

ABSTRACT
Efficient duplication of genomes depends on reactivation of replication forks outside the origin. Replication restart can be facilitated by recombination proteins, especially if single- or double-strand breaks form in the DNA. Each type of DNA break is processed by a distinct pathway, though both depend on the RecA protein. One common obstacle that can stall forks, potentially leading to breaks in the DNA, is transcription. Though replication stalling by transcription is prevalent, the nature of DNA breaks and the prerequisites for replication restart in response to these encounters remain unknown. Here, we used an engineered site-specific replication-transcription conflict to identify and dissect the pathways required for the resolution and restart of replication forks stalled by transcription in Bacillus subtilis. We found that RecA, its loader proteins RecO and AddAB, and the Holliday junction resolvase RecU are required for efficient survival and replication restart after conflicts with transcription. Genetic analyses showed that RecO and AddAB act in parallel to facilitate RecA loading at the site of the conflict but that they can each partially compensate for the other's absence. Finally, we found that RecA and either RecO or AddAB are required for the replication restart and helicase loader protein, DnaD, to associate with the engineered conflict region. These results suggest that conflicts can lead to both single-strand gaps and double-strand breaks in the DNA and that RecA loading and Holliday junction resolution are required for replication restart at regions of replication-transcription conflicts.
IMPORTANCE Head-on conflicts between replication and transcription occur when a gene is expressed from the lagging strand. These encounters stall the replisome and potentially break the DNA. We investigated the necessary mechanisms for Bacillus subtilis cells to overcome a site-specific engineered conflict with transcription of a protein-coding gene. We found that the recombination proteins RecO and AddAB both load RecA onto the DNA in response to the head-on conflict. Additionally, RecA loading by one of the two pathways was required for both replication restart and efficient survival of the collision. Our findings suggest that both single-strand gaps and double-strand DNA breaks occur at head-on conflict regions and demonstrate a requirement for recombination to restart replication after collisions with transcription.
INTRODUCTION
Bacterial chromosomal replication initiates from a single origin of replication and proceeds bidirectionally until completion at the terminus. Replication forks regularly encounter obstacles and need to be restarted as they traverse the chromosome (1, 2). The essentiality of restart proteins supports the notion that every single fork initiated from oriC will be disrupted at least once, if not more often, before reaching the terminus (3, 4). Transcription frequently impedes replication in bacteria, necessitating numerous factors to resolve conflicts between the two machineries (5). Head-on collisions between replication and transcription, which occur when a gene is carried on the lagging strand, cause mutagenesis, genomic instability, single-stranded DNA (ssDNA) accumulation, and potentially double-strand DNA breaks (6–9).
Previous studies investigating the consequences and coping strategies of cells with transcription from the lagging strand have relied on the use of strains harboring large chromosomal inversions that oriented the highly transcribed, repetitive rRNA gene operons head-on to replication (10–12). In Escherichia coli, recombination-mediator proteins were required for efficient survival of rRNA gene inversions, though the repair protein RecA itself did not contribute to viability in these experiments (11, 12). Inversion of the rRNA genes leads to RecA-green fluorescent protein (GFP) focus formation in Bacillus subtilis (10). Subsequent studies in B. subtilis demonstrated that RecA localizes to loci containing highly expressed head-on protein-coding genes (6). DNA processing by recombination proteins and/or break repair is likely important for replication-transcription conflict resolution. However, the type of DNA damage occurring upon collisions between replication and transcription and the role, if any, that RecA plays at these regions remain elusive.
RecA catalyzes strand invasion as the first step in DNA break repair by homologous recombination (13). Depending on the type of DNA break, RecA is loaded onto the DNA (1) by either the RecFOR or AddAB pathway (14–17). RecFOR recognizes single-strand gaps in the DNA (18), likely through an interaction with the single-stranded DNA binding protein (SSB) (19). AddAB, in contrast, processes double-stranded ends to generate 3′ overhangs (20, 21) in a manner analogous to the action of RecBCD in E. coli (22). Whether AddAB itself loads RecA onto DNA in B. subtilis (an activity mediated by RecB in E. coli [23]) is an ongoing topic of debate.
RecA loading and strand invasion lead to the formation of four-way Holliday junction DNA intermediates (24, 25). Cells rely on resolvases, such as the RuvC protein in E. coli, to cleave four-way Holliday junction DNA and separate linked sister chromosomes (26–28). RecU, the homologue of E. coli RuvC, carries out this function in B. subtilis (29–31). When recombination occurs at stalled replication forks, restart proteins associate with resolved recombination intermediates for growth to continue (32–34).
Restart in B. subtilis involves an ordered association of essential primosomal proteins, PriA and DnaD, with the DNA, followed by recruitment of helicase loader proteins, DnaB and DnaI. DnaB and DnaI then load the helicase, DnaC, onto the DNA, allowing replication to proceed (35–38). Purified PriA protein can bind both stalled fork structures and D-loops in vitro, though it has much higher affinity for the latter (32, 39, 40). Consistent with this, genetic analyses indicated that recombination proteins remodel stalled forks for PriA binding to occur (41). Reconstituted restart reactions in vitro demonstrated that both RecBCD and RecOR facilitate restart (42). However, the requirements for PriA binding to DNA in vivo remain poorly studied.
We set out to determine if RecA played a role in resolving conflicts between replication and transcription. To address this question, we inserted a single protein-coding gene into the chromosome of B. subtilis, oriented either head-on or codirectionally to replication. We find that when the engineered construct is transcribed in the head-on, but not the codirectional, orientation, the presence of at least one RecA loading mediator, RecO or AddAB, and the Holliday junction resolvase, RecU, is required for efficient survival. The transcription and orientation dependence of these phenotypes suggests that replication-transcription conflicts are the underlying cause of the observed survival defects. Epistasis analyses indicate that RecO, AddAB, and RecU act in the same pathway as RecA to promote survival in response to, specifically, head-on transcription. These data suggested that recombination is required for resolution of replication-transcription conflicts. Consistent with this, we find that both AddAB and RecO load RecA onto DNA at the conflict region in a transcription-dependent manner, as indicated by chromatin immunoprecipitations (ChIPs) of RecA. Additionally, we found that RecA is required for the restart protein, DnaD, to associate with the engineered head-on conflict region. Our findings demonstrate that resumption of replication upon conflicts with transcription depends primarily on a classical recombination-mediated and RecA-dependent replication restart pathway in B. subtilis.
MATERIALS AND METHODS
Bacterial culture conditions.
Cultures were initiated from single-colony isolates grown on solid agar-containing plates supplemented with the appropriate antibiotic. Starter cultures were grown to an optical density (OD) of 0.03 to 0.5 in LB medium at 37°C with vigorous shaking and then diluted back to an OD of 0.05 in an appropriate volume of LB for experimental purposes.
Strain constructions.
Gene deletions were introduced by standard transformation techniques as described previously (67). ΔrecO and ΔrecU strains were constructed using genomic DNA from deletion mutants obtained from the Bacillus Genetic Stock Center (BGSC). Markerless deletions were made by evicting erythromycin resistance cassettes via flanking loxP sites and a plasmid-encoded Cre recombinase (pDR224 carries the cre gene and a temperature-sensitive origin of replication), obtained from the BGSC. A detailed list of strains used in this study is in Table S1 in the supplemental material.
