

Abstract
The Corynebacterium alkanolyticum xylEFGD gene cluster comprises the xylD gene that encodes an intracellular β-xylosidase next to the xylEFG operon encoding a substrate-binding protein and two membrane permease proteins of a xyloside ABC transporter. Cloning of the cluster revealed a recombinant β-xylosidase of moderately high activity (turnover for p-nitrophenyl-β-d-xylopyranoside of 111 ± 4 s−1), weak α-l-arabinofuranosidase activity (turnover for p-nitrophenyl-α-l-arabinofuranoside of 5 ± 1 s−1), and high tolerance to product inhibition (Ki for xylose of 67.6 ± 2.6 mM). Heterologous expression of the entire cluster under the control of the strong constitutive tac promoter in the Corynebacterium glutamicum xylose-fermenting strain X1 enabled the resultant strain X1EFGD to rapidly utilize not only xylooligosaccharides but also arabino-xylooligosaccharides. The ability to utilize arabino-xylooligosaccharides depended on cgR_2369, a gene encoding a multitask ATP-binding protein. Heterologous expression of the contiguous xylD gene in strain X1 led to strain X1D with 10-fold greater β-xylosidase activity than strain X1EFGD, albeit with a total loss of arabino-xylooligosaccharide utilization ability and only half the ability to utilize xylooligosaccharides. The findings suggest some inherent ability of C. glutamicum to take up xylooligosaccharides, an ability that is enhanced by in the presence of a functional xylEFG-encoded xyloside ABC transporter. The finding that xylEFG imparts nonnative ability to take up arabino-xylooligosaccharides should be useful in constructing industrial strains with efficient fermentation of arabinoxylan, a major component of lignocellulosic biomass hydrolysates.
INTRODUCTION
Hemicellulose, a heteropolymer of various pentose (d-xylose and l-arabinose) and hexose (d-glucose, d-mannose, and d-galactose) sugars, is a major component of plant cell walls (1). Its most common constituent is the polymer xylan, a linear backbone of β-1,4-linked d-xylose residues branched with various side groups such as acetyl, l-arabinofuranose, and O-methyl-d-glucuronic acid (2). Complete biological degradation of xylan necessitates removal of the side groups by accessory enzymes such as α-l-arabinofuranosidases, α-d-glucuronidase, and acetylxylan esterases for the linear backbone of d-xylose residues to be accessible to hydrolysis by endo-β-1,4-xylanases and β-xylosidases (2). Xylanolytic microorganisms produce some or all of the enzymes necessary to utilize xylan as a carbon source. Fungal xylanolytic enzymes are usually secreted, whereas bacterial α-l-arabinofuranosidases, α-d-glucuronidase, and β-xylosidases are intracellular enzymes. In xylanolytic bacteria, extracellular xylooligosaccharides such as xylobiose and xylotriose resulting from the action of endo-β-1,4-xylanases must be transported into the cells by carrier proteins prior to hydrolysis to xylose by intracellular β-xylosidases. Bacterial xylooligosaccharide transporters are either xyloside/Na+ (H+) symporters or xyloside ABC transporters. Xyloside/Na+ (H+) symporters are in essence multipass transmembrane proteins that transport xylooligosaccharides by sodium (proton) motive force. Xylooligosaccharide symporters able to transport as many as six (3) or as few as three (4) xylosyl units have been confirmed in Klebsiella spp. In contrast to symporter structure and mechanism, each xyloside ABC transporter is a complex of a substrate-binding protein, a membrane permease protein, and an ATP-binding protein that combine to facilitate the active transport of xylooligosaccharides across the cell membrane. To date, the only two confirmed xyloside ABC transporters are from Streptomyces thermoviolaceus BxlEFG (5) and Geobacillus stearothermophilus XynEFG (6). S. thermoviolaceus BxlE and G. stearothermophilus XynE, the respective substrate-binding proteins of the transporters, have confirmed high affinities toward xylooligosaccharides, but their substrate specificities in vivo remain uncharacterized.
Corynebacteria are high-G+C-content Gram-positive actinomycetes that are widely distributed in nature in soil, water, and plants. Though no reported molecular characterization of xylanolytic enzymes and related transporters in corynebacteria is known, glutamate- and biosurfactant-producing Corynebacterium alkanolyticum (7, 8) grows on xylooligosaccharide as a sole carbon source. In this study, we clone β-xylosidase and xylooligosaccharide transporter genes from C. alkanolyticum and express them in the heterologous xylose-fermenting host Corynebacterium glutamicum (9). We consequently confirm the ability of the respective xylooligosaccharide transporter to transport both arabino-xylooligosaccharides and xylooligosaccharides. This is the first confirmation of a bacterial arabino-xylooligosaccharide transporter, to the best of our knowledge. Given the efficacy of C. glutamicum to produce a variety of chemicals, including succinate (10), ethanol (11), isobutanol (12), alanine (13, 14), and valine (15), this study could contribute to better understanding and optimization of utilization of the arabinoxylan component of lignocellulosic biomass by C. glutamicum and further enhance the utility of the microorganism in the production of more valuable products from lignocellulosic biomass.
MATERIALS AND METHODS
Bacterial strains, medium, and cultivation conditions.
All bacterial strains and plasmids used in this study are listed in Table 1. For genetic manipulations, Escherichia coli strains were grown at 33°C in Luria-Bertani medium (16). For aerobic growth conditions, C. alkanolyticum and C. glutamicum R (JCM18229) (17) and recombinant strains were precultured at 33°C overnight in nutrient-rich medium (A medium) containing (per liter) 2 g of urea, 2 g of yeast extract, 7 g of Casamino Acids, 7 g of (NH4)2SO4, 0.5 g of KH2PO4, 0.5 g of K2HPO4, 0.5 g of MgSO4 · 7H2O, 6 mg of FeSO4 · 7H2O, 4.2 mg of MnSO4 · H2O, 0.2 mg of biotin, and 0.2 mg of thiamine, supplemented with 4% (wt/vol) glucose (10). Where appropriate, the medium was supplemented with antibiotics. The final antibiotic concentrations were as follows: for E. coli, 50 μg ml−1 of ampicillin and 50 μg ml−1 of kanamycin, and for C. glutamicum, 50 μg ml−1 of kanamycin.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s) | Reference or source |
|---|---|---|
| Strains | ||
| E. coli | ||
| HST02 | F′ [traD36 proA+B+ lacIq lacZΔM15] D(lac-proAB) recA endA1 gyrA96 thi e14 mutant (mcrA mutant), supE44 relA1 ΔdeoR Δ(mrr-hsdRMS-mcrBC) | TaKaRa |
| SCS110 | rpsL (Strr) thr leu endA thi-1 lacY galK galT ara tonA tsx dam dcm supE44 Δ(lac-proAB) [F′ traD36 proAB lacIqZΔM15] | Agilent |
| BL21(DE3) | F− ompT hsdSB(rB− mB−) gal dcm (DE3) | TaKaRa |
| C. alkanolyticum ATCC 21511 | ||
| C. glutamicum | ||
| R | JCM18229 | 17 |
| X1 | Markerless Ptrc-xylA and Ptrc-xylB gene recombinant integrated into Xyl1 of strain R | 9 |
| X1D | Kmr; C. glutamicum X1 bearing pCRF100 | This work |
| X1EFGD | Kmr; C. glutamicum X1 bearing pCRF101 | This work |
| X1(Δ2369) | C. glutamicum X1 ΔcgR_2369 | This work |
| X1(Δ2369)D | Kmr; C. glutamicum X1Δ2369 bearing pCRF100 | This work |
| X1(Δ2369)EFGD | Kmr; C. glutamicum X1Δ2369 bearing pCRF101 | This work |
| X1(Δ2369)EFGDK | Kmr; C. glutamicum X1Δ2369 bearing pCRF102 | This work |
| X1(Δ2369)EFGDM | Kmr; C. glutamicum X1Δ2369 bearing pCRF103 | This work |
| BsrAXH | Kmr; C. glutamicum R bearing pCRF104 | This work |
| Plasmids | ||
| pCRB52T | Kmr; Ptac promoter; E. coli-Corynebacterium sp. shuttle vector derived from pHSG298 and pBY503 | This work |
| pCRF100 | Kmr; pCRB52T with a 2.4-kb NdeI-KpnI PCR fragment of the xylD gene | This work |
| pCRF101 | Kmr; pCRB52T with a 5.6-kb NdeI-KpnI PCR fragment of the xylEFGD gene cluster | This work |
| pCRF102 | Kmr; pCRF101 with a 1.1-kb KpnI PCR fragment of the cgR_2369 gene | This work |
| pCRF103 | Kmr; pCRF101 with a 1.1-kb KpnI PCR fragment of the msiK gene | This work |
| pCRF104 | Kmr; pCRA725 with a 3.0-kb XbaI PCR fragment of the target sequence for deletion of the cgR_2369 gene | This work |
| pCRA725 | Kmr; pHSG298 with Ptac-sacR-sacB genes | |
| pCRF105 | Kmr; pCRB52T with a 1.5-kb NdeI-BglII PCR fragment of the axh gene | This work |
| pCRF106 | Amr; pColdII with a 2.4-kb NdeI-BglII PCR fragment of the xylD gene | This work |
To investigate the growth performance of C. glutamicum under aerobic conditions, recombinant strains were harvested by centrifugation (5,000 × g at room temperature for 5 min). Cell pellets were subsequently washed twice with mineral medium (BT medium) containing (per liter) 7 g of (NH4)2SO4, 0.5 g of KH2PO4, 0.5 g of K2HPO4, 0.5 g of MgSO4 · 7H2O, 6 mg of FeSO4 · 7H2O, 4.2 mg of MnSO4 · H2O, 0.2 mg of biotin, and 0.2 mg of thiamine. After the second wash, cells were resuspended in 10 ml of BT medium containing an appropriate concentration of sugars. The resulting mixture was incubated at 33°C with constant agitation (200 rpm) in a test tube.
