-speed centrifugation. For purification of total virions (i.e., extra- and intracellular virions), infected cells were subjected to three rounds of freezing and thawing, and supernatants were harvested as described above. Extracellular and total virions were purified from the virion-containing supernatants as described previously (48).
Generation of recombinant retroviruses and establishment of cell lines stably expressing shRNA against p32 and firefly luciferase.
Recombinant retroviruses were generated as described previously (53). Briefly, sh-p32-HEp-2 and sh-Luc-HEp-2 cells were isolated from HEp-2 cells that had been infected with retrovirus-containing supernatants of Plat-GP cells that had been transfected with pSSCH-p32 and pSSCH-Luc, respectively, and selected with 50 μg of hygromycin B/ml.
Assay for cell viability.
The viability of sh-Luc-HEp-2 and sh-p32-HEp-2 cells was determined using a Cell Counting Kit-8 (Dojindo) according to the manufacturer's instructions.
Establishment of sh-p32-HEp-2 cells expressing p32 exogenously and their control cells.
sh-p32-HEp-2/p32(+) and sh-p32-HEp-2/Ct cells were isolated from sh-p32-HEp-2 cells that had been infected with retrovirus-containing supernatants of Plat-GP cells that had been transfected with pMXs-p32 or pMXs, respectively, and selected with 2 μg of puromycin/ml.
Electron microscopic analysis.
sh-Luc-HEp-2 and sh-p32-HEp-2 cells infected with wild-type HSV-1(F) at an MOI of 5 for 24 h were examined by ultrathin-section electron microscopy as described previously (56). Immunoelectron microscopy to detect p32 using anti-p32 rabbit polyclonal antibody was performed as described previously (8).
RESULTS
Identification of cell proteins that interacted with UL47 and confirmation of UL47 interaction with p32.
To identify host cell proteins that interacted with UL47, we used tandem affinity purification coupled with mass spectrometry-based proteomics analysis. These experiments identified 14 cell proteins that coimmunoprecipitated with transiently expressed UL47 fused to an MEF tag with Myc and Flag epitopes and a TEV protease cleavage site (MEF-UL47) (data not shown). Of these proteins, we focused on p32 in this study. To verify and extend the interaction data obtained with mass spectrometry-based proteomics screening, we performed two series of experiments. In the first series of experiments, 293T cells were mock transfected or transfected with pCMV-p32(F) expressing Flag-tagged p32 alone, pcDNA-MEF-UL47 expressing MEF-tagged UL47 alone, or pCMV-p32(F) in combination with either pcDNA-MEF-UL47 or pcDNA-MEF-gB expressing MEF-tagged UL47 or gB, respectively. The cells were lysed 2 days posttransfection and immunoprecipitated with anti-Myc antibody, and the immunoprecipitates were analyzed by immunoblotting with anti-Flag antibody. As shown in Fig. 3A, anti-Myc antibody coprecipitated Flag-tagged p32 from cells cotransfected with MEF-tagged UL47, but not with MEF-tagged gB. These results not only confirmed the interaction between UL47 and p32 in transfected cells, but also indicated that UL47 interacted with p32 in the absence of other HSV-1 proteins. In the second series of experiments, Vero cells infected with YK536 (MEF-UL47) encoding MEF-tagged UL47 (Fig. 1) or wild-type HSV-1(F) were lysed at 18 h postinfection and immunoprecipitated with anti-Flag antibody, and the immunoprecipitates were analyzed by immunoblotting with anti-p32 antibody. We previously reported that MEF tagging of UL47 had little effect on viral growth in cell cultures (8). As shown in Fig. 3B, anti-Flag antibody coprecipitated endogenous p32 with MEF-tagged UL47 from lysates of YK536 (MEF-UL47)-infected Vero cells, but not from lysates of wild-type HSV-1(F)-infected cells. These results indicated that UL47 interacted with p32 in HSV-1-infected cells.
FIG 3.
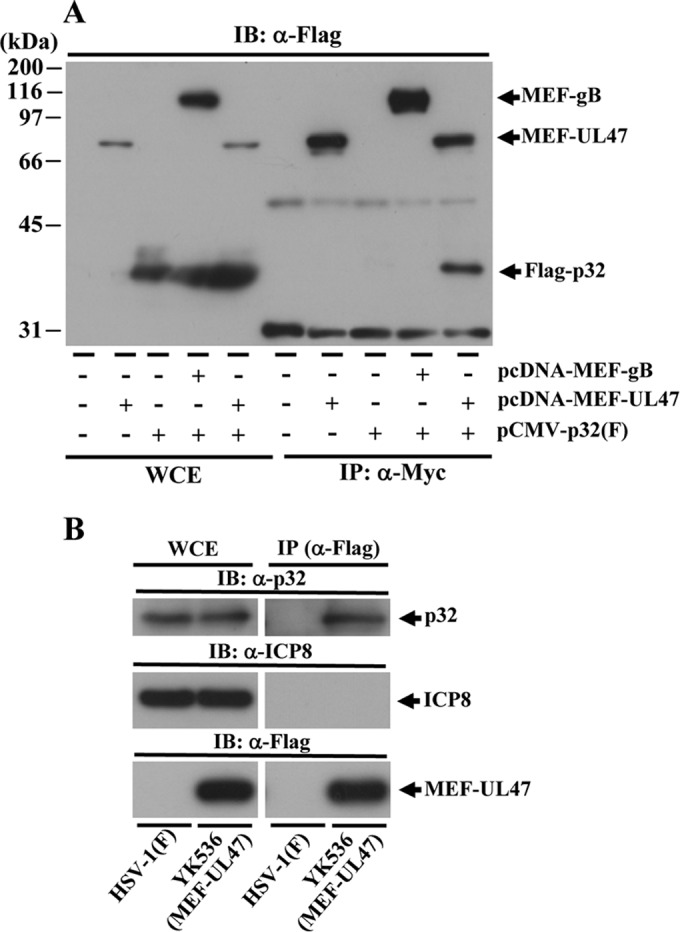
Interaction of UL47 with p32. (A) 293T cells were mock transfected or transfected with pcDNA-MEF-UL47 alone, pCMV-p32(F) encoding Flag-tagged p32 alone, or pCMV-p32(F) in combination with either pcDNA-MEF-UL47 or pcDNA-MEF-gB. At 2 days posttransfection, cells were harvested, immunoprecipitated (IP) with anti-Myc antibody, and analyzed by immunoblotting (IB) with anti-Flag antibody. WCE, whole-cell extract. Molecular mass markers are indicated on the left. (B) Vero cells infected with wild-type HSV-1(F) or YK536 (MEF-UL47) at an MOI of 5 for 18 h were harvested, immunoprecipitated with anti-Flag antibody, and analyzed by immunoblotting with anti-p32 antibody, anti-ICP8 antibody, or anti-Flag antibody.
UL47 was required for the accumulation of p32 at the nuclear rim in HSV-1-infected cells.
To investigate the interaction between UL47 and p32 in HSV-1-infected cells, we examined the subcellular localization of p32 in the presence or absence of UL47 in infected cells. For these studies, Vero cells were mock infected or infected with wild-type HSV-1(F), the UL47-null mutant virus YK545 (ΔUL47), its repaired virus YK546 (ΔUL47-repair), or R7356 (ΔUL13), which has a null mutation in the HSV-1 UL13 protein kinase, and p32 localization in the infected cells was analyzed by immunofluorescence microscopy. As reported previously (42), p32 was detected predominantly in the cytoplasm of mock-infected cells (Fig. 4), whereas in wild-type HSV-1(F) and YK546 (ΔUL47-repair)-infected cells, although p32 was still present in the cytoplasm, it was predominantly at the nuclear rim (Fig. 4A) and colocalized with lamins A and C (Fig. 4B), which are markers for nuclear lamina. Similarly, in Vero cells infected with R7356 (ΔUL13), p32 was predominantly localized at the nuclear rim (Fig. 4C). In contrast, p32 was detected predominantly in the cytoplasm of cells infected with YK545 (ΔUL47) (Fig. 4A). We noted that p32 appeared to be distributed more diffusely in the cytoplasm of cells infected with YK545 (ΔUL47) than in mock-infected cells (Fig. 4A). These results indicated that UL47 was specifically required for the accumulation of p32 at the nuclear rim in HSV-1-infected cells.
FIG 4.

Localization of p32 in HSV-1-infected cells and effect of UL47 and UL13 on p32 localization. (A) Vero cells mock infected or infected with wild-type HSV-1(F), YK545 (ΔUL47), or YK546 (ΔUL47-repair) at an MOI of 3 were fixed at 18 h postinfection, permeabilized, stained with anti-p32 antibody or anti-ICP8 antibody, and examined by confocal microscopy. (B) Vero cells mock infected or infected with wild-type HSV-1(F) at an MOI of 3 were fixed at 18 h postinfection, permeabilized, stained with anti-p32 antibody or anti-lamin A/C antibody, and examined by confocal microscopy. (C) Experiments were done by the same procedure as for panel A), except R7356 (DUL13) was used instead of YK545 (ΔUL47) and YK546 (ΔUL47-repair). Scale bar, 5 μm.
Interaction of p32 with components of the HSV-1 NEC in infected cells.