ChIPs.
Starter LB cultures were initiated from isolated colonies grown on solid agar plates supplemented with the appropriate antibiotic and grown at 37°C, with shaking, to an OD of 0.3 to 0.4. Starter cultures were diluted to an OD of 0.05 in 25 ml LB and grown again at 30°C, with shaking, to an OD of 0.3. Upon reaching an OD of 0.3, cultures were harvested into 0.1% formaldehyde and processed as described previously (43). DnaD IPs were performed essentially as described previously (43). GFP IPs were performed using an anti-GFP antibody purchased from Abcam (ab290) as described by Million-Weaver et al. (6). Quantitative PCRs (qPCRs) to amplify central regions of the yhaX and lacZ genes were performed as described previously (43), using the oligonucleotides HM192 and -193 (CCGTCTGACCCGATCTTTTA and GTCATGCTGAATGTCGTGCT), and HM188 and -189 (GGCTTTCGCTACCTGGAGAG and GACGAAGCCGCCCTGTAAAC), respectively.
Microscopy.
Microscopy was performed as described previously by Million-Weaver et al. (6). To summarize, cultures were grown in LB supplemented with the appropriate antibiotic at 37° with aeration (260 rpm) to exponential phase (OD = 0.3 to 0.8). Cells were then fixed by formaldehyde treatment, stained with DAPI (4′,6′-diamidino-2-phenylindole), and transferred to agarose pads for visualization by microscopy. DAPI fluorescence and GFP fluorescence were used to quantify total cells and total RecA-GFP foci, respectively. Data analysis was conducted using ImageJ.
Plating efficiency.
Cultures were grown in LB supplemented with the appropriate antibiotic at 37° with aeration (260 rpm) to exponential phase (OD = 0.3). Exponential-phase cultures that grew to higher optical densities were diluted to an OD of 0.3. Serial dilutions were plated on LB-agar medium, and total viable cells were quantified for each experimental condition. The plating efficiency ratio for a given genetic background was determined by dividing the viable colonies arising from strains expressing a gene by those arising from strains repressed for transcription of the reporter gene.
RESULTS
RecA, along with RecO and AddAB, promotes survival of cells experiencing severe head-on replication-transcription conflicts.
Previous studies in E. coli indicated that head-on encounters between replication and transcription from inverted rRNA gene operons caused RecA-independent replication fork reversal (RFR) (11, 12). We wondered if RecA played a role in overcoming conflicts between replication and transcription of a protein-coding gene in B. subtilis. There is precedence for this hypothesis, given that RecA-GFP localization has been observed in in B. subtilis strains harboring rRNA gene inversions (10) and that RecA association with regions of head-on transcription has been detected by ChIP (6).
To address if and how RecA contributes to survival of replication-transcription collisions, we utilized a previously described site-specific inducible conflict (6, 43). Strains harboring this reporter carry a copy of the lacZ gene under the control of the ICEBs1 promoter, Pxis, in the head-on orientation with respect to replication at the thrC locus of the B. subtilis chromosome. In strain backgrounds cured of the ICEBs1 element, the Pxis promoter is highly expressed, whereas in the presence of this element, the promoter is tightly repressed (44), leading to minimal expression of the lacZ gene. We grew cultures of B. subtilis, either expressing or repressed for transcription of the Pxis-lacZ gene, to similar optical densities, spotted serial dilutions onto LB agar plates, and enumerated CFU arising under both conditions (Fig. 1A). We then quantified any survival defects associated with deleting genes for homologous recombination under both conditions. To determine the specific contribution of each protein of interest to survival of the highly expressed head-on gene, we took ratios (expressed as plating efficiency percentages) of CFU arising in the presence of transcription of lacZ to CFU in strains where lacZ was repressed (Fig. 1B).
FIG 1.
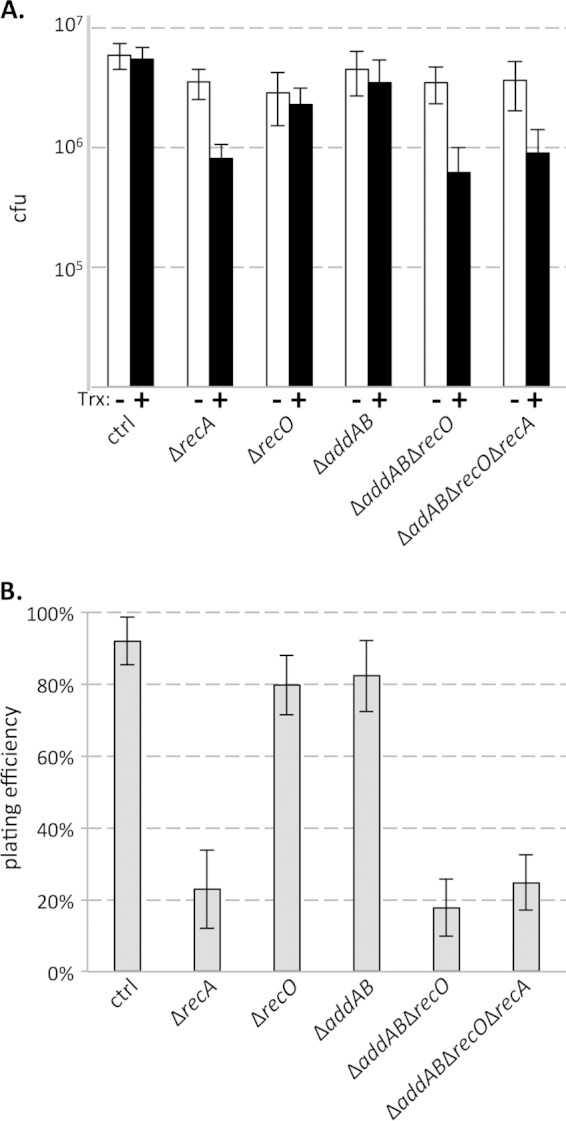
(A) CFU arising from exponentially growing cultures at an OD600 of 0.3 were enumerated in the presence (+) and absence (−) of transcription (Trx) from Pxis-lacZ. (B) Plating efficiencies (transcription-specific survival) were determined by enumerating the ratio of CFU arising from cultures of strains expressing Pxis-lacZ to that from strains repressed for transcription of the construct in a given mutant background. Data shown are averages from 8 to 12 biological replicates per strain. Error bars represent standard error of the mean (A) or standard deviations (B).
