

ABSTRACT
Bacterial type IV coupling proteins (T4CPs) bind and mediate the delivery of DNA substrates through associated type IV secretion systems (T4SSs). T4CPs consist of a transmembrane domain, a conserved nucleotide-binding domain (NBD), and a sequence-variable helical bundle called the all-alpha domain (AAD). In the T4CP structural prototype, plasmid R388-encoded TrwB, the NBD assembles as a homohexamer resembling RecA and DNA ring helicases, and the AAD, which sits at the channel entrance of the homohexamer, is structurally similar to N-terminal domain 1 of recombinase XerD. Here, we defined the contributions of AADs from the Agrobacterium tumefaciens VirD4 and Enterococcus faecalis PcfC T4CPs to DNA substrate binding. AAD deletions abolished DNA transfer, whereas production of the AAD in otherwise wild-type donor strains diminished the transfer of cognate but not heterologous substrates. Reciprocal swaps of AADs between PcfC and VirD4 abolished the transfer of cognate DNA substrates, although strikingly, the VirD4-AADPcfC chimera (VirD4 with the PcfC AAD) supported the transfer of a mobilizable plasmid. Purified AADs from both T4CPs bound DNA substrates without sequence preference but specifically bound cognate processing proteins required for cleavage at origin-of-transfer sequences. The soluble domains of VirD4 and PcfC lacking their AADs neither exerted negative dominance in vivo nor specifically bound cognate processing proteins in vitro. Our findings support a model in which the T4CP AADs contribute to DNA substrate selection through binding of associated processing proteins. Furthermore, MOBQ plasmids have evolved a docking mechanism that bypasses the AAD substrate discrimination checkpoint, which might account for their capacity to promiscuously transfer through many different T4SSs.
IMPORTANCE For conjugative transfer of mobile DNA elements, members of the VirD4/TraG/TrwB receptor superfamily bind cognate DNA substrates through mechanisms that are largely undefined. Here, we supply genetic and biochemical evidence that a helical bundle, designated the all-alpha domain (AAD), of T4SS receptors functions as a substrate specificity determinant. We show that AADs from two substrate receptors, Agrobacterium tumefaciens VirD4 and Enterococcus faecalis PcfC, bind DNA without sequence or strand preference but specifically bind the cognate relaxases responsible for nicking and piloting the transferred strand through the T4SS. We propose that interactions of receptor AADs with DNA-processing factors constitute a basis for selective coupling of mobile DNA elements with type IV secretion channels.
INTRODUCTION
Bacterial type IV secretion systems (T4SSs) translocate DNA and protein substrates to target cells, generally by a mechanism requiring direct cell-to-cell contact (1). These systems are phylogenetically widely distributed among many Gram-negative and -positive bacterial species (2, 3). In both cell types, the T4SSs function as conjugation machines by delivering DNA substrates to bacterial recipient cells (3–5). Many Gram-negative pathogens additionally have adapted T4SSs for translocation of effector proteins to eukaryotic target cells during the course of infection (6, 7). Nearly all T4SSs are composed of a transenvelope channel and a cytoplasmic membrane ATPase, termed a type IV coupling protein (T4CP), that functions as the receptor for DNA or protein substrates (8, 9). Structural studies have identified striking architectural features of conjugation channels elaborated by model Gram-negative bacterial systems. The plasmid pKM101 conjugation system, for example, encodes 3 subunits which together assemble in 14 copies each as a large ∼1-MDa barrel-shaped substructure termed the core complex (10, 11). This complex spans the cell envelope and has been postulated to function as a structural scaffold for the translocation channel (12). Very recently, a massive 3.2-MDa substructure, comprised of nearly the entire plasmid R388 conjugation system, presented as two distinct subassemblies, the outer-membrane-associated core complex (also termed the outer membrane complex [OMC]) and an even larger inner membrane complex (IMC) (13). The T4CP of this system, TrwB, was not part of this large structure, but several lines of genetic and biochemical evidence suggest it associates with the IMC through a combination of N-terminal transmembrane domain (NTD) and extramembranous contacts (14–17).
Despite these elegant new findings, fundamental questions remain about early-stage substrate recruitment and docking reactions, as well as the route of substrate passage across the cell envelope. A crystal structure exists for the soluble domain of one member of the T4CP receptor superfamily, R388-encoded TrwB. The protomer consists of two domains, a nucleotide-binding domain (NBD) similar to RecA and DNA ring helicases and a 7-helix bundle termed the all-alpha domain (AAD). When assembled as the homohexamer, the soluble domain presents as a sphere with overall dimensions of 110 Å in diameter and 90 Å high; a central channel 20 Å wide extends from the cytoplasmic base to the NTD (18). The NTD was shown by electron microscopy to form a 25-Å-wide appendix that, when modeled with the soluble domain's X-ray structure, gives rise to an overall F1F0-ATPase-like ball-stem structure with a central channel traversing its entire length (19, 20). The T4CPs are phylogenetically closely related, and therefore, the TrwB hexamer is considered a structural prototype for the receptor superfamily (8). T4CPs are also related to the FtsK and SpoIIIE DNA translocases, leading to a proposal that they might pump DNA substrates through their central channels across the cytoplasmic membrane (21).
The AAD is of special interest with respect to a possible role in substrate engagement. The AAD of TrwB bears structural similarity to N-terminal domain 1 (the N domain) of the site-specific recombinase XerD (18). In XerD, the N domain is partly responsible for binding DNA and coordinating the DNA cleavage reaction by the C-terminal domain (CTD) (22, 23). In TrwB, the AAD is situated at the entrance of the channel at the cytoplasmic pole of the hexamer and forms a constriction of 8 Å at the opening of the channel (21). At this position, the AAD might bind DNA substrates as a prerequisite for passage directly through the T4CP lumen or delivery to one of the other VirB4- or VirB11-like T4SS ATPases for further substrate processing prior to translocation (24–26). NBDs are highly conserved among the T4CP superfamily members, but the AADs vary considerably in size, sequence composition, and numbers of predicted α-helical folds (Fig. 1) (27).
FIG 1.

Sequence alignments and structures of AADs from the TrwBR388, PcfCpCF10, and VirD4At T4CPs. (A) Schematic showing the domain architectures of VirD4 and PcfC, together with the structural prototype TrwB, with boundaries derived from sequence alignments and Phyre 2 structural modeling (27, 51). Domain boundaries are denoted by residue numbers, and the sizes of the AADs in residues are indicated. (B) Sequence alignments of the three AADs generated by ClustalW, with α-helices shown by colored shading corresponding to the colors in panel A, based on secondary-structure predictions using the Phyre 2 Web tool (51). Weakly (periods) or strongly (colons) conserved or identical (asterisks) residues are indicated. (C) Structures of the AADs solved by X-ray crystallography for TrwB (18) or predicted by Phyre 2 structural modeling (51) for PcfC and VirD4.
Our laboratory is investigating the mechanisms of action of paradigmatic T4SSs, including the Agrobacterium tumefaciens VirB/VirD4 system, which delivers transfer DNA (T-DNA) and effector proteins to susceptible plant cells, and the Enterococcus faecalis Prg/Pcf system, which mediates conjugative transfer of the pheromone-responsive plasmid pCF10 to recipient enterococci (28). Like other conjugation machines, these systems translocate their cargo substrates through formation of mating junctions with target cells. However, they exhibit important differences both in early-stage substrate-processing/docking reactions and in overall T4SS machine architectures, the latter due in large part to the diderm (two-membrane) versus monoderm (one-membrane) nature of the Gram-negative versus Gram-positive cell envelopes (2, 29, 30). Given that type IV secretion is a major underlying mechanism for both widespread dissemination of antibiotic resistance and virulence genes among bacterial populations and interkingdom translocation of effector proteins during infection, it is imperative to define key substeps of the translocation pathway. To this end, we focused here on the substrate receptor with a goal of testing the hypothesis that the AAD imparts specificity to the DNA substrate docking reaction.
VirD4 and PcfC are the receptors of the A. tumefaciens and E. faecalis systems (Fig. 1). Previously, we showed that VirD4 and PcfC form formaldehyde (FA)-cross-linkable complexes with DNA substrates in vivo independently of other channel subunits, confirming their roles as substrate receptors (25, 26, 30). Detection of substrate-receptor cross-linking in both A. tumefaciens and E. faecalis requires the DNA transfer and replication (Dtr) proteins, which assemble as the relaxosome at the origin-of-transfer (oriT) sequences and catalyze nicking of the DNA strand (T-strand) destined for transfer. In A. tumefaciens, these Dtr proteins include the VirD2 relaxase and accessory factor VirD1, which together bind and cleave the oriT-like sequences located within the T-DNA border repeats (31, 32). Two other accessory proteins, VirC1 and VirC2, bind near the nic site and stimulate the nicking reaction (33). VirC1, a member of the ParA/MinD ATPase superfamily, and another protein termed the VirD2-binding protein (Vbp) additionally function as spatial determinants to enhance the efficiency of the T-DNA substrate–VirD4 docking reaction (29, 34, 35). In E. faecalis, the PcfG relaxase and accessory factor PcfF suffice for nicking at the pCF10 oriT sequence in vitro (36) and FA cross-linking of the pCF10 substrate with PcfC in vivo (30). A soluble form of PcfC lacking its NTD (PcfCΔN103) binds single-stranded DNA (ssDNA) and double-stranded DNA (dsDNA) substrates without sequence or strand preference and also binds PcfG and PcfF in vitro (30).
We defined the contributions of the AADs of both T4CPs to DNA substrate transfer through phenotypic studies of AAD deletion and domain-swapped variants in vivo and by assaying for AAD binding of DNA and Dtr proteins in vitro. Our findings strongly support a model in which the AAD contributes to DNA substrate recognition through selective binding of cognate Dtr proteins. We also present evidence that the non-self-transmissible MOBQ plasmid RSF1010 has evolved a mechanism to bypass the AAD substrate discrimination checkpoint, which might account for the capacity of the plasmid to translocate through many different T4SSs of Gram-negative bacteria.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
A. tumefaciens strain A348 corresponds to strain C58 carrying the octopine-type pTiA6NC plasmid (37, 38). Strain KA2000 contains an in-frame deletion of virD4 from strain A348 obtained by marker exchange eviction mutagenesis (26). C58C1 Strr Rifr (39) served as the recipient in mating experiments. Strain LBA4404 has the oncogenic T-DNA deleted (40), and At12516 is a virE2 mutant (41). A. tumefaciens strains were grown at 28°C in MG/L medium, consisting of LB broth supplemented with mannitol and glutamate (42). The A. tumefaciens vir genes were induced at 22°C in AB inducing medium (ABIM) (glucose-containing minimal medium [pH 5.5], 1 mM phosphate, 200 μM acetosyringone [AS]), as previously described (42). When necessary, the medium was supplemented with the following antibiotics: gentamicin (100 μg/ml), kanamycin (100 μg/ml), tetracycline (5 μg/ml), carbenicillin (100 μg/ml), spectinomycin (500 μg/ml), streptomycin (500 μg/ml), and rifampin (300 μg/ml). All antibiotics were obtained from Sigma Chemical Co. E. faecalis OG1RF carrying plasmids of interest served as the donor strain and OG1SSp or OG1ES served as the recipient in mating experiments (43, 44). E. faecalis strains were cultured in brain heart infusion (BHI) at 37°C without shaking. E. faecalis was grown in the presence of the following antibiotics as needed: erythromycin (100 μg/ml final concentration for plasmid markers; 10 μg/ml for chromosomal markers), fusidic acid (25 μg/ml), rifampin (200 μg/ml), spectinomycin (1,000 μg/ml for plasmid markers; 250 μg/ml for chromosomal markers), streptomycin (1,000 μg/ml), and tetracycline (10 μg/ml). Escherichia coli strain DH5α served as the host for plasmid construction (Gibco-BRL), and E. coli strain BL21(DE3) was used for protein production (Novagen). E. coli strains were grown at 37°C in LB broth unless otherwise indicated.
