

Abstract
Rotavirus, the leading cause of diarrheal diseases in children under the age of five, is often resistant to conventional wastewater treatment and thus can remain infectious once released into the aquatic environment. Solar and heat treatments can inactivate rotavirus, but it is unknown how these treatments inactivate the virus on a molecular level. To answer this question, our approach was to correlate rotavirus inactivation with the inhibition of portions of the virus life cycle as a means to identify the mechanisms of solar or heat inactivation. Specifically, the integrity of the rotavirus NSP3 gene, virus-host cell interaction, and viral RNA synthesis were examined after heat (57°C) or solar treatment of rotavirus. Only the inhibition of viral RNA synthesis positively correlated with a loss of rotavirus infectivity; 57°C treatment of rotavirus resulted in a decrease of rotavirus RNA synthesis at the same rate as rotavirus infectivity. These data suggest that heat treatment neutralized rotaviruses primarily by targeting viral transcription functions. In contrast, when using solar disinfection, the decrease in RNA synthesis was responsible for approximately one-half of the decrease in infectivity, suggesting that other mechanisms, including posttranslational, contribute to inactivation. Nevertheless, both solar and heat inactivation of rotaviruses disrupted viral RNA synthesis as a mechanism for inactivation.
INTRODUCTION
Of all waterborne microbes that can cause pathogenic diseases, viruses remain the most challenging for study in aquatic environments (1–4). This is for several reasons. First, compared to bacterial pathogens, viruses are more resistant to disinfection by standard water disinfection treatments (2, 5, 6). Second, for those conditions that do indeed neutralize viruses, the molecular mechanism(s) responsible for inactivation is unclear (7, 8). Third, it is only recently that technologies such as quantitative PCR (qPCR) and deep genome sequencing have allowed for the detection of viral genomes in water samples (1, 4, 9, 10).
Enteric viruses include rotavirus, adenovirus, coxsackievirus, and norovirus (4). Rotavirus is the leading cause of diarrhea hospitalizations for children under 5 years of age and is responsible for the deaths of an estimated 453,000 children each year (11). Despite this impact on human health, little is known as to what conditions result in the best disinfection of rotavirus in wastewater and drinking water. In addition, it is unclear how a disinfectant functions on a molecular level to inactivate rotavirus.
Rotavirus is a nonenveloped virus with a double-stranded, segmented RNA genome. Six structural proteins make up the capsid, including VP4 and VP7. For rotavirus to attach to its host cell, VP4 binds to the host cell receptors, and after a multistep process involving the VP4, VP7, and other cellular proteins, rotavirus enters the host cell through endocytosis. Upon penetration, rotavirus loses its outer capsid (VP7 and VP4), and the double-layered particle is transported to the cell cytoplasm. Transcription, translation, and replication of the viral genome, which includes the NSP3 gene, occur, and then mature virions are assembled and released by cell lysis (12–15).
Rotavirus is inactivated effectively by solar disinfection and by heat treatment (16, 17). Inactivation of viruses by solar UV (280 to 400 nm) occurs through direct and indirect damage to virus components, whereby only the virus and light are involved (5); this mechanism of inactivation is referred to as endogenous in this text (16, 18). Human rotavirus is readily inactivated by UVB wavelengths from 280 to 320 nm (16). Alternatively, when organic sensitizers present in solutions are exposed to solar UVA and visible light (320 to 700 nm), transient species capable of photooxidizing virus components are formed (16, 18–20). Of these transient species, hydroxyl radicals and triplet state species have been implicated with the inactivation of human rotavirus at 25°C; this mechanism of inactivation is referred to as exogenous (16). Based on work with MS2 bacteriophage and adenovirus type 2, a reasonable hypothesis is that solar treatment damages the rotavirus genome and capsid proteins, preventing virus attachment and viral genome replication (21, 22). In contrast, heat treatment is expected to destabilize the rotavirus capsid, as shown with heated MS2 bacteriophage, adenovirus type 5 proteins, parvoviruses, human picornavirus, and feline calicivirus, preventing the initial virus-host cell interactions that are required for a productive virus infection (21, 23–25). However, neither hypothesis has been proven formally for rotaviruses.
The goal of this study is to define the conditions that result in rotavirus disinfection and to identify potential mechanisms of disinfection by heat or solar treatment. Here, we examined trends between the rate constants of virus inactivation, as measured by an infectivity focus-forming unit (FFU) assay, the integrity of a single virus gene, and two virus life cycle events: virus binding to host cell and viral gene synthesis. This approach allowed us to identify how heat or solar treatment disinfects rotavirus particles on a molecular level. We argue that the identification of these molecular mechanisms will be the basis for evaluation of the improvement of current technologies to detect and control rotaviruses in water.
MATERIALS AND METHODS
Human rotavirus propagation and purification and cell lines.
Amplification of human rotavirus Wa occurred in the monkey kidney MA104 cell line, and the FFU assay was used to quantify infectious rotavirus particles as described previously (16, 17). All purified rotavirus stocks were adjusted such that the final concentration was 106 FFU/ml.
Disinfection treatment of rotaviruses.
Inactivation experiments were conducted with a virus concentration of 2 × 105 FFU/ml in 50-μl samples containing 1 mM NaHCO3 buffer. Each disinfection experiment consisted of at least four treatment time points, and each treatment time point corresponded to two 50-μl samples of virus. All experiments were performed in duplicate. Two types of disinfection were conducted: solar treatment and 57°C treatment in the dark.
Solar treatment of viruses.
All solar irradiation tests were performed in 96-well plates that were incubated in a 20°C water bath. The 96-well plate containing the rotavirus samples was irradiated with an Atlas Suntest XLS+ photosimulator (Chicago, IL). Full-spectrum and UVA and visible light irradiating conditions were accomplished as described in detail previously (16, 17), and the spectra are shown in Fig. S1 in the supplemental material. Full-spectrum irradiation tests were performed to primarily investigate the direct and endogenous mechanisms, whereas UVA and visible light irradiation tests were conducted to assess the exogenous effects. Sample evaporation over the course of the solar experiments was <5%.
Four different conditions were used for solar treatments of rotaviruses: full-spectrum irradiation of solutions containing 20 mg C/liter of the organic sensitizer (full spectrum + sensitizer), full-spectrum irradiation of a sensitizer-free buffer solution (full spectrum), UVA and visible light irradiation of solutions containing 20 mg C/liter of the organic sensitizer (UVA+vis + sensitizer), and UVA and visible light irradiation of a sensitizer-free buffer solution (UVA+vis). The model organic sensitizer used in this study was the Suwannee River natural organic matter (SRNOM; International Humic Substances Society) and was prepared as detailed elsewhere (17). The transient species produced by 20 mg C/liter of SRNOM at 25°C ± 1°C upon UVA and visible light exposure have been characterized in our previous study, in which the steady-state concentrations of OH radical and singlet oxygen concentrations were 1.7 × 10−16 ± 0.6 M and 2.5 × 10−13 ± 0.3 M, respectively (17). Under the same conditions, the hydrogen peroxide accumulation rate was 1.7 ± 0.6 M · h−1 (17).