We next investigated whether p32 interacted with components of the HSV-1 NEC other than UL47, including UL31, UL34, Us3, and ICP22, all of which have been reported to be important for viral nuclear egress at the nuclear rim (4, 7, 8, 57), and performed two series of experiments. In the first series of experiments, Vero cells were infected with wild-type HSV-1(F), YK536 (MEF-UL47), YK538 (MEF-UL34) encoding MEF-tagged UL34, or YK539 (MEF-UL31) encoding MEF-tagged UL31 (Fig. 1). At 18 h postinfection, infected cells were lysed and immunoprecipitated with anti-Myc antibody, and the immunoprecipitates were analyzed by immunoblotting with antibodies to the viral and cellular proteins shown in Fig. 5. We previously reported that MEF tagging of UL31 and UL34 had little effect on viral growth in cell cultures (8). As shown in Fig. 5A, anti-Myc antibody coprecipitated UL31, UL34, Us3, ICP22, and p32 with MEF-tagged UL47 from lysates of YK536 (MEF-UL47)-infected Vero cells but did not coprecipitate VP23. In contrast, the anti-Myc antibody did not immunoprecipitate any of these viral and cellular proteins from lysates of wild-type HSV-1(F)-infected cells (Fig. 5A). These results indicated that UL47 formed a complex(es) with UL31, UL34, Us3, ICP22 and/or p32 in HSV-1-infected cells. Similarly, anti-Myc antibody coprecipitated UL34, Us3, UL47, ICP22, and p32 but not VP23, with MEF-tagged UL31 from lysates of YK539 (MEF-UL31)-infected cells (Fig. 5B), and coprecipitated UL31, Us3, UL47, ICP22, and p32, but not VP23, with MEF-tagged UL34 from lysates of YK538 (MEF-UL34)-infected cells (Fig. 5C). These results indicated that UL31 formed a complex(es) with UL34, Us3, UL47, ICP22, and/or p32, and UL34 formed a complex(es) with UL31, Us3, UL47, ICP22, and/or p32, in HSV-1-infected cells.
FIG 5.

Interactions among UL47, p32, UL31, UL34, ICP22, and Us3 in HSV-1-infected cells. Vero cells infected with wild-type HSV-1(F) (A to C) and YK536 (MEF-UL47) (A), YK539 (MEF-UL31) (B), or YK538 (MEF-UL34) (C) at an MOI of 5 for 18 h were harvested, immunoprecipitated with anti-Myc antibody, and analyzed by immunoblotting with the indicated antibodies.
Us3 has been reported to regulate localization of UL31, UL34, UL47 and ICP22 at the nuclear rim in HSV-1-infected cells (3, 7, 8). In the absence of Us3 protein or its catalytic activity, these viral proteins were shown to accumulate aberrantly in punctate structures at the nuclear rim in HSV-1-infected cells (3, 7, 8). Therefore, in the second series of experiments, we investigated the localization of p32 in the absence of Us3 catalytic activity in infected cells. Vero cells were infected with wild-type HSV-1(F), the Us3 kinase-dead mutant YK511 (Us3K220M), or its repaired virus YK513 (Us3K220M-repair), fixed at 18 h postinfection, and then stained with anti-p32 antibody in combination with anti-UL34 or anti-UL31 antibody; localization of these viral and cellular proteins was examined by confocal microscopy. As shown in Fig. 6, p32 was predominantly localized at the nuclear rim in Vero cells infected with wild-type HSV-1(F) or YK513 (Us3K220M-repair), in agreement with the results shown in Fig. 4, and colocalized with UL34 and UL31 at the nuclear rim with a uniform distribution. In contrast, p32 was detected in punctate structures at the nuclear rim in Vero cells infected with YK511 (Us3K220M) and colocalized with UL31 and UL34 in these structures (Fig. 6). Next, Vero cells were infected with YK524 (mRFP1-UL47) encoding mRFP1-UL47, YK527 (mRFP1-UL47/Us3K220M) encoding mRFP1-UL47 and Us3 with the kinase-dead K220M mutation, or YK528 (mRFP1-UL47/Us3K220M-repair) in which the Us3 K220M mutation in YK527 was repaired (Fig. 1). At 18 h postinfection, infected cells were fixed and then stained with anti-p32 antibody, and localization of p32 with mRFP1-UL47 was examined by confocal microscopy. It has been noted that the anti-UL47 antibodies reported to date were not useful for immunofluorescence assays because they showed nonspecific staining (20, 58). Therefore, we used YK524 (mRFP1-UL47), YK527 (mRFP1-UL47/Us3K220M), and YK528 (mRFP1-UL47/Us3K220M-repair) expressing UL47 tagged with the fluorescent protein mRFP1 (20). As shown in Fig. 7, mRFP1-UL47 was localized throughout the nuclei of Vero cells infected with YK524 (mRFP1-UL47) or YK528 (mRFP1-UL47/Us3K220M-repair), in agreement with our previous reports (8, 20), and was colocalized at the nuclear rim with p32, distributed smoothly around the nuclear rim. In contrast, in Vero cells infected with YK527 (mRFP1-UL47/Us3K220M), p32 accumulated in punctate structures at the nuclear rim, colocalized with mRFP1-UL47 (Fig. 7). Taken together, these results indicated that p32 colocalized with UL31, UL34, and UL47 at the nuclear rim in the presence or absence of Us3 catalytic activity in HSV-1-infected cells, and that Us3 catalytic activity was required for proper localization of p32 at the nuclear rim in HSV-1-infected cells.
FIG 6.

Effect of Us3 kinase activity on localization of p32, UL31, and UL34 in HSV-1-infected cells. Vero cells were infected with wild-type HSV-1(F), YK511 (Us3K220M), or YK513 (Us3K220M-repair) at an MOI of 3, fixed at 18 h postinfection, permeabilized, stained with anti-p32 antibody in combination with anti-UL34 (A) or anti-UL31 (B) antibody, and examined by confocal microscopy. Scale bar, 5 μm.
FIG 7.
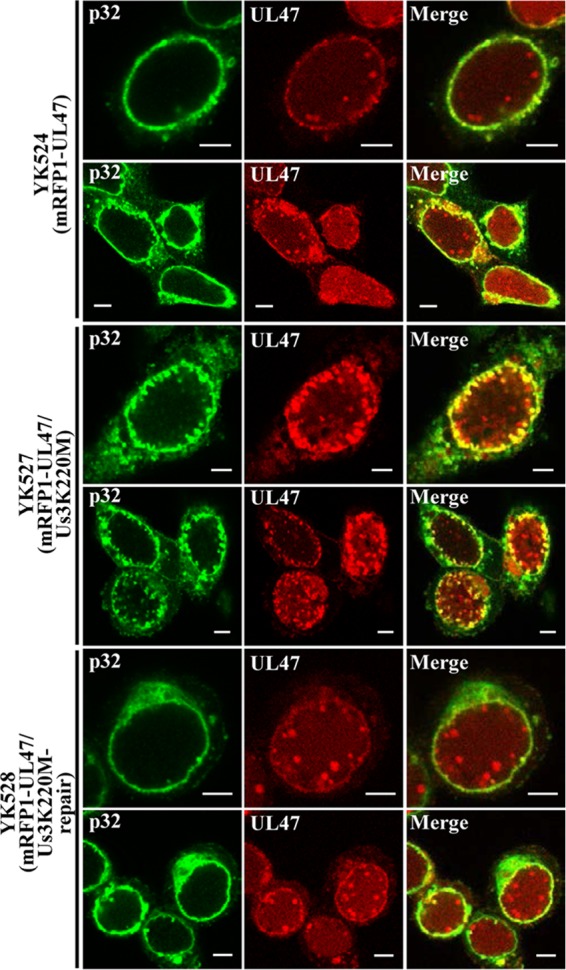
Effect of Us3 kinase activity on localization of p32 and mRFP1-UL47 in HSV-1-infected cells. Vero cells were infected with YK524 (mRFP1-UL47), YK527 (mRFP1-UL47/Us3K220M), or YK528 (mRFP1-UL47/Us3K220M-repair) at an MOI of 3, fixed at 18 h postinfection, permeabilized, stained with anti-p32 antibody, and examined by confocal microscopy. Scale bar, 5 μm.
Localization of p32 in infected cells by immunoelectron microscopy.
Components of the HSV-1 NEC, including UL31, UL34, Us3, and UL47, have been reported to be incorporated into primary enveloped virions in the perinuclear space (8, 12). To localize p32 in HSV-1-infected cells at the ultrastructural level, Vero cells infected with wild-type HSV-1(F) were examined by immunoelectron microscopy using anti-p32 serum to detect p32. Preimmune serum of the rabbit used to generate anti-p32 serum was used as a negative control and was barely detected by immunoelectron microscopy of mock- and wild-type HSV-1(F)-infected cells (data not shown). As shown in Fig. 8A, the p32-specific antibody localized at the nuclear membrane in wild-type HSV-1(F)-infected cells, in agreement with the immunofluorescence results described above (Fig. 4, 6, and 7). p32 was also found in areas in the nucleus with many capsids and on a significant fraction of capsids in the nucleus (Fig. 8B and H). In contrast, much less p32 was detected in areas in the nucleus without capsids (Fig. 8H). These results suggested that p32 was specifically recruited to capsids in the nucleus in wild-type HSV-1(F) infected cells. In addition, p32 was detected on most primary enveloped virions in the perinuclear space (Fig. 8C and D) and on capsids in the cytoplasm (Fig. 8E and I), but it was barely detected on secondary enveloped cytoplasmic and extracellular virions (Fig. 8F, G, I, and J). In agreement with these results, p32 was not detected on purified extracellular virions by immunoblotting, whereas it was detected on purified cell-associated virions, i.e., nucleocapsids, primary enveloped virions and secondary enveloped virions (Fig. 9). These results suggested that p32 was a component of primary enveloped virions and that HSV-1 virions acquired p32 in the nucleus prior to primary envelopment and subsequently lost p32 during secondary envelopment.
FIG 8.