Plasmid construction.
The plasmids and oligonucleotides used in this study are listed in Tables 1 and 2, respectively. When necessary for clarification, proteins are identified by their source in subscript, e.g., TrwBR388, and domains by their protein origin, e.g., AADTrwB.
TABLE 1.
Plasmids used in this study
| Plasmid | Description and/or relevant featuresa | Source or reference |
|---|---|---|
| Vectors | ||
| pPC914KS | Crbr; PvirB expression vector | 48 |
| pPC914KS.NcoI | Crbr; PvirB expression vector | 74 |
| pGEX6P-1 | Crbr; expression vector for GST tagging | GE Healthcare |
| pMALc2x | Crbr; expression vector for MBP tagging | NEB |
| pET28b(+) | Kanr; expression vector for His tagging | Novagen |
| pAP1 | Kanr; pET28a(+) derivative | 75 |
| pML122 | Genr Kanr; broad-host-range, mobilizable RSF1010 (MOBQ) | 76 |
| pAJ1 | Spcr; mobA1 derivative of RSF1010 (MOBQ) | 76 |
| pAJ6 | Spcr; oriT1 derivative of RSF1010 (MOBQ) | 76 |
| pXZ153 | Kanr; broad-host-range IncP cloning vector | 77 |
| pBBRMCS2 | Genr; broad-host-range cloning vector | 46 |
| pDL278p23 | Spcr; pDL278 shuttle vector with L. lactis P23 promoter | 36 |
| pCJK21 | Ermr Spcr; shuttle vector with inducible Pnis promoter | 78 |
| pCIp23 | Camr; pCI372 shuttle vector with P23 promoter | 30 |
| E. coli expression plasmids | ||
| pPC2004 | pGEX6P-1 with Ptac::GST-AADVirD4; VirD4 residues R201 to R346 | This work |
| pPC2013 | pET28b(+) with PT7::His6-AADVirD4 | This work |
| pPC2009 | pMALc2x with Ptac::mbp-NBD/CTDVirD4 (ΔN87/ΔAADVirD4) | This work |
| pPC2039 | pET28b(+) with PT7::His6-NBD/CTDVirD4 | This work |
| pPC2044 | pGEX6P-1 with Ptac::GST-NBD/CTDVirD4 | This work |
| pPC2038 | pET28b(+) with PT7::His6-VirD4ΔAADVirD4 | This work |
| pPC2071 | pAPI with PT7::His6-virD2 | This work |
| pPC2072 | pAPI with PT7::His6-virD2ΔC35 | This work |
| pPC2214 | pET28b(+) with PT7::His6-virD1 | This work |
| pPC2101 | pGEX6P-1 with Ptac::GST-AADPcfC; PcfC residues L204 to N360 | This work |
| pPC2100 | pET28b(+) with PT7::His6-AADPcfC | This work |
| pPC2102 | pMALc2x with Ptac::mbp-NBD/CTDPcfC (ΔN103/ΔAADPcfC) | This work |
| pPC2016 | pET28b(+) with PT7::His6-NBD/CTDPcfC | This work |
| pPC2027 | pET28b(+) with PT7::His6-PcfCΔAADPcfC | This work |
| pCY33 | pET28b(+) with PT7::His6-pcfF | 30 |
| pCY36 | pET28b(+) with PT7::pcfG-His6 | 30 |
| pPC67 | pET28b(+) with PT7::prgJ-His6 | 50 |
| A. tumefaciens expression plasmids | ||
| pKA9 | pPC914KS with PvirB::virD4 | 64 |
| pPC2011 | pPC914KS with PvirB::AADVirD4 | This work |
| pPC2078 | pPC914KS with PvirB::AADPcfC | This work |
| pPC2020 | pPC914KS with PvirB::VirD4ΔAAD | This work |
| pPC2022 | pPC914KS with PvirB::virD4-AADPcfC chimera | This work |
| pPC2024 | pPC914KS with PvirB::NBD/CTDVirD4 (ΔN87/ΔAADVirD4) | This work |
| E. faecalis expression plasmids | ||
| pCY25 | pCIp23 with P23::pcfC | 30 |
| pPC2104 | pDL278p23 with P23::AADPcfC; PcfC residues L204 to N360 | This work |
| pPC2105 | pDL278p23 with P23::pcfC-AADVirD4 chimera | This work |
| pPC2106 | pDL278p23 with P23::AADVirD4; VirD4 residues R201 to R346 | This work |
| pPC2107 | pDL278p23 with P23::NBD/CTDPcfC (ΔN103/ΔAADPcfC) | This work |
| pPC2108 | pDL278p23 with P23::PcfCΔAAD | This work |
| pPC2109 | pCJK21 with Pnis::AADPcfC | This work |
Vector plasmids conferred resistance to the following antibiotics: carbenicillin (Crb), kanamycin (Kan), gentamicin (Gen), spectinomycin (Spc), chloramphenicol (Cam), and erythromycin (Erm).
TABLE 2.
Oligonucleotides used in this study
| Purpose and oligonucleotide | Sequence | Reference |
|---|---|---|
| Cloning | ||
| D4F | 5′ CGGTGAACATATGAATTCCAGCAA 3′ | |
| D4-SD-F | 5′ CTCATCATACATATGCGCAATC 3′ | |
| D4R | 5′ TCACTCGAGGCATCAGCCTG 3′ | |
| AAD_Nde | 5′ AATACATATGGAGCGGAAGACTCATTGT 3′ | |
| D4AAD_For_Bam | 5′ AATAGGATCCGGAGCGGAAGACTCATTGT 3′ | |
| D4AAD_For_Sac | 5′ AATAGAGCTCGGAGCGGAAGACTCATTGT 3′ | |
| D4AAD_Rev | 5′ AAACTCGAGTCATAGAGCTCCCCTCCGGAGATCGTAAAC 3′ | |
| D4AAD_Rev_Sac | 5′ AATAGAGCTCCGCCTCCGGAGATCGTAAAC 3′ | |
| ΔAAD_Rev_Sac | 5′ TGAAGAGCTCTCAGAATCTAGG 3′ | |
| ΔAAD_For | 5′ AATAGAGCTCCAAGAAGACCTGCATTTAT 3′ | |
| CAAD-5 | 5′ GGAATTCCATATGTTGTCAAATAGTGATACG 3′ | |
| CAAD-3 | 5′ CCGCTCGAGTTAATTCCAGGAGTCAATATC 3′ | |
| CAAD-D1 | 5′ CGGAGCTCCGCGTGTTTAAGTCTAAGACCTT 3′ | |
| CAAD-D2 | 5′ GCGAGCTCCGAAGAAAAAACAGCAGTG 3′ | |
| pcfC_F_5 | 5′ AAACTGTCATATGCAGACGTAC 3′ | |
| pcfCΔN103-F | 5′ GCTCGTTTTCATATGGCTACACCTGC 3′ | |
| pcfC-R-XhoI | 5′ AACGCACGTCCTCGAGCTATTAGGCTAGAACGTGTGTTTC 3′ | |
| ΔN103R | 5′ ACATGCATGCCTGCAGCTCGAGTTAAACGTGTGTTTCTTCTT 3′ | |
| GST-pcfC-Δ103-F | 5′ CATGGTCTCGGATCCGCTCGTTTTGCTACACCTGC 3′ | |
| CAAD_For_Sac | 5′ CGGAGCTCGTTGTCAAATAGTGATACG 3′ | |
| CAAD_Rev_Sac | 5′ GCGAGCTCGAATTCCAGGAGTCAATATC 3′ | |
| D4AAD_For_Sac | 5′ AATAGAGCTCGGAGCGGAAGACTCATTGT 3′ | |
| D4AAD_Rev_Sac | 5′ AAACTCGAGTCATAGAGCTCCCCTCCGGAGATCGTAAAC 3′ | |
| EMSAs | ||
| pCF10-S | 5′ AAATTCGCAACATGCTAGCATGTTGCTCCGCTTGCAAAAAGAAAGCCG3′ | 30 |
| pCF10-AS | 5′GATCCGGCTTTCTTTTTGCAAGCGGAGCAACATGCTAGCATGTTGCG 3′ | 30 |
| RB-S | 5′ AGCTCAAATTACAACGGTATATATCCTGCCAGTCAGCATCATCACACCAA 3′ | |
| RB-AS | 5′TTGGTGTGATGATGCTGACTGGCAGGATATATACCGTTGTAATTTGAGCT 3′ |
(i) E. coli expression plasmids.