Heat treatment of viruses.
For all treatments, inactivation experiments were conducted with 2 × 105 FFU/ml virus stock in 50 μl of virus solution containing 1 mM NaHCO3 buffer. Virus aliquots were placed in a 200-μl plastic tube and incubated in a 57°C water bath for treatment times as indicated in the figure legends. For these inactivation assays, tests were performed in the dark to minimize disinfection due to light exposure.
Infectivity/FFU assay.
The infectivity assay was performed as described previously (16, 17). Briefly, after each disinfection treatment, each virus sample was incubated with 10 μg/ml trypsin from porcine pancreas (Sigma-Aldrich) for 30 min at 37°C. Next, virus samples were added to confluent MA104 cell monolayers grown in 24-well plates with minimum essential medium (MEM) (MA104 cell growth medium; Gibco) at 37°C in a 5% CO2 incubator. Cells and viruses were incubated for 30 min in an incubator with 5% CO2 set to 37°C. Subsequently, cellular monolayers were washed twice with MEM and incubated 37°C for 15 to 18 h. At 15 to 18 h postinfection (hpi), the FFU assay was used to quantify rotavirus infectivity decay rate constants (17). Infectivity rate constants were expressed as kinfectivity and were obtained from the first-order kinetics plot of ln(FFUt/FFU0) versus treatment time (hours), where FFUt refers to the focus-forming units measured for the treated virus samples and FFU0 is for nontreated virus samples. Each independent experiment included duplicate samples of MEM lacking rotaviruses as a negative control. The detection limit of the FFU assay was ∼2 × 103 FFU/ml.
To better understand how solar or 57°C treatment inactivated human rotavirus inactivation, we assessed the function of specific portions of the rotavirus life cycle over treatment time. A schematic summarizing the approaches taken is shown in Fig. 1, which includes a diagram that highlights the portion of the life cycle that we investigated and a brief description of the experimental approach used for each portion of the life cycle.
FIG 1.

Summary of the rotavirus replication cycle and our experimental approaches for investigating the inactivation mechanisms.
RT-qPCR to detect rotavirus NSP3 RNA from rotavirus virions.
Reverse transcriptase quantitative PCR (RT-qPCR) was used to quantify rotavirus NSP3 gene segments isolated either from purified virions or from cells inoculated with viruses. All viruses used for RT-qPCR experiments were treated as described above or left untreated. RT-qPCR tests were performed separately from the infectivity tests. The rotavirus concentration used for the RT-qPCR tests was 6 × 104 FFU/ml in 50-μl samples, while the rotavirus concentration for the FFU tests was 2 × 105 FFU/ml in 50-μl samples. Due to the high detection limit of the FFU infectivity assay, ∼ 2 × 103 FFU/ml, the higher initial human rotavirus concentration was necessary to capture a 2-log10 unit reduction.
Total RNA was extracted from purified virions using the E.Z.N.A. total RNA kit I (Omega Bio-Tek) according to the manufacturer's instructions. For assays in which cells were inoculated with viruses, cells were lysed in 350 μl of E.Z.N.A. total RNA kit I lysis buffer and total RNA was extracted as described above. Next, the extracted RNA was quantified using a Qubit 2.0 fluorometer (Life Technologies) and the Qubit RNA BR assay kit (Life Technologies). For all samples, RNA was eluted in 30 μl of molecular-grade water and stored at −80°C. To preserve the stability of a sample, RNA samples underwent no more than two freeze/thaw cycles.
RT-qPCRs were performed in 48 low-profile tube strips (Bio-Rad, CA) using the iTaq universal SYBR green one-step kit (Bio-Rad, Hercules, CA) and a MiniOpticon RT-PCR system (Bio-Rad). A total reaction volume of 10 μl was prepared, consisting of 3 μl of RNA template and 7 μl of RT-qPCR mastermix. The mastermix consisted of a mixture of 1× one-step SYBR green reaction mix, 1× iScript RT enzyme mix, molecular-grade water, and 300 nM each forward (JVKF) and reverse (JVKR) primers, which were obtained from Jothikumar et al. and Mattioli et al. (26, 27) and are listed in Table S1 in the supplemental material. Cycling parameters were as follows: 10-min reverse transcription step at 50°C; 1-min denaturation step at 95°C; 35 cycles of 95°C for 10 s, 54°C for 15 s, and 60°C for 30 s. A dissociation cycle of 15 s at 95°C, 15 s at 54°C, and 95°C for 15 s was performed to assess for nonspecific amplification. Each RNA sample was evaluated for NSP3 RNA twice using RT-qPCR.
In a separate reaction performed in parallel, a cDNA plasmid standard containing the cDNA of rotavirus A isolate L26 NSP3 gene nucleotides 1 to 206 (synthesized by IDT, Coralville, IA) was also tested in the qRT-PCR assay mentioned above to allow for the calculation of the number of NSP3 amplicons present in experimental samples (22). The cDNA plasmid standard concentrations were converted to copy numbers using the following formula: (moles/gram) × (molecules/mole) = molecules/gram. This was done via a DNA copy number calculator, which can be found at http://cels.uri.edu/gsc/cndna.html. The amount (in nanograms) of double-stranded DNA (dsDNA) was measured with a Qubit 2.0 fluorometer (Life Technologies) and Qubit DNA HS assay kit (Life Technologies). A calibration curve was generated with six 10-fold cDNA plasmid standard dilutions ranging from 2 × 106 to 2 copies/μl. This calibration curve was then used to calculate the number of NSP3 amplicons generated when genomic rotavirus RNA was amplified by RT-qPCR. A 100-μl sample of 1 × 106 FFU/ml resulted in 1.4 × 106 genomic NSP3 copies/μl. Multiple aliquots of the extracted and diluted genomic RNA (1.4 × 106 − 14 genomic NSP3 copies/μl) were stored at −80°C and used in every RT-qPCR assay. The freeze/thaw cycles for the RNA did not exceed three cycles. This number of freeze/thaw cycles had no effect on the RT-qPCR efficiency calculated from the linear regression of both the log cDNA plasmid standard and genomic rotavirus RNA versus threshold cycle (CT), where the efficiency was calculated using the formula 10(−1/slope) − 1 (in this study, 83% ± 2%). Human rotavirus RNA, and not the cDNA plasmid standard, was used as a standard both to serve as a positive control and to monitor the performance of the RT-qPCR assay from reverse transcription to amplification. Using these standardized curves, the number of NSP3 amplicons generated during RT-qPCR was calculated. Based on the lower NSP3 copies measured consistently (100% of RT-qPCR runs) using genomic RNA, the detection limit was 14 copies/μl. In addition, two samples without an RNA template were included in each RT-qPCR as a negative control.