Localization of p32 in HSV-1-infected cells by immunoelectron microscopy. Vero cells were infected with wild-type HSV-1(F) at an MOI of 5, fixed at 18 h postinfection, embedded, sectioned, stained with rabbit anti-p32 polyclonal antibody followed by goat anti-rabbit IgG conjugated to 10-nm gold particles, and examined by transmission electron microscopy. Nu, nucleus; Cy, cytoplasm; NM, nuclear membrane; INM, inner nuclear membrane; ONM, outer nuclear membrane; PM, plasma membrane. p32 was detected along the nuclear membrane (A), on capsids in the nucleus (A to C and H) and cytoplasm (E and I), and on primary enveloped virions in the perinuclear space (C and D), but it was barely detectable on secondary enveloped virions in the cytoplasm (F and I) and in the extracellular space (G and J). Bars, 200 nm.
FIG 9.
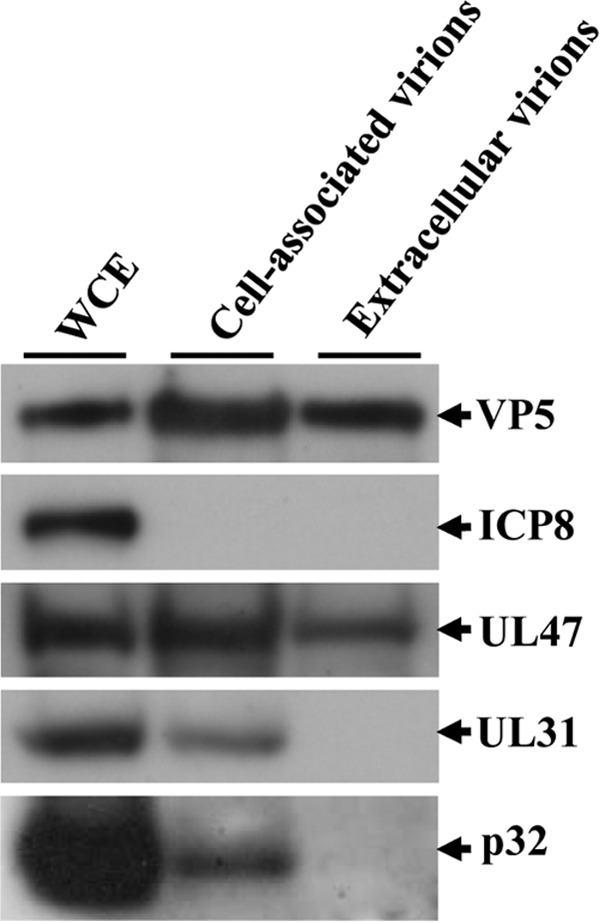
Detection of p32 in cell-associated and extracellular virions. Cell-associated and extracellular virions were purified and analyzed by immunoblotting with antibodies to the indicated proteins.
Effect of p32 on HSV-1 nuclear egress.
To directly examine whether p32 functioned in HSV-1 nuclear egress, we generated HEp-2 cell lines stably expressing shRNA against the 3′ UTR of p32 mRNA (sh-p32-HEp-2) to knock down p32 expression and a control cell line (sh-Luc-HEp-2) expressing shRNA against firefly luciferase mRNA. As shown in Fig. 10A, considerably less endogenous p32 protein was expressed in sh-p32-HEp-2 cells than in sh-Luc-HEp-2 cells. In contrast, the viability of sh-p32-HEp-2 cells was similar to that of sh-Luc-HEp-2 cells, indicating that p32 knockdown had no effect on HEp-2 cell viability (Fig. 10B). In addition, to examine whether the phenotype(s) observed in sh-p32-HEp-2 cells was due to a nonspecific effect(s) of the shRNA, we generated sh-p32-HEp-2/p32(+) cells, in which p32 was expressed exogenously by transduction of sh-p32-HEp-2 cells with a retrovirus vector expressing p32, and control sh-p32-HEp-2/Ct cells, in which sh-p32-HEp-2 cells were transduced by the empty retrovirus vector (Fig. 10C). sh-p32-HEp-2, sh-Luc-HEp-2, sh-p32-HEp-2/Ct, and/or sh-p32-HEp-2/p32(+) cells were infected with wild-type HSV-1(F), YK545 (ΔUL47), or YK546 (ΔUL47-repair) at an MOI of 5 for 24 h, and the effect of the p32 knockdown on viral morphogenesis was examined by quantitating the number of virus particles at different morphogenetic stages by electron microscopy in the presence or absence of UL47. As shown in Fig. 11, membranous invagination structures containing primary enveloped virions were observed in the nucleoplasm adjacent to the nuclear membrane in sh-p32-HEp-2 cells infected with wild-type HSV-1(F), but few of these structures were seen in HSV-1(F)-infected sh-Luc-HEp-2 cells. Quantitation of these data showed a 21-fold increase in membranous invaginations and a 25-fold increase in enveloped virions in membranous invaginations in HSV-1(F)-infected sh-p32-HEp-2cells compared to the quantity in HSV-1(F)-infected sh-Luc-HEp-2 cells (Table 1). The fractions of total virus particles that were enveloped virions in the perinuclear space in HSV-1(F)-infected sh-Luc-HEp-2 (13.0%) and sh-p32-HEp-2 (14.8%) cells were similar (Table 1), indicating that primary enveloped virions accumulated in the invagination structures in sh-p32-HEp-2 cells. Similar results were found in HSV-1(F)-infected sh-p32-HEp-2/Ct and sh-p32-HEp2/p32(+) cells (Table 2). In particular, the membranous invagination structures were barely detectable in YK545 (ΔUL47)-infected sh-p32-HEp-2 cells (Table 1). In contrast, in sh-p32-HEp-2 cells infected with YK546 (ΔUL47-repair), the invagination structures were induced as observed in wild-type HSV-1(F)-infected sh-p32-HEp-2 cells (Table 1).
FIG 10.
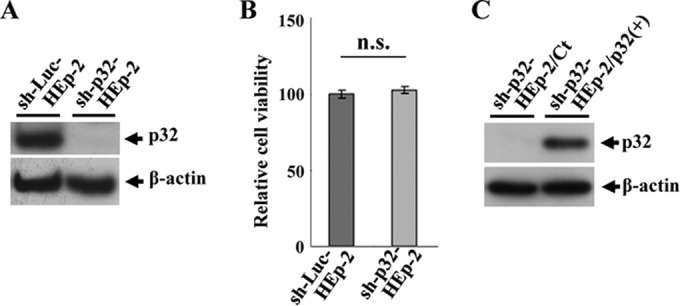
Characterization of sh-Luc-HEp-2, sh-p32-HEp-2, sh-p32-HEp-2/Ct, and sh-p32-HEp-2/p32(+) cells. (A) Expression of p32 in sh-Luc-HEp-2 and sh-p32-HEp-2 cells analyzed by immunoblotting with anti-p32 (top) and anti-β-actin (bottom) antibodies. (B) Cell viability of sh-Luc-HEp-2 and sh-p32-HEp-2 cells assayed 24 h after 2 × 104 cells were seeded on 96-well plates. Each value is the mean ± standard error of the results of triplicate experiments and is expressed relative to the mean for sh-Luc-HEp-2 cells, which was normalized to 100%. n.s., not statistically significant. Data are representative of three independent experiments. (C) Expression of p32 in sh-p32-HEp-2/Ct and sh-p32-HEp-2/p32(+) cells analyzed by immunoblotting with anti-p32 (top) and anti-β-actin (bottom) antibodies.
FIG 11.

Ultrastructural analysis of the effect of p32 on HSV-1 nuclear egress. sh-Luc-HEp-2 (A) and sh-p32-HEp-2 cells (B) infected with wild-type HSV-1(F) at an MOI of 5 were fixed at 24 h postinfection, embedded, sectioned, stained, and examined by transmission electron microscopy. (B-a and B-b) Higher magnifications of the corresponding boxed areas in panel B showing invagination structures containing primary enveloped virions. Nu, nucleus; Cy, cytoplasm; NM, nuclear membrane.
TABLE 1.
Effect of p32 knockdown on distribution of progeny virus particles in infected HEp-2 cells
| Cell type | Virus | No. of intranuclear invaginations | No. of virus particles in morphogenetic stagea |
Total counted (particles/cells) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Nucleocapsids in the nucleus | Enveloped virions in intranuclear invaginations | Enveloped virions in the perinuclear space | Nucleocapsids in the cytoplasm | Enveloped virions in the cytoplasm | Extracellular enveloped virions | ||||
| sh-Luc-HEp-2 | HSV-1 (F) | 1 | 639 (22.3) | 4 (0.1) | 372 (13.0) | 228 (7.9) | 678 (23.7) | 947 (33.0) | 2,868/22 |
| sh-Luc-HEp-2 | YK545 (ΔUL47) | 0 | 600 (56.4) | 0 (0) | 22 (2.1) | 97 (9.1) | 46 (4.3) | 299 (28.1) | 1,064/22 |
| sh-Luc-HEp-2 | YK546 (ΔUL47-repair) | 0 | 755 (28.9) | 0 (0) | 285 (10.9) | 210 (8.0) | 563 (21.5) | 801 (30.7) | 2,813/22 |
| sh-p32-HEp-2 | HSV-1 (F) | 21 | 607 (30.5) | 101 (5) | 295 (14.8) | 268 (13.5) | 324 (16.3) | 396 (19.9) | 1,991/22 |
| sh-p32-HEp-2 | YK545 (ΔUL47) | 0 | 449 (61.8) | 0 (0) | 0 (0) | 71 (9.8) | 55 (7.6) | 151 (20.8) | 726/22 |
| sh-p32-HEp-2 | YK546 (ΔUL47-repair) | 17 | 552 (29.4) | 103 (5.5) | 181 (9.7) | 274 (14.6) | 366 (19.5) | 400 (21.3) | 1,876/22 |
Numbers in parentheses are the percentages of virus particles in the morphogenetic stage.