Plasmid pPC2100 produces N-terminally His6-tagged AADPcfC (residues Leu204 to Asn360 of PcfC) from the PT7 promoter. It was constructed by PCR amplification of bp 610 to 1080 of pcfC using primers CAAD-5 and CAAD-3, digestion of the PCR fragment with NdeI and XhoI, and introduction of the digested fragment into similarly digested vector pET28b(+). Plasmid pPC2101, producing glutathione S-transferase (GST)–AADPcfC, was constructed by excision of the AADPcfC fragment from pPC2100 by NdeI/XhoI digestion and introduction into similarly digested pCY34 (pGEX6p-pcfF [36]), substituting for the pcfF fragment. Plasmid pPC2016, producing an N-terminally His6-tagged variant of PcfC lacking its NTD and AAD (here designated NBD/CTDPcfC [Fig. 1]), was constructed by PCR amplification of bp 310 to 609 with primers pcfCΔN103-F and CAAD-D1 to generate the ΔAAD-up fragment and amplification of bp 1081 to 1830 using primers CAAD-D2 and pcfC-R-XhoI to generate the ΔAAD-down fragment. The ΔAAD-up fragment was digested with NdeI and SacI, the ΔAAD-down fragment was digested with SacI and XhoI, and both fragments were introduced into pET28b(+) digested with NdeI and XhoI. Plasmid pPC2027, producing His6-PcfCΔAAD, was constructed by replacement of the ΔAAD-up fragment in PC16 with the full-length upstream region of pcfC (from nucleotide positions 1 to 609) generated by PCR amplification with primers pcfC-F-5 and CAAD-D1 and subsequent digestion with NdeI and SacI. Plasmid pPC2102, producing maltose-binding protein (MBP) fused to NBD/CTDPcfC, was constructed by PCR amplification of NBD/CTDPcfC from pPC2016 using primers GST-pcfC-Δ103-F and ΔN103R. The resulting PCR product was digested with BsaI and PstI and then cloned into vector pMALc-2x digested with BamHI and PstI. All PCR amplifications of virD4 fragments were performed using pMY1153 as the template (31). Plasmid pPC4, producing GST-AADVirD4, was constructed by amplification of the sequence encoding AADVirD4 (residues Arg201 to Arg346 of VirD4) using primers AAD_Nde and D4AAD_Rev, digestion with NdeI and XhoI, and introduction into similarly digested pPC2101, substituting for AADPcfC. Plasmid pPC2013, producing His6-AADVirD4, was constructed by amplification of AADVirD4 using primers D4AAD_For_Bam and D4AAD_Rev, digestion with BamHI and XhoI, and introduction into similarly digested pET28b. Plasmid pPC2039, producing His6-NBD/CTDVirD4 (VirD4 with its 87-residue NTD and AAD deleted) was constructed by amplifying virD4 sequences located upstream and downstream of the AAD sequence with primers D4-SD-F and ΔAAD_Rev_Sac and ΔAAD_For and D4R, respectively. The upstream and downstream fragments were digested with NdeI/SacI and SacI/XhoI, respectively, and together introduced into pET28b(+). Plasmid pPC2038, producing His6-VirD4ΔAAD, was constructed as described for pPC2039 except that the upstream fragment was amplified with primers D4F and ΔAAD_Rev_Sac. Plasmid pPC2044, producing GST-NBD/CTDVirD4, was constructed by substituting the NdeI/XhoI fragment carrying NBD/CTDVirD4 from plasmid pPC2039 for the AADVirD4 fragment of pPC4. Plasmid pPC2009, producing MBP-NBD/CTDVirD4, was constructed by substituting a BamHI/XhoI fragment carrying NBD/CTDVirD4 from pPC2044 for the AADVirD4 fragment of pMALc2x-AADVirD4 (pPC43). Plasmids pPC2071 and pPC2072, producing His6-VirD2 and His6-VirD2ΔC35, respectively, were constructed by introducing NdeI/XhoI fragments from pKA205 (29) and pKA172 (15) into similarly digested pAP1. Plasmid pPC2214, producing His6-VirD1, was produced by introducing an NdeI/XhoI fragment from pKA204 (29) into similarly digested pET15b(+).
(ii) A. tumefaciens expression plasmids.
Plasmid pPC2011, producing AADVirD4 from the PvirB promoter, was obtained by substituting an NdeI/XhoI fragment carrying AADVirD4 from pPC2013 for an NdeI/XhoI fragment carrying GST in pZZ11 (45). Plasmids pPC2020 and pPC2024, producing VirD4ΔAAD and NBD/CTDVirD4 from the PvirB promoter, respectively, were constructed by introducing NdeI/XhoI fragments from pPC2038 and pPC2039 into similarly digested pPC914KS. Plasmid pPC2078, producing AADPcfC from the PvirB promoter, was constructed by introducing the AADPcfC fragment from pPC2100 into similarly digested pPC914KS.NdeI. Plasmid pPC2022, producing VirD4-AADPcfC from the PvirB promoter, was constructed by amplification of the AADPcfC fragment with primers CAAD_For_Sac and CAAD_Rev_Sac, digestion with SacI, and introduction into SacI-digested pPC2038. From the resulting plasmid, an NdeI/XhoI fragment carrying the virD4-AADPcfC chimera was isolated and introduced into similarly digested pPC914KS. ColE1 plasmids expressing alleles of interest were introduced into A. tumefaciens by ligation to broad-host-range (BHR) plasmids pXZ153 (Kanr) or pBBR1MCS2 (Genr) (46). Such cointegrants retained the ColE1 plasmid name plus “B” for BHR. For introduction of two plasmids into the same cell, the plasmids were first ligated to compatible BHR vectors, and the resulting cointegrant plasmids were introduced into A. tumefaciens with appropriate antibiotic selection.
(iii) E. faecalis expression plasmids.
Plasmid pPC2104, producing AADPcfC from the P23 promoter, was obtained by introducing an NdeI/XhoI fragment carrying AADPcfC from plasmid pPC2100 into similarly digested pCY31 (30) to substitute for pcfCΔN103. Similarly, plasmids pPC2108, pPC2107, and pPC2106, producing PcfCΔAAD, NBD/CTDPcfC, and AADVirD4 from the P23 promoter, respectively, were constructed by introducing the corresponding fragments from plasmids pPC2027, pPC2016, and pPC13, respectively, into pPC31. Plasmid pPC2105, producing PcfC-AADVirD4 from the P23 promoter, was constructed by amplification of the AADVirD4 sequence with primers D4AAD_For_Sac and D4AAD_Rev_Sac, digestion with SacI, and introduction into SacI-digested pPC2027. From the resulting plasmid, an NdeI/XhoI fragment carrying the pcfC-AADVirD4 chimera was isolated and introduced into similarly digested pCY31. Plasmid pPC2109, producing His6-AADPcfC from the Pnis promoter, was obtained by introducing an NcoI-XhoI fragment carrying AADPcfC from pPC2100 into similarly digested pMSP3545 (47). The integrity of all cloned fragments was confirmed by DNA sequence analysis.
Conjugation assays.
A. tumefaciens donor strains carrying plasmid pML122Kanr or pML122Genr were mated with the recipient strain C58C1RS as described previously (26). Briefly, exponential-phase cells (optical density at 600 nm [OD600], ∼0.5) were harvested and diluted in induction medium (ABIM) for expression of vir genes for 8 h at 22°C. After induction, the cells were adjusted to an OD600 of ∼0.5 before mixing donors and recipient in a ratio of 1:5. The mating mixtures were spotted onto nitrocellulose filters placed on an ABIM agar plate. After 4 days of growth at 18°C, cells were harvested from the filters, and serial dilutions were plated onto MG/L medium with appropriate antibiotic selection for transconjugant and donor cells. E. faecalis 2-h filter and broth matings were carried out as previously described (30). Briefly, donors (OG1RF background) and recipients (OG1ES or OG1SSp) were inoculated in BHI and grown overnight without shaking. Cultures were diluted 1:10 in BHI and grown for 1 h at 37°C without shaking. The donor and recipient strains were mixed in a ratio of 1:10 and allowed to mate in liquid without shaking, or 10 μl of the mating mixture was spotted onto nitrocellulose on BHI plates. After 2 h of mating, the filters were resuspended in 1 ml of 1× PBS. Mating mixtures from the liquid and solid-surface matings were serially diluted in 1× PBS, and the numbers of donors and transconjugants were obtained by plating on selective BHI agar plates. For both A. tumefaciens and E. faecalis matings, transfer efficiencies were determined by the transconjugant/donor (Tc/D) ratio. Conjugation experiments were repeated in triplicate at least three times, and the results are presented as averages of three experiments.
Plant virulence assays.
A. tumefaciens strains were tested for virulence by inoculating wound sites of Kalanchoe daigremontiana leaves as previously described (48). A348 and avirulent strain KA2000 (ΔvirD4) served as controls for the tumorigenesis assay. The virulence of strains or mixed infections was assayed at least three times on three separate leaves each time. Mixed infections were performed by coinoculation of two strains of interest on the same wound (49). Tumor formation was analyzed 6 weeks after inoculation.
Protein production and purification.
For detection of protein production in A. tumefaciens, cultures were induced in ABIM for 12 h and normalized to an OD600 of 0.5, and cells from equivalent culture volumes were harvested by centrifugation at 5,000 × g for 10 min at 4°C. Cell pellets were resuspended in lysis buffer A (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 20% sucrose, 10 mg/ml lysozyme, 1× complete protease inhibitor cocktail [Roche]) and incubated for 20 min at room temperature, and the cells were lysed by sonication (Branson Sonifier 250). The cell lysates were recovered by two rounds of centrifugation at 15,700 × g for 30 min to remove unbroken cells and cell debris. For corresponding analyses of protein abundance in E. faecalis, strains of interest were induced with 20 ng/ml of cCF10 peptide or nisin for 1.5 h, and cells were harvested by centrifugation. The cell pellet was frozen at −20°C, thawed to facilitate cell breakage, and then resuspended in lysis buffer A supplemented with 10 μg/ml mutanolysin and incubated for 20 min at 37°C. The suspension was sonicated and centrifuged twice at 15,700 × g for 30 min to remove unbroken cells and cell debris. Soluble lysates of A. tumefaciens and E. faecalis were mixed with 5× Laemmli buffer, boiled for 5 min, and loaded on SDS-polyacrylamide gels on a per cell (CFU) equivalent basis. The proteins were transferred onto nitrocellulose membranes, and blots were developed with anti-VirD4 or -PcfC antibodies and goat anti-rabbit secondary antibodies conjugated to alkaline phosphatase for visualization (Bio-Rad). Molecular mass markers were from Gibco-BRL (Grand Island, NY). AADVirD4 and AADPcfC were highly enriched as follows. Overnight cultures (2.5 ml) of strains carrying pPC2004 (GST-AADVirD4), pPC2101 (GST-AADPcfC), or pGEX6p (GST only) were inoculated into 250 ml fresh LB medium. The cultures were incubated with shaking at 37°C to an OD600 of 0.4, induced with 0.8 mM (final concentration) IPTG (isopropyl-β-d-thiogalactopyranoside), and incubated with shaking at room temperature (RT) for 3 h. The cells were pelleted by centrifugation at 6,000 × g for 10 min. The pellet was resuspended in 2 ml of lysis buffer A, incubated at 37°C for 10 min, and lysed by sonication. The lysates were clarified by two rounds of centrifugation at 15,000 × g at 4°C for 30 min, and the resulting supernatant was mixed with 100 μl of glutathione-Sepharose beads and gently shaken for 1 h at 4°C. The mixture was loaded onto a 1.0- by 10-cm column, and the beads were washed 5 times with wash buffer (50 mM Tris-HCl [pH 7.4], 0.15 M NaCl, 1 mM EDTA, 1 mM dithiothreitol [DTT]) to remove unbound protein. Bound GST-AAD protein or GST alone was eluted from the column with buffer A supplemented with 10 mM glutathione. Alternatively, the GST tag was cleaved from the AADs by treatment with PreScission enzyme (2 units) at 4°C for 6 h. The protein-Sepharose mixture was centrifuged for 5 min at 0.5 × g, and the supernatant was mixed with 50 μl of glutathione-Sepharose beads for 2 h at 4°C to remove residual GST and PreScission enzyme. The purified AADPcfC or AADVirD4 fragments were recovered by centrifugation for 5 min at 500 × g. His6-tagged forms of PcfF, PcfG, VirD1, VirD2, VirD2ΔC35, and PrgJ were highly enriched by Co2+ affinity chromatography as previously described (30, 50). Briefly, 500-ml cultures of BL21(DE3) strains carrying the relevant plasmids were grown at 37°C to an OD600 of 0.4, induced with IPTG (0.1 mM final concentration), and incubated at room temperature for 3 h. Cells were harvested by centrifugation at 6,000 × g for 10 min, resuspended in 5 ml buffer A, and incubated for 10 min at 37°C. The cells were sonicated, and the lysate was centrifuged at 40,000 × g at 4°C for 30 min. The supernatant was mixed with 1 ml of Co2+ (Talon; Clontech) affinity resin, washed extensively with buffer A containing 10 mM imidazole, and incubated for 12 h at 4°C. The bound proteins were eluted with buffer A containing 200 mM imidazole. Soluble domains of VirD4 and PcfC lacking their NTDs and AADs (designated NBD/CTDs) were enriched as described above for enrichment of the GST-AADs, except that cell lysates were mixed with 100 μl of amylose resin and gently shaken for 1 h at 4°C. The beads were washed 5 times with wash buffer (50 mM Tris-HCl [pH 7.4], 0.15 M NaCl, 1 mM EDTA, 1 mM DTT) to remove unbound protein, and the bound MBP-NBD/CTD protein or MBP alone was eluted from the column with buffer A (minus lysozyme) supplemented with 10 mM maltose.