We found that cells expressing Pxis-lacZ in otherwise wild-type backgrounds did not display significant reductions in plating efficiency, consistent with the presence of multiple pathways to resolve the negative consequences of head-on collisions between replication and transcription in bacteria (5). Cells lacking recA displayed significant survival defects associated with transcription from the lacZ gene, leading to a 5-fold reduction in plating efficiency (Fig. 1A and B). Neither deletion of recO nor deletion of addAB individually caused significant transcription-specific survival defects (Fig. 1B). However, lacZ expression significantly impaired survival of cells lacking both recO and addAB (Fig. 1B). To confirm that the survival defects we observed were specific to head-on conflicts with transcription and not simply lacZ expression itself, we also performed plating efficiency assays in strains where the gene was oriented codirectionally with replication (i.e., on the leading strand) (see Fig. S1 in the supplemental material). Although codirectional transcription does impede the replisome (43, 45), these encounters are much less disruptive than when replication and transcription meet head-on. When lacZ was expressed from the leading strand, deletion of recA or recO and addAB did not cause any significant reduction in plating efficiencies, arguing against transcription of lacZ alone causing survival defects. The orientation specificity implies that head-on conflicts with replication upon lacZ transcription necessitate RecA and its loader proteins for efficient survival. The lack of a significant phenotype for either addAB or recO single mutants, combined with the synergistic effect of combining the two mutations, indicates one of two possibilities: either each individual pathway alone is not important, or one compensates for the other's absence.
We wondered if AddAB and RecO contributed to survival by loading RecA at regions of head-on transcription. To determine whether recO and addAB act in the same pathway as recA, we constructed strains lacking all three genes. We found that combining deletions of recA with the recO and addAB deletions did not lead to any additional survival defects compared to those of the parent strains (Fig. 1A and B). The epistatic relationship of recA, addAB, and recO indicates that both RecO and AddAB facilitate survival of obstacles to replication through RecA.
Both RecO and AddAB load RecA at head-on conflict regions.
Two pathways potentially load RecA onto DNA in B. subtilis: RecFOR at single-strand gaps or AddAB at double-strand breaks. Previous studies indicated that RecO is required for RecA localization in response to treatment with DNA-damaging agents in cells grown in minimal medium (16). However, the potential contribution of either pathway to RecA loading in cells grown in rich medium without exogenous damage is unclear. The plating efficiency data indicated that both pathways contribute to survival of head-on conflicts through a genetic interaction with RecA. We set out to determine if RecFOR and/or AddAB loads RecA onto the DNA at a specific replication-transcription conflict region in B. subtilis.
To determine which RecA loading pathway, RecFOR and/or AddAB, contributed to RecA localization in response to head-on transcription, we first quantified RecA-GFP focus formation in response to transcription of the Pxis-lacZ reporter using microscopy. In the absence of transcription, we found that RecA-GFP formed foci in roughly 10% of cells, consistent with previous reports (46) (Fig. 2A). In the backgrounds where the lacZ reporter was not expressed, cells lacking RecO did not show a reduction in RecA-GFP focus formation, whereas cells without AddAB displayed reduced RecA-GFP localization. When the reporter gene was transcribed, we observed RecA-GFP foci in 20% of cells in otherwise wild-type backgrounds. Deletion of either recO or addAB led to reduced RecA-GFP localization in cells expressing the lacZ gene. Combining the two mutations (recO addAB double mutant) had an additive effect (Fig. 2A).
FIG 2.
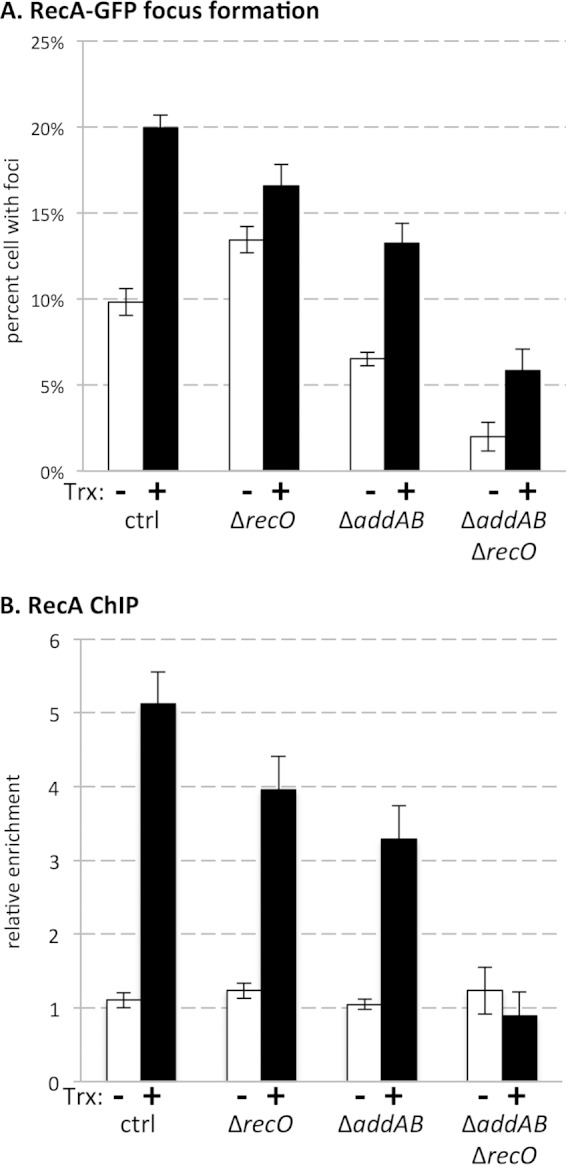
(A) RecA-GFP focus formation was quantified by microscopy in the absence (−) and presence (+) of transcription (Trx) of the Pxis-lacZ gene. (B) Relative association of RecA with the head-on gene-containing region was measured by ChIP-qPCR in the absence and presence of transcription of the Pxis-lacZ gene. Data shown represent averages from 6 to 16 biological replicates. Error bars represent standard error of the mean.
The results of the microscopy experiments suggested that AddAB, and not RecO, is the major mediator required for RecA localization during growth in rich media. Cells lacking RecO did show a modest increase in RecA-GFP localization in the absence of transcription of the reporter, relative to control cells. Therefore, RecO may, to some degree, also load RecA at single-strand gaps under these conditions. The increase observed in RecO-deficient strains probably reflects unresolved single-strand gaps being converted to double-strand breaks due to runoff replication (47).
RecA-GFP focus formation cannot distinguish between general replication stress (i.e., RecA localizing to the DNA elsewhere in the genome) and the specific effects of transcription of the reporter gene. We set out to directly determine the contribution of each pathway to RecA localization specifically at the collision region. To do this, we used ChIPs to measure the relative association of RecA with the replication roadblock, compared to the control locus yhaX (other control loci produce similar results to yhaX [6]), in the presence and absence of transcription. When the reporter gene was repressed, we did not detect preferential association of RecA with the region in any genetic background. In strains expressing the Pxis-lacZ construct, we observed a 5-fold enrichment of RecA at the region relative to the control locus (Fig. 2B). RecA association with the region was reduced in strain backgrounds deficient for recO or addAB. Similar to the results of the microscopy experiments, combining the recO and addAB deletions had an additive effect; we no longer detected any preferential RecA association at the replication roadblock when lacZ was expressed in cells lacking both RecO and AddAB (Fig. 2B). The RecA ChIPs suggest that, consistent with the results of the plating efficiency assays, at least RecO or AddAB is required for RecA to associate with regions of head-on transcription, but one pathway may compensate for the other's absence.