TABLE 2.
Effect of p32 knock-in on distribution of progeny virus particles in HSV-1(F)-infected HEp-2 cells
| Cell type | No. of intranuclear invaginations | No. of virus particles in morphogenetic stagea |
Total counted (particles/cells) | |||||
|---|---|---|---|---|---|---|---|---|
| Nucleocapsids in the nucleus | Enveloped virions in intranuclear invaginations | Enveloped virions in the perinuclear space | Nucleocapsids in the cytoplasm | Enveloped virions in the cytoplasm | Extracellular enveloped virions | |||
| sh-p32-HEp-2/Ct | 23 | 616 (31.5) | 130 (6.7) | 234 (12.0) | 257 (13.1) | 314 (16.0) | 406 (20.7) | 1,957/22 |
| sh-p32-HEp-2/p32(+) | 2 | 633 (23.3) | 7 (0.3) | 345 (12.7) | 199 (7.4) | 655 (24.1) | 874 (32.2) | 2,713/22 |
Numbers in parentheses are the percentages of virus particles in the morphogenetic stage.
We also examined the effect of p32 knockdown on localization of UL31 and UL34 by immunofluorescence microscopy. As shown in Fig. 12A, in sh-p32-HEp-2 cells infected with wild-type HSV-1(F), UL31 and UL34 colocalized at the nuclear rim and, in a fraction of the cells, in the punctate structures adjacent to the nuclear rim that protruded into the nucleoplasm. The punctate structures were induced in similar fractions of sh-p32-HEp-2 and sh-p32-HEp-2/Ct cells infected with wild-type HSV-1(F) (27.7% and 28.4%, respectively) but were barely detectable in HSV-1(F)-infected sh-Luc-HEp-2 and sh-p32-HEp-2/p32(+) cells (Fig. 12B and C). In contrast, punctate structures were barely detectable in sh-p32-HEp-2 cells infected with YK545 (ΔUL47) (Fig. 13).
FIG 12.
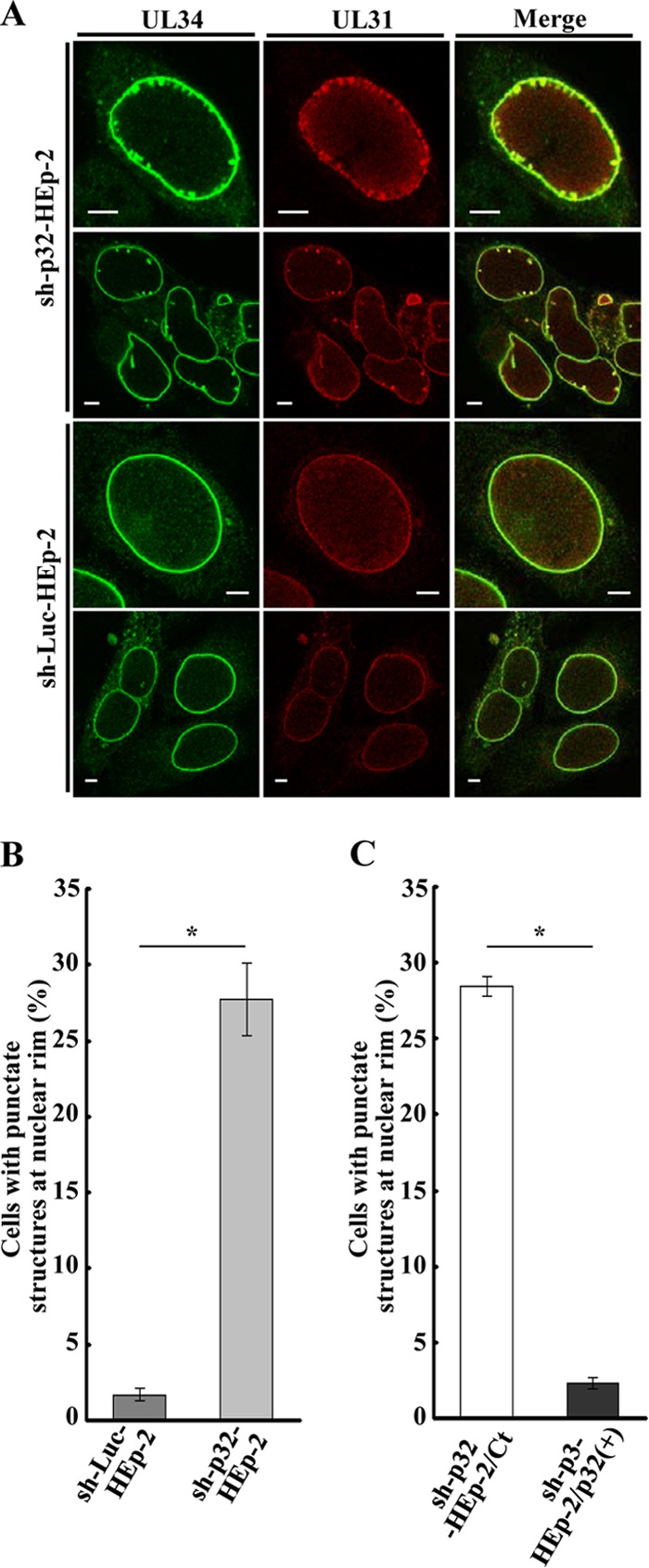
Effect of p32 on localization of UL34 and UL31 in HSV-1-infected cells. (A) sh-p32-HEp-2 and sh-Luc-HEp-2 cells were infected with wild-type HSV-1(F) at an MOI of 5, fixed at 24 h postinfection, permeabilized, stained with anti-UL34 and anti-UL31 antibodies, and examined by confocal microscopy. (B and C) Quantification of infected cells showing aberrant punctate structures at the nuclear rim. Infected sh-p32-HEp-2 and sh-Luc-HEp-2 cells (B) and infected sh-p32-HEp-2/Ct and sh-p32-HEp-2/p32(+) cells (C) were examined by confocal microscopy as described for panel A, and the percentage of cells with aberrant punctate structures at the nuclear rim was determined for 100-cell samples. Each value is the mean ± standard error of the results of triplicate experiments. Asterisks indicate statistically significant differences (*, P < 0.05). Data are representative of results from three independent experiments.
FIG 13.
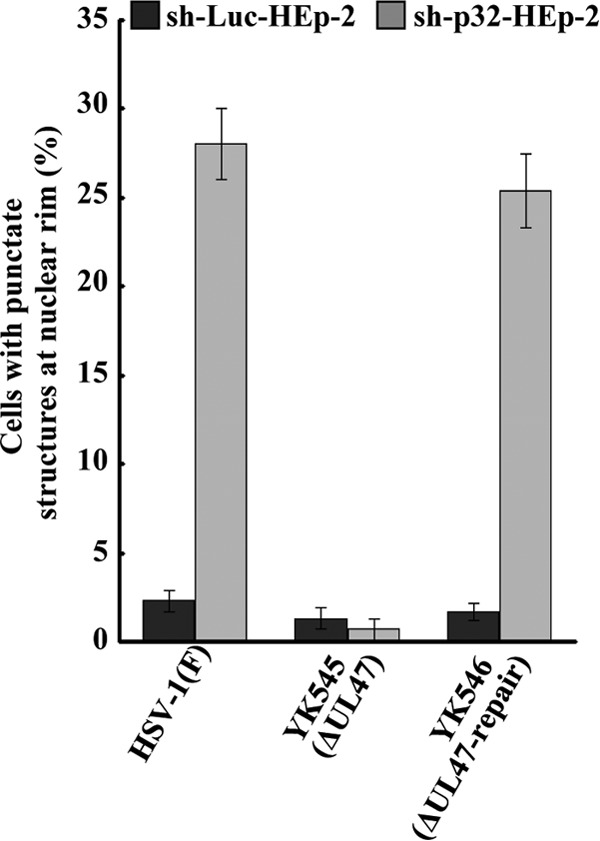
Effect of p32 and UL47 on localization of UL34 and UL31 in HSV-1-infected cells. sh-Luc-HEp-2 and sh-p32-HEp-2 cells were infected with wild-type HSV-1(F), YK545 (ΔUL47) or YK546 (ΔUL47-repair) at an MOI of 5, fixed at 24 h postinfection, permeabilized, stained with anti-UL34 and anti-UL31 antibodies, and examined by confocal microscopy. The percentage of cells with aberrant punctate structures at the nuclear rim was determined for 100-cell samples. Each value is the mean ± standard error of the results of triplicate experiments. Asterisks indicate statistically significant differences (*, P < 0.05). Data are representative of results from three independent experiments.
In sh-Luc-HEp-2 cells infected with wild-type HSV-1(F) or YK546 (ΔUL47-repair), 13.0 and 10.9%, respectively, of the total number of virus particles were primary enveloped virions in the perinuclear space. However, in sh-Luc-HEp-2 cells infected with YK545 (ΔUL47), only 2.1% were primary enveloped virions in the perinuclear space (Table 1). In contrast, in sh-Luc-HEp-2 cells infected with wild-type HSV-1(F) and YK546 (ΔUL47-repair), 22.3 and 28.9%, respectively, of total virus particles were nucleocapsids in the nucleus, but 56.4% of total virus particles were nucleocapsids in the nucleus in sh-Luc-HEp-2 cells infected with YK545 (ΔUL47) (Table 1). These results were in agreement with our previous report (8). Similarly, in sh-p32-HEp-2 cells, the UL47-null mutation reduced the fraction of virus particles in primary enveloped virions in the perinuclear space, but the fraction of nucleocapsids in the nucleus increased (Table 1).