EMSAs.
Binding of purified AADs and NBD/CTDs to DNA substrates was assessed by electrophoretic mobility shift assays (EMSAs) using ssDNA and dsDNA substrates. The ssDNA substrates included 50-mers corresponding to the sense or antisense strands of the minimal oriT region of plasmid pCF10 (30) or the nic region of the T-DNA right-border repeat (38). The ssDNA oligonucleotides were synthesized and purified by PAGE (Sigma-Aldrich). A 39-bp fragment provided in the DIG gel shift kit (Roche) was used as a short, nonspecific dsDNA substrate. ssDNA and dsDNA substrates were labeled at the 3′ ends with digoxigenin (DIG)-11-ddUTP using the DIG gel shift kit (Roche) according to the manufacturer's instructions. EMSAs were carried out in 20-μl reaction mixtures containing the recombinant proteins, DIG-labeled ssDNA or dsDNA, 0.2 μg of poly(dA-dT), 0.1 μg of poly-l-lysine, and 1× reaction buffer (20 mM Tris-HCl [pH 8.0], 0.1 M KCl, 10% glycerol, 5 mM EDTA). The reaction mixtures were incubated at RT for 15 min and loaded onto 8% polyacrylamide gels in 0.5× Tris-borate-EDTA buffer (pH 7.9). After electrophoresis at 100 V for 1.5 h, the gels were transferred to a nylon membrane using the Genie electrophoretic blotter (Idea Scientific). DIG-labeled DNA was visualized by an enzyme immunoassay according to the manufacturer's instructions (DIG gel shift kit; Roche).
Protein interaction assays.
For GST pulldown assays, soluble fractions of E. coli cells producing GST-AADVirD4, GST-AADPcfC, or GST alone were mixed with 100 μl of glutathione-Sepharose beads for 1 h at RT. The Sepharose beads were pelleted by centrifugation at 500 × g for 5 min and washed three times with 1× physiologically buffered saline (PBS). The beads were resuspended in 500 μl of binding buffer (20 mM Tris-HCl, pH 8.0, 0.1 M NaCl, 10 μg bovine serum albumin). Enriched His6-VirD1, His6-VirD2, His6-PcfF, PcfG-His6, or PrgJ-His6 (20 μg) protein, prepared as described above, was incubated with the beads prebound with GST or the GST-AAD fusions for 3 h at RT. The beads were pelleted, and extensively washed beads were eluted twice with elution buffer (10 mM glutathione, 0.1 M NaCl). Cell lysates containing the His6-tagged proteins were prebound with 100 μl of Co2+ resin as described above, and the beads with bound proteins recovered by centrifugation were mixed with enriched forms of GST-AADVirD4, GST-AADPcfC, or GST alone. After incubation at 4°C for 3 h, the resins were washed extensively with binding buffer with 20 mM imidazole, and the bound proteins were eluted with buffer A containing 250 mM imidazole. The eluted fractions were assayed for PcfC, PcfF, and PcfG proteins by SDS-PAGE and immunostaining. For the MBP pulldowns, cell lysates containing the MBP-NBD/CTDVirD4 and MBP-NBD/CTDPcfC proteins, or MBP alone, were mixed with 100 μl of amylose resin, and the resin with bound proteins recovered by centrifugation was mixed with enriched forms of the His6-tagged Dtr proteins. After incubation at 4°C for 3 h, the resins were washed extensively with binding buffer, and the bound proteins were eluted with binding buffer containing 10 mM maltose. The eluates from the pulldown assays were analyzed by SDS-PAGE and immunostaining.
RESULTS
The all-alpha-domains of VirD4 and PcfC are essential for T4CP function and exert negative dominance.
We identified domains of VirD4 and PcfC by protein sequence alignments and structure predictions based on the X-ray structure of TrwB and by using the Phyre 2 algorithm (51) (Fig. 1A). The AADs of VirD4 and PcfC share a low degree of primary sequence similarity with each other and with that of TrwB (Fig. 1B). AADVirD4 and AADPcfC also are larger and are predicted to adopt distinct α-helical bundle folds in comparison to the 7-helix bundle of AADTrwB (Fig. 1C).
To evaluate the functional importance of the AADs, we deleted the AADs of both receptors and assayed for effects on substrate transfer (Fig. 2). Native VirD4, as well as a VirD4ΔAAD variant, accumulated at comparable levels when produced from the AS-inducible PvirB promoter, yet only the native protein supported T-DNA transfer and tumor production on wounded K. daigremontiana leaves (Fig. 2A). Similarly, although native PcfC and the PcfCΔAAD mutant protein accumulated at high levels in E. faecalis donors, only the former supported high-frequency plasmid transfer in both liquid and solid-surface (filter) matings (Fig. 2B). We also tested whether separate production of the AAD and ΔAAD might reconstitute a functional T4CP by production of both protein pairs in A. tumefaciens and E. faecalis, but neither of these strains transferred their cognate DNA substrates at detectable levels (Fig. 2).
FIG 2.
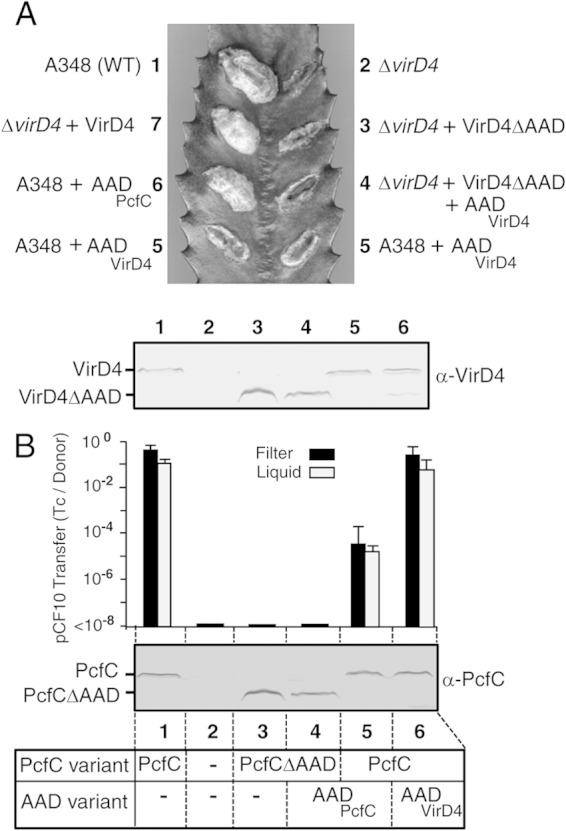
Contributions of the AADs of VirD4 and PcfC to T4CP function in vivo. (A) Virulence of A. tumefaciens strains producing variant forms of VirD4 on K. daigremontiana. (Top) Leaf wound sites were inoculated with A. tumefaciens A348 (WT) producing native VirD4 or the ΔvirD4 mutant KA2000 (ΔvirD4). Where indicated, strains carried broad-host-range plasmids producing native VirD4, VirD4ΔAAD, AADVirD4, or AADPcfC. The indicated wound sites were inoculated with the following strain (the plasmid names correspond to those of the ColE1 plasmids plus “B” for BHR): 1, A348; 2, KA2000; 3, KA2000(pPCB2020); 4, KA2000(pPCB2020, pPCB2011); 5, A348(pPCB2011); 6, A348(pPCB2078); 7, KA2000(pKA9). (Bottom) Immunoblot showing steady-state levels of VirD4 (72 kDa) and VirD4ΔAAD (56 kDa) produced in A. tumefaciens A348 (WT) or KA2000 (ΔvirD4) strains. The lane numbers of the analyzed strains correspond to those presented in the virulence assay. (B) (Top) Histogram showing transfer frequencies of pCF10 from OG1RF donors producing variant forms of PcfC in 2-h filter and liquid matings, presented as the number of transconjugants per donor cell (Tc/Donor). Experiments were repeated at least 3 times, and the histogram depicts the average values with standard deviations. (Middle) Immunoblot showing steady-state levels of PcfC (67 kDa) and PcfCΔAAD (50 kDa) in E. faecalis OG1RF strains. Lanes: 1, OG1RF(pCF10); 2, OG1RF; 3, OG1RF(pCF10ΔpcfC, pPC2108); 4, OG1RF(pCF10ΔpcfC, pPC2108, pPC2109); 5, OG1RF(pCF10, pPC2104); 6, OG1RF(pCF10, pPC2106). (Bottom) Schematic showing analyzed OG1RF strains that produced combinations of native PcfC or PcfCΔAAD with the AADPcfC or AADVirD4 variants shown.
If the AAD functions in substrate binding, its production in an otherwise wild-type (WT) background might exert negative dominant effects on transfer, for example, through substrate binding and sequestration. To examine this possibility, we assayed for inhibitory effects accompanying AAD production on DNA transfer. Strikingly, A. tumefaciens A348 expressing PvirB::AADVirD4 from a BHR plasmid was strongly suppressed in its capacity to induce tumor formation on susceptible plants (Fig. 2A). Similarly, E. faecalis OG1RF(pCF10) donors engineered to express P23::AADPcfC transferred pCF10 at frequencies 3 orders of magnitude lower than WT donors in both liquid and solid-surface matings (Fig. 2B). Our antibodies to VirD4 and PcfC did not react against the respective AADs, preventing assessments of steady-state accumulation of these domains. Next, we tested whether the AADs inhibited transfer of DNA substrates in the heterologous species. We expressed PvirB::AADPcfC in A. tumefaciens WT A348 and, conversely, P23::AADVirD4 in E. faecalis strain OG1RF(pCF10). Neither domain exerted negative dominance, as monitored by A. tumefaciens-mediated T-DNA transfer to plants or conjugative transfer of pCF10 to E. faecalis recipients (Fig. 2).