DNA manipulations.
Plasmid DNA was isolated by using a NucleoSpin plasmid kit (TaKaRa) according to the manufacturer's instructions. PCR was performed with KAPA Taq Extra polymerase (Nippon Genetics) or PrimeStar GXL DNA polymerase (TaKaRa) using a GeneAmp PCR system (Applied Biosystems, Foster City, CA). Oligonucleotide primers used in this study are listed in Table 2. The resultant PCR fragments were purified with NucleoSpin Gel and PCR Clean-Up kit (TaKaRa) according to the manufacturer's instructions.
TABLE 2.
Oligonucleotides used in this study
| Primer name | Sequence (5′–3′)a | Cohesive end |
|---|---|---|
| DPXF | CACGAGGA(A/G)TG(C/G)CT(C/G)(A/G)C(C/G)GG | |
| DPXR | CG(G/C)TG(G/C)GA(G/C)TT(T/C)GTGAAGC(G/A)G | |
| DPMF | CG(C/G)GACATCGC(A/C)ATGGT(G/T)TTCCAGA | |
| DPMR | CAGCG(A/G)CCGAAGTAGCCGAG | |
| APF | TCCGAGCGTTGCATGACCACAGTAAGCTATCACGTAC | |
| AP-NotIR | GGCCGTACGTGATAGCTTAC | |
| AP-BamHIR | GATCGTACGTGATAGCTTAC | |
| AP-MluIR | CGCGGTACGTGATAGCTTAC | |
| A P1 | CCGAGCGTTGCATGACCACAGTAAGCT | |
| AP2 | CGTTGCATGACCACAGTAAGCTATCACGTA | |
| GWR1-R2 | CGCCCATCTCGGCGACGAGGTC | |
| GWR1-R1 | AGCCCCTGGTGGATCCCGAGCTC | |
| GWF1-F2 | CGTCGACGAGTGCATCGCCGAGGA | |
| GWF1-F1 | GCCGCGATCGGCGACACGATGC | |
| GWR2-R2 | CGATTGAAACACGGCCGTGTCCGACC | |
| GWR2-R1 | GCGAAGACGAGAATGTCATCGCTCAC | |
| CAmsi-R2 | GGCGATCTTGAGCGCGAAGCCCATG | |
| CAmsi-R1 | ACCCGAGAGGGCCTTCGGCTTGC | |
| CAmsi-F2 | CGACCGCATCGCGGTCCTCAAGGA | |
| CAmsi-F1 | CCTCGGCGTCACCACGGTCTACG | |
| Primer 1 | GCCGGCAACCATATGACCGAACTGCCCCCTCT | NdeI |
| Primer 2 | GGGGTACCTCAGCTGACGGGTGCCTCGGCC | KpnI |
| Primer 3 | GGGAATTCCATATGAGAGCACACAGGATCCTGACG | NdeI |
| Primer 4 | GGGGTACCACGGATCGTTCGGCACGTAC | KpnI |
| Primer 5 | GGGGTACCTTAAGGGAGGCTCTTGCCAG | KpnI |
| Primer 6 | GCGGTACCACTACGGATCGTCCGGCACGTA | KpnI |
| Primer 7 | CCGGTACCTTACGCCGAGACGATCGCCTTG | KpnI |
| Primer 8 | GGAATTCCATATGAGGAAAAAGTGTAGCGTATGTTTA | NdeI |
| Primer 9 | GAAGATCTCTATCTTTGCGTAAACTGCCAG | BglII |
| Primer 10 | AGGAAGATCTCAGCTGACGGGTGCCTCGGCC | BglII |
| CgR2369_d1F | GCTCTAGAAAGGAACATCGTCATCGACCTTG | XbaI |
| CgR2369_d1R | GACAAAGCCCGGAAGCCATG | |
| CgR2369_d2F | CATGGCTTCCGGGCTTTGTCCTACCCCGAGGCACGTAATG | |
| CgR2369_d2R | GCTCTAGAGGCGGCAGTAACGACCTTGAC | XbaI |
The restriction site overhangs used in the cloning procedure are underlined.
Corynebacteria were transformed by electroporation as previously described (18). Transformation of E. coli was performed by the CaCl2 procedure (16).
DNA sequencing.
All sequencing was performed by the dideoxy chain termination method as previously described (19) with an ABI Prism 3100 genetic analyzer (Applied Biosystems) using a BigDye Terminator, version 3.1, cycle sequencing kit (Applied Biosystems). DNA sequence data were analyzed with the Genetyx program (Software Development, Tokyo, Japan). Database searches were performed using the BLAST server of National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov).
Degenerate PCR and adaptor ligation PCR.
Chromosomal DNA was isolated from C. alkanolyticum by using Wizard SV Mini Preps (Promega) according to the manufacturer's instructions. Degenerate nested PCRs were performed using C. alkanolyticum chromosomal DNA as the template with the degenerate primers to generate core DNA fragments which were homologous to known bacterial β-xylosidase genes or msiK genes. The degenerate PCR primers for the β-xylosidase gene were DPXF and DPXR, and those for the msiK gene were DPMF and DPMR (Table 2). The resultant core fragments were ligated to pGEM-T Easy vector (Promega), and their nucleotide sequences were analyzed. The nucleotide sequences upstream and downstream of the core fragments were obtained by chromosome walking using the adaptor ligation PCR method (20). C. alkanolyticum chromosomal DNA was digested with a single restriction enzyme, such as BglII, MluI, or NotI, followed by ligation with the adaptor DNA constituted by oligonucleotides APF and AP-BamHR, AP-MluIR, or AP-NotIR (Table 2), which contained PCR primer sequences. After ligation, nested PCR was performed using the ligation product as the template with the gene-specific primers and the adaptor primers AP1 for the first PCR and AP2 for the nested PCR (Table 2). The gene-specific primers GWF1-F1, GWF1-F2, GWR1-R1, GWR1-R2, GWR2-R1, and GWR2-R2 for cloning of the xylEFGDHR gene cluster and the gene-specific primers CAmsi-F1, CAmsi-F2, CAmsi-R1, and CAmsi-R2 for cloning the msiK gene are listed in Table 2. Resultant PCR fragments were inserted into pGEM-T Easy vector (Promega), and their nucleotide sequences were analyzed.
Vectors.
E. coli-C. glutamicum shuttle vector pCRB52T was constructed as follows. Ltac5 vector derived from pHSG298 and pCASE1 harboring the tac promoter was digested with BamHI, and the 0.7-kb fragment containing the tac promoter and terminator was ligated to BamHI- and BglII-digested pCRB52G vector derived from pHSG298 and pBY503. The resultant vector was digested with NdeI and ligated with a linker DNA containing a KpnI site, yielding vector pCRB52T (Table 1).