Taken together, these results indicated that (i) p32 knockdown induced membranous invagination structures containing primary enveloped virions adjacent to the nuclear rim and primary enveloped virions accumulated in these structures, (ii) p32 knockdown induced aberrant localization of UL34 and UL31 in punctate structures adjacent to the nuclear rim, (iii) UL47 was required for the effects of p32 knockdown on HSV-1 nuclear egress, and (iv) the effects of UL47 on HSV-1 nuclear egress were independent of the presence of p32.
Effect of p32 on HSV-1 replication in the presence or absence of UL47.
To investigate the effect of p32 knockdown on HSV-1 replication, sh-p32-HEp-2, sh-Luc-HEp-2, sh-p32-HEp-2/Ct, and sh-p32-HEp-2/p32(+) cells were infected with wild-type HSV-1(F) at an MOI of 0.01 and viral titers were assayed at various times postinfection. Progeny virus titers in sh-p32-HEp-2 cells were significantly (12.9- to 33.3-fold) lower than in sh-Luc-HEp-2 cells at all postinfection times studied (Fig. 14A), but the progeny virus titers in sh-p32-HEp-2/p32(+) cells were restored to the titers in sh-Luc-Hp-2 cells (Fig. 14B). Similar results were also found with these cells infected at an MOI of 5 (data not shown). These results were in agreement with the previous report by Wang et al. (41) and confirmed their observation that p32 was required for efficient HSV-1 replication in cell cultures.
FIG 14.
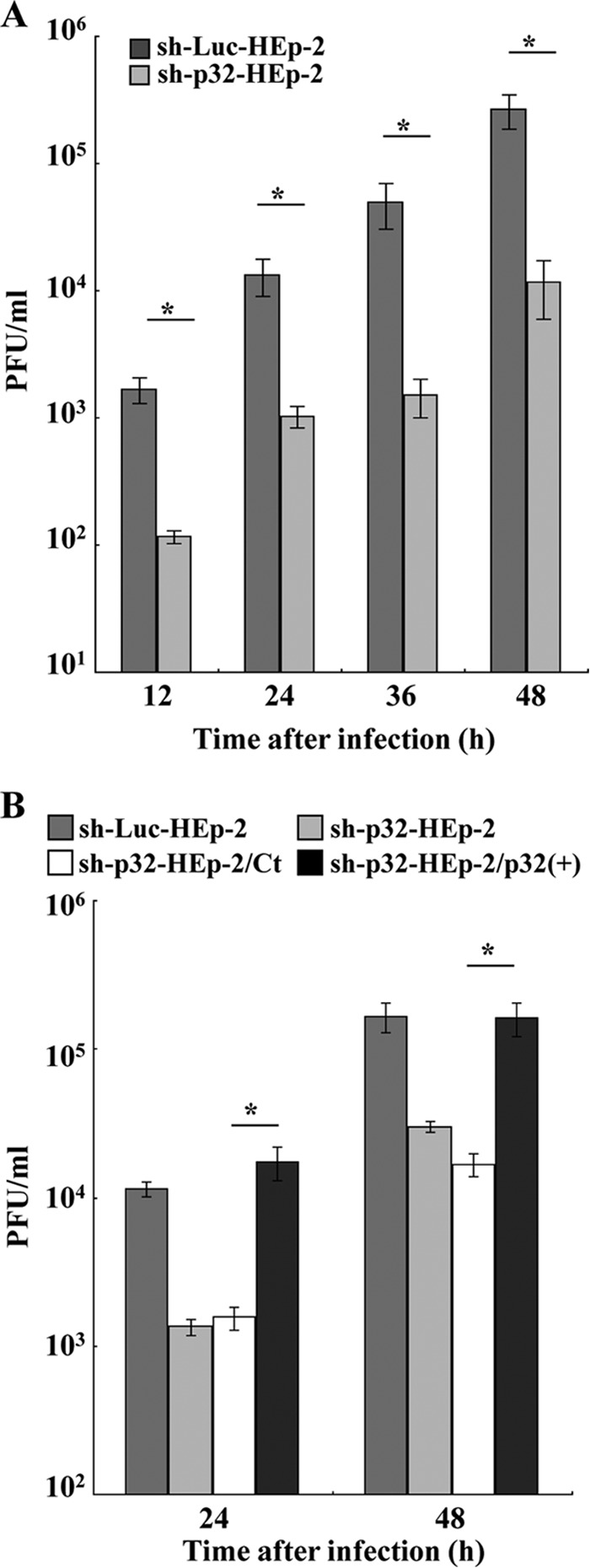
Effect of p32 on HSV-1 replication in cell cultures. (A) sh-Luc-HEp-2 and sh-p32-HEp-2 cells were infected with wild-type HSV-1(F) at an MOI of 0.01. At the indicated times postinfection, total virus from cell culture supernatants and infected cells was harvested and assayed on Vero cells. Each value is the mean ± the standard error of the results of triplicate experiments. Asterisks indicate statistically significant differences (*, P < 0.05). Data are representative of results from three independent experiments. (B) sh-Luc-HEp-2, sh-p32-HEp-2, sh-p32-HEp-2/Ct, and sh-p32-HEp-2/p32(+) cells were infected with wild-type HSV-1(F) at an MOI of 0.01. At 24 and 48 h postinfection, total virus from cell culture supernatants and infected cells was harvested and assayed on Vero cells. Each value is the mean ± standard error of the results of triplicate experiments. Asterisks indicate statistically significant differences (*, P < 0.05). Data are representative of results from three independent experiments.
Next, we compared the effects of p32 knockdown on viral replication in the presence and absence of UL47. For these studies, sh-p32-HEp-2 and sh-Luc-HEp-2 cells were infected with wild-type HSV-1(F), YK545 (ΔUL47), or YK546 (ΔUL47-repair) at an MOI of 0.01, and viral titers were assayed at 48 h postinfection. In agreement with the results in Fig. 14, p32 knockdown reduced progeny virus titers in wild-type HSV-1(F)- and YK546 (ΔUL47-repair)-infected cells 19.0- and 15.0-fold, respectively, but reduced the progeny virus titer in YK545 (ΔUL47)-infected cells only 2-fold (Fig. 15). Also, in agreement with previous reports showing that the UL47-null mutation reduced viral replication in cell cultures (20), YK545 (ΔUL47) had a progeny virus titer 25.0- and 22.5-fold lower than those of wild-type HSV-1(F) and YK546 (ΔUL47-repair), respectively, in sh-Luc-HEp-2 cells (Fig. 15). However, YK545 (ΔUL47) had a progeny virus titer only 2.7- and 3.1-fold lower than those of wild-type HSV-1(F) and YK546 (ΔUL47-repair), respectively, in sh-p32-HEp-2 cells (Fig. 15). Similar results were also found with these cells infected at an MOI of 5 (data not shown). These results indicated that the effects of p32 and UL47 on HSV-1 replication were mutually dependent.
FIG 15.
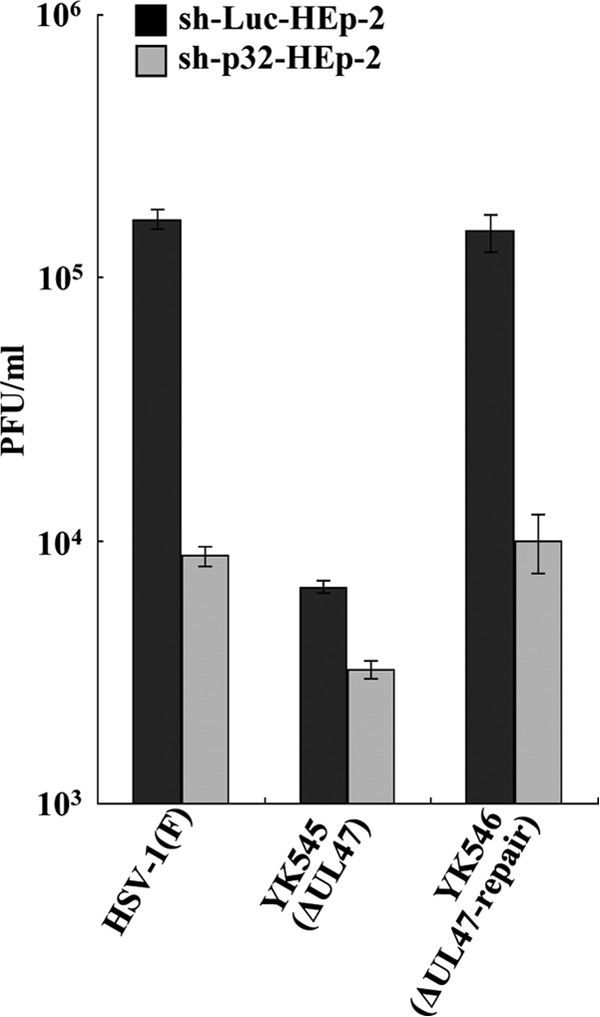
Effect of p32 and UL47 on HSV-1 replication in cell cultures. sh-Luc-HEp-2 and sh-p32-HEp-2 cells were infected with wild-type HSV-1(F), YK545 (ΔUL47), or YK546 (ΔUL47-repair) at an MOI of 0.01. At 24 h postinfection, total virus from cell culture supernatants and infected cells was harvested and assayed on Vero cells. Each value is the mean ± standard error of the results of triplicate experiments. Asterisks indicate statistically significant differences (*, P < 0.05). Data are representative of results from three independent experiments.