A VirD4-AADPcfC chimera supports transfer of a mobilizable MOBQ plasmid through the A. tumefaciens VirB/VirD4 T4SS.
The above-mentioned findings were consistent with a role for receptor AADs in binding of cognate DNA substrates. To further test this model, we reciprocally swapped the AADs of VirD4 and PcfC and assayed for functionality of the chimeric proteins. The chimeric proteins VirD4-AADPcfC and PcfC-AADVirD4 accumulated at detectable levels in A. tumefaciens and E. faecalis donor strains but did not support T-DNA transfer to plant cells or plasmid transfer to E. faecalis recipients (Fig. 3A and B). Strikingly, however, A. tumefaciens donors producing VirD4-AADPcfC transferred the mobilizable plasmid pML122 (RSF1010 derivative; MOBQ incompatibility group) through the VirB channel to A. tumefaciens recipients, establishing that this chimeric protein engages productively with the plasmid transfer intermediate and the VirB channel (Fig. 3C). As expected, VirD4-AADPcfC did not support the transfer of RSF1010 derivatives sustaining deletions of the oriT sequence (pAJ6) or the mobA relaxase (pAJ1) (Fig. 3C). VirD4 with its AAD deleted also did not mobilize pML122 transfer at detectable frequencies, which confirms the requirement for an AAD—from either VirD4 or PcfC—for transmissibility of the MOBQ plasmid through the VirB/VirD4 T4SS. We also tested whether production of AADVirD4 or AADPcfC disrupted pML122 transfer through the native VirB/VirD4 T4SS, but neither AAD exerted dominant-negative effects (Fig. 3). These findings suggest that the MOBQ plasmid substrate is capable of docking with VirD4 through a mechanism that bypasses an AAD substrate discrimination checkpoint.
FIG 3.
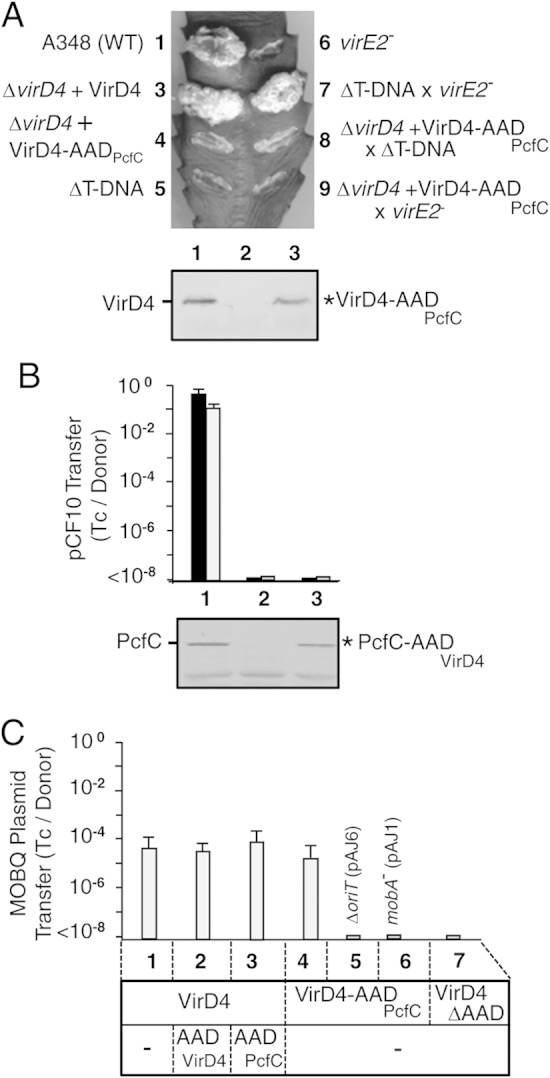
Functionality of T4CP chimeras in A. tumefaciens and E. faecalis. (A) Virulence of A. tumefaciens strains producing variant forms of VirD4 on K. daigremontiana. (Top) Leaf wound sites were inoculated with A. tumefaciens A348 (WT) producing native VirD4 or the ΔvirD4 mutant KA2000 (ΔD4). Where indicated, KA2000 was engineered to produce native VirD4 or the VirD4-AADPcfC (D4-AADC) chimera. Mixed infections were carried out using strains Mx358 (virE2 mutant; translocates T-DNA) and LBA4404 (ΔT-DNA; translocates VirE2). (Bottom) Immunoblot showing steady-state levels of VirD4 (72 kDa) and VirD4-AADPcfC (73 kDa), labeled - and *, respectively, detected by development with anti-VirD4 antibodies. The strains for the virulence assays and immunoblot are numbered as follows: 1, A348; 2, KA2000 (ΔvirD4); 3, KA2000(pKA9); 4, KA2000(pPCB2022); 5, LBA4404 (ΔT-DNA); 6, Mx358 (virE2 mutant); 7, LBA4404 × Mx358 coinoculation; 8, KA2000(pPCB2022) × LAB4404 coinoculation; 9, KA2000(pPCB2022) × Mx358 coinoculation. (B) (Top) Transfer frequencies of pCF10 from donors producing variant forms of PcfC in 2-h filter and liquid matings, presented as the number of transconjugants per donor cell (Tc/Donor), with average values and standard deviations shown. 1, OG1RF(pCF10); 2, OG1RF(pCF10ΔpcfC); 3, OG1RF(pCF10ΔpcfC, pPC2105). (Bottom) Immunoblot showing steady-state levels of PcfC (67 kDa) and PcfC-AADVirD4 (66 kDa), labeled as - and *, respectively, detected by development with anti-PcfC antibodies. The lower bands correspond to an unknown cross-reactive species. (C) (Top) Transfer of the MOBQ plasmid pML122 or, where indicated, the ΔoriT (pAJ6) or mobA mutant (pAJ1) variant through the A. tumefaciens VirB/VirD4 T4SS. 1, A348(pML122); 2, A348(pML122, pPCB2011); 3, A348(pML122, pPCB2078); 4, KA2000(pML122, pPCB2022); 5, KA2000(pAJ6, pPCB2022); 6, KA2000(pAJ1, pPCB2022); 7, KA2000(pML122, pPCB2020). (Bottom) Analyzed A. tumefaciens strains that produced combinations of VirD4, VirD4-AADPcfC, or VirD4ΔAAD with the AADVirD4 or AADPcfC variants shown.
In the A. tumefaciens transfer system, the efficiency of T-DNA delivery to the plant nucleus is significantly enhanced by cotranslocation of effector proteins, such as the VirE2 single-stranded DNA-binding protein (SSB). Upon cotransfer of the T-DNA transfer intermediate (VirD2–T-strand) and effector proteins into a plant cell, the latter bind and facilitate delivery of the T-DNA intermediate to the nucleus (52). Interestingly, coinoculation on the same wound site of two avirulent strains, a ΔT-DNA mutant and a virE2 mutant, incites WT tumor production as a result of VirE2 transfer by the former strain and T-DNA transfer by the latter into the same plant cell (Fig. 3A) (49, 53). To determine if VirD4-AADPcfC was selectively impaired in mediating T-DNA or VirE2 effector translocation, we coinoculated plant wound sites with a strain producing the chimeric receptor with either the ΔT-DNA mutant or the virE2 mutant. Neither mixed infection incited tumor production (Fig. 3A), indicating that the chimeric protein fails to engage productively with either substrate despite its capacity to support translocation of the MOBQ plasmid.
The AADs of VirD4 and PcfC bind DNA substrates without sequence specificity.
We next assayed for binding of the AADs to DNA and Dtr components of the relaxosome in vitro. Previously, we showed that a His6-tagged soluble form of PcfC, His6-PcfCΔN103, binds ssDNA and dsDNA substrates without sequence preference in vitro (30). We were unable to enrich His6-tagged forms of AADVirD4 and AADPcfC due to problems of insolubility, but the addition of a GST tag followed by removal of the tag by PreScission protease cleavage yielded highly enriched soluble forms of both domains (Fig. 4A).
FIG 4.
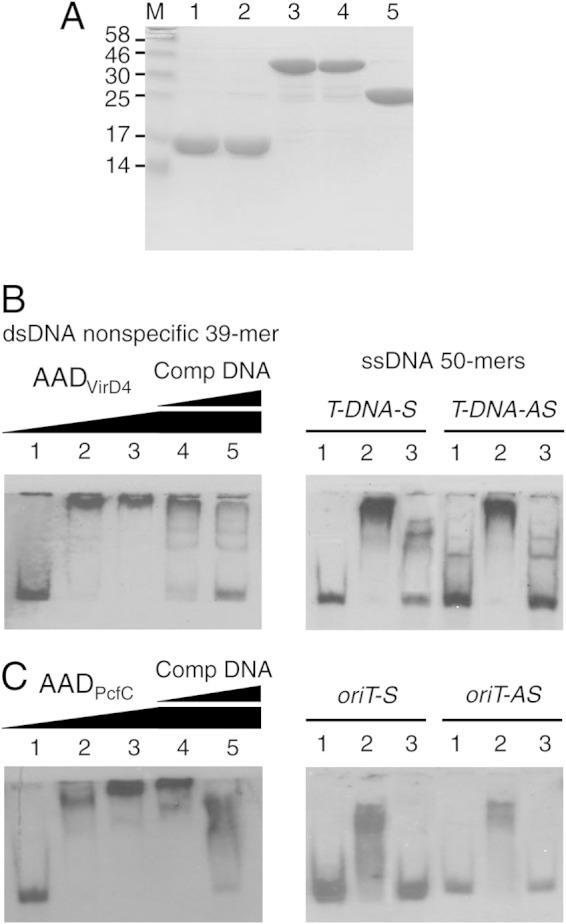
DNA-binding activities of AADVirD4 and AADPcfC. (A) Enrichment of GST-tagged and untagged AADVirD4 and AADPcfC by glutathione affinity chromatography, as described in the text. Lanes: 1 and 2, AADVirD4 and AADPcfC following removal of GST tags; 3 and 4, GST-tagged AADVirD4 and AADPcfC; 5, GST alone; M, molecular size markers (in kilodaltons). (B) EMSAs testing for binding of AADVirD4 to the 3′-end-DIG-labeled, dsDNA and ssDNA substrates listed above each gel. Lanes for the dsDNA 39-mer: 1, DNA probe only; 2 to 5, DNA probe plus AADVirD4 at concentrations of 72 nM (2), 144 nM (3), and 504 nM (3 to 5). In the last two lanes, AADVirD4 was preincubated with unlabeled dsDNA competitor at concentrations 10- and 100-fold higher than that of the DIG-labeled substrate. Lanes for the ssDNA probes: 1, DNA probe only; 2, DNA probe plus AADVirD4 (576 nM); 3, same as lane 2 except that AADVirD4 was preincubated with unlabeled competitor DNA at a 100-fold-higher concentration than the DIG-labeled substrate. (C) EMSAs testing for binding of AADPcfC to the 3′-end-DIG-labeled dsDNA and ssDNA substrates listed above each gel. The lanes are as described for panel B using the same AADPcfC protein concentrations.