Construction of recombinant plasmids.
The 2.4-kb C. alkanolyticum xylD gene was amplified using C. alkanolyticum chromosomal DNA as the template and the oligonucleotides primer 1 and primer 2 (Table 2) to generate a DNA fragment with NdeI and KpnI cohesive ends. The PCR amplicon was subsequently ligated to NdeI- and KpnI-digested pCRB52T, yielding plasmid pCRF100 (Table 1). The 5.6-kb C. alkanolyticum xylEFGD operon was amplified using the oligonucleotides primer 2 and primer 3 (Table 2) to generate a DNA fragment with NdeI and KpnI cohesive ends. The PCR amplicon was subsequently ligated to NdeI- and KpnI-digested pCRB52T, yielding plasmid pCRF101 (Table 1). The 1.1-kb C. glutamicum cgR_2369 gene was amplified using the oligonucleotides primer 4 and primer 5 (Table 2) to generate a DNA fragment with KpnI cohesive ends. The PCR amplicon was subsequently ligated to KpnI-digested pCRF101, yielding plasmid pCRF102 (Table 1). The 1.1-kb C. alkanolyticum msiK gene was amplified using the oligonucleotides primer 6 and primer 7 (Table 2) to generate a DNA fragment with KpnI cohesive ends. The PCR amplicon was subsequently ligated to KpnI-digested pCRF101, yielding plasmid pCRF103 (Table 1). The 1.5-kb arabinoxylan arabinofuranohydrolase (AXH) gene of Bacillus subtilis was amplified using the oligonucleotides primer 8 and primer 9 (Table 2) to generate a DNA fragment with NdeI and BglII cohesive ends. The PCR amplicon was subsequently ligated to NdeI- and BglII-digested pCRB52T, yielding plasmid pCRF105 (Table 1). To express the His6-tagged XylD protein, the 2.4-kb xylD gene was amplified using C. alkanolyticum chromosomal DNA as the template and the oligonucleotides primer 1 and primer 10 (Table 2) to generate a DNA fragment with NdeI and BglII cohesive ends. The PCR amplicon was subsequently ligated to NdeI- and BamHI-digested pColdII vector (TaKaRa), yielding plasmid pCRF106 (Table 1).
Purification and kinetic analysis of the XylD protein.
To express the His6-tagged XylD protein, E. coli BL21(DE3) strain was transformed with the plasmid pCRF106. The resultant strain was first grown at 37°C in LB medium containing 50 μg ml−1 of ampicillin to an optical density at 600 nm (OD600) of 0.5 and then grown at 15°C in the presence of 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for induction for another 24 h. The cells were collected by centrifugation and resuspended with the lysis buffer (100 mM Tris-HCl [pH 8.0], 0.5 M NaCl, 25 mM imidazole, 10 mM 2-mercaptoethanol), followed by sonication. The cell debris was removed by centrifugation, and the cell lysate was applied on a nickel-nitrilotriacetic acid (NTA) column. The column was washed with the wash buffer (100 mM Tris-HCl [pH 8.0], 0.5 M NaCl, 25 mM imidazole), and then protein was eluted with the elution buffer (100 mM Tris-HCl [pH 8.0], 0.5 M NaCl, 250 mM imidazole). The eluent was buffer exchanged to buffer A (20 mM NaPi [pH 7.0], 1 mM EDTA, 5 mM MgCl2) and subsequently applied to a 1-ml HiTrap Q HP column. The protein was eluted in a linear gradient of sodium chloride to 1.0 M. The purification procedure on the HiTrap Q HP column was performed using an AKTA fast-protein liquid chromatography (FPLC) system (GE Healthcare). The purified XylD protein was subjected to SDS-polyacrylamide gel electrophoresis (PAGE), and its homogeneity was confirmed. The protein concentration was determined using a Bio-Rad protein assay kit.
Kinetic analysis was performed at 33°C in 1 ml of 100 mM HEPES (pH 7.0) solution containing p-nitrophenyl-β-d-xylopyranoside (0.04, 0.1, 0.4, 0.8, or 2 mM) or p-nitrophenyl-α-l-arabinofuranoside (0.2, 0.4, 1.0, 2.0, or 4.0 mM). Reactions were initiated by the addition of 1 μg of the purified enzyme, and absorbance was measured at 410 nm using a Beckman DU800 spectrophotometer (Beckman Coulter, Inc., Fullerton, CA). To determine the Ki value for xylose, assays were performed at 33°C in 1 ml of 100 mM HEPES (pH 7.0) containing p-nitrophenyl-xylopyranoside (0.4, 0.8, 1.2, or 2.0 mM) solution in the presence of xylose (40, 60, 80, or 100 mM). Reactions were initiated by the addition of 1 μg of the purified enzyme, and absorbance was measured at 410 nm. To examine the inhibitory effects of glucose, xylose, and arabinose on XylD activity, assays were performed at room temperature in 0.1 ml of 100 mM HEPES (pH 7.0) containing 1 mM p-nitrophenyl-xylopyranoside solution in the presence of each sugar (0, 50, 100, 150, or 200 mM) on a 96-well microtiter plate. Reactions were initiated by the addition of 0.1 μg of the purified enzyme, and absorbance was measured at 410 nm a using SpectraMax M2e (Molecular Devices).
Enzyme assays.
For enzyme assay of recombinant strains, cultures were harvested by centrifugation, and cell pellets were washed twice with extraction buffer (100 mM HEPES, pH 7.0). The resulting cell suspensions were homogenized with glass beads for 20 30-s periods, interrupted by 30-s cooling intervals. Cell debris was removed by centrifugation, and the cell lysates were subsequently used as crude extracts for enzyme assays. Enzyme assays were performed in 0.5 ml of reaction mixture (50 mM HEPES [pH 7.0], 1 mM p-nitrophenyl-xylopyranoside) at 33°C for 10 min. Reactions were stopped by the addition of 0.5 ml of 50 mM NaOH, and enzyme activities were measured at 410 nm. One unit of enzyme activity was defined as the amount of activity necessary to release 1 μmol of p-nitrophenol per min.
Construction of the cgR_2369 deletion mutant.
Deletion of the cgR_2369 gene was achieved via a markerless system using suicide vector pCRA725 carrying the sacB gene (11). A 1.5-kb DNA fragment just upstream of the start codon of the cgR_2369 gene and a 1.5-kb DNA fragment just downstream of the stop codon were amplified by PCR using the primer pair CgR2369_d1F/CgR2369_d1R and the pair CgR2369_d2F/CgR2369_d2R, respectively. Both fragments were combined by overlap extension PCR. The resultant fragment was digested with XbaI and cloned into pCRA725, yielding pCRF1104. C. glutamicum X1 was transformed by electroporation with pCRF104, and screening for the deletion mutants was performed as described previously (21). Deletion of the cgR_2369 gene was checked by PCR. The mutant strain obtained, X1 ΔcgR_2369, was designated X1(Δ2369).
Analytical procedure.
Samples were centrifuged (10,000 × g at 4°C for 10 min), and the resulting supernatants were analyzed for the presence of sugars. Sugar concentrations were determined by high-performance liquid chromatography (HPLC) using an apparatus (8020; Tosoh Corporation) equipped with a refractive index detector and an HPX-87P column (Bio-Rad) operating at 85°C with a water mobile phase at a flow rate of 0.6 ml min−1. Cell mass was determined by measuring the optical density at 610 nm (OD610) using a spectrophotometer (DU800; Beckman Coulter, Inc.).
Oligosaccharide profiles in culture medium and cell extract were determined by liquid chromatography-electrospray ionization-mass spectrometry (LC-ESI-MS). For analysis of culture medium, samples were centrifuged (10,000 × g at 4°C for 10 min), and the supernatants were diluted 10-fold, followed by mixing with four volumes of acetonitrile. After centrifugation again, the resultant supernatants were analyzed. For analysis of cell extract, 300-samples were centrifuged (10,000 × g at 4°C for 10 min), and the cell pellets were washed with 1 ml of water twice and then resuspended with 50 μl of water and extracted by the addition of 200 μl of acetonitrile. After a 30-min incubation on ice, samples were centrifuged (10,000 × g at 4°C for 10 min), and the resultant supernatants were analyzed. The chromatographic separations were performed using an apparatus (Shimazu Corporation) and a TSKgel Amide-80 column (4.5 cm by 0.2 mm; Tosoh Corporation) operating at 60°C with a linear gradient of 75 to 35% buffer B (buffer A, 10 mM ammonium acetate, aqueous; buffer B, acetonitrile) at a flow rate of 0.2 ml min−1. LC and MS analyses were controlled using an AB SCIEX QTRAP 5500 LC-tandem MS (LC-MS/MS) system (Applied Biosystems/MDS Analytical Technologies). Negative ions were detected. Authentic xylooligosaccharides were purchased from Megazyme.