DISCUSSION
Tandem affinity purification of transiently expressed HSV-1 UL47 in 293T cells coupled with mass spectrometry-based proteomics technology identified a putative interaction between UL47 and cell protein p32, which was verified by coimmunoprecipitation studies with cells transiently overexpressing MEF-tagged UL47 and/or Flag-tagged p32. The interaction of UL47 with p32 was confirmed in HSV-1-infected cells: p32 coprecipitated with MEF-tagged UL47 in lysates of cells infected with recombinant virus YK536 (MEF-UL47). p32 is a ubiquitous protein in most cell types and a multifunctional protein regulating various cellular functions, including apoptosis, RNA splicing, mitochondrial translation and metabolism, and autophagy (27, 59–62). We also found that p32 accumulated at the nuclear rim in HSV-1-infected cells and that UL47 was required for the p32 accumulation. These features of p32 and UL47 are in agreement with previous reports that (i) HSV-1 infection causes translocation of p32 from the cytoplasm to the nucleus and/or nuclear rim (41, 42), (ii) herpesvirus proteins that interact with p32, including HSV-1 ICP27 and ICP34.5, EBV EBNA-1, HVS ORF63, and MHV-68 M2, can cause the redistribution of p32 (40–42, 46, 47), and (iii) UL47 regulates localization of HSV-1 and cellular proteins that interact with pUL47, including ICP27, Us3, and PABC1 (20, 22). It has been reported that viruses regulated the mitochondrial membrane potential and the mitochondrial membrane permeability and induced the release of mitochondrial proteins from the mitochondria (63, 64). In agreement with these previous reports, HSV-1 has been reported to change the integrity of the mitochondria and the membrane potential in HSV-1-infected cells (65). Therefore, HSV-1 infection may induce the release of p32 from the mitochondria, and pUL47 may take p32 to the nuclear rim by binding to it and anchor it at the nuclear membrane.
We have shown here that MEF-tagged UL47 coimmunoprecipitated with UL34, UL31, Us3, ICP22, and p32; MEF-tagged UL31 coimmunoprecipitated with UL34, Us3, UL47, ICP22, and p32; and MEF-tagged UL34 coimmunoprecipitated with UL31, UL47, Us3, ICP22, and p32. These results indicated that UL47 formed a complex with p32, UL31, UL34, UL47, Us3, and/or ICP22 in HSV-1-infected cells. At present, it remains to be determined whether UL31, UL34, UL47, Us3, ICP22, and p32 form a high-order complex in HSV-1-infected cells. However, the reciprocal coimmunoprecipitation experiments in this study, together with previous studies (3, 7, 8, 20) showing coimmunoprecipitation of (i) UL31 and UL34, (ii) UL47, UL31, and UL34, (iii) Us3 and UL47, and (iv) UL31, UL34, UL47, Us3, and ICP22 strongly suggested that a high-order complex is formed. Since UL31 and UL34 have been shown to be mostly localized at the nuclear rim in wild-type HSV-1-infected cells by immunofluorescence microscopy (3), it is likely that the interactions of UL31, UL34, UL47, Us3, ICP22, and p32 observed in this study were mainly at the nuclear rim in HSV-1-infected cells. Taken together, these observations were in agreement with the hypothesis, based on our previous reports (7, 8), that the HSV-1 NEC contained UL31, UL34, UL47, Us3, and ICP22 and suggested that p32 is a novel component of the HSV-1 NEC. In support of these hypotheses, we showed in this study that like other components of the HSV-1 NEC, p32 was aberrantly localized and colocalized with other components of the HSV-1 NEC, including UL31, UL34, and UL47, in punctate structures at the nuclear rim of infected cells in the absence of Us3 kinase activity and was incorporated into primary enveloped virions.
Electron microscopic analysis of HSV-1-infected sh-p32-HEp-2 and sh-Luc-HEp-2 cells showed that p32 knockdown significantly induced membranous invagination structures adjacent to the nuclear membrane and that primary enveloped virions accumulated in these invagination structures. Immunofluorescence analysis of these infected cells also showed that p32 knockdown resulted in aberrant localization of UL31 and UL34 in punctate structures adjacent to the nuclear rim, which protruded into the nucleoplasm and appeared to correspond to the invaginations at the nuclear membrane detected by electron microscopy. The membranous invaginations adjacent to the nuclear membrane and the accumulation of primary enveloped virions in these structures in p32 knockdown cells observed in this study have also been reported to be induced by a mutation(s) that blocks Us3 kinase activity, expression of both gB and gH, Us3 phosphorylation of UL31, and Us3 phosphorylation of gB together with expression of gH (9–12, 66). Aberrant virion accumulation in membranous invagination structures has been suggested to reflect an imbalance between the rate of virion delivery into the perinuclear space and the rate of egress from this space: the rate of virion egress from the perinuclear space may have decreased, while the rate of egress from the nucleoplasm may have not changed or not decreased as much. Therefore, it has been hypothesized that Us3, gB, gH, and UL31 were regulators for HSV-1 de-envelopment. In this model, p32 was suggested to regulate HSV-1 de-envelopment during viral nuclear egress.
At present, the mechanism by which p32 acts in HSV-1 de-envelopment remains uncertain. One possibility is that p32 may be involved in regulation of the fusogenic activity of primary enveloped virions by Us3 phosphorylation of UL31 and gB. As described above, it has been reported that Us3 phosphorylation of gB and UL31 regulated HSV-1 de-envelopment (9, 10). We have shown here that p32 was a component of primary enveloped virions, which would enable p32 to interact with UL31 and the cytoplasmic domain of gB, which contains the Us3 phosphorylation site in primary enveloped virions. Interestingly, p32 is known to bind to arginine-rich regions of its target proteins (46) and the consensus Us3 target sequence is the arginine-rich sequence RnX(S/T)YY, where n is ≥2, X can be Arg, Ala, Val, Pro, or Ser, and Y can be any amino acid except an acidic residue (67–69). Therefore, p32 may regulate Us3 phosphorylation of UL31 and gB by binding to the phosphorylation sites in gB and UL31 to promote perinuclear fusion activity for HSV-1 de-envelopment.
In this study, we showed that in HSV-1-infected cells, the invagination structures containing primary enveloped virions and the UL31/UL34 punctate structures induced by p32 knockdown were not observed in the absence of UL47. However, p32 knockdown had no effect on the reduction in primary enveloped virions in the perinuclear space or accumulation of capsids in the nucleus induced by the UL47-null mutation. These results suggested that both UL47 and p32 regulated the HSV-1 nuclear egress pathway and that UL47 acted in the HSV-1 nuclear egress pathway before the process regulated by p32. The data also supported the conclusions in our previous and present reports that UL47 and p32 played roles in primary envelopment and de-envelopment, respectively (8). In addition, our results suggested that p32 accumulation at the nuclear rim, which probably required binding to UL47 as described above, was necessary for p32 to function in de-envelopment at the nuclear membrane.
Wang et al. recently reported that p32 interacts with ICP34.5 to facilitate HSV-1 nuclear egress (41). In that study, both p32 knockdown and an ICP34.5-null mutation were shown to reduce phosphorylation of nuclear lamina and to localize aberrantly in infected HeLa cells, suggesting that p32 interacted with ICP34.5 to promote HSV-1 nuclear egress by disintegration of the nuclear lamina (41). Disintegration of the nuclear lamina by phosphorylation during herpesvirus infection has been proposed to dissolve the nuclear lamina, to facilitate herpesvirus nucleocapsid access to the INM, thereby promoting HSV-1 primary envelopment (1, 2). However, in this study, we did not observe any defects in HSV-1 primary envelopment in p32 knockdown HEp-2 cells, including accumulation of capsids in the nucleus or reduction of primary enveloped virions in the perinuclear space as previously reported for mutations in HSV-1 regulatory proteins, including UL31, UL34, UL47, and ICP22 (4, 7, 8, 57). A possible explanation of this discrepancy may be if the effect of p32 on primary envelopment was dependent on cell type. As shown in this study, p32 appeared to be a component of the viral NEC, and therefore, p32 may be able to regulate HSV-1 primary envelopment, like other NEC components, including UL31, UL34, UL47, and ICP22.
In agreement with the previous report by Wang et al. (41), we showed that p32 knockdown significantly reduced HSV-1 replication, confirming that p32 was required for efficient HSV-1 replication. Interestingly, p32 knockdown and the UL47-null mutation reduced HSV-1 replication to similar levels, the effect of p32 knockdown on HSV-1 replication was considerably reduced in the absence of UL47, and the effect of the UL47-null mutation on HSV-1 replication was reduced in p32 knockdown cells. These results suggested two possibilities: (i) the interaction between UL47 and p32 may be important in HSV-1 replication, and (ii) UL47 and p32 may function in the same pathway in HSV-1 replication. These possibilities appear to be in agreement with our hypotheses described above that UL47 and p32 functioned differently in HSV-1 nuclear egress and that the interaction between UL47 and p32 caused the accumulation of p32 at the nuclear rim, which was necessary for p32 to function in HSV-1 de-envelopment. Therefore, the regulatory roles of UL47 and p32 found in our studies may contribute to efficient HSV-1 replication, although we cannot eliminate the possibility that these viral and cellular proteins played a role(s) in HSV-1 replication other than in HSV-1 nuclear egress.
It has recently been reported that p32 is a components of the HCMV NEC together with UL50 (a homolog of HSV-1 UL34), UL53 (a homolog of HSV-1 UL31), and HCMV protein kinase UL97 (a homolog of HSV-1 UL13), emerin and protein kinase C α (70). Although a direct role of p32 in HCMV nuclear egress has not been determined, these observations raised the interesting possibility that p32 may play a conserved role in the nuclear egress of alpha- and betaherpesviruses. This may also be the case in gammaherpesviruses, based on the observations that infection of cells with gammaherpesviruses HVS and murid herpesvirus 68 (MHV-68) translocated p32 to the nuclear membrane (46, 47), as was observed with HSV-1 infection in this study.