Integrity of the genomic NSP3 RNA in treated and untreated purified virions.
When assaying purified virions for NSP3 RNA, total RNA was extracted from treated or untreated rotaviruses as described above. Three microliters of extracted RNA was analyzed by RT-qPCR to quantify the NSP3 copies in the treated samples (RT-qPCRt) and nontreated samples (RT-qPCR0). The NSP3 gene amplification decay rate constant, kdamage, as an indirect measure of NSP3 gene damage, was calculated from the negative slope of the ln(RT-qPCRt/RT-qPCR0) versus treatment time (hours) plot.
NSP3 RNA RT-qPCR to detect rotavirus binding to host cells.
Using the rotavirus NSP3 gene as a marker of rotavirus particles, we attempted to measure whether solar or heat treatment of rotaviruses would compromise the ability of rotavirus to bind to cell receptors (Fig. 1). As explained further in “Limitations of the RT-qPCR binding assay and its use in assessing rotavirus binding to host cells upon 57°C or solar treatments” below (in Results), as a first step in establishing if RT-qPCR could be used to detect bound rotaviruses by tracking the NSP3 gene, a calibration curve was needed. To generate the calibration curve, 6 dilutions of untreated particles ranging from 3 × 103 to 3 × 105 FFU/ml of untreated rotavirus particles were incubated with MA104 cellular monolayers and allowed to adsorb at 4°C for 1 h. Monolayers were occasionally rocked during a 1-h incubation at 4°C, conditions known to allow virus-host interactions but prevent virus internalization (28–30). Cells were washed three times with 500 μl of ice-cold phosphate-buffered saline (PBS) to remove unbound rotaviruses. Next, cellular monolayers were lysed, and total cellular and viral RNAs were isolated as described above. Three microliters of extracted RNA from each sample was evaluated by RT-qPCR to quantify rotavirus NSP3 RNA. The results of the calibration curve are presented in “Limitations of the RT-qPCR binding assay and its use in assessing rotavirus binding to host cells upon 57°C or solar treatments” below.
The effect of disinfection on the loss of binding was evaluated by comparing the number of NSP3 copies of untreated (RT-qPCR0) and treated (RT-qPCRt) rotaviruses. First, treated or untreated rotavirus samples were incubated with 10 μg/ml of trypsin for 30 min at 37°C. These rotavirus samples (50 μl of 6 × 104 FFU/ml) were diluted with MEM to a final volume of 150 μl. Next, the virus samples were inoculated onto confluent MA104 cells (∼5 × 105 cells) grown in 24-well plates with a multiplicity of infection (MOI) of 0.006. A low MOI was desired to increase the likelihood that no more than one rotavirus adsorbed to a given MA104 cell (21). Unbound viruses were removed by washing cellular monolayers, and total RNA was extracted from cellular monolayers that would have bound virions. Next, RNA was analyzed for the rotavirus NSP3 gene by RT-qPCR, and the numbers of bound rotaviruses (or NSP3 copies) were calculated. A rate constant for the loss of rotavirus binding ability, kobserved_binding, was calculated from the negative slope of the ln(RT-qPCRt/RT-qPCR0) versus treatment time (hours) plot. The kobserved_binding rate constant was calculated to obtain a corrected binding rate constant, k′binding, as follows: k′binding = kobserved_binding − kdamage. The kdamage rate constant was accounted for to distinguish between the decrease in the NSP3 gene PCR signal due to NSP3 gene damage by the treatment or decrease in NSP3 gene PCR signal due to loss of binding function by the same treatment. For the treatments where kobserved_binding and kdamage rate constants were not statistically different (P > 0.05), the k′binding rate constant was set to zero.
RT-qPCR to detect NSP3 RNA synthesis.
As illustrated in Fig. 1, after binding and penetration, the rotavirus dsRNA genome is transcribed to positive-sense RNA (+RNA) transcripts. Positive-sense RNAs encode the rotaviral proteins and function as the templates for production of negative strands to make dsRNA (14, 15). Thus, our initial hypothesis was that during infection, the number of synthesized +RNA copies, including NSP3, would exceed the number of genomic RNA copies stemming from the viruses initiating the infection. To test this hypothesis, confluent cellular MA104 monolayers were inoculated with virus samples and incubated for 1 h at 4°C (identical to the binding assay procedure). Next, cell monolayers were washed three times with 500 μl of PBS and allowed to internalize the cells by incubating at 37°C. At various times ranging from 0 to 22 hpi, monolayers were washed once with PBS and lysed, and total RNA was extracted. Three microliters of RNA was used for RT-qPCR analysis. Figure S2 in the supplemental material shows that the relative NSP3 gene copies (CT/C0) synthesized during MA104 cell infection from 0 (C0) to 22 h (CT) reached and exceeded a factor of 20 for incubation times greater than 9 h. For consistency with the FFU assay incubation period, detection of NSP3 gene synthesis for the disinfection experiments occurred at 15 to 18 hpi as further explained in “Influence of 57°C or solar treatments on rotavirus RNA synthesis” below.
The effect of disinfection on the loss on NSP3 gene synthesis was evaluated by comparing the number of NSP3 copies of untreated (RT-qPCR0) and treated (RT-qPCRt) rotaviruses. Untreated (RT-qPCR0) or treated (RT-qPCRt) viruses were incubated at 4°C with cellular MA104 monolayers for 1 h. Cell monolayers were washed three times with PBS prior to incubation at 37°C. At 15 to 18 h postinfection, monolayers were washed once with PBS and lysed, and total RNA was extracted. Three microliters of RNA was used for RT-qPCR analysis. The first-order decay rate constant of RNA synthesis, kobserved_synthesis, was calculated from the negative slope of the ln(RT-qPCRt/RT-qPCR0) versus treatment time (hours) plot.
Data analysis.