RecU acts in the same pathway as RecO, AddAB, and RecA to help cells survive head-on conflicts.
The localization of RecA at the conflict region and contribution to efficient survival suggested that strand invasion at the site of the conflict helped cells tolerate head-on encounters between replication and transcription. Strand invasion by RecA leads to the formation of four-way Holliday junction intermediates, which are cleaved by resolvases to separate the interlinked chromosomes (24, 25). We hypothesized that resolution of recombination intermediates would therefore contribute to cellular survival of replication-transcription conflicts. To test this, we extended our epistasis analysis to determine the effect of the B. subtilis Holliday junction resolvase, RecU, which binds Holliday junctions with subnanomolar affinity (31), on plating efficiencies for cells harboring the highly expressed head-on lacZ gene.
Deletion of recU alone led to reduced plating efficiencies, suggesting that cleavage of four-way junctions contributes to survival of head-on conflicts (Fig. 3). No additional reductions in plating efficiencies were observed when the recU and recA deletions were combined, indicating that these genes function in the same pathway, likely by forming and resolving four-way junctions, respectively. However, because of the potential limited sensitivity of the plating efficiency assays, we cannot exclude the possibility that RecU performs another RecA-independent DNA processing function at head-on conflict regions.
FIG 3.
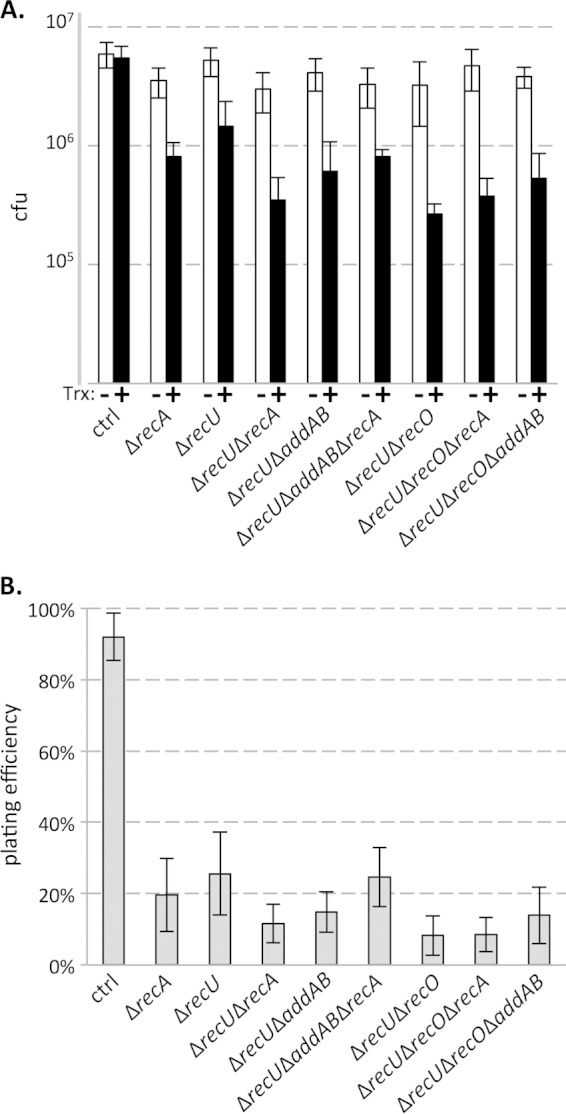
(A) CFU arising from exponentially growing cultures at an OD600 of 0.3 were enumerated in the presence (+) and absence (−) of transcription (Trx) from Pxis-lacZ. (B) Plating efficiencies (transcription-specific survival) were determined by enumerating the ratio of CFU arising from cultures of strains expressing Pxis-lacZ to that from strains repressed for transcription of the construct in a given mutant background. Data shown are averages from 8 to 14 biological replicates per strain. Error bars represent standard error of the mean (A) or standard deviation (B).
Because single deletions of recO or addAB did not cause significant survival defect phenotypes, the genetic interactions between recU and either individual pathway cannot be inferred from cellular survival data. However, deleting recU in backgrounds lacking both recO and addAB did not lead to any additional survival defects (Fig. 3). The observed epistasis between all three genes suggests that RecU acts in the same pathway as both RecO and AddAB, as well as RecA, to promote survival of cells upon head-on collisions between replication and transcription. These genetic interactions are consistent with RecU's classically defined function to resolve Holliday junction intermediates arising from homologous recombination.
RecA loading is required for the restart protein, DnaD, to associate with head-on conflict regions.
Based on the results presented above, we hypothesized that RecA aids replication reactivation in response to conflicts by facilitating the formation of D-loops, which can promote association of replication restart proteins with stalled forks (41, 42). To test this model, we used ChIP to measure the relative association of the restart protein, DnaD, with the roadblock-containing region compared to that for the control locus, yhaX, in the presence and absence of transcription in strains deficient for each recombination protein. During primosome assembly, PriA facilitates DnaD loading in order to reload the replicative helicase, DnaC.
Expression of the reporter gene in recombination-proficient backgrounds led to significantly increased relative DnaD association with the lacZ-containing region. In strains lacking RecA, we no longer detected preferential DnaD association with the region when the reporter gene was expressed (Fig. 4). Individual deletions of either recO or addAB reduced DnaD association with the lacZ locus (Fig. 4). Similar to the results obtained with the recA deletion, in backgrounds lacking both recO and addAB, we no longer observed preferential DnaD association with the highly expressed head-on gene (Fig. 4).
FIG 4.
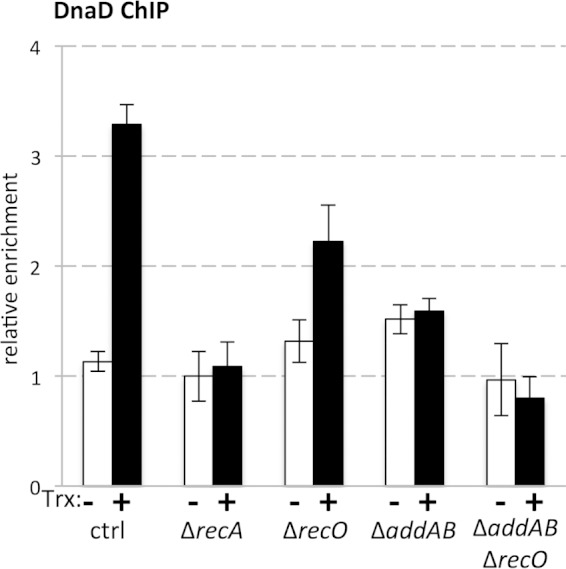
Relative association of DnaD with the head-on gene-containing region was measured by ChIP-qPCR in the absence (−) and presence (+) of transcription (Trx) of the Pxis-lacZ gene. Data shown represent averages from 12 biological replicates. Error bars represent standard error of the mean.