ACKNOWLEDGMENTS
We thank Tomoko Ando, Shihoko Koyama, and Atsuko Minowa for excellent technical assistance.
This study was supported by the Funding Program for Next Generation World-Leading Researchers, Grants for Scientific Research from the Japan Society for the Promotion of Science (JSPS), a contract research fund for the Program of Japan Initiative for Global Research Network on Infectious Diseases (J-GRID), a grant for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports and Technology (MEXT) of Japan, and a grant from the Takeda Science Foundation.
REFERENCES
- 1.Mettenleiter TC, Muller F, Granzow H, Klupp BG. 2013. The way out: what we know and do not know about herpesvirus nuclear egress. Cell Microbiol 15:170–178. doi: 10.1111/cmi.12044. [DOI] [PubMed] [Google Scholar]
- 2.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 3.Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ. 2001. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J Virol 75:8803–8817. doi: 10.1128/JVI.75.18.8803-8817.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roller RJ, Zhou Y, Schnetzer R, Ferguson J, DeSalvo D. 2000. Herpes simplex virus type 1 U(L)34 gene product is required for viral envelopment. J Virol 74:117–129. doi: 10.1128/JVI.74.1.117-129.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 6.Bigalke JM, Heuser T, Nicastro D, Heldwein EE. 2014. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat Commun 5:4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maruzuru Y, Shindo K, Liu Z, Oyama M, Kozuka-Hata H, Arii J, Kato A, Kawaguchi Y. 2014. Role of herpes simplex virus 1 immediate early protein ICP22 in viral nuclear egress. J Virol 88:7445–7454. doi: 10.1128/JVI.01057-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Z, Kato A, Shindo K, Noda T, Sagara H, Kawaoka Y, Arii J, Kawaguchi Y. 12 February 2014. Herpes simplex virus 1 UL34 interacts with critical viral nuclear egress factors UL31, UL34 and Us3, and regulates viral nuclear egress. J Virol doi: 10.1128/JVI.00137-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wisner TW, Wright CC, Kato A, Kawaguchi Y, Mou F, Baines JD, Roller RJ, Johnson DC. 2009. Herpesvirus gB-induced fusion between the virion envelope and outer nuclear membrane during virus egress is regulated by the viral Us3 kinase. J Virol 83:3115–3126. doi: 10.1128/JVI.01462-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mou F, Wills E, Baines JD. 2009. Phosphorylation of the U(L)31 protein of herpes simplex virus 1 by the U(S)3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J Virol 83:5181–5191. doi: 10.1128/JVI.00090-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ryckman BJ, Roller RJ. 2004. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the Us3-UL34 catalytic relationship. J Virol 78:399–412. doi: 10.1128/JVI.78.1.399-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and Us3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J Virol 76:8939–8952. doi: 10.1128/JVI.76.17.8939-8952.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mou F, Forest T, Baines JD. 2007. Us3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J Virol 81:6459–6470. doi: 10.1128/JVI.00380-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mou F, Wills EG, Park R, Baines JD. 2008. Effects of lamin A/C, lamin B1, and viral US3 kinase activity on viral infectivity, virion egress, and the targeting of herpes simplex virus UL34-encoded protein to the inner nuclear membrane. J Virol 82:8094–8104. doi: 10.1128/JVI.00874-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park R, Baines JD. 2006. Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J Virol 80:494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muranyi W, Haas J, Wagner M, Krohne G, Koszinowski UH. 2002. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 297:854–857. doi: 10.1126/science.1071506. [DOI] [PubMed] [Google Scholar]
- 17.Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol 82:8605–8618. doi: 10.1128/JVI.00904-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donnelly M, Verhagen J, Elliott G. 2007. RNA binding by the herpes simplex virus type 1 nucleocytoplasmic shuttling protein UL47 is mediated by an N-terminal arginine-rich domain that also functions as its nuclear localization signal. J Virol 81:2283–2296. doi: 10.1128/JVI.01677-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donnelly M, Elliott G. 2001. Nuclear localization and shuttling of herpes simplex virus tegument protein VP13/14. J Virol 75:2566–2574. doi: 10.1128/JVI.75.6.2566-2574.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato A, Liu Z, Minowa A, Imai T, Tanaka M, Sugimoto K, Nishiyama Y, Arii J, Kawaguchi Y. 2011. Herpes simplex virus 1 protein kinase Us3 and major tegument protein UL47 reciprocally regulate their subcellular localization in infected cells. J Virol 85:9599–9613. doi: 10.1128/JVI.00845-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Sirko DA, McKnight JL. 1991. Role of herpes simplex virus type 1 UL46 and UL47 in alpha Tif-mediated transcriptional induction: characterization of three viral deletion mutants. J Virol 65:829–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dobrikova E, Shveygert M, Walters R, Gromeier M. 2010. Herpes simplex virus proteins ICP27 and UL47 associate with polyadenylate-binding protein and control its subcellular distribution. J Virol 84:270–279. doi: 10.1128/JVI.01740-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shu M, Taddeo B, Zhang W, Roizman B. 2013. Selective degradation of mRNAs by the HSV host shutoff RNase is regulated by the UL47 tegument protein. Proc Natl Acad Sci U S A 110:E1669–E1675. doi: 10.1073/pnas.1305475110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Leeuwen HC, O'Hare P. 2001. Retargeting of the mitochondrial protein p32/gC1Qr to a cytoplasmic compartment and the cell surface. J Cell Sci 114:2115–2123. [DOI] [PubMed] [Google Scholar]
- 25.Brokstad KA, Kalland KH, Russell WC, Matthews DA. 2001. Mitochondrial protein p32 can accumulate in the nucleus. Biochem Biophys Res Commun 281:1161–1169. doi: 10.1006/bbrc.2001.4473. [DOI] [PubMed] [Google Scholar]
- 26.Itahana K, Zhang Y. 2008. Mitochondrial p32 is a critical mediator of ARF-induced apoptosis. Cancer Cell 13:542–553. doi: 10.1016/j.ccr.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sunayama J, Ando Y, Itoh N, Tomiyama A, Sakurada K, Sugiyama A, Kang D, Tashiro F, Gotoh Y, Kuchino Y, Kitanaka C. 2004. Physical and functional interaction between Bh3-only protein Hrk and mitochondrial pore-forming protein p32. Cell Death Differ 11:771–781. doi: 10.1038/sj.cdd.4401418. [DOI] [PubMed] [Google Scholar]
- 28.Rubinstein DB, Stortchevoi A, Boosalis M, Ashfaq R, Ghebrehiwet B, Peerschke EI, Calvo F, Guillaume T. 2004. Receptor for the globular heads of C1q (gC1q-R, p33, hyaluronan-binding protein) is preferentially expressed by adenocarcinoma cells. Int J Cancer 110:741–750. doi: 10.1002/ijc.20105. [DOI] [PubMed] [Google Scholar]
- 29.Robles-Flores M, Rendon-Huerta E, Gonzalez-Aguilar H, Mendoza-Hernandez G, Islas S, Mendoza V, Ponce-Castaneda MV, Gonzalez-Mariscal L, Lopez-Casillas F. 2002. p32 (gC1qBP) is a general protein kinase C (Pkc)-binding protein; interaction and cellular localization of p32-PKC complexes in ray hepatocytes. J Biol Chem 277:5247–5255. doi: 10.1074/jbc.M109333200. [DOI] [PubMed] [Google Scholar]
- 30.Lu PD, Galanakis DK, Ghebrehiwet B, Peerschke EI. 1999. The receptor for the globular “heads” of C1q, gC1q-R, binds to fibrinogen/fibrin and impairs its polymerization. Clin Immunol 90:360–367. doi: 10.1006/clim.1998.4660. [DOI] [PubMed] [Google Scholar]
- 31.Lim BL, White RA, Hummel GS, Schwaeble W, Lynch NJ, Peerschke EI, Reid KB, Ghebrehiwet B. 1998. Characterization of the murine gene of gC1qBP, a novel cell protein that binds the globular heads of C1q, vitronectin, high molecular weight kininogen and factor Xii. Gene 209:229–237. doi: 10.1016/S0378-1119(98)00055-9. [DOI] [PubMed] [Google Scholar]
- 32.Yu L, Loewenstein PM, Zhang Z, Green M. 1995. In vitro interaction of the human immunodeficiency virus type 1 Tat transactivator and the general transcription factor Tfiib with the cellular protein Tap. J Virol 69:3017–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simos G, Georgatos SD. 1994. The lamin B receptor-associated protein p34 shares sequence homology and antigenic determinants with the splicing factor 2-associated protein p32. FEBS Lett 346:225–228. doi: 10.1016/0014-5793(94)00479-X. [DOI] [PubMed] [Google Scholar]
- 34.Luo Y, Yu H, Peterlin BM. 1994. Cellular protein modulates effects of human immunodeficiency virus type 1 Rev. J Virol 68:3850–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berro R, Kehn K, de la Fuente C, Pumfery A, Adair R, Wade J, Colberg-Poley AM, Hiscott J, Kashanchi F. 2006. Acetylated Tat regulates human immunodeficiency virus type 1 splicing through its interaction with the splicing regulator p32. J Virol 80:3189–3204. doi: 10.1128/JVI.80.7.3189-3204.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matthews DA, Russell WC. 1998. Adenovirus core protein v interacts with p32—a protein which is associated with both the mitochondria and the nucleus. J Gen Virol 79(Part 7):1677–1685. [DOI] [PubMed] [Google Scholar]