Nucleotide sequence accession numbers.
The nucleotide sequences of the C. alkanolyticum msiK gene, the gene cluster containing the xylH and xylEFGD genes, and the xylR gene have been deposited in the GenBank database under the accession numbers KM066602, KM066603, and KM066604, respectively.
RESULTS
The C. alkanolyticum xylEFGD gene cluster for xylooligosaccharide metabolism is widely conserved.
Bacterial xylooligosaccharide metabolism involves sugar uptake by xylooligosaccharide transporters prior to hydrolysis by intracellular β-xylosidases, but functional characterization of the genes involved in xylooligosaccharide metabolism in corynebacteria is not known. Nevertheless, knowing that Corynebacterium alkanolyticum ATCC 21511 (7, 8) is able to grow in the presence of xylooligosaccharide as the sole carbon source facilitated the cloning of β-xylosidase and xylooligosaccharide transporter genes using degenerate primers, DPXF and DPXR (Table 2), designed from highly conserved regions of β-xylosidases of actinobacteria. Subsequent degenerate PCR using C. alkanolyticum chromosomal DNA as the template revealed a 360-bp core DNA fragment encoding a polypeptide homologous to bacterial β-glycosidase. DNA fragments upstream (GWR1 and GWR2) and downstream (GWF1) of this core fragment (Fig. 1A) were obtained by chromosome walking based on the adaptor ligation PCR method (20). An approximately 9-kb region of the C. alkanolyticum chromosome containing the gene fragment homologous to β-glycosidase was thus sequenced to reveal a G+C content of 71.4%, which is well above the genomic G+C content of corynebacteria of 51 to 65%. The region encompasses six predicted complete open reading frames (ORFs), xylH, xylR, xylE, xylF, xylG, and xylD, with each of xylE, xylF, and xylG overlapping its neighbor (Fig. 1B). A segment not exceeding 100 bp between xylD and xylR could not be sequenced, suggesting the possibility of a long palindromic sequence. A BLAST search (Fig. 1B) strongly suggested that xylE encodes a putative sugar ABC transporter solute-binding protein, whereas xylF and xylG encoded putative sugar ABC transporter permeases, with the trio collectively comprising a sugar ABC transporter. The putative solute-binding protein encoded by xylE showed highest homology to the extracellular solute-binding protein of Cellulomonas flavigena but weak homology (27.9% identity at the amino acid level) to Streptomyces thermoviolaceus BxlE (5), a confirmed solute-binding protein of a xylooligosaccharide ABC transporter. xylD encoded a putative glycoside hydrolase family 3 protein, with highest homology to that of Isoptericola variabilis but lower homology (26.5% identity at the amino acid level) to S. thermoviolaceus β-xylosidase BxlA (22). As no signal sequence associated with the putative xylD-encoded protein was detected, the gene was predicted to encode an intracellular enzyme. xylH and xylR encoded putative proteins bearing high homology to the LacI type transcription factor and ROK (repressor or kinase) family transcription factor, respectively.
FIG 1.

The C. alkanolyticum gene cluster for xylooligosaccharide utilization. (A) Physical map of the cluster indicating restriction sites for MluI, NotI, and BglII used in construction of a genomic DNA library for chromosome walking. The three fragments determined by the chromosome walking (thin solid bars) are depicted relative to the core fragment (thick solid bar). (B) Organization of genes on the cluster. The region whose nucleotide sequence could not be determined (dotted line) is located between xylD and xylR. Accession numbers are given at right.
To determine the enzymatic properties of the xylD-encoded protein, the protein was fused with a His6 tag at its N terminus, expressed in E. coli, purified by a Ni-NTA-Sepharose column and anion exchange chromatography, and confirmed to be homogeneous by SDS-PAGE. XylD's optimal pH for activity at 33°C was 7.0. Its Michaelis constant (Km) was 2.4 ± 0.1 mM toward p-nitrophenyl-β-d-xylopyranoside (pNPX) and 44.0 ± 9.0 mM toward p-nitrophenyl-α-d-arabinofuranoside (pNPA). Its maximum velocity (Vmax) was 0.06 ± 0.01 μmol s−1 μg−1 toward pNPX and 1.3 ± 0.1 μmol s−1 μg−1 toward pNPA. Its turnover number was 111 ± 4 s−1 toward pNPX and 5 ± 1 s−1 toward pNPA. The XylD activity toward pNPX was inhibited by xylose and arabinose but not glucose (Fig. 2). Xylose was a more potent inhibitor of XylD than arabinose. Arabinose at 200 mM reduced XylD activity by approximately 40%, while 200 mM xylose reduced XylD activity by approximately 70%. End-product inhibition of activity in the presence of pNPX as the substrate was competitive, with an inhibition constant (Ki) for xylose of 67.6 ± 2.6 mM.
FIG 2.
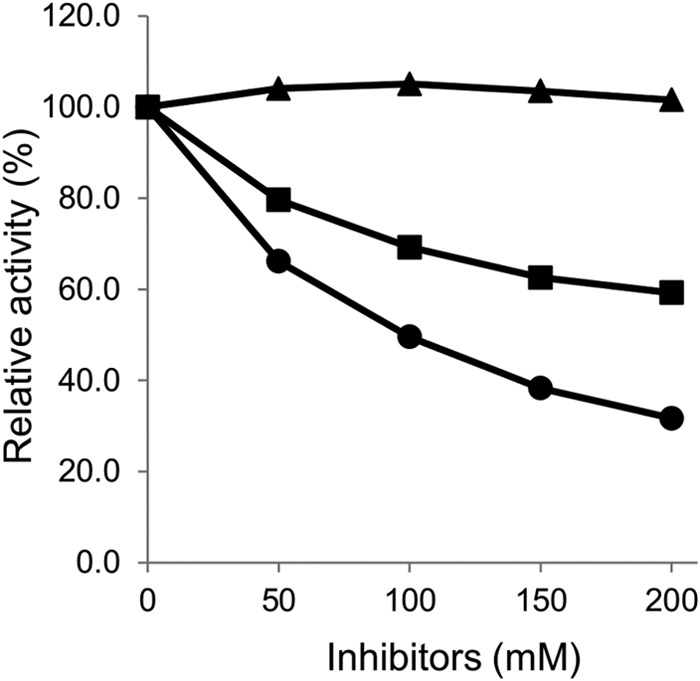
Inhibitory effects of various sugars on XylD activity. XylD was incubated with pNPX in the presence of each sugar (0, 50, 100, 150, and 200 mM), and β-xylosidase activities were compared by initial velocity. The data represent the averages of triplicate experiments. ▲, glucose; ●, xylose; ■, arabinose.
C. glutamicum X1EFGD xylosidase activity and xylooligosaccharide transport are independent.
C. glutamicum X1 is a xylose-utilizing strain by virtue of harboring E. coli xylA encoding xylose isomerase and xylB encoding xylulokinase (9). The well-acknowledged merits that C. glutamicum possesses as a mixed-sugar-utilizing bacterium provided motivation for heterologous expression of the xylEFGD gene cluster in strain X1 in order to determine the functional characteristics of the cluster. Strain X1 transformed with plasmid pCRF100 harboring the xylD gene was designated X1D, whereas strain X1 transformed with plasmid pCRF101 harboring the xylEFGD gene cluster was designated X1EFGD. A cgR_2369 disruption strain of X1 similarly transformed with pCRF100 yielded X1(Δ2369)D, whereas X1 transformed with pCRF101 yielded X1(Δ2369)EFGD. C. glutamicum cgR_2369 exhibits high homology to the Streptomyces msiK gene encoding a multitask ATP-binding protein that assists different oligosaccharide ABC transporters (23). The genes carried by each plasmid used in the transformation were expressed under the control of the strong constitutive tac promoter (Fig. 3A). As per expectations, no xylosidase activity was detected with strain X1 due to its lack of an xylD gene. Less obvious was the observation that the β-xylosidase activities of strains transformed with pCRF101 were consistently 10-fold weaker than those of corresponding strains transformed with pCRF100, irrespective of the presence of the cgR_2369 gene (Table 3). It follows that in strains transformed with pCRF101, XylD translation from xylEFGD mRNA exhibiting a stable secondary structure predicted around the translation initiation site of the xylD gene (Fig. 3B) was not efficient.