To rule out the possibility that altered transcription of the Pxis-lacZ construct in backgrounds lacking recA eliminated the obstacle to replication in these strains, we used ChIP to measure the association of RNA polymerase (RNAP) beta subunit (RpoB) with the lacZ-containing region with and without transcription from Pxis. We found that deletion of recA did not alter RNA polymerase occupancy of the reporter gene compared to that observed in wild-type backgrounds (see Fig. S2A in the supplemental material). Furthermore, to rule out the possibility that pleiotropic effects of the recA deletion on replication somehow reduced the severity of the effects of conflicts with transcription in these strains, we measured the relative association of the replicative helicase, DnaC, with the region in the presence and absence of transcription. The replicative helicase is expected to localize evenly along the chromosome in an asynchronous population of cells in the absence of specific replication stress. As such, preferential association relative to a control locus probably indicates replication stalling at that region. We found that, as we have reported previously, active transcription from Pxis led to a significant increase in the relative association of DnaC with the region (6). This association, however, was not affected by deletion of recA (see Fig. S2B in the supplemental material), indicating that severe conflicts between transcription and replication still occur even in recA deletion backgrounds and that the absence of RecA specifically inhibits replication restart.
Taken together, these results suggest that a RecA-dependent mechanism is needed for restart proteins to localize to head-on conflict regions. Though RecO and AddAB each partially contribute, at least one pathway is necessary for DnaD association with highly expressed head-on genes.
DISCUSSION
Our results demonstrate that RecA contributes to efficient survival of head-on replication-transcription conflicts, likely by promoting replication restart. The plating efficiencies and ChIP data strongly suggest that both single-strand gaps and double-strand breaks occur at head-on conflict regions, both of which are acted upon by RecA (Fig. 5). The survival defects that we observe are modest, indicating that not every conflict causes catastrophic consequences for the cell. However, the requirement for RecA loading for replication restart proteins to associate with the conflict region suggests that when severe collisions do occur, replication requires RecA to proceed. Our results establish RecA-dependent recombination as a prerequisite for the reactivation of replication upon stalling at transcription units in B. subtilis.
FIG 5.
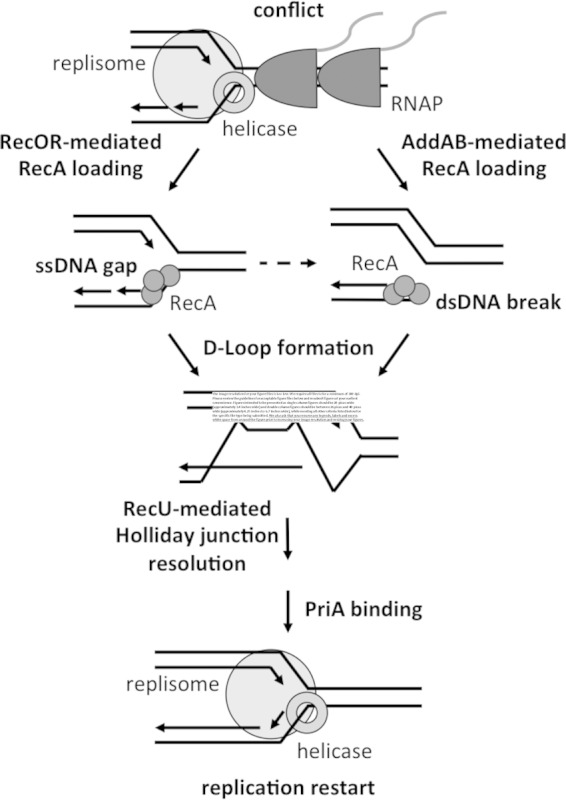
Model for restart upon replication-transcription collision in B. subtilis. RecFOR may recognize single-stranded DNA at stalled forks, leading to RecA loading, recombination, and restart. Alternatively, double-strand breaks in the DNA at stalled forks may be processed by AddAB, leading to RecA loading, recombination, and restart.
RecO- and AddAB-mediated conflict resolution.
RecA binds single-stranded DNA with 3′ overhangs (48). RecA rapidly polymerizes on DNA to form nucleoprotein filaments, the functional complexes that catalyze strand invasion (49). However, RecA monomers alone have low affinity for Ssb-coated DNA (50). In order for RecA filaments to form, monomers require nucleation onto DNA, a process called RecA loading (51). Two pathways exist for RecA loading in B. subtilis, depending on the type of DNA break that occurs.
RecOR loads RecA onto single-stranded DNA in B. subtilis. RecO is likely recruited to ssDNA by an interaction with the C-terminal tail of Ssb, as has been demonstrated in E. coli (52). AddAB processes double-strand breaks to generate 3′ overhangs (22). Previous work suggested that RecO is required to load RecA onto the DNA regardless of the type of damage, even at double-strand breaks (16). These investigations used RecA-GFP focus formation as a readout for RecA loading in cells growing in minimal medium. We observed RecA-GFP focus formation and DNA association by ChIP even in cells lacking RecO, suggesting that other pathways, such as AddAB, also facilitate loading of RecA at stalled replication forks. The differences between our results and the previous study could arise from the different growth conditions (minimal versus rich medium) or some undetermined cofactor present at replication forks stalled due to transcription that is absent when forks stall in response to chemical DNA damage.
Our results also indicate that one pathway may act in the other's absence, which suggests that AddAB can activate RecA loading independently of RecO. The ability of AddAB to compensate for RecO likely arises because gaps formed at stalled replication forks are converted to double-strand breaks during subsequent rounds of replication (47). RecO may compensate for the absence of AddAB if other nucleases within the cell, such as RecJ in concert with RecQ or RecS (53), resect the double-stranded ends to generate 3′ overhangs for RecA loading. Studies in Deinococcus radiodurans have demonstrated that RecFOR can play a predominant role in repairing double-strand breaks (54), suggesting that the absence of AddAB does not necessarily preclude this type of damage repair. Regardless of the mechanism or type of damage, however, in our system, both RecO and AddAB act through RecA.
Sources of single-strand gaps and double-strand breaks at head-on conflicts.
The results of the plating efficiency assays and ChIPs showing that both RecO and AddAB contribute to RecA loading and restart suggest that both single-strand gaps and double-strand breaks occur at head-on conflict regions. Our assays lack the sensitivity to directly determine which type of damage predominates; however, it is clear that RecA is important for replication restart at both gaps and breaks. The initial encounter between replication and transcription likely generates a single-strand gap, which would be recognized by RecO. However, because both pathways contribute to tolerating collisions, some double-strand breaks may occur.
We envision three possible scenarios (as illustrated in Fig. 5): direct RecA loading by RecO at single-strand gaps, end processing and RecA loading by AddAB at any breaks that do occur, and, occasionally, gaps being converted into double-strand breaks due to runoff replication.
Single-strand gaps may arise because both RNA polymerase and DNA polymerase unzip the double-helical DNA template, causing negative supercoils and underwound DNA accumulation behind each of the machineries (55–57). Negative supercoiling behind RNAP contributes to R-loop formation, which may maintain one strand of the DNA duplex in a single-stranded state (58). At the site of the collision itself, persistent gaps on the lagging-strand template, such as observed in restart-deficient PriA mutants (59), could necessitate RecFOR-mediated repair.