The negative slopes, first-order decay rate constants, and their standard errors were calculated from the ln(FFUt/FFU0) or ln(RT-qPCRt/RT-qPCR0) versus time (hours) plots of data aggregated from at least duplicate experiments. To account for the changes of the NSP3 gene segment signal due to treatment alone, the kobserved_binding rate constants were corrected by subtracting the corresponding kdamage rate constant. The law of error propagation for simple average errors was used to propagate the standard error to the corrected rate constants. Linear regression and multiple linear regression tests were used to assess if the slopes of the lines were statistically different from a slope of 0 or if the slopes of two lines were statistically different, respectively. P values of <0.05 were considered significant.
RESULTS
Loss of human rotavirus infectivity upon 57°C or solar treatment.
Inactivation of human rotavirus Wa due to 57°C treatment was compared with inactivation by solar irradiation in the presence and absence of reactive-radical-producing organic sensitizer. The inactivation kinetics and the first-order decay rate constants obtained from the negative slope of the ln(FFUt/FFU0) versus treatment time (h−1) plots were calculated and are reported as kinfectivity rate constants (see Fig. 3 and 5, respectively). Notably, no significant decay in infectivity was observed for the dark control, as shown in Fig. 2. Consistent with our previous findings (16), 57°C treatment of viruses resulted in the fastest inactivation rate constant (kinfectivity ± standard error, 57.4 ± 11.6 h−1). Treatments by full-spectrum light irradiation of solutions containing or lacking the sensitizer (24.0 ± 3.0 h−1 and 12.8 ± 0.59 h−1, respectively) resulted in the next-highest kinfectivity rate constants. The rate constants for solutions irradiated with UVA and visible light with (5.91 ± 0.26 h−1) and without (2.56 ± 0.19 h−1) the organic sensitizer were lower than for the full-spectrum irradiation conditions. Rate constants for each treatment were statistically different from the other rate constants (P < 0.01). Finally, in agreement with our previous findings (16), transient radicals produced through the irradiation of the organic sensitizer enhanced human rotavirus inactivation compared to the sensitizer-free buffer control.
FIG 3.

First-order rate constants for RT-qPCR amplification of the NSP3 gene (kdamage) associated with binding (both corrected and uncorrected k′binding and kobserved_binding, respectively) and viral NSP3 gene synthesis (kobserved_synthesis) and rotavirus infectivity (kinfectivity) upon exposure to the indicated treatments. Data from at least two separate experiments were aggregated and plotted as ln(FFUt or RT-qPCRt/FFU0 or RT-qPCR0) versus treatment time (hours), and the negative slope and the standard error are the first-order rate constants and error bars, respectively, shown in the bar chart. The k′binding rate constant was corrected to account for the rate constant stemming from treatment alone. One asterisk (*) denotes that kdamage did not deviate from 0 (P > 0.05); two asterisks (**) denote that k′binding was assigned a value of zero because there was no statistical difference between kdamage and kobserved_binding (P > 0.05).
FIG 5.
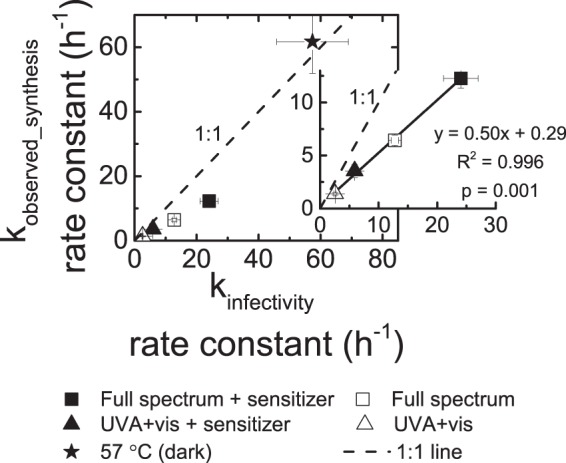
First-order decay rate constants (kobserved_synthesis) of viral RNA synthesis in infected cells with rotaviruses treated by heat at 57°C in the dark or simulated sunlight irradiation as a function of infectivity decay rate constants (kinfectivity). Data from at least two separate experiments were aggregated and plotted as ln(RT-qPCRt or FFUt/RT-qPCR0 or FFU0) versus treatment time (hours), and the negative slope and the standard error were calculated from linear regression tests. For each treatment, the calculated negative slopes are represented here as the rate constants in viral RNA synthesis and the standard errors are represented by error bars. The insets show the significant linear correlations (P < 0.01) between kobserved_synthesis and kinfectivity for solar treatments. The data for treatment at 57°C were not used in the linear regression analysis because their trend was different from that of the solar treatment data.
FIG 2.
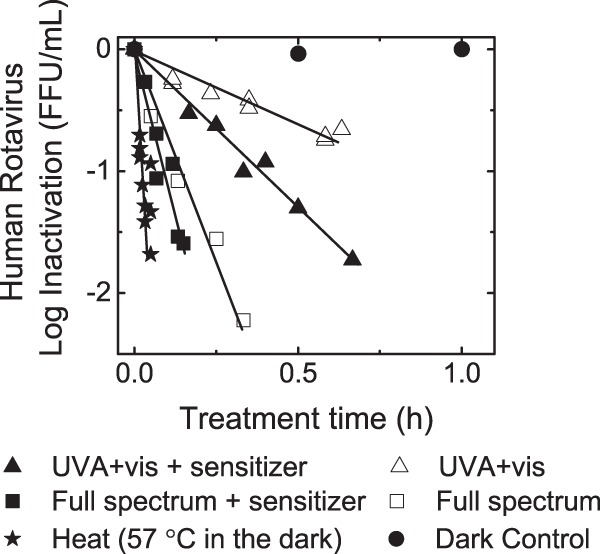
Rotavirus infectivity after solar and 57°C treatments. The infectivity data from at least two independent experiments were aggregated and plotted here as log10(FFUt/FFU0) versus treatment time (hours). Inactivation experiments were conducted in solutions containing 1 mM NaHCO3 buffer (pH 8), 0 or 20 mg C/liter of an organic sensitizer, and an initial rotavirus concentration of 2 × 105 FFU/ml. Samples of treated or untreated rotaviruses were inoculated onto MA104 cell monolayers. At 15 to 18 h postinfection, cells were fixed and immunostained, and FFUs were quantified using an inverted microscope.
After determining the inactivation rate constants, the extent to which each treatment affected specific portions of the rotavirus life cycle were investigated using RT-qPCR. All data used in this study are summarized in Table S2 in the supplemental material. No loss of signal was measured in dark controls conducted at room temperature for any of the rotavirus RNA measurement assays (see Fig. S3D in the supplemental material).
Integrity of NSP3 gene upon 57°C or solar treatments.