By EMSAs (30, 50), we determined that both AADVirD4 and AADPcfC bound ssDNA and dsDNA substrates without sequence preference (Fig. 4B and C). Both AADs formed complexes with a 39-bp nonspecific dsDNA substrate and with ssDNA 50-mers corresponding to the sense or antisense strands of the pTiA6NC oriT-like right-border repeat (for AADVirD4) or the pCF10 oriT sequence (for AADPcfC). Although we could detect binding of the AADs to longer 186- to 200-bp dsDNA substrates spanning the pCF10 oriT or A. tumefaciens T-DNA right-border repeat, these results were not reproducible, possibly because of weak-affinity interactions (data not shown). For all tested substrates, DNA binding was inhibited, at least to some extent, by preincubation with unlabeled competitor DNAs (Fig. 4B and C).
The AADs of VirD4 and PcfC bind only cognate Dtr processing factors.
VirD4 and PcfC form FA-cross-linkable complexes with DNA substrates in vivo, but only in strains competent for assembly of the relaxosome at the T-DNA border or oriT sequences (25, 30). Furthermore, purified His6-PcfCΔN103 binds the relaxosome components PcfF and PcfG, which together catalyze nicking within the pCF10 oriT sequence (30). To test whether the AADs of VirD4 and PcfC bind one or more of the Dtr processing proteins in vitro, we obtained highly enriched GST-tagged forms of the AADs (Fig. 4A) and His6-tagged forms of PcfF and PcfG, as well as the corresponding His6-tagged forms of A. tumefaciens VirD1 and VirD2 required for nicking at T-DNA border sequences (Fig. 5). For pulldown assays, GST-AADVirD4 or GST alone was prebound to glutathione affinity resin, the protein-bound resin was mixed with the His-tagged Dtr factors, and complexes were eluted with glutathione. As shown in Fig. 5A, His6-VirD2 coeluted with GST-AADVirD4 but not GST alone. In reciprocal pulldown assays, His6-VirD2 prebound to Co2+ resin formed a complex with GST-AADVirD4 but not GST alone. Despite repeated efforts, we were unable to detect an interaction between His6-VirD1 and GST-AADVirD4 (Fig. 5A). In contrast, both His6-PcfF and PcfG-His6 from E. faecalis formed complexes with GST-AADPcfC but not GST alone (Fig. 5C).
FIG 5.

Interactions between the AADs and Dtr proteins. (A) (Top) Cobalt affinity pulldown assays testing for GST-AADVirD4 interactions with Dtr proteins. His6-VirD1, His6-VirD2, or His6-VirD2ΔC35 was highly enriched by cobalt affinity chromatography (lanes labeled “load”) and bound to Co2+ beads, and the protein-bead complexes were mixed with GST-AADVirD4 (second lane) or GST alone (third lane). Following extensive washes, the bound proteins were eluted from the beads with imidazole, and proteins recovered in the eluates were identified by immunostaining with anti-GST and anti-His antibodies (shown on the left of each gel). (Bottom) Reciprocal GST affinity pulldowns. Enriched GST-AADVirD4 or GST alone was bound to glutathione beads, and the protein-bead complexes were mixed with the His-tagged proteins shown at the top. Following extensive washes, the bound proteins were eluted from the beads with glutathione and identified by immunostaining with anti-GST and anti-His antibodies (shown on the left of each gel). (B) Cobalt and GST affinity pulldown assays testing for GST-AADPcfC interaction with His6-VirD2, as described for panel A. (C) Cobalt and GST affinity pulldown assays testing for GST-AADPcfC interaction with His6-PcfF and PcfG-His6, as described for panel A. (D) Cobalt and GST affinity pulldown assays testing for GST-AADVirD4 interaction with PcfG-His6, as described for panel A.
Full-length VirD2, and just a C-terminal fragment of the relaxase, mediate intercellular transfer of Cre when fused to the recombinase, as monitored by excision of a lox cassette in engineered plant cells using the Cre recombinase assay for translocation (CRAfT) (54, 55). Conversely, C-terminal-truncation mutations block relaxase-mediated T-DNA transfer to plant cells (53, 55). Recently, we further showed that, although VirD2 with its C-terminal 35 residues deleted (VirD2ΔC35) exhibits wild-type relaxase activity, the processed T-DNA substrate fails to interact productively with VirD4 (15). To examine whether VirD2's C terminus contributes to the VirD2-AADVirD4 interaction, we repeated the pulldown assays using a His6-VirD2ΔC35 variant. We were unable to detect an interaction between the ΔC35 relaxase mutant and GST-AADVirD4 (Fig. 5A), suggesting that VirD2's C terminus participates directly or indirectly in mediating the AADVirD4 interaction.
Taken together, the above-mentioned findings suggest that the AADs of PcfC and VirD4 participate in a network of interactions with DNA and relaxosome components and also might contribute to substrate discrimination through binding of cognate relaxases. To further test for a possible discrimination function, we assayed for binding of relaxases with the heterologous GST-AADPcfC or GST-AADVirD4 variant. Although His6-VirD2 bound GST-AADVirD4 (Fig. 5A), we were unable to detect binding with GST-AADPcfC (Fig. 5B). Similarly, His6-PcfF and PcfG-His6 bound GST-AADPcfC (Fig. 5C), but PcfG-His6 failed to bind GST-AADPcfC (Fig. 5D). The AADs of VirD4 and PcfC thus selectively bound only their cognate relaxases, and AADPcfC additionally bound the accessory factor PcfF.
VirD4 and PcfC soluble domains lacking the AADs exhibit nonspecific substrate binding.
Our data indicated that receptor AADs confer specificity for binding of DNA substrates through Dtr protein interactions but did not exclude the possibility that other T4CP domains, including the conserved NBDs, as well as the sequence-variable CTDs present in a large number of T4CPs (8), also contribute to the substrate docking reaction (Fig. 1). We tested the potential for soluble domains of VirD4 and PcfC lacking their AADs, designated NBD/CTDVirD4 and NBD/CTDPcfC, respectively, to bind substrates. In the in vivo genetic tests for negative dominance, neither NBD/CTDVirD4 nor NBD/CTDPcfC suppressed substrate transfer when produced in otherwise wild-type A. tumefaciens A348 or E. faecalis OG1RF(pCF10) strains, respectively (Fig. 6). These truncation mutants thus failed to sequester substrate or otherwise impair T4SS machine function, despite accumulating at levels comparable to or greater than those of the native T4CP in the merodiploid strains (Fig. 6).
FIG 6.
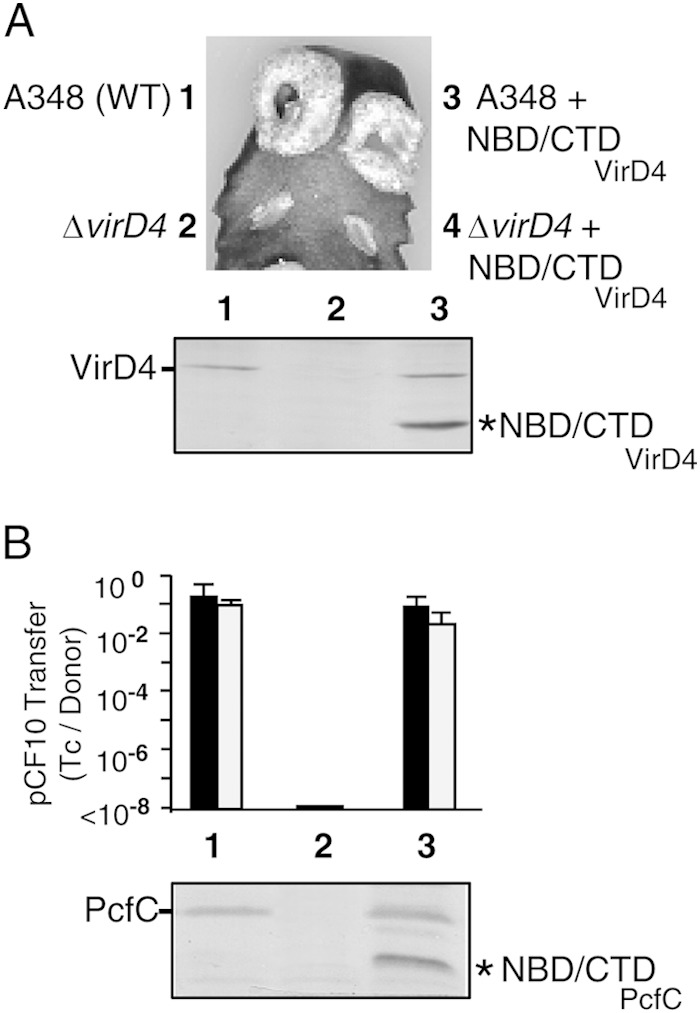
Effects of producing NBD/CTD truncation mutants on native VirD4 and PcfC function in vivo. (A) Virulence of A. tumefaciens strains producing VirD4 variants on K. daigremontiana. (Top) Leaf wound sites were inoculated with A. tumefaciens A348 producing native VirD4 (WT) or the ΔvirD4 mutant KA2000 (ΔvirD4) without or with the NBD/CTDVirD4 expression plasmid. (Bottom) Immunoblot showing steady-state levels of VirD4 (72 kDa) and NBD/CTDVirD4 (46 kDa) produced in the strains listed and detected by development with anti-VirD4 antibodies. Strains for virulence assays and immunoblot: 1, A348; 2, KA2000 (ΔvirD4); 3, A348(pPCB2024); 4, KA2000(pPCB2024). (B) (Top) Transfer frequencies of pCF10 from donors producing variant forms of PcfC in 2-h filter and liquid matings, presented as the number of transconjugants per donor cell (Tc/Donor), with average values and standard deviations shown. 1, OG1RF(pCF10); 2, OG1RF(pCF10ΔpcfC); 3, OG1RF(pCF10, pPC2107). (Bottom) Immunoblot showing steady-state levels of PcfC (67 kDa) and NBD/CTDPcfC (39 kDa) detected by development with anti-PcfC antibodies.