It is unclear if head-on collisions between replication and transcription directly cause double-strand breaks in the DNA. The torsional strain that accumulates due to excessive positive supercoiling between the two machineries might be sufficient to break the ester linkages of the sugar-phosphate backbone. However, evidence of chromosome breaks arising directly due to a collision (as opposed to endonucleolytic processing of the DNA in the region) has not been demonstrated.
Any single-strand gaps in the template can be converted into double-strand breaks through replication runoff as forks converge. Direct restart from these structures would likely result in aberrant chromosomes. Therefore, double-strand break repair may be required even if the initial insult to the DNA generated only a single-strand gap or nick.
The role of D-loops in replication restart.
PriA allows for origin-independent replication initiation by recruiting helicase loader proteins to the DNA in B. subtilis. Purified PriA binds DNA bubble structures with low affinity (dissociation constant [Kd] = 150 nM) yet displays 10-fold-enhanced binding kinetics to D-loop structures (Kd = 15 nM) (40). Previous studies of primosome assembly have relied extensively on reconstituted systems in vitro because of the essential nature of most replication restart proteins. Our system allowed us to investigate the requirements for restart in vivo. Because RecA and RecA loading were required for DnaD to associate with the conflict-containing region, we hypothesize that a RecA-mediated D loop is required for restart at conflict regions. Additionally, the contribution of RecU to efficient survival suggests a requirement for Holliday junction resolution at head-on conflicts. Consistent with this observation, PriA binds four-way DNA junctions with extremely low affinity (40), and RecU does not cleave D-loops in B. subtilis (60). Therefore, although it is speculative, we hypothesize that RecU remodels interlinked chromosomes after strand invasion, generating a D-loop structure which is recognized by PriA.
Differences between replication fork reactivation mechanisms in E. coli and B. subtilis.
Head-on collisions between replication and transcription at inverted rRNA gene operons in E. coli are overcome through a RecA-independent replication fork reversal (RFR) mechanism (11, 12). During RFR, the nascent DNA strands dissociate from their templates, regress, and reanneal to form a four-way DNA junction with a double-stranded end. Processing of the regressed fork by RecBCD allows for replication restart subsequent to these encounters. Alternatively, the Holliday junction resolvase, RuvC, may cleave the reversed fork, causing chromosome breakage (61).
Our results suggest that at head-on conflicts, RecA-independent replication fork reversal (RFR) does not occur, at least in B. subtilis. In RFR in E. coli, RecA is dispensable but RecBCD is essential for viability. Additionally, the RFR model predicts that cleavage of the reversed fork by RuvC (RecU in Bacillus) leads to chromosome breakage in RecBCD-deficient cells. Our results clearly demonstrate that RecA associates with head-on conflict regions and establish that RecA loading and Holliday junction resolution reestablish the fork at conflict regions. Additionally, RecA and RecU act in the same pathway to promote conflict survival, suggesting that RecU alone does not play a role in processing the forks such as in the proposed RFR reaction. Nevertheless, we cannot rule out RFR at the conflict region by a RecA-dependent process. Indeed, RecA regresses forks in vitro (62), and studies in eukaryotic cells demonstrate that RAD51 promotes fork reversal in response to a broad array of genotoxic stresses (63).
The observed discrepancies between the RecA-independent RFR model and our results could arise from either the nature of the head-on conflict investigated or the model organism studied. The E. coli studies were based on rRNA gene inversions, whereas our results rely on a protein-coding gene. rRNA gene operons are repetitive and significantly longer than the single head-on protein-coding gene we used in our study. The impact of gene length on conflict severity and cellular strategies for resolution remains an understudied question. Additionally, rRNA gene transcription is distinct from expression of a protein-coding gene. Antitermination factors such as NusB stabilize RNA polymerase in rRNA genes (64), and high expression from rRNA gene promoters maintains the DNA in these regions in a persistently underwound state behind the transcription apparatus, whereas positive supercoils accumulate ahead of RNAP. Potentially the DNA topology innate to ribosomal DNA contributes to catalyzing fork reversal when operons are inverted head-on to replication.
The fundamental differences between the gammaproteobacteria E. coli and the firmicute B. subtilis may also account for an absence of detectable RecA-independent RFR in our experiments. B. subtilis uses two polymerases for chromosomal replication, DnaE and PolC (65), whereas E. coli employs only one (66). Additionally, E. coli and B. subtilis overcome replication-transcription conflicts by distinct strategies. E. coli harbors two accessory helicases, UvrD and Rep, which cooperate to facilitate replication progression across inverted rRNA gene operons, whereas B. subtilis appears to harbor only one essential accessory helicase (PcrA), which is probably also important for conflict resolution. B. subtilis displays a more significant leading-strand bias (74%) in the organization of its genome than observed in E. coli (55%) (5). Finally, inverting the rRNA gene causes cell death in 10% of cells in B. subtilis, whereas these genomic rearrangements are not significantly detrimental to survival of E. coli cells (10, 11). The results we present here, together with the prior investigations on replication-transcription conflicts in bacteria, highlight the significant differences in resolution mechanisms used by these two model organisms. Therefore, a deep understanding of the mechanisms employed by cells to facilitate accurate and timely DNA replication requires investigations in more than one species.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the Grossman lab for providing anti-DnaD antibody.
H.M. was supported by an NIGMS Award (DP2GM110773), and S.M.-W. was supported by a National Research Service Award (T32 GM07270).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00237-15.