The integrity of NSP3 gene for human rotavirus exposed to 57°C or solar treatments was determined to explore whether these treatments damaged encapsidated NSP3 gene segments. If the NSP3 genome segment of rotavirus were indeed amplified by RT-qPCR, then we interpreted this to mean that a disinfecting treatment did not damage the NSP3 gene sufficiently to prevent its interaction with NSP3-specific primers or amplification of the gene. The NSP3 segment decay rate constants were higher for full-spectrum (kdamage ± standard error, ≥0.69 ± 0.12 h−1) than for UVA and visible light (≤0.41 ± 0.04 h−1) treatments (Fig. 3), independent of the presence or absence of the organic sensitizer. The kdamage decay rate constants resulting from either full-spectrum or UVA and visible light irradiation were not statistically different (P > 0.05) in the presence or absence of the organic sensitizer (Fig. 3B to D). Conversely, no significant decay in NSP3 gene signal was detected for the 57°C treatment of rotaviruses (P > 0.05). Overall, solar treatments induced NSP3 gene segment damage, but 57°C treatment did not.
Limitations of the RT-qPCR binding assay and its use in assessing rotavirus binding to host cells upon 57°C or solar treatments.
An initial hypothesis was that solar or heat treatment of rotaviruses would alter the structure of the rotavirus capsid, such that rotavirus-host cell interactions would be compromised. An approach to test this hypothesis was to use RT-qPCR to detect the rotavirus NSP3 gene as a marker of rotavirus particles bound to cells under various conditions and to use this approach to quantify the loss of binding damage upon treatment, if any. To validate the binding assay, it was key to assess the following: (i) there was a significant relationship between initial untreated rotavirus concentrations and NSP3 gene copies associated with the bound rotaviruses, and the assay was both reproducible and sensitive; and (ii) the assay was specific.
First, to establish if the RT-qPCR binding assay was sensitive and reproducible and whether there was a linear relationship between the recovered NSP3 copies and untreated rotaviruses, six rotavirus concentrations ranging from 3 × 103 to 3 × 105 FFU and a fixed number of host cells (∼5 × 105 cells) were used to generate a calibration curve. Each sample was used in duplicate. As shown in Fig. 4, there was a linear relationship for rotavirus concentrations less than ∼2 × 105 FFU/ml, at which virus binding began to plateau. The plateau was probably the result of saturating the binding sites of the MA104 cell monolayer (31–33). Based on this finding, the binding assay and other RT-qPCR assays were carried out with rotavirus concentrations of 6 × 104 FFU/ml to ensure that they were working within the linear portion of the binding curve. It should be noted that the NSP3 copy numbers obtained for the binding assay ranged between 200 and ∼2,200, which was within the range (126 to ∼2,200 NSP3 gene copies/μg total RNA) of the calibration curve. Table S2 in the supplemental material summarizes the data used for the binding calibration curve.
FIG 4.
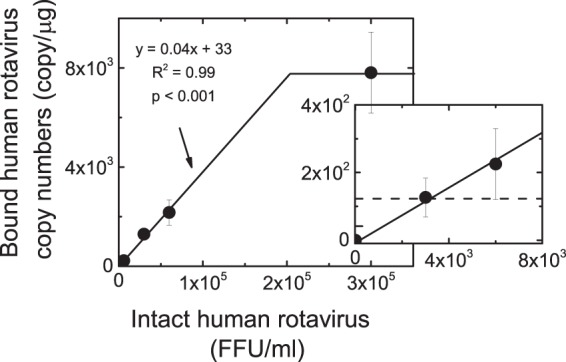
A quantification of the population of intact rotavirus particles that bind with host cells. Binding experiments were conducted with the following intact rotavirus concentrations (FFU/ml): 3 × 103, 6 × 103, 3 × 104, 6 × 104, and 3 × 105. Human rotavirus samples were inoculated onto MA104 cell monolayers (∼5 × 105 cells) and incubated at 4°C for 1 h. Next, monolayers were washed and lysed, and total RNA was extracted from lysates. Total RNA was subjected to RT-qPCR to amplify and quantify the viral NSP3 gene, which is present as a single copy in each rotavirus virion. The error bars denote the range of duplicate samples tested. The dashed line in the inset shows the lowest copy number measured with the binding assay, about 126 copies.
Second, it is known that rotavirus binding depends on virus and host cell concentrations (32). Yet, it is unknown how or if modified binding proteins, due to disinfection treatment, would result in nonspecific binding of the virus to the host cells. The three-time wash with PBS after cell and virus incubation at 4°C, an approach employed here and by others (21, 31, 32, 34), may not have removed all nonspecifically bound viruses. Still, we were able to detect binding inhibition. Binding inhibition was determined based on the comparison between kdamage and kobserved_binding rate constants for each treatment. If kdamage and kobserved_binding rate constants were significantly different (P < 0.05), then a corrected k′binding rate constant was calculated by subtracting kdamage from the kobserved_binding rate constant. Based on the k′binding rate constants, binding inhibition was detected when rotaviruses were treated with solar light in the presence of the organic sensitizer and when treated at 57°C (Fig. 3). For solar treatments without an organic sensitizer, no binding inhibition was detected (Fig. 3).
Influence of 57°C or solar treatments on rotavirus RNA synthesis.
We developed an RT-qPCR-based assay that quantified the rate constant at which viral RNA synthesis occurred in cells inoculated with untreated or treated viruses by measuring NSP3 gene levels in infected cells. Unlike the FFU assay, which detects the completion of the rotavirus life cycle from binding through cell lysis, viral RNA synthesis captures binding through RNA transcription but does not detect posttranscriptional virus events such as protein translation (schematic shown in Fig. 1). When cells were inoculated with untreated rotavirus, there was a 20-fold increase in NSP3 amplicons at 15 to 18 h postinfection (see Fig. S2 in the supplemental material). The first-order decay rate constants in rotavirus RNA synthesis were reported as kobserved_synthesis rate constants (shown in Fig. 3).
In this study, rotavirus RNA synthesis decreased at a rate similar to that of infectivity for all treatments tested (Fig. 3 and 5). The kobserved_synthesis and kinfectivity rate constants were the highest for 57°C treatment and lowest for the UVA and visible light irradiation treatment. Rotavirus treatment at 57°C resulted in kobserved_synthesis and kinfectivity rate constants of 61.6 ± 9.7 h−1 and 57.4 ± 11.6 h−1, respectively, both of which are not statistically different (P > 0.05). For solar treatments, the kobserved_synthesis and kinfectivity rate constants significantly (P = 0.001) correlated, but the magnitude of kobserved_synthesis rate constants was one-half of the magnitude of kinfectivity rate constants (Fig. 5).