We were unable to enrich His6- or GST-tagged forms of NBD/CTDVirD4 or NBD/CTDPcfC due to aggregation, but MBP fusions were soluble in E. coli and amenable to enrichment (Fig. 7A). Interestingly, both MBP-tagged NBD/CTDs shifted all DNA substrates tested in the EMSAs, including the 39-mer nonspecific dsDNA and short ssDNA substrates corresponding to both strands of the pCF10 oriT or A. tumefaciens T-DNA region (Fig. 7B). In control experiments, enriched MBP (Fig. 7A) did not detectably bind these DNA substrates (Fig. 7B). Thus, the NBDs or CTDs of both T4CPs bind DNA without sequence or strand preference.
FIG 7.
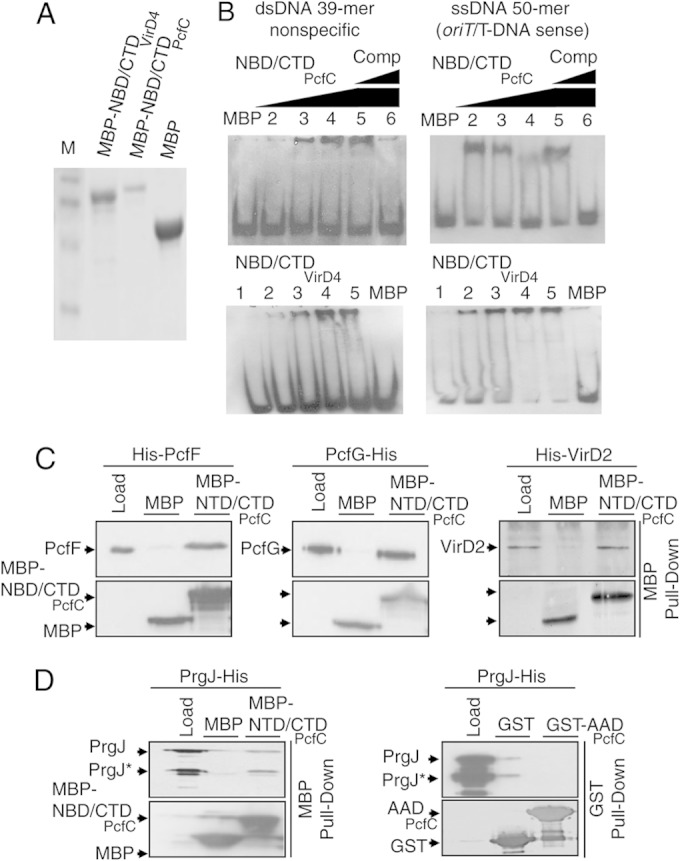
In vitro binding activities of NBD/CTDVirD4 and NBD/CTDPcfC. (A) Coomassie-stained protein gel showing enrichments of MPB-NBD/CTDVirD4 (82-kDa), MBP-NBD/CTDPcfC (88-kDa), and MBP alone (migrating at ∼48-kb) by amylose affinity chromatography. M, molecular size markers. (B) EMSAs showing binding of MBP-NBD/CTDPcfC (top) and MBP-NBD/CTDVirD4 (bottom) to the 3′-end-DIG-labeled nonspecific dsDNA 39-mer and the ssDNA 50-mer substrates encompassing the nicked strands of pCF10 oriT (for MBP-NBD/CTDPcfC) or T-DNA right-border repeat (for MBP-NBD/CTDVirD4). NBD/CTDPcfC EMSA lanes: DNA probe plus MBP or NBD/CTDPcfC (1) at 84 nM (2), 168 nM (3), 336 nM (4), or 504 nM (5 and 6). In lanes 5 and 6, the protein was preincubated with unlabeled 39-mer dsDNA competitor or 50-mer ssDNAs at concentrations 10- and 100-fold higher than that of the DIG-labeled substrate. NBD/CTDVirD4 EMSA lanes: (1) DNA probe alone or (2 to 5) plus NBD/CTDVirD4 at the same concentrations as the corresponding lanes of NBD/CTDPcfC EMSAs or (6) plus MBP. (C) MBP affinity pulldowns assaying for interactions of NBD/CTDPcfC with Dtr proteins. Enriched MBP-NBD/CTDPcfC or MBP alone (lanes labeled “load”) was bound to amylose beads, and the protein-bead complexes were mixed with enriched His6-PcfF, PcfG-His6, or His6-VirD2. Following extensive washes, the bound proteins were eluted from the beads with maltose, and proteins recovered in the eluates were identified by immunostaining with anti-MBP or anti-His antibodies (shown on the left of each gel). (D) MBP or GST affinity pulldowns assaying for interactions of PrgJ-His6 with MBP-NTD/CTDPcfC or GST-AADPcfC, as described for panel C and Fig. 5A, respectively.
We were unable to detect binding of the MBP-NBD/CTDVirD4 variant with cognate VirD1 or VirD2 or heterologous PcfF and PcfG (data not shown). In contrast, MBP-NBD/CTDPcfC bound not only the cognate Dtr proteins His6-PcfF and PcfG-His6 but also heterologous His6-VirD2 (Fig. 7C). MBP alone did not detectably bind these His-tagged Dtr proteins (Fig. 7C). Although MBP-NBD/CTDVirD4 bound DNA substrates, we cannot exclude the possibility that this variant fails to bind Dtr proteins because of a folding defect. Even so, our in vivo and in vitro findings together suggest that the AADs and not the NBDs or CTDs of VirD4 and PcfC confer DNA substrate recognition through selective binding of cognate Dtr proteins.
Finally, once PcfC binds the pCF10 substrate, the receptor delivers its cargo to the VirB4-like PrgJ ATPase, possibly for further processing prior to translocation (50). Soluble PcfCΔN103 binds PrgJ in vitro (50), prompting us to test whether AADPcfC or NBD/CTDPcfC participates in PrgJ binding. As shown in Fig. 7D, PrgJ-His6 coeluted with MBP-NBD/CTDPcfC but not MBP alone. In E. coli, PrgJ accumulates as both a full-length protein and a presumptive degradation product (50), and the NBD/CTDPcfC variant bound both forms of this ATPase. In contrast, in the corresponding GST pulldown assays, PrgJ-His6 did not form a detectable complex with GST-AADPcfC (Fig. 7D). These findings suggest that PcfC's NBD or CTD, or both domains, mediates receptor-PrgJ contacts necessary for the first step of substrate transfer through the Prg/Pcf T4SS.
DISCUSSION
In this study, we defined the contributions of the AADs of T4CPs from the A. tumefaciens VirB/VirD4 and E. faecalis Prg/Pcf systems to binding of DNA substrates. We presented evidence that the AADs of A. tumefaciens VirD4 (VirD4At) and E. faecalis pCF10-encoded PcfC (PcfCpCF10) participate in selection of T-DNA and pCF10 substrates, respectively, through binding of the associated Dtr processing proteins. Other domains of these T4CPs, including the NBDs and CTDs, also might participate in substrate binding but lack the discriminating function of the AAD for cognate DNA substrates. Furthermore, we identified differences between T4CP domain-Dtr protein contacts in the A. tumefaciens and E. faecalis transfer systems that might be explained by variations in early-stage substrate processing and recruitment reactions evolved by these two systems. Finally, our analyses of chimeric T4CPs suggested that MOBQ plasmids have evolved the capacity for promiscuous transfer through different T4SSs, at least in part by evolving a mechanism to bypass the T4CP AAD specificity checkpoint. We discuss our findings in the context of a new model (Fig. 8) depicting the AAD as a key regulatory domain required not only for docking of cognate substrates but also for substrate-driven channel activation.
FIG 8.

Model depicting proposed contributions of the AADs of VirD4 and PcfC to the substrate docking reaction. (Left) Schematic of the VirB/VirD4 T4SS spanning the Gram-negative A. tumefaciens cell envelope. The relaxosome assembles at the oriT-like nic site within the right T-DNA border sequence, and VirD2 relaxase nicks and remains covalently bound to the 5′ end of the T-strand. The spatial adaptors VirC1 and Vbp significantly enhance the efficiency of VirD2–T-strand docking with VirD4 (brown hexamer); adaptor interactions with VirD2 and VirD4 are not yet specified. VirD2 binds the AADVirD4 substrate specificity determinant by a mechanism dependent on the relaxase's C-terminal translocation signal. The docked substrate then transfers through the lumen of the VirD4 hexamer or, more likely, to one or more of the VirB4 or VirB11 ATPases for further processing and substrate transfer through a channel formed by the IMC. The promiscuous MOBQ plasmid RSF1010 bypasses the AADVirD4 substrate discrimination checkpoint by MobB-mediated spatial colocalization of the RSF1010 relaxosome with the VirB/VirD4 T4SS and one or more unspecified contacts between MobA's internal or C-terminal translocation signals and VirD4. (Right) Schematic of the Prg/Pcf T4SS spanning the Gram-positive E. faecalis cell envelope. The accessory factor PcfF recruits PcfG to the pCF10 oriT sequence, and the two Dtr factors mediate docking of the relaxosome through direct contacts with the AAD of PcfC. PcfG nicks the pCF10 substrate, remains covalently bound to the 5′ end of the transferred strand, and pilots the DNA substrate across the cytoplasmic membrane either via the lumen of the PcfC hexamer or through a translocation channel formed by the IMC.
The T4CP AAD is a substrate specificity checkpoint.
The notion that the AAD participates in DNA substrate binding originated from X-ray crystallography studies showing that the AAD of TrwBR388 is positioned at the cytoplasmic entrance of the lumen of the NBD hexamer and exhibits structural similarity to a DNA-binding domain of XerD recombinase (18). Mutational studies then supplied evidence for the functional importance of AADs associated with two T4CPs, TraGRP4 and TrwBR388. Specifically, insertions of a 31-residue peptide (i31) in the AAD of TraG abrogated RP4 plasmid transfer (56), and substitutions of AAD residues predicted by the X-ray structure to be surface exposed or to line the cytoplasmic entrance of the hexamer lumen similarly abolished TrwBR388 function (57).
We extended these earlier findings by showing that VirD4At and PcfCpCF10 with their AADs deleted are nonfunctional and production of these domains in trans in otherwise WT strains inhibited substrate transfer (Fig. 2). Importantly, negative dominance observed in the merodiploid strains was substrate specific, as evidenced by selective inhibition of pCF10 transfer by AADpCF10 in E. faecalis and of T-DNA transfer by AADVirD4 in A. tumefaciens. We confirmed the prediction (18) that T4CP AADs bind DNA (Fig. 4); however, binding lacked detectable oriT/T-DNA sequence or strand preference, arguing against the notion that these domains contribute to substrate selection solely through their DNA-binding activities (Fig. 4). Interestingly, an earlier study showed that the TrwBR388 receptor preferentially binds potential G-quadruplex (G4) DNA sequences and also that G4 DNA stimulates TrwBR388 ATPase activity to a greater extent than other DNA sequences (58, 59). These findings, coupled with the results of the present studies and additional observations discussed below, support a proposal that AAD binding of DNA is not required for substrate docking per se but nevertheless plays an important role in activation of the T4CP as a prerequisite for subsequent substrate transfer through the T4SS channel.