REFERENCES
- 1.Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. 2000. The importance of repairing stalled replication forks. Nature 404:37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- 2.Mirkin EV, Mirkin SM. 2007. Replication fork stalling at natural impediments. Microbiol Mol Biol Rev 71:13–35. doi: 10.1128/MMBR.00030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cox MM. 2001. Historical overview: searching for replication help in all of the rec places. Proc Natl Acad Sci U S A 98:8173–8180. doi: 10.1073/pnas.131004998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cox MM. 1998. A broadening view of recombinational DNA repair in bacteria. Genes Cells 3:65–78. doi: 10.1046/j.1365-2443.1998.00175.x. [DOI] [PubMed] [Google Scholar]
- 5.Merrikh H, Zhang Y, Grossman AD, Wang JD. 2012. Replication-transcription conflicts in bacteria. Nat Rev Microbiol 10:449–458. doi: 10.1038/nrmicro2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Million-Weaver S, Samadpour AN, Moreno-Habel DA, Nugent P, Brittnacher MJ, Weiss E, Hayden HS, Miller SI, Liachko I, Merrikh H. 2015. An underlying mechanism for the increased mutagenesis of lagging-strand genes in Bacillus subtilis. Proc Natl Acad Sci U S A 112:E1096-1105. doi: 10.1073/pnas.1416651112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin YL, Pasero P. 2012. Interference between DNA replication and transcription as a cause of genomic instability. Curr Genomics 13:65–73. doi: 10.2174/138920212799034767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim N, Jinks-Robertson S. 2012. Transcription as a source of genome instability. Nat Rev Genet 13:204–214. doi: 10.1038/nrg3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Helmrich A, Ballarino M, Nudler E, Tora L. 2013. Transcription-replication encounters, consequences and genomic instability. Nat Struct Mol Biol 20:412–418. doi: 10.1038/nsmb.2543. [DOI] [PubMed] [Google Scholar]
- 10.Srivatsan A, Tehranchi A, MacAlpine DM, Wang JD. 2010. Co-orientation of replication and transcription preserves genome integrity. PLoS Genet 6:e1000810. doi: 10.1371/journal.pgen.1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boubakri H, de Septenville AL, Viguera E, Michel B. 2010. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J 29:145–157. doi: 10.1038/emboj.2009.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Septenville AL, Duigou S, Boubakri H, Michel B. 2012. Replication fork reversal after replication-transcription collision. PLoS Genet 8:e1002622. doi: 10.1371/journal.pgen.1002622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cox MM. 1991. The RecA protein as a recombinational repair system. Mol Microbiol 5:1295–1299. doi: 10.1111/j.1365-2958.1991.tb00775.x. [DOI] [PubMed] [Google Scholar]
- 14.Morimatsu K, Kowalczykowski SC. 2003. RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange: a universal step of recombinational repair. Mol Cell 11:1337–1347. doi: 10.1016/S1097-2765(03)00188-6. [DOI] [PubMed] [Google Scholar]
- 15.Arnold DA, Kowalczykowski SC. 2000. Facilitated loading of RecA protein is essential to recombination by RecBCD enzyme. J Biol Chem 275:12261–12265. doi: 10.1074/jbc.275.16.12261. [DOI] [PubMed] [Google Scholar]
- 16.Lenhart JS, Brandes ER, Schroeder JW, Sorenson RJ, Showalter HD, Simmons LA. 2014. RecO and RecR are necessary for RecA loading in response to DNA damage and replication fork stress. J Bacteriol 196:2851–2860. doi: 10.1128/JB.01494-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alonso JC, Luder G, Tailor RH. 1991. Characterization of Bacillus subtilis recombinational pathways. J Bacteriol 173:3977–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fernandez S, Kobayashi Y, Ogasawara N, Alonso JC. 1999. Analysis of the Bacillus subtilis recO gene: RecO forms part of the RecFLOR function. Mol Gen Genet 261:567–573. doi: 10.1007/s004380051002. [DOI] [PubMed] [Google Scholar]
- 19.Manfredi C, Carrasco B, Ayora S, Alonso JC. 2008. Bacillus subtilis RecO nucleates RecA onto SsbA-coated single-stranded DNA. J Biol Chem 283:24837–24847. doi: 10.1074/jbc.M802002200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yeeles JT, Dillingham MS. 2010. The processing of double-stranded DNA breaks for recombinational repair by helicase-nuclease complexes. DNA Repair (Amst) 9:276–285. doi: 10.1016/j.dnarep.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 21.Dillingham MS, Kowalczykowski SC. 2008. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol Mol Biol Rev 72:642–671. doi: 10.1128/MMBR.00020-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wigley DB. 2013. Bacterial DNA repair: recent insights into the mechanism of RecBCD, AddAB and AdnAB. Nat Rev Microbiol 11:9–13. doi: 10.1038/nrmicro2917. [DOI] [PubMed] [Google Scholar]
- 23.Churchill JJ, Kowalczykowski SC. 2000. Identification of the RecA protein-loading domain of RecBCD enzyme. J Mol Biol 297:537–542. doi: 10.1006/jmbi.2000.3590. [DOI] [PubMed] [Google Scholar]
- 24.Kuzminov A. 1999. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol Mol Biol Rev 63:751–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Courcelle J, Hanawalt PC. 2003. RecA-dependent recovery of arrested DNA replication forks. Annu Rev Genet 37:611–646. doi: 10.1146/annurev.genet.37.110801.142616. [DOI] [PubMed] [Google Scholar]
- 26.Grompone G, Sanchez N, Dusko Ehrlich S, Michel B. 2004. Requirement for RecFOR-mediated recombination in priA mutant. Mol Microbiol 52:551–562. doi: 10.1111/j.1365-2958.2004.03997.x. [DOI] [PubMed] [Google Scholar]
- 27.Iwasaki H, Takahagi M, Shiba T, Nakata A, Shinagawa H. 1991. Escherichia coli RuvC protein is an endonuclease that resolves the Holliday structure. EMBO J 10:4381–4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dunderdale HJ, Benson FE, Parsons CA, Sharples GJ, Lloyd RG, West SC. 1991. Formation and resolution of recombination intermediates by E. coli RecA and RuvC proteins. Nature 354:506–510. doi: 10.1038/354506a0. [DOI] [PubMed] [Google Scholar]
- 29.Sanchez H, Kidane D, Reed P, Curtis FA, Cozar MC, Graumann PL, Sharples GJ, Alonso JC. 2005. The RuvAB branch migration translocase and RecU Holliday junction resolvase are required for double-stranded DNA break repair in Bacillus subtilis. Genetics 171:873–883. doi: 10.1534/genetics.105.045906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carrasco B, Cozar MC, Lurz R, Alonso JC, Ayora S. 2004. Genetic recombination in Bacillus subtilis 168: contribution of Holliday junction processing functions in chromosome segregation. J Bacteriol 186:5557–5566. doi: 10.1128/JB.186.17.5557-5566.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ayora S, Carrasco B, Doncel-Perez E, Lurz R, Alonso JC. 2004. Bacillus subtilis RecU protein cleaves Holliday junctions and anneals single-stranded DNA. Proc Natl Acad Sci U S A 101:452–457. doi: 10.1073/pnas.2533829100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu J, Xu L, Sandler SJ, Marians KJ. 1999. Replication fork assembly at recombination intermediates is required for bacterial growth. Proc Natl Acad Sci U S A 96:3552–3555. doi: 10.1073/pnas.96.7.3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kreuzer KN. 2005. Interplay between DNA replication and recombination in prokaryotes. Annu Rev Microbiol 59:43–67. doi: 10.1146/annurev.micro.59.030804.121255. [DOI] [PubMed] [Google Scholar]
- 34.McGlynn P, Lloyd RG. 2002. Recombinational repair and restart of damaged replication forks. Nat Rev Mol Cell Biol 3:859–870. doi: 10.1038/nrm951. [DOI] [PubMed] [Google Scholar]