DISCUSSION
In this study, we assessed the integrity of virus binding and the fidelity of viral NSP3 gene transcription as a means to determine the portion of the virus life cycle inhibited by disinfection. These data would, in turn, identify how a disinfection treatment inactivates rotaviruses. The implications of our findings for each RT-qPCR-based assay developed here are discussed next.
Our results for the decay rate constants of the NSP3 target segment, kdamage, due to solar and heat treatments are in agreement with previous findings (22, 35, 36). We observed higher kdamage rate constants when viruses were treated with full-spectrum than with UVA and visible light, consistent with previous findings that UVB causes more-extensive damage than UVA irradiation (35). The presence of organic sensitizers did not significantly affect kdamage rate constants, suggesting that damage resulting from the exogenous mechanism minimally affected NSP3 gene integrity. Similar results regarding the effect of a sensitizer were reported when disinfecting human adenovirus type 2 (22). While NSP3 gene damage was detected for solar treatment of viruses, no significant decay in NSP3 gene signal was detected during 57°C treatment of rotaviruses (P > 0.05), even though it resulted in a high kinfectivity rate constant. The lack of NSP3 gene damage at 57°C may be explained by the high resistance of viral nucleic acids to heat treatment (36).
The decay rate constant of the NSP3 gene segment, kdamage, serves as evidence for genome damage. A damaged genome can hinder genome transcription and other posttranscriptional functions such as protein translation and genome replication. Though kdamage rate constants described the damage to a single gene segment comprising 0.7% of the entire rotavirus genome, it is impracticable to extrapolate these kdamage values to the entire genome. A previous study targeting six genome segments and 50% genome coverage found that damage due to several treatments, including UV254 nm and UVA and visible light in the presence of a sensitizer, was not consistent across the monitored regions of the MS2 RNA genome (21). Some regions were more susceptible to the reactive oxygen species and others to UV254 nm radiation (21). In this study, kdamage rate constants served to show that genome damage occurred, which in turn suggested that damage to other genome-mediated life cycle steps were possible.
Just as kdamage rate constants served as evidence for genome damage, evidence of binding inhibition, as expressed by k′binding rate constants, was also indicative of protein damage. Though the binding assay performed in this study may not have discriminated against nonspecific binding of treated or untreated viruses, k′binding rate constants served to indicate the occurrence of binding inhibition in three of the five treatments tested: heat treatment at 57°C and the two solar treatments in which the organic sensitizer was present. Because proteins are the only rotavirus components able to attach to the host receptors, binding inhibition may also indicate inhibition of rotavirus entry, as structural proteins VP4 and VP7 are responsible for both binding and entry (13, 37, 38). It should be noted that protein conformations have been associated with heat treatment of viruses (21, 24). In addition, reactive radicals produced through UVA and visible light irradiation in the presence of sensitizers resulted in protein damage and loss of binding ability in human adenovirus type 2 and MS2 bacteriophage, respectively (21, 22).
Both kdamage and k′binding rate constants provided evidence for genome and protein damage, respectively, but on their own neither decreased at the same rate constant as inactivation for all treatments tested. The kobserved_synthesis rate constants, which comprise the decay in RT-qPCR signal arising from damages in the rotavirus NSP3 gene, binding to host cells, and RNA synthesis, predicted kinfectivity rate constants, which in turn captured all rotavirus life cycles from binding through cell lysis. The correlation between kobserved_synthesis and kinfectivity for solar treatments and the similarity in the magnitudes of kobserved_synthesis and kinfectivity rate constants indicated that rotavirus inactivation was largely (for solar treatments) or entirely (for 57°C treatment) linked to a defect in RNA synthesis. The trends of the kobserved_synthesis, k′binding, and kdamage rate constants aided in recognizing possible virus components that are targets for each treatment. For example, because kobserved_synthesis fully contributed to kinfectivity and binding inhibition was observed upon treatment at 57°C, it is likely that 57°C treatment primarily targets rotavirus components that mediate early life cycle steps involved in viral RNA synthesis, such as binding and entry proteins. For solar treatments, kobserved_synthesis rate constants accounted for about 50% of the kinfectivity rate constants, suggesting that while viral components involved in early life cycle steps were significant targets, those components involved in posttranscriptional functions (e.g., genome) were equally important.
Assessment of viral functions that can be directly associated with inactivation is essential in determining which components are the principal targets of a treatment. Often, the detection and quantification of isolated protein or gene damages cannot be directly linked to virus inactivation (22, 39, 40). An approach that combines virus functionality and specific virus structures is needed to identify the specific targets for solar and heat treatments. To our knowledge, this study is the first to investigate and correlate human rotavirus Wa inactivation by 57°C and solar treatments with a defect in viral RNA synthesis.
We have shown that the type of treatment influenced the kobserved_synthesis and kinfectivity rate constants. Because the approach using detection of specific viral genes with RT-qPCR after inoculating cell cultures did not capture posttranscriptional damages, the kobserved_synthesis rate constants were lower than the kinfectivity rate constants. These findings may provide further insight into the inconsistencies reported between cell culture and integrated cell culture (ICC)-RT-qPCR inactivation data. For example, inactivation data obtained with the ICC-RT-qPCR method showed that a much higher UV dose was needed to achieve a 3-log rotavirus reduction than the previously reported dose with data determined by a cell culture assay (41). In a study by Blackmer and Reynolds, the contact time for chlorine disinfection of poliovirus was found to be about five times greater when employing the ICC-RT-qPCR assay than when employing a conventional cell culture assay (42). Inactivation data obtained with the ICC-RT-qPCR approach could overestimate the treatment dose needed to achieve a desired log viral reduction, resulting in unnecessarily high treatment costs. We suggest that understanding inactivation mechanisms of pathogenic viruses with various treatments is crucial for the improvement or design of technologies used to control or detect viruses in water.
Recently, several studies have probed the life cycle of rotavirus infection (43–45). Interestingly, some rotavirus strains (including human rotavirus Wa) undergo internalization via clathrin-mediated endocytosis to enter an endosomal compartment and then a late endosome, while other rotavirus strains can enter the host cell via a clathrin-independent process (43). This internalization process is VP4 dependent. Regardless, cleavage of VP4 into VP5 and VP8 occurs as the virus changes from its triple-layer form to its double-layer form. Here, we investigated the replication of the human rotavirus NSP3 gene as a means to determine the portion of the virus life cycle blocked by heat or solar treatment. Thus, one limitation with this approach is that, while it is quantitative, it does not detect the steps in the life cycle that range from virus attachment to progression through the maturing endosomes. A future direction is to ask whether either solar or heat treatment would affect the stability of VP4 protein such that it could not be cleaved or if either treatment affects the ability of the virus to enter into the cell or traffic to endosomal compartments. Such approaches would rely heavily on microscopy and subcellular fractionation.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the financial support of an NSF Career grant to T.H.N. (0954501) and NSF GRF DGE 07-15088 FLW to O.C.R.-M. This publication was made possible by research partially supported by grant RD83582601-0 from the U.S. Environmental Protection Agency (EPA).