- 37.Suppiah S, Mousa HA, Tzeng WP, Matthews JD, Frey TK. 2012. Binding of cellular p32 protein to the rubella virus P150 replicase protein via Pxxpxr motifs. J Gen Virol 93:807–816. doi: 10.1099/vir.0.038901-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beatch MD, Everitt JC, Law LJ, Hobman TC. 2005. Interactions between rubella virus capsid and host protein P32 are important for virus replication. J Virol 79:10807–10820. doi: 10.1128/JVI.79.16.10807-10820.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kittlesen DJ, Chianese-Bullock KA, Yao ZQ, Braciale TJ, Hahn YS. 2000. Interaction between complement receptor gC1qR and hepatitis C virus core protein inhibits T-lymphocyte proliferation. J Clin Invest 106:1239–1249. doi: 10.1172/JCI10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Scoy S, Watakabe I, Krainer AR, Hearing J. 2000. Human p32: a coactivator for Epstein-Barr virus nuclear antigen-1-mediated transcriptional activation and possible role in viral latent cycle DNA replication. Virology 275:145–157. doi: 10.1006/viro.2000.0508. [DOI] [PubMed] [Google Scholar]
- 41.Wang YYY, Wu S, Pan S, Zhou C, Ma Y, Ru Y, Dong S, He B, Zhang C, Cao Y. 2014. p32 is a novel target for viral protein ICP34.5 of herpes simplex virus type 1 and facilitates viral nuclear egress. J Biol Chem 289:35795–35805. doi: 10.1074/jbc.M114.603845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bryant HE, Matthews DA, Wadd S, Scott JE, Kean J, Graham S, Russell WC, Clements JB. 2000. Interaction between herpes simplex virus type 1 IE63 protein and cellular protein p32. J Virol 74:11322–11328. doi: 10.1128/JVI.74.23.11322-11328.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bruni R, Roizman B. 1996. Open reading frame P—a herpes simplex virus gene repressed during productive infection encodes a protein that binds a splicing factor and reduces synthesis of viral proteins made from spliced mRNA. Proc Natl Acad Sci U S A 93:10423–10427. doi: 10.1073/pnas.93.19.10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Milbradt J, Auerochs S, Sticht H, Marschall M. 2009. Cytomegaloviral proteins that associate with the nuclear lamina: components of a postulated nuclear egress complex. J Gen Virol 90:579–590. doi: 10.1099/vir.0.005231-0. [DOI] [PubMed] [Google Scholar]
- 45.Marschall M, Marzi A, aus dem Siepen P, Jochmann R, Kalmer M, Auerochs S, Lischka P, Leis M, Stamminger T. 2005. Cellular p32 recruits cytomegalovirus kinase pUL97 to redistribute the nuclear lamina. J Biol Chem 280:33357–33367. doi: 10.1074/jbc.M502672200. [DOI] [PubMed] [Google Scholar]
- 46.Hall KT, Giles MS, Calderwood MA, Goodwin DJ, Matthews DA, Whitehouse A. 2002. The herpesvirus saimiri open reading frame 73 gene product interacts with the cellular protein p32. J Virol 76:11612–11622. doi: 10.1128/JVI.76.22.11612-11622.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liang X, Shin YC, Means RE, Jung JU. 2004. Inhibition of interferon-mediated antiviral activity by murine gammaherpesvirus 68 latency-associated M2 protein. J Virol 78:12416–12427. doi: 10.1128/JVI.78.22.12416-12427.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sugimoto K, Uema M, Sagara H, Tanaka M, Sata T, Hashimoto Y, Kawaguchi Y. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J Virol 82:5198–5211. doi: 10.1128/JVI.02681-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arii J, Uema M, Morimoto T, Sagara H, Akashi H, Ono E, Arase H, Kawaguchi Y. 2009. Entry of herpes simplex virus 1 and other alphaherpesviruses via the paired immunoglobulin-like type 2 receptor alpha. J Virol 83:4520–4527. doi: 10.1128/JVI.02601-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J Virol 82:6172–6189. doi: 10.1128/JVI.00044-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kato A, Hirohata Y, Arii J, Kawaguchi Y. 2014. Phosphorylation of herpes simplex virus 1 dutpase upregulated viral dutpase activity to compensate for low cellular dutpase activity for efficient viral replication. J Virol 88:7776–7785. doi: 10.1128/JVI.00603-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawaguchi Y, Van Sant C, Roizman B. 1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J Virol 71:7328–7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arii J, Goto H, Suenaga T, Oyama M, Kozuka-Hata H, Imai T, Minowa A, Akashi H, Arase H, Kawaoka Y, Kawaguchi Y. 2010. Non-muscle myosin IIa is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862. doi: 10.1038/nature09420. [DOI] [PubMed] [Google Scholar]
- 54.Maruzuru Y, Fujii H, Oyama M, Kozuka-Hata H, Kato A, Kawaguchi Y. 2013. Roles of P53 in herpes simplex virus 1 replication. J Virol 87:9323–9332. doi: 10.1128/JVI.01581-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kato A, Yamamoto M, Ohno T, Kodaira H, Nishiyama Y, Kawaguchi Y. 2005. Identification of proteins phosphorylated directly by the Us3 protein kinase encoded by herpes simplex virus 1. J Virol 79:9325–9331. doi: 10.1128/JVI.79.14.9325-9331.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morimoto T, Arii J, Tanaka M, Sata T, Akashi H, Yamada M, Nishiyama Y, Uema M, Kawaguchi Y. 2009. Differences in the regulatory and functional effects of the Us3 protein kinase activities of herpes simplex virus 1 and 2. J Virol 83:11624–11634. doi: 10.1128/JVI.00993-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang YE, Van Sant C, Krug PW, Sears AE, Roizman B. 1997. The null mutant of the U(L)31 gene of herpes simplex virus 1: construction and phenotype in infected cells. J Virol 71:8307–8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scholtes LD, Yang K, Li LX, Baines JD. 2010. The capsid protein encoded by U(L)17 of herpes simplex virus 1 interacts with tegument protein Vp13/14. J Virol 84:7642–7650. doi: 10.1128/JVI.00277-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiao H, Su GQ, Dong W, Zhang L, Xie W, Yao LM, Chen P, Wang ZX, Liou YC, You H. 24 April 2015. Chaperone-like protein P32 regulates Ulk1 stability and autophagy. Cell Death Differ doi: 10.1038/cdd.2015.34. [DOI] [PubMed] [Google Scholar]
- 60.Yagi M, Uchiumi T, Takazaki S, Okuno B, Nomura M, Yoshida S, Kanki T, Kang D. 2012. P32/Gc1qr is indispensable for fetal development and mitochondrial translation: importance of its RNA-binding ability. Nucleic Acids Res 40:9717–9737. doi: 10.1093/nar/gks774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fogal V, Richardson AD, Karmali PP, Scheffler IE, Smith JW, Ruoslahti E. 2010. Mitochondrial P32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol Cell Biol 30:1303–1318. doi: 10.1128/MCB.01101-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Petersen-Mahrt SK, Estmer C, Ohrmalm C, Matthews DA, Russell WC, Akusjarvi G. 1999. The splicing factor-associated protein, P32, regulates RNA splicing by inhibiting Asf/Sf2 RNA binding and phosphorylation. EMBO J 18:1014–1024. doi: 10.1093/emboj/18.4.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anand SK, Tikoo SK. 2013. Viruses as modulators of mitochondrial functions. Adv Virol 2013:738794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G. 2008. Viral control of mitochondrial apoptosis. PLoS Pathog 4:e1000018. doi: 10.1371/journal.ppat.1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Murata T, Goshima F, Daikoku T, Inagaki-Ohara K, Takakuwa H, Kato K, Nishiyama Y. 2000. Mitochondrial distribution and function in herpes simplex virus-infected cells. J Gen Virol 81:401–406. [DOI] [PubMed] [Google Scholar]
- 66.Farnsworth A, Wisner TW, Webb M, Roller R, Cohen G, Eisenberg R, Johnson DC. 2007. Herpes simplex virus glycoproteins Gb and Gh function in fusion between the virion envelope and the outer nuclear membrane. Proc Natl Acad Sci U S A 104:10187–10192. doi: 10.1073/pnas.0703790104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Leader DP. 1993. Viral protein kinases and protein phosphatases. Pharmacol Ther 59:343–389. doi: 10.1016/0163-7258(93)90075-O. [DOI] [PubMed] [Google Scholar]
- 68.Leader DP, Deana AD, Marchiori F, Purves FC, Pinna LA. 1991. Further definition of the substrate specificity of the alpha-herpesvirus protein kinase and comparison with protein kinases A and C. Biochim Biophys Acta 1091:426–431. [DOI] [PubMed] [Google Scholar]
- 69.Purves FC, Deana AD, Marchiori F, Leader DP, Pinna LA. 1986. The substrate specificity of the protein kinase induced in cells infected with herpesviruses: studies with synthetic substrates [corrected] indicate structural requirements distinct from other protein kinases. Biochim Biophys Acta 889:208–215. doi: 10.1016/0167-4889(86)90106-0. [DOI] [PubMed] [Google Scholar]
- 70.Milbradt J, Kraut A, Hutterer C, Sonntag E, Schmeiser C, Ferro M, Wagner S, Lenac T, Claus C, Pinkert S, Hamilton ST, Rawlinson WD, Sticht H, Coute Y, Marschall M. 2014. Proteomic analysis of the multimeric nuclear egress complex of human cytomegalovirus. Mol Cell Proteomics 13:2132–2146. doi: 10.1074/mcp.M113.035782. [DOI] [PMC free article] [PubMed] [Google Scholar]