FIG 3.
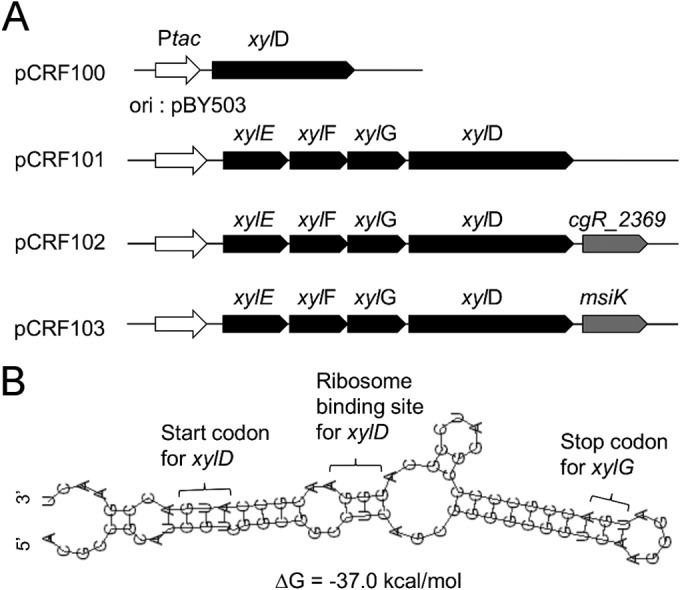
Plasmids for heterologous expression of xylEFGD genes in C. glutamicum. (A) Architecture of expression cassettes on xylD-harboring plasmid pCRF100, xylEFGD-harboring plasmid pCRF101, xylEFGD- and cgR_2369-harboring plasmid pCRF102, and xylEFGD- and msiK-harboring plasmid pCRF103. All plasmid genes are under the control of the strong constitutive tac promoter. Each expression cassette is on the C. glutamicum-E. coli shuttle vector where the corynebacterial replication of origin is from plasmid pBY503. (B) Secondary structure of the translation initiation region of the xylD gene as predicted by the RNAfold WebServer (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi).
TABLE 3.
b-Xylosidase activities in C. glutamicum recombinant strains
| Strain | Specific activity (U/mg of protein)a |
|---|---|
| X1 | ND |
| X1D | 0.2561 ± 0.0026 |
| X1EFGD | 0.0282 ± 0.0003 |
| X1(Δ2369)D | 0.2565 ± 0.0015 |
| X1(Δ2369)EFGD | 0.0274 ± 0.0002 |
| X1(Δ2369)EFGDK | 0.0292 ± 0.0001 |
| X1(Δ2369)EFGDM | 0.0284 ± 0.0002 |
Cells were grown aerobically to late log phase in A medium containing 40 g liter−1 glucose with 50 mg ml−1 kanamycin. The reported data represent the averages calculated from triplicate measurements. ND, no activity detected.
The relative abilities of strains X1, X1D, and X1EFGD to utilize xylooligosaccharides was assayed by monitoring growth and sugar consumption in rich medium supplemented with a 0.25% (wt/vol) commercial xylooligosaccharide mixture. The HPX-87P HPLC column employed permitted resolution of xylose and xylobiose but not higher xylooligosaccharides. The maximum cell density (OD610) was 2.4 with strain X1, 3.9 with strain X1D, and 4.3 with strain X1EFGD (Fig. 4A). Given the earlier mentioned inability of any of the strains to grow in minimal medium, the growth of strain X1 in rich medium observed here must be attributed to carbon sources other than the xylooligosaccharide mixture. HPLC analysis confirmed that strain X1 did not utilize xylobiose, and no xylose was detected in its culture medium (Fig. 4B). In contrast, rapid consumption of 30% of the initial xylobiose by strain X1D and of 95% by strain X1EFGD in 12 h was mirrored by comparative differences in xylose accumulation in both strains. Thereafter, xylobiose consumption by strain X1D decreased to such an extent that after 24 h about 55% of the xylobiose remained. Xylose, too, was gradually utilized by both strains during the latter phase. The higher proficiency of strain X1EFGD with respect to xylobiose utilization must reside in the role played by an xylEFG gene product.
FIG 4.

Growth and pentose sugar consumption of C. glutamicum recombinant strains precultured in nutrient-rich A medium containing 40 g liter−1 glucose, inoculated to an initial OD610 of 0.5 in A medium containing 2.5 g liter−1 xylooligosaccharide mixture. Xylobiose consumption levels in the recombinant strains were analyzed by HPLC. (A) Aerobic growth of strain X1 (⧫), strain X1D (■), and strain X1EFGD (▲). (B) Sugar consumption of strain X1 (⧫), strain X1D (■), and strain X1EFGD (▲). Solid and dashed lines indicate the concentrations of xylobiose and xylose, respectively. (C) Comparison of xylobiose consumption levels of strain X1(Δ2369)D (□) and X1(Δ2369)EFGD (△) with those of strains X1D (■) and X1EFGD (▲). (D) Comparison of xylobiose consumption levels of strains X1(Δ2369)EFGDK (○) and X1(Δ2369)EFGDM (◊) with those of strains X1EFGD (▲) and X1(Δ2369)EFGD (△). Xylobiose concentration was represented as the concentration of xylose per unit. The data represent the averages of triplicate experiments.
C. glutamicum cgR_2369 gene product facilitates xylooligosaccharide transport.
The expression of the xylEFGD operon alone was enough to affect efficient uptake of xylooligosaccharides in C. glutamicum. This suggested that an ATP binding protein known to assist the XylEFG activity is expressed independently in C. glutamicum. The gene organization of the xylEFG cluster is conserved in many species (5, 6, 24). In Streptomyces species, msiK encodes a multitask ATP-binding protein that assists different oligosaccharide ABC transporters (23). The C. glutamicum R cgR_2369 putative gene product highly homologous to the msiK product (58.9% amino acid identity to Streptomyces reticuli MsiK) is located apart from any ABC transporter-encoding gene cluster on the chromosome, as is characteristic of msiK genes. The effect, if any, of the cgR_2369 gene product on XylEFG was assayed using a cgR_2369 deletion mutant. The cgR_2369 gene was deleted from strain X1 using a markerless system to yield strain X1(Δ2369). Strain X1(Δ2369) transformed with pCRF100 was designated X1(Δ2369)D, whereas X1(Δ2369) transformed with pCRF101 was designated X1(Δ2369)EFGD (Table 1). The β-xylosidase activities of strain X1(Δ2369)D and X1(Δ2369)EFGD were unchanged from those of strain X1D and X1EFGD, respectively (Table 4). However, whereas strain X1(Δ2369)D consumed xylobiose at the same rate as strain X1D, strain X1(Δ2369)EFGD's initial rate of xylobiose consumption was only 17% of the 0.344 mM/h rate observed with strain X1EFGD (Fig. 4C).
To confirm the role of cgR_2369, the cgR_2369 gene with the 5′ untranslated region was inserted just downstream of xylD on plasmid pCRF101 so as to express the xylEFGD and cgR_2369 genes polycistronically under the control of the tac promoter, and strain X1(Δ2369)EFGD was complemented with the cgR_2369 gene of C. glutamicum. The resultant plasmid pCRF102 (Fig. 2) was used to transform strain X1(Δ2369) to strain X1(Δ2369)EFGDK (Table 1). The β-xylosidase activity of strain X1(Δ2369)EFGDK was comparable to the activities of strains X1EFGD and X1(Δ2369)EFGD (Table 4), but its xylooligosaccharide consumption was restored to the levels observed with strain X1EFGD (Fig. 4D).