The contents of this article are solely the responsibility of the grantee and do not necessarily represent the official views of the EPA. Further, the EPA does not endorse the purchase of any commercial products or services mentioned in the publication.
We kindly thank Alexandra B. Boehm and Mia Mattioli from Stanford University for providing us with the design of the rotavirus cDNA plasmid standard for the NSP3 gene used in this study. We also thank Hanting Wang for helping with experiments and Peter A. Maraccini for insightful discussions.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00027-15.
REFERENCES
- 1.Metcalf TG, Melnick JL, Estes MK. 1995. Environmental virology: from detection of virus in sewage and water by isolation to identification by molecular biology—a trip of over 50 years. Annu Rev Microbiol 49:461–487. [DOI] [PubMed] [Google Scholar]
- 2.Fong T, Lipp E. 2005. Enteric viruses of humans and animals in aquatic environments: health risks, detection, and potential water quality assessment tools. Microbiol Mol Biol Rev 69:357–371. doi: 10.1128/MMBR.69.2.357-371.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wyn-Jones AP, Sellwood J. 2001. Enteric viruses in the aquatic environment. J Appl Microbiol 91:945–962. doi: 10.1046/j.1365-2672.2001.01470.x. [DOI] [PubMed] [Google Scholar]
- 4.Bosch A. 1998. Human enteric viruses in the water environment: a minireview. Int Microbiol 1:191–196. [PubMed] [Google Scholar]
- 5.Davies-Colley R, Donnison A, Speed D, Ross C, Nagels J. 1999. Inactivation of faecal indicator micro-organisms in waste stabilisation ponds: interactions of environmental factors with sunlight. Water Res 33:1220–1230. doi: 10.1016/S0043-1354(98)00321-2. [DOI] [Google Scholar]
- 6.Payment P, Trudel M, Plante R. 1985. Elimination of viruses and indicator bacteria at each step of treatment during preparation of drinking water at seven water treatment plants. Appl Environ Microbiol 49:1418–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wigginton KR, Kohn T. 2012. Virus disinfection mechanisms: the role of virus composition, structure, and function. Curr Opin Virol 2:84–89. doi: 10.1016/j.coviro.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Page MA, Shisler JL, Mariñas BJ. 2010. Mechanistic aspects of adenovirus serotype 2 inactivation with free chlorine. Appl Environ Microbiol 76:2946–2954. doi: 10.1128/AEM.02267-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodríguez RA, Pepper IL, Gerba CP. 2009. Application of PCR-based methods to assess the infectivity of enteric viruses in environmental samples. Appl Environ Microbiol 75:297–307. doi: 10.1128/AEM.01150-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bae J, Schwab KJ. 2008. Evaluation of murine norovirus, feline calicivirus, poliovirus, and MS2 as surrogates for human norovirus in a model of viral persistence in surface water and groundwater. Appl Environ Microbiol 74:477–484. doi: 10.1128/AEM.02095-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parashar U, Steele D, Neuzil K, De Quadros C, Tharmaphornpilas P, Serhan F, Santosham M, Patel M, Glass R. 2013. Progress with rotavirus vaccines: summary of the Tenth International Rotavirus Symposium. Expert Rev Vaccines 12:113–117. doi: 10.1586/erv.12.148. [DOI] [PubMed] [Google Scholar]
- 12.Desselberger U. 2000. Rotaviruses: basic facts, p 1–8. In Gray J, Desselberger U (ed), Rotaviruses: methods and protocols. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 13.Lopez S, Arias CF. 2006. Early steps in rotavirus cell entry, p 39–66. In Roy P. (ed), Reoviruses: entry, assembly and morphogenesis, 1st ed Springer-Verlag, Berlin, Germany. [DOI] [PubMed] [Google Scholar]
- 14.Pesavento JB, Crawford SE, Estes MK, Prasad BVV. 2006. Rotavirus proteins: structure and assembly. Curr Top Microbiol Immunol 309:189–219. [DOI] [PubMed] [Google Scholar]
- 15.Patton JT, Silvestri LS, Tortorici MA, Vasquez-Del Carpio R, Taraporewala ZF. 2006. Rotavirus genome replication and morphogenesis: role of the viroplasm. Curr Top Microbiol Immunol 309:169–187. doi: 10.1007/3-540-30773-7_6. [DOI] [PubMed] [Google Scholar]
- 16.Romero-Maraccini OC, Sadik J, Rosado-Lausell SL, Pugh CR, Niu X-Z, Croue J-P, Nguyen TH. 2013. Sunlight-induced inactivation of human Wa and porcine OSU rotaviruses in the presence of exogenous photosensitizers. Environ Sci Technol 47:11004–11012. doi: 10.1021/es402285u. [DOI] [PubMed] [Google Scholar]
- 17.Romero OC, Straub AP, Kohn T, Nguyen TH. 2011. Role of temperature and Suwannee River natural organic matter on inactivation kinetics of rotavirus and bacteriophage MS2 by solar irradiation. Environ Sci Technol 45:10385–10393. doi: 10.1021/es202067f. [DOI] [PubMed] [Google Scholar]
- 18.Silverman AI, Peterson BM, Boehm AB, McNeill K, Nelson KL. 2013. Sunlight inactivation of human viruses and bacteriophages in coastal waters containing natural photosensitizers. Environ Sci Technol 47:1870–1878. doi: 10.1021/es3036913. [DOI] [PubMed] [Google Scholar]
- 19.Kohn T, Nelson KL. 2007. Sunlight-mediated inactivation of MS2 coliphage via exogenous singlet oxygen produced by sensitizers in natural waters. Environ Sci Technol 41:192–197. doi: 10.1021/es061716i. [DOI] [PubMed] [Google Scholar]
- 20.Rosado-Lausell SL, Wang H, Gutiérrez L, Romero-Maraccini OC, Niu X-Z, Gin KYH, Croué J-P, Nguyen TH. 2013. Roles of singlet oxygen and triplet excited state of dissolved organic matter formed by different organic matters in bacteriophage MS2 inactivation. Water Res 47:4869–4879. doi: 10.1016/j.watres.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 21.Wigginton KR, Pecson BM, Bosshard F, Kohn T, Sigstam T. 2012. Virus inactivation mechanisms: impact of disinfectants on virus function and structural integrity. Environ Sci Technol 46:12069–12078. doi: 10.1021/es3029473. [DOI] [PubMed] [Google Scholar]