- 35.Smits WK, Goranov AI, Grossman AD. 2010. Ordered association of helicase loader proteins with the Bacillus subtilis origin of replication in vivo. Mol Microbiol 75:452–461. doi: 10.1111/j.1365-2958.2009.06999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marsin S, McGovern S, Ehrlich SD, Bruand C, Polard P. 2001. Early steps of Bacillus subtilis primosome assembly. J Biol Chem 276:45818–45825. doi: 10.1074/jbc.M101996200. [DOI] [PubMed] [Google Scholar]
- 37.Polard P, Marsin S, McGovern S, Velten M, Wigley DB, Ehrlich SD, Bruand C. 2002. Restart of DNA replication in Gram-positive bacteria: functional characterisation of the Bacillus subtilis PriA initiator. Nucleic Acids Res 30:1593–1605. doi: 10.1093/nar/30.7.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bruand C, Farache M, McGovern S, Ehrlich SD, Polard P. 2001. DnaB, DnaD and DnaI proteins are components of the Bacillus subtilis replication restart primosome. Mol Microbiol 42:245–255. doi: 10.1046/j.1365-2958.2001.02631.x. [DOI] [PubMed] [Google Scholar]
- 39.Heller RC, Marians KJ. 2005. The disposition of nascent strands at stalled replication forks dictates the pathway of replisome loading during restart. Mol Cell 17:733–743. doi: 10.1016/j.molcel.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 40.McGlynn P, Al-Deib AA, Liu J, Marians KJ, Lloyd RG. 1997. The DNA replication protein PriA and the recombination protein RecG bind D-loops. J Mol Biol 270:212–221. doi: 10.1006/jmbi.1997.1120. [DOI] [PubMed] [Google Scholar]
- 41.Gregg AV, McGlynn P, Jaktaji RP, Lloyd RG. 2002. Direct rescue of stalled DNA replication forks via the combined action of PriA and RecG helicase activities. Mol Cell 9:241–251. doi: 10.1016/S1097-2765(02)00455-0. [DOI] [PubMed] [Google Scholar]
- 42.Xu L, Marians KJ. 2003. PriA mediates DNA replication pathway choice at recombination intermediates. Mol Cell 11:817–826. doi: 10.1016/S1097-2765(03)00061-3. [DOI] [PubMed] [Google Scholar]
- 43.Merrikh H, Machon C, Grainger WH, Grossman AD, Soultanas P. 2011. Co-directional replication-transcription conflicts lead to replication restart. Nature 470:554–557. doi: 10.1038/nature09758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Auchtung JM, Lee CA, Garrison KL, Grossman AD. 2007. Identification and characterization of the immunity repressor (ImmR) that controls the mobile genetic element ICEBs1 of Bacillus subtilis. Mol Microbiol 64:1515–1528. doi: 10.1111/j.1365-2958.2007.05748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dutta D, Shatalin K, Epshtein V, Gottesman ME, Nudler E. 2011. Linking RNA polymerase backtracking to genome instability in E. coli. Cell 146:533–543. doi: 10.1016/j.cell.2011.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simmons LA, Grossman AD, Walker GC. 2007. Replication is required for the RecA localization response to DNA damage in Bacillus subtilis. Proc Natl Acad Sci U S A 104:1360–1365. doi: 10.1073/pnas.0607123104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuzminov A. 2001. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc Natl Acad Sci U S A 98:8241–8246. doi: 10.1073/pnas.131009198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eggleston AK, Kowalczykowski SC. 1991. An overview of homologous pairing and DNA strand exchange proteins. Biochimie 73:163–176. doi: 10.1016/0300-9084(91)90199-B. [DOI] [PubMed] [Google Scholar]
- 49.Lusetti SL, Cox MM. 2002. The bacterial RecA protein and the recombinational DNA repair of stalled replication forks. Annu Rev Biochem 71:71–100. doi: 10.1146/annurev.biochem.71.083101.133940. [DOI] [PubMed] [Google Scholar]
- 50.Kowalczykowski SC, Krupp RA. 1987. Effects of Escherichia coli SSB protein on the single-stranded DNA-dependent ATPase activity of Escherichia coli RecA protein. Evidence that SSB protein facilitates the binding of RecA protein to regions of secondary structure within single-stranded DNA. J Mol Biol 193:97–113. [DOI] [PubMed] [Google Scholar]
- 51.Cox MM. 2007. Regulation of bacterial RecA protein function. Crit Rev Biochem Mol Biol 42:41–63. doi: 10.1080/10409230701260258. [DOI] [PubMed] [Google Scholar]
- 52.Ryzhikov M, Gupta R, Glickman M, Korolev S. 2014. RecO protein initiates DNA recombination and strand annealing through two alternative DNA binding mechanisms. J Biol Chem 289:28846–28855. doi: 10.1074/jbc.M114.585117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sanchez H, Kidane D, Castillo Cozar M, Graumann PL, Alonso JC. 2006. Recruitment of Bacillus subtilis RecN to DNA double-strand breaks in the absence of DNA end processing. J Bacteriol 188:353–360. doi: 10.1128/JB.188.2.353-360.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bentchikou E, Servant P, Coste G, Sommer S. 2010. A major role of the RecFOR pathway in DNA double-strand-break repair through ESDSA in Deinococcus radiodurans. PLoS Genet 6:e1000774. doi: 10.1371/journal.pgen.1000774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Champoux JJ. 2001. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem 70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 56.Booker BM, Deng S, Higgins NP. 2010. DNA topology of highly transcribed operons in Salmonella enterica serovar Typhimurium. Mol Microbiol 78:1348–1364. doi: 10.1111/j.1365-2958.2010.07394.x. [DOI] [PubMed] [Google Scholar]
- 57.Krasilnikov AS, Podtelezhnikov A, Vologodskii A, Mirkin SM. 1999. Large-scale effects of transcriptional DNA supercoiling in vivo. J Mol Biol 292:1149–1160. doi: 10.1006/jmbi.1999.3117. [DOI] [PubMed] [Google Scholar]
- 58.Aguilera A, Garcia-Muse T. 2012. R loops: from transcription byproducts to threats to genome stability. Mol Cell 46:115–124. doi: 10.1016/j.molcel.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 59.Grompone G, Bidnenko V, Ehrlich SD, Michel B. 2004. PriA is essential for viability of the Escherichia coli topoisomerase IV parE10(Ts) mutant. J Bacteriol 186:1197–1199. doi: 10.1128/JB.186.4.1197-1199.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carrasco B, Ayora S, Lurz R, Alonso JC. 2005. Bacillus subtilis RecU Holliday-junction resolvase modulates RecA activities. Nucleic Acids Res 33:3942–3952. doi: 10.1093/nar/gki713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grompone G, Ehrlich D, Michel B. 2004. Cells defective for replication restart undergo replication fork reversal. EMBO Rep 5:607–612. doi: 10.1038/sj.embor.7400167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robu ME, Inman RB, Cox MM. 2001. RecA protein promotes the regression of stalled replication forks in vitro. Proc Natl Acad Sci U S A 98:8211–8218. doi: 10.1073/pnas.131022698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M. 2015. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208:563–579. doi: 10.1083/jcb.201406099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Torres M, Balada JM, Zellars M, Squires C, Squires CL. 2004. In vivo effect of NusB and NusG on rRNA transcription antitermination. J Bacteriol 186:1304–1310. doi: 10.1128/JB.186.5.1304-1310.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sanders GM, Dallmann HG, McHenry CS. 2010. Reconstitution of the B. subtilis replisome with 13 proteins including two distinct replicases. Mol Cell 37:273–281. doi: 10.1016/j.molcel.2009.12.025. [DOI] [PubMed] [Google Scholar]
- 66.O'Donnell M. 2006. Replisome architecture and dynamics in Escherichia coli. J Biol Chem 281:10653–10656. doi: 10.1074/jbc.R500028200. [DOI] [PubMed] [Google Scholar]
- 67.Harwood CR, Cutting SM (ed). 1990. Molecular biological methods for Bacillus. Wiley, Chichester, United Kingdom. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.