To obtain a native msiK gene of C. alkanolyticum, degenerate primers DPMF and DPMR (Table 2) were designed from the highly conserved region of actinobacterial msiK genes. Degenerate PCR using C. alkanolyticum chromosomal DNA as the template gave a 480-bp core DNA fragment encoding a polypeptide highly homologous to actinobacteria msiK. Subsequent DNA fragments upstream and downstream of the core fragment were obtained by chromosome walking, and the complete ORF of msiK was determined. The protein deduced thereof shared 83% amino acid identity in the N-terminal 240-amino-acid portion and 67% amino acid identity overall with S. reticuli MsiK. To coexpress msiK with the xylEFGD cluster, the msiK gene with the ribosome binding site was inserted just downstream of xylD on plasmid pCRF101 so as to express the xylEFGD and the msiK genes polycistronically under the control of the tac promoter. The resultant plasmid pCRF103 (Fig. 2) was used to transform strain X1(Δ2369) to strain X1(Δ2369)EFGDM (Table 1). The β-xylosidase activity of strain X1(Δ2369)EFGDM was comparable to the activities strains X1EFGD and X1(Δ2369)EFGD (Table 4), but its xylooligosaccharide consumption was restored to the levels observed with strain X1EFGD (Fig. 4D).
The xylEFG product transports arabino-xylooligosaccharides as well as xylooligosaccharides.
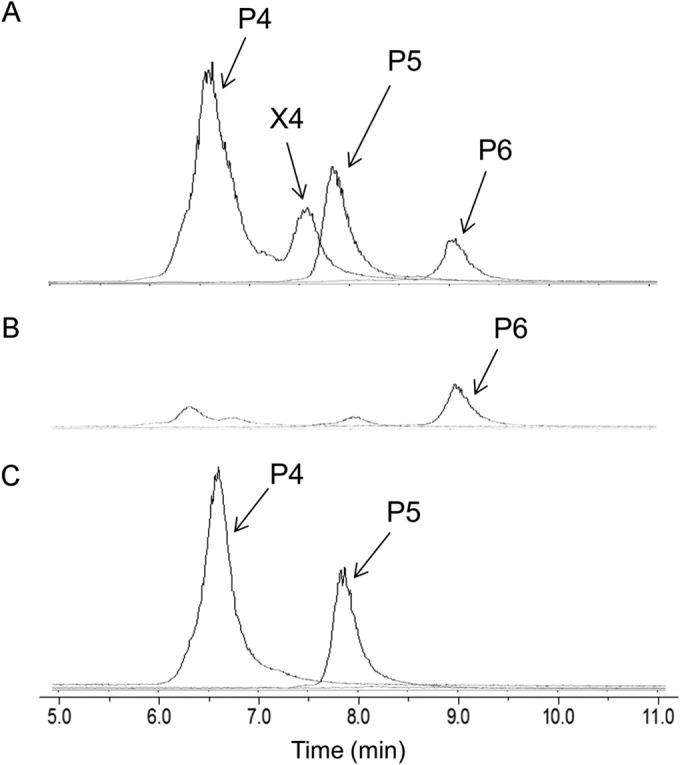
LC-ESI-MS analysis with a TSKgel Amide-80 column showed that the commercial xylooligosaccharide mixture used in this study contained xylobiose (X2), xylotriose (X3), xylotetraose (X4), and unknown pentose oligosaccharides with degrees of polymerization of 4, 5, and 6 (P4, P5, and P6, respectively) (Fig. 5A, row a). The culture medium of each of strain X1D and X1EFGD grown in A medium supplemented with 0.25% (wt/vol) xylooligosaccharide mixture and analyzed by LC-ESI-MS at 24 h or 120 h revealed that, whereas strain X1EFGD was able to completely consume all but P6 (Fig. 5A, row b), strain X1D completely consumed only xylobiose, xylotriose, and xylotetraose (Fig. 5A, row c). This ability of strain X1EFGD to utilize P4 and P5 must therefore be attributed to the xylEFG product.
FIG 5.

Comparison of substrate utilization range of C. glutamicum recombinant strains. Strain X1EFGD and strain X1D were first grown aerobically to late log phase in A medium containing 40 g liter−1 of glucose and then inoculated to an initial OD610 of 0.5 into A medium containing 2.5 g liter−1 of xylooligosaccharide mixture. (A) Culture medium of the recombinant strains was analyzed by LC-ESI-MS at 0 h (a), at 30 h for strain X1EFGD (b), and at 120 h for strain X1D (c) after the start of cultivation. (B) Characterization of oligosaccharides P4 and P5. The culture medium of strain X1D from the experiment represented in panel A, row c, was incubated with or without the cells of strain BsrAXH expressing the extracellular arabinoxylan arabinofuranohydrolase (AXH). After incubation, the samples were centrifuged, and the supernatants were analyzed by LC-ESI-MS. The lower spectrum was from the sample treated with (+) AXH, and the upper one was from the sample without (−) AXH. Compounds P4, P5, and P6 were oligosaccharides whose molecular weights corresponded to pentose-oligosaccharides composed of 4 to 6 pentose units. X2, xylobiose; X3, xylotriose; X4, xylotetraose.
The commercial xylooligosaccharide mixture used in this study is a product of enzymatic hydrolysis of arabinoxylan, implying that the three unknown fractions could be arabino-xylotriose (P4), arabino-xylotetraose (P5), and arabino-xylopentaose (P6). Confirming the identity of the fractions may indicate why P6 was not consumed, thus revealing the substrate range that the product of xylEFG binds. To this end, a strain X1D digest of the xylooligosaccharide mixture (used for the experiment shown in Fig. 5A) was incubated with cells of the C. glutamicum recombinant strain BsrAXH (Table 1), a recombinant R strain harboring the gene encoding the extracellular arabinoxylan arabinofuranohydrolase (AXH) from B. subtilis (25). A supernatant of the digest thereof analyzed by LC-ESI-MS revealed that P4 and P5 were decomposed to xylotriose and xylotetraose with the release of a pentose monomer (Fig. 5B). Arabinose, being indistinguishable from xylose by TSKgel Amide-80 chromatography, was confirmed by HPLC on an HPX-87P column (data not shown). These results suggested that P4 and P5 were arabino-xylotriose and arabino-xylotetraose, respectively. P6 was not decomposed by the treatment with strain BsrAXH, and its chemical structure is still unknown.
Arabino-xylooligosaccharides were accumulated in the cell in strain X1EFGD.
In strain X1EFGD, uptake of arabino-xylooligosaccharides was not followed by release and accumulation of arabinose although strain X1EFGD had none of the arabinose metabolism genes, such as araBAD (arabinose isomerase, ribulokinase, and ribulose-5-phosphate 4-epimerase). When the xylooligosaccharide mixture used in this study was incubated with the purified XylD under the same conditions as used in the enzyme assays, oligosaccharides P4, P5, and P6 were hardly decomposed while xylooligosaccharides were completely decomposed to xylose (data not shown). These results suggested that most of arabino-xylooligosaccharides taken up were accumulated in the cell without any decomposition. To examine the hypothesis, the cell pellets of strain X1EFGD grown in A medium supplemented with 0.25% (wt/vol) xylooligosaccharide mixture were extracted with an acetonitrile-water solution after extensive washing at 24 h. The culture medium and cell extract at 24 h analyzed by LC-ESI-MS revealed that in strain X1EFGD, oligosaccharides P4 and P5 (arabino-xylotriose and arabino-xylotetraose) absent in culture medium were detected in cell extract, while xylooligosaccharides were fully metabolized, and oligosaccharide P6 present in culture medium was absent from the cell extract (Fig. 6B and C), which indicated that arabino-xylotriose and arabino-xylotetraose were taken up but not metabolized efficiently. The arabino-xylooligosaccharide composition in the experiment shown in Fig. 6A was different from that in the experiment shown in Fig. 5 as the production lot of the xylooligosaccharide mixture used in this experiment was different from that used in Fig. 5.
FIG 6.
Detection of intracellular arabino-xylooligosaccharides. Strain X1EFGD was first grown aerobically to late log phase in A medium containing 40 g liter−1 glucose and then inoculated to an initial OD610 of 0.5 into A medium containing 2.5 g liter−1 xylooligosaccharide mixture. (A) Culture medium analyzed by LC-ESI-MS at 0 h. (B) Culture medium analyzed by LC-ESI-MS at 24 h. (C) Cell extract analyzed by LC-ESI-MS at 24 h. For each analysis, negative ions corresponding to pentose oligosaccharides composed of 4 to 6 pentose units were detected. Compounds P4, P5, and P6 were oligosaccharides whose molecular weights corresponded to pentose oligosaccharides composed of 4 to 6 pentose units. X4, xylotetraose.