- 22.Bosshard F, Armand F, Hamelin R, Kohn T. 2013. Mechanisms of human adenovirus inactivation by sunlight and UVC light as examined by quantitative PCR and quantitative proteomics. Appl Environ Microbiol 79:1325–1332. doi: 10.1128/AEM.03457-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiethoff CM, Wodrich H, Gerace L, Nemerow GR. 2005. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J Virol 79:1992–2000. doi: 10.1128/JVI.79.4.1992-2000.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nelson CDS, Minkkinen E, Bergkvist M, Hoelzer K, Fisher M, Bothner B, Parrish CR. 2008. Detecting small changes and additional peptides in the canine parvovirus capsid structure. J Virol 82:10397–10407. doi: 10.1128/JVI.00972-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nuanualsuwan S, Cliver DO. 2003. Capsid functions of inactivated human picornaviruses and feline calicivirus. Appl Environ Microbiol 69:350–357. doi: 10.1128/AEM.69.1.350-357.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mattioli MC, Pickering AJ, Gilsdorf RJ, Davis J, Boehm AB. 2013. Hands and water as vectors of diarrheal pathogens in Bagamoyo, Tanzania. Environ Sci Technol 47:355–363. doi: 10.1021/es303878d. [DOI] [PubMed] [Google Scholar]
- 27.Jothikumar N, Kang G, Hill VR. 2009. Broadly reactive TaqMan assay for real-time RT-PCR detection of rotavirus in clinical and environmental samples. J Virol Methods 155:126–131. doi: 10.1016/j.jviromet.2008.09.025. [DOI] [PubMed] [Google Scholar]
- 28.Kaljot K, Shaw R. 1988. Infectious rotavirus enters cells by direct cell membrane penetration, not by endocytosis. J Virol 62:1136–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Méndez E, López S, Cuadras MA, Romero P, Arias CF. 1999. Entry of rotaviruses is a multistep process. Virology 263:450–459. doi: 10.1006/viro.1999.9976. [DOI] [PubMed] [Google Scholar]
- 30.Isa P, Realpe M, Romero P, López S, Arias CF. 2004. Rotavirus RRV associates with lipid membrane microdomains during cell entry. Virology 322:370–381. doi: 10.1016/j.virol.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 31.Zárate S, Espinosa R, Romero P, Guerrero CA, Arias CF, López S. 2000. Integrin α2β1 mediates the cell attachment of the rotavirus neuraminidase-resistant variant nar3. Virology 278:50–54. doi: 10.1006/viro.2000.0660. [DOI] [PubMed] [Google Scholar]
- 32.Rolsma MD, Gelberg HB, Kuhlenschmidt MS. 1994. Assay for evaluation of rotavirus-cell interactions: identification of an enterocyte ganglioside fraction that mediates group A porcine rotavirus recognition. J Virol 68:258–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zárate S, Espinosa R, Romero P. 2000. The VP5 domain of VP4 can mediate attachment of rotaviruses to cells. J Virol 74:593–599. doi: 10.1128/JVI.74.2.593-599.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haselhorst T, Fleming FE, Dyason JC, Hartnell RD, Yu X, Holloway G, Santegoets K, Kiefel MJ, Blanchard H, Coulson BS, von Itzstein M. 2009. Sialic acid dependence in rotavirus host cell invasion. Nat Chem Biol 5:91–93. doi: 10.1038/nchembio.134. [DOI] [PubMed] [Google Scholar]
- 35.Schuch AP, Menck CFM. 2010. The genotoxic effects of DNA lesions induced by artificial UV-radiation and sunlight. J Photochem Photobiol B 99:111–116. doi: 10.1016/j.jphotobiol.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 36.Li D, Gu AZ, Yang W, He M, Hu X-H, Shi H-C. 2010. An integrated cell culture and reverse transcription quantitative PCR assay for detection of infectious rotaviruses in environmental waters. J Microbiol Methods 82:59–63. doi: 10.1016/j.mimet.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 37.Díaz-Salinas M, Romero P. 2013. The spike protein VP4 defines the endocytic pathway used by rotavirus to enter MA104 cells. J Virol 87:1658–1663. doi: 10.1128/JVI.02086-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martínez M, López S, Arias C, Isa P. 2013. Gangliosides have a functional role during rotavirus cell entry. J Virol 87:1115–1122. doi: 10.1128/JVI.01964-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eischeid AC, Meyer JN, Linden KG. 2009. UV disinfection of adenoviruses: molecular indications of DNA damage efficiency. Appl Environ Microbiol 75:23–28. doi: 10.1128/AEM.02199-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pecson BM, Martin LV, Kohn T. 2009. Quantitative PCR for determining the infectivity of bacteriophage MS2 upon inactivation by heat, UV-B radiation, and singlet oxygen: advantages and limitations of an enzymatic treatment to reduce false-positive results. Appl Environ Microbiol 75:5544–5554. doi: 10.1128/AEM.00425-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li D, Gu AZ, He M, Shi H-C, Yang W. 2009. UV inactivation and resistance of rotavirus evaluated by integrated cell culture and real-time RT-PCR assay. Water Res 43:3261–3269. doi: 10.1016/j.watres.2009.03.044. [DOI] [PubMed] [Google Scholar]
- 42.Blackmer F, Reynolds K. 2000. Use of integrated cell culture-PCR to evaluate the effectiveness of poliovirus inactivation by chlorine. Appl Environ Microbiol 66:2267–2269. doi: 10.1128/AEM.66.5.2267-2268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Diaz-Salinas MA, Silva-Ayala D, Lopez S, Arias CF. 2014. Rotaviruses reach late endosomes and require the cation-dependent mannose-6-phosphate receptor and the activity of cathepsin proteases to enter the cell. J Virol 88:4389–4402. doi: 10.1128/JVI.03457-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin S, Lorrot M, Alaoui M, Azher E, Vasseur M. 2002. Ionic strength- and temperature-induced KCa shifts in the uncoating reaction of rotavirus strains RF and SA11: correlation with membrane permeabilization. J Virol 76:552–559. doi: 10.1128/JVI.76.2.552-559.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silva-Ayala D, López T. 2013. Genome-wide RNAi screen reveals a role for the ESCRT complex in rotavirus cell entry. Proc Natl Acad Sci U S A 110:10270–10275. doi: 10.1073/pnas.1304932110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.