DISCUSSION
Xylooligosaccharides are β-1,4-linked xylose oligomers derived from the hydrolysis of xylan, an important component of cellulosic biomass. Xylanolytic microorganisms secrete enzymes that hydrolyze xylan to its constituent xylooligosaccharides that can then be taken up by cells. In bacteria the xylooligosaccharides are transported into cells by the action of xyloside transporters before subsequent hydrolysis to xylose by the intracellular 1,4-β-xylosidases. Most bacterial β-xylosidases and xyloside transporters characterized to date are from Firmicutes and Proteobacteria (3, 20, 26, 27). Homologues of β-xylosidase and xyloside transporter genes are extensively evident among completely sequenced actinobacterial genomes, but functional characterization of the encoded enzymes is not known except in Streptomycineae (5, 22, 28). In this study, the β-xylosidase and xyloside transporter genes of the glutamate- and biosurfactant-producing corynebacterium C. alkanolyticum (7, 8) were shown to form a functional cluster exhibiting a 71.4% G+C content that is markedly higher than the typical 51 to 65% of genomes of corynebacteria. The proteins encoded by xylD and xylE showed high homology to Cellulomonas flavigena glycoside hydrolase family 3 proteins and the extracellular solute-binding protein, respectively. Moreover, a partial sequence of 16S rRNA of C. alkanolyticum showed the highest homology to that of Microbacterium testaceum. Given that both C. flavigena and M. testaceum are Micrococcineae, these similarities betray a closer relationship of C. alkanolyticum to Micrococcineae than to Corynebacterineae.
The xylD-encoded protein was a typical bacterial β-xylosidase with a moderately high turnover number and high Ki value for xylose, suggesting an inherent potential for industrial application in a suitable industrial strain. In C. glutamicum β-xylosidase activity derived from the unmodified xylEFGD cluster was suppressed owing to reduced translation efficiency from a stable secondary structure within the translation initiation region. A similar stable stem-loop structure within the translation initiation region in the β-xylosidase-encoding xylC of Thermoanaerobacterium saccharolyticum resulting in comparable suppression of activity has been removed by site-directed mutagenesis (29). A similar approach to enhance the xylEFGD cluster-derived xylosidase activity may lead to more productive industrial strains. The significance of the translational modification of the xylD gene in xylooligosaccharide utilization by C. alkanolyticum ought to be explored.
The similar capacity of strain X1D not expressing xylEFG to utilize xylobiose suggests the presence of an inherent xylobiose uptake mechanism independent of the xylEFG-encoded transporter in C. glutamicum. It is clear that a doubling of this capacity occurs only in strains expressing the transporter in the presence of a functional multitask ATP-binding protein. Strains not expressing the transporter were able to utilize xylobiose, xylotriose, and xylotetraose, but utilization of the more complex arabino-xylotriose and arabino-xylotetraose required the xylEFG-encoded transporter. In strain X1EFGD, arabino-xylotriose and arabino-xylotetraose taken up by the xylEFG-encoded transporter were hardly metabolized in the cell because of low activity of the xylD-encoded β-xylosidase toward them. For efficient utilization of arabino-xylooligosaccharides, coexpression of arabinofuranosidase must be required.
The two other xylooligosaccharide ABC transport systems reported to date have not been functionally characterized beyond demonstration of their substrate binding proteins to exhibit high affinity toward xylooligosaccharides (5, 6). Nevertheless, similarly organized ABC transport-encoding gene clusters, including S. reticuli cebEFG for cellobiose (24) and B. subtilis araNPQ for arabino-oligosaccharides (30), do require multitask ATP-binding proteins to function. These ATP-binding proteins are invariably encoded by genes distant from the clusters: msiK in S. reticuli and msmX in B. subtilis. The successful expression of the xylooligosaccharide ABC transporter gene cluster in a heterologous host in this study demonstrates the central role of the cluster in uptake of arabino-xylooligosaccharides. C. glutamicum has been engineered and improved to be an effective platform for pentose fermentation (31–33), producing ethanol (34), succinic acid (35), 1,5-diaminopentane (36), and amino acids (37, 38) from pentose sugars. The findings described here could offer faster, broader, and more efficient pentose sugar utilization to such potential industrial strains, and could contribute significantly to advancement of microbial lignocellulosic biomass exploitation.
ACKNOWLEDGMENT
We thank Crispinus A. Omumasaba (Research Institute of Innovative Technology for the Earth) for helpful comments on the manuscript.
REFERENCES
- 1.Aristidou A, Penttilä M. 2000. Metabolic engineering applications to renewable resource utilization. Curr Opin Biotechnol 11:187–198. doi: 10.1016/S0958-1669(00)00085-9. [DOI] [PubMed] [Google Scholar]
- 2.Dodd D, Cann I. 2009. Enzymatic deconstruction of xylan for biofuel production. Glob Change Biol Bioenergy 1:2–17. doi: 10.1111/j.1757-1707.2009.01004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qian Y, Yomano L, Preston J, Aldrich H, Ingram L. 2003. Cloning, characterization, and functional expression of the Klebsiella oxytoca xylodextrin utilization operon (xynTB) in Escherichia coli. Appl Environ Microbiol 69:5957–5967. doi: 10.1128/AEM.69.10.5957-5967.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shin H, McClendon S, Vo T, Chen R. 2010. Escherichia coli binary culture engineered for direct fermentation of hemicellulose to a biofuel. Appl Environ Microbiol 76:8150–8159. doi: 10.1128/AEM.00908-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsujibo H, Kosaka M, Ikenishi S, Sato T, Miyamoto K, Inamori Y. 2004. Molecular characterization of a high-affinity xylobiose transporter of Streptomyces thermoviolaceus OPC-520 and its transcriptional regulation. J Bacteriol 186:1029–1037. doi: 10.1128/JB.186.4.1029-1037.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shulami S, Zaide G, Zolotnitsky G, Langut Y, Feld G, Sonenshein A, Shoham G. 2007. A two-component system regulates the expression of an ABC transporter for xylo-oligosaccharides in Geobacillus stearothermophilus. Appl Environ Microbiol 73:874–884. doi: 10.1128/AEM.02367-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crosman J, Pinchuk R, Cooper D. 2002. Enhanced biosurfactant production by Corynebacterium alkanolyticum ATCC 21511 using self-cycling fermentation. J Am Oil Chem Soc 79:467–472. doi: 10.1007/s11746-002-0507-5. [DOI] [Google Scholar]
- 8.Nakao Y, Kanamaru T, Kikuchi M, Yamatoda S. 1973. Action of penicillin on membrane-permeability barrier to l-glutamic acid.1. Extracellular accumulation of phospholipids, UDP-N-acetylhexosamine derivatives and l-glutamic acid by penicillin-treated Corynebacterium alkanolyticum. Agric Biol Chem 37:2399–2404. [Google Scholar]
- 9.Sasaki M, Jojima T, Inui M, Yukawa H. 2008. Simultaneous utilization of d-cellobiose, d-glucose, and d-xylose by recombinant Corynebacterium glutamicum under oxygen-deprived conditions. Appl Microbiol Biotechnol 81:691–699. doi: 10.1007/s00253-008-1703-z. [DOI] [PubMed] [Google Scholar]
- 10.Inui M, Murakami S, Okino S, Kawaguchi H, Vertès A, Yukawa H. 2004. Metabolic analysis of Corynebacterium glutamicum during lactate and succinate productions under oxygen deprivation conditions. J Mol Microbiol Biotechnol 7:182–196. doi: 10.1159/000079827. [DOI] [PubMed] [Google Scholar]
- 11.Inui M, Kawaguchi H, Murakami S, Vertès A, Yukawa H. 2004. Metabolic engineering of Corynebacterium glutamicum for fuel ethanol production under oxygen-deprivation conditions. J Mol Microbiol Biotechnol 8:243–254. doi: 10.1159/000086705. [DOI] [PubMed] [Google Scholar]
- 12.Yamamoto S, Suda M, Niimi S, Inui M, Yukawa H. 2013. Strain optimization for efficient isobutanol production using Corynebacterium glutamicum under oxygen deprivation. Biotechnol Bioeng 110:2938–2948. doi: 10.1002/bit.24961. [DOI] [PubMed] [Google Scholar]
- 13.Jojima T, Fujii M, Mori E, Inui M, Yukawa H. 2010. Engineering of sugar metabolism of Corynebacterium glutamicum for production of amino acid l-alanine under oxygen deprivation. Appl Microbiol Biotechnol 87:159–165. doi: 10.1007/s00253-010-2493-7. [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto S, Gunji W, Suzuki H, Toda H, Suda M, Jojima T, Inui M, Yukawa H. 2012. Overexpression of genes encoding glycolytic enzymes in Corynebacterium glutamicum enhances glucose metabolism and alanine production under oxygen deprivation conditions. Appl Environ Microbiol 78:4447–4457. doi: 10.1128/AEM.07998-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hasegawa S, Suda M, Uematsu K, Natsuma Y, Hiraga K, Jojima T, Inui M, Yukawa H. 2013. Engineering of Corynebacterium glutamicum for high-yield l-valine production under oxygen deprivation conditions. Appl Environ Microbiol 79:1250–1257. doi: 10.1128/AEM.02806-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 17.Yukawa H, Omumasaba CA, Nonaka H, Kos P, Okai N, Suzuki N, Suda M, Tsuge Y, Watanabe J, Ikeda Y, Vertès AA, Inui M. 2007. Comparative analysis of the Corynebacterium glutamicum group and complete genome sequence of strain R. Microbiology 153:1042–1058. doi: 10.1099/mic.0.2006/003657-0. [DOI] [PubMed] [Google Scholar]
- 18.Vertès AA, Inui M, Kobayashi M, Kurusu Y, Yukawa H. 1993. Presence of mrr- and mcr-like restriction systems in coryneform bacteria. Res Microbiol 144:181–185. doi: 10.1016/0923-2508(93)90043-2. [DOI] [PubMed] [Google Scholar]
- 19.Sanger F, Nicklen S, Coulson A. 1977. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riley J, Butler R, Ogilvie D, Finniear R, Jenner D, Powell S, Anand R, Smith J, Markham A. 1990. A novel, rapid method for the isolation of terminal sequences from yeast artificial chromosome (YAC) clones. Nucleic Acids Res 18:2887–2890. doi: 10.1093/nar/18.10.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arai K, Fukushima T, Tomiya M, Mitsuhashi S, Sasaki T, Toyo'oka T. 2008. Simultaneous determination of N-acetylaspartylglutamate and N-acetylaspartate in rat brain homogenate using high-performance liquid chromatography with pre-column fluorescence derivatization. J Chromatogr B Analyt Technol Biomed Life Sci 875:358–362. doi: 10.1016/j.jchromb.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 22.Morioka H, Miki Y, Saito K, Tomoo K, Ishida T, Hasegawa T, Yamano A, Takada C, Miyamoto K, Tsujibo H. 2010. Crystallization and preliminary X-ray crystallographic analysis of BxlA, an intracellular beta-d-xylosidase from Streptomyces thermoviolaceus OPC-520. Acta Crystallogr Sect F Struct Biol Cryst Commun 66:791–793. doi: 10.1107/S1744309110013400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schlosser A, Kampers T, Schrempf H. 1997. The Streptomyces ATP-binding component MsiK assists in cellobiose and maltose transport. J Bacteriol 179:2092–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schlosser A, Jantos J, Hackmann K, Schrempf H. 1999. Characterization of the binding protein-dependent cellobiose and cellotriose transport system of the cellulose degrader Streptomyces reticuli. Appl Environ Microbiol 65:2636–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vandermarliere E, Bourgois T, Winn MD, van Campenhout S, Volckaert G, Delcour J, Strelkov S, Rabijns A, Courtin C. 2009. Structural analysis of a glycoside hydrolase family 43 arabinoxylan arabinofuranohydrolase in complex with xylotetraose reveals a different binding mechanism compared with other members of the same family. Biochem J 418:39–47. doi: 10.1042/BJ20081256. [DOI] [PubMed] [Google Scholar]
- 26.Kim YA, Yoon KH. 2010. Characterization of a Paenibacillus woosongensis beta-xylosidase/alpha-arabinofuranosidase produced by recombinant Escherichia coli. J Microbiol Biotechnol 20:1711–1716. [PubMed] [Google Scholar]
- 27.Czjzek M, Bravman T, Henrissat B, Shoham Y. 2004. Crystallization and preliminary crystallographic analysis of a thermostable family 52 beta-d-xylosidase from Geobacillus stearothermophilus T-6. Acta Crystallogr D Biol Crystallogr 60:1461–1463. doi: 10.1107/S0907444904013320. [DOI] [PubMed] [Google Scholar]
- 28.Pinphanichakarn P, Tangsakul T, Thongnumwon T, Talawanich Y, Thamchaipenet A. 2004. Purification and characterization of beta-xylosidase from Streptomyces sp. CH7 and its gene sequence analysis. World J Microbiol Biotechnol 20:727–733. doi: 10.1007/s11274-004-4513-1. [DOI] [Google Scholar]
- 29.Shao W, Xue Y, Wu A, Kataeva I, Pei J, Wu H, Wiegel J. 2011. Characterization of a novel beta-xylosidase, XylC, from Thermoanaerobacterium saccharolyticum JW/SL-YS485. Appl Environ Microbiol 77:719–726. doi: 10.1128/AEM.01511-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferreira M, Sá-Nogueira I. 2010. A multitask ATPase serving different ABC-type sugar importers in Bacillus subtilis. J Bacteriol 192:5312–5318. doi: 10.1128/JB.00832-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawaguchi H, Vertes AA, Okino S, Inui M, Yukawa H. 2006. Engineering of a xylose metabolic pathway in Corynebacterium glutamicum. Appl Environ Microbiol 72:3418–3428. doi: 10.1128/AEM.72.5.3418-3428.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kang MK, Lee J, Um Y, Lee TS, Bott M, Park SJ, Woo HM. 2014. Synthetic biology platform of CoryneBrick vectors for gene expression in Corynebacterium glutamicum and its application to xylose utilization. Appl Microbiol Biotechnol 98:5991–6002. doi: 10.1007/s00253-014-5714-7. [DOI] [PubMed] [Google Scholar]
- 33.Radek A, Krumbach K, Gatgens J, Wendisch VF, Wiechert W, Bott M, Noack S, Marienhagen J. 2014. Engineering of Corynebacterium glutamicum for minimized carbon loss during utilization of D-xylose containing substrates. J Biotechnol 192:156–160. doi: 10.1016/j.jbiotec.2014.09.026. [DOI] [PubMed] [Google Scholar]
- 34.Jojima T, Noburyu R, Sasaki M, Tajima T, Suda M, Yukawa H, Inui M. 2015. Metabolic engineering for improved production of ethanol by Corynebacterium glutamicum. Appl Microbiol Biotechnol 99:1165–1172. doi: 10.1007/s00253-014-6223-4. [DOI] [PubMed] [Google Scholar]
- 35.Wang C, Zhang H, Cai H, Zhou Z, Chen Y, Ouyang P. 2014. Succinic acid production from corn cob hydrolysates by genetically engineered Corynebacterium glutamicum. Appl Biochem Biotechnol 172:340–350. doi: 10.1007/s12010-013-0539-x. [DOI] [PubMed] [Google Scholar]
- 36.Buschke N, Becker J, Schafer R, Kiefer P, Biedendieck R, Wittmann C. 2013. Systems metabolic engineering of xylose-utilizing Corynebacterium glutamicum for production of 1,5-diaminopentane. Biotechnol J 8:557–570. doi: 10.1002/biot.201200367. [DOI] [PubMed] [Google Scholar]
- 37.Meiswinkel TM, Gopinath V, Lindner SN, Nampoothiri KM, Wendisch VF. 2013. Accelerated pentose utilization by Corynebacterium glutamicum for accelerated production of lysine, glutamate, ornithine and putrescine. Microb Biotechnol 6:131–140. doi: 10.1111/1751-7915.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gopinath V, Meiswinkel TM, Wendisch VF, Nampoothiri KM. 2011. Amino acid production from rice straw and wheat bran hydrolysates by recombinant pentose-utilizing Corynebacterium glutamicum. Appl Microbiol Biotechnol 92:985–996. doi: 10.1007/s00253-011-3478-x. [DOI] [PubMed] [Google Scholar]