Both AADPcfC and AADVirD4 selectively bound the cognate Dtr proteins required for processing of pCF10 and T-DNA (Fig. 5). Structural resolution of the AAD-Dtr protein interface(s) requires further work; however, an accumulating body of evidence points to the importance of VirD2's C-terminal translocation signal for contact with VirD4 in the A. tumefaciens system. We (15, 25, 29) and others (55) have supplied evidence that VirD2's C terminus is dispensable for nicking at T-DNA border sequences but is required for docking of the T-DNA transfer intermediate with native VirD4 and for successful transfer of T-DNA to plants. Furthermore, we recently reported that VirD2-mediated docking of the T-DNA substrate with the VirD4 T4CP corresponds to one of two important intracellular signals required for activation of the VirB/VirD4 channel (15). The second is represented by the ATP hydrolysis activities of the VirD4 and VirB11 ATPases (60). Together, these intracellular signals induce a conformational change in the channel subunit VirB10, which is necessary for substrate passage through the VirB channel to the cell surface (15, 60). Importantly, a VirD2ΔC35 truncation mutant catalyzes formation of a VirD2ΔC35–T-strand complex, but this is a dead-end transfer intermediate, as shown by its failure to induce the VirB10 conformational switch (15). These observations, combined with our present finding that the VirD2ΔC35 mutant fails to bind AADVirD4 in vitro (Fig. 5), suggest that VirD2's C-terminal domain is essential not only for VirD2-AADVirD4 binding but also for coupling a substrate-binding signal to conformationally driven changes in the VirB/VirD4 T4SS required for channel activation and substrate passage (Fig. 8).
Distinguishing features of the DNA substrate docking reactions in the A. tumefaciens VirB/VirD4 and E. faecalis Pcf/Prg transfer systems.
Although the VirD2-AADVirD4 interaction is a proposed early step of T-DNA transfer, the A. tumefaciens VirB/VirD4 system has evolved additional mechanisms to optimize the efficiency of substrate engagement and delivery to plant cells. VirD2, for example, carries a second internal translocation signal between residues 205 and 264 that is important for efficient T-DNA transfer to plants (55). Further studies are needed to determine if or how this signal coordinates with the C-terminal domain to mediate substrate-VirD4 docking. Additionally, two accessory factors that enhance the efficiency of T-DNA transfer have been identified. One, the ParA/MinD-like ATPase VirC1, assembles as a component of the relaxosome and stimulates VirD2 nicking at T-DNA borders (33). VirC1 also spatially couples the VirD2–T-strand transfer intermediate with the VirB/VirD4 channel through a combination of partner interactions with VirD2 and VirD4 and an intrinsic affinity for the membrane at the cell poles (29). The second factor, termed VirD2-binding protein (Vbp), also promotes coupling of the VirD2–T-strand complex and VirD4 through binary contacts (34, 35).
The importance of these accessory factors for efficient T-DNA substrate docking potentially derives from an unusual feature of this “conjugation” system. In other well-characterized conjugation systems, such as those encoded by the E. coli F, RP4, and R388 plasmids, the early-stage processing reactions, including relaxosome assembly and oriT cleavage, occur concomitantly with binding of the relaxosome with the T4CP (4, 61). In striking contrast, in the A. tumefaciens system, processing of the T-DNA substrate is physically uncoupled from the docking and translocation reactions. Indeed, upon induction of the vir regulon encoding this transfer system with plant inducing signals, an individual A. tumefaciens cell can accumulate as many as 50 copies of the processed VirD2–T-strand complex over the ensuing postinduction period (15, 29). In nature, preloading of the T-DNA substrate in induced A. tumefaciens cells might have evolved as a mechanism to enhance the efficiency or amount of T-DNA substrate delivered once bacterial donors are able to form productive contacts with plant target cells. Accordingly, such a system would necessitate an efficient substrate recruitment system, and VirC1 and Vbp might have been coopted for this purpose (29, 34, 35).
At this time, the network of contacts formed between these adaptors and the VirD proteins for substrate-receptor docking is not completely defined. We were unable to detect VirD1 contacts with any of the VirD4 domains (Fig. 5 and 7), suggesting that this accessory factor does not contribute to the docking reaction or, alternatively, is dissociated from the transfer intermediate prior to substrate docking. We also have not been able to detect a direct interaction between VirD2 and VirD4 in A. tumefaciens cell extracts through standard pulldown assays (29, 34), in spite of our ability here to detect VirD2-AADVirD4 complex formation in vitro (Fig. 5). The VirC1 and Vbp adaptors might contribute not only to substrate recruitment but also to stabilization of the T-DNA substrate–VirD4 interaction in vivo.
In the E. faecalis pCF10 system, in contrast, several lines of evidence suggest that the relaxosome binds directly to the PcfC receptor for coupled substrate processing and translocation. This system transfers pCF10 to neighboring enterococci at extremely high frequencies in less than 15 to 30 min following exposure of pheromone sensing by donor cells. Indeed, nearly identical kinetics profiles have been reported for accumulation of prg and pcf transcripts and Prg and Pcf proteins as well for plasmid transfer, highly suggestive of spatiotemporal coupling of these reactions (30, 50, 62). Studies also have established that the Dtr proteins PcfF and PcfG both suffice for and impart specificity to the pCF10 processing and receptor-binding reactions (36). We extended these findings, first, by showing that the pCF10 DNA substrate binds the PcfC receptor only in cells competent for assembly of the catalytically competent relaxosome, that is, cells harboring a plasmid with an intact oriT sequence and producing both PcfF and PcfG (30). Furthermore, we showed that PcfF and PcfG bind a purified soluble form of PcfC (30). Finally, here we demonstrated binding of these Dtr proteins with the AADPcfC in vitro (Fig. 5). Although we also detected binding of both Dtr factors to the NBD/CTDpCF10 fragment, this fragment also bound nonspecifically to the VirD2 relaxase (Fig. 7), implicating the AAD and not the NBD or CTD of PcfC in substrate discrimination. Thus, we propose that upon binding of PcfF and PcfG at pCF10's oriT sequence, both Dtr factors form a ternary complex with the AADpCF10, thereby physically coupling the pCF10 relaxosome to the PcfC receptor. Further definition of these Dtr-receptor contacts is of special interest in view of the fact that the C termini of PcfF and PcfG lack clusters of positively charged or hydrophobic residues characteristically found in the C-terminal translocation signals of VirD2 and other T4SS substrates (54, 63).
Contributions of other T4CP domains to substrate binding.
As noted above, the NBD/CTD truncation mutants of both VirD4 and PcfC bound DNA nonspecifically, and NBD/CTDpCF10 also nonselectively bound Dtr proteins (Fig. 7). The biological relevance of these findings remains to be evaluated, but it should be noted that several studies have identified functions of the various T4CP domains that could contribute—directly or indirectly—to substrate binding. The NTDs, for example, are unlikely to directly bind substrates, but studies have identified several NTD functions, including binding interactions with other channel subunits, stimulatory effects on T4CP oligomerization and catalytic activity, and contributions to T4CP spatial positioning in the cell (17, 64–67), that could indirectly affect substrate docking. By virtue of their possible accessibility to secretion substrates in the cytoplasm, the NBDs and CTDs might coordinate with the AAD to recruit substrates through direct substrate engagement. In fact, the best-characterized substrate-T4CP contact to date is that between the CTD of TraD encoded by F-like plasmids and TraM, a component of the F relaxosome. Structural studies have determined that the last 7 amino acids (V712EPGDDF717) of TraD's CTD fit into a positively charged groove of TraM. Further mutational analyses of this partner interaction confirmed that the TraD CTD-TraM interaction is required for highly efficient F plasmid transfer and, conversely, is strongly inhibitory for F plasmid mobilization of MOBQ plasmids (68, 69). These findings warrant further studies exploring the importance of the CTDs of PcfCpCF10, VirD4At, and other T4CPs that carry such domains for possible DNA or effector protein contacts.
MOBQ plasmid mobilization bypasses the AAD specificity checkpoint.
Finally, our finding that the VirD4-AADPcfC chimera supported transfer of the MOBQ plasmid pML122 raises the intriguing question of how the MOBQ plasmid transfer intermediate bypasses the proposed AAD substrate specificity checkpoint. We envision two scenarios that are not mutually exclusive. First, MobA relaxases of MOBQ plasmids might carry distinct translocation signals capable of mediating relaxosome docking with heterologous AADs, including that of AADPcfC. Indeed, MobA has been shown to carry a negatively charged translocation motif reminiscent of that of VirD2 at its C terminus (54), as well as two large internal translocation signals, each capable of mediating substrate transfer (70). The internal signals are unrelated to each other, with the exception of a shared 7-residue motif (G[E/D]R[L/M]R[V/F]T) that, interestingly, is also present in internal translocation signals recently identified in the TraIR16 and TrwCR388 relaxases (71–73). Such signals might conform to a general motif that for MobA enables binding to heterologous AADs.
The second docking scenario derives from earlier work establishing that another MOBQ-encoded factor termed MobB plays an important role in recruitment of the MOBQ relaxosome to different T4SSs. Interestingly, MobB was also shown to be important for the functionality of both of MobA's internal translocation signals (70). MobB has an intrinsic affinity for the cytoplasmic membrane, which led to a proposal that MobB functions to colocalize the MOBQ plasmid relaxosome at the membrane in spatial juxtaposition with T4SSs. In this capacity, MobB would contribute to the stabilization of inherently weak MobA relaxase contacts with heterologous T4CPs (70). In the A. tumefaciens chimeric system, MobB might stabilize weak contacts between MobApML122 and the AADPcfC or mediate MOBQ relaxosome docking with the chimeric receptor completely independently of an AADPcfC contact (Fig. 8). Our finding that the VirD4ΔAAD mutant did not support pML122 transfer (Fig. 2) does not preclude the latter bypass mechanism, since, as noted above, the AAD likely contributes in ways other than binding of cognate Dtr factors to overall T4CP function.
ACKNOWLEDGMENTS
We thank members of the Christie laboratory for helpful discussions.
Studies in the Christie laboratory were supported by NIH R01GM48476 and R21AI105454.
REFERENCES
- 1.Christie PJ, Whitaker N, Gonzalez-Rivera C. 2014. Mechanism and structure of the bacterial type IV secretion systems. Biochim Biophys Acta 1843:1578–1591. doi: 10.1016/j.bbamcr.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhatty M, Laverde Gomez JA, Christie PJ. 2013. The expanding bacterial type IV secretion lexicon. Res Microbiol 164:620–639. doi: 10.1016/j.resmic.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goessweiner-Mohr N, Arends K, Keller W, Grohmann E. 2013. Conjugative type IV secretion systems in Gram-positive bacteria. Plasmid 70:289–302. doi: 10.1016/j.plasmid.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zechner EL, Lang S, Schildbach JF. 2012. Assembly and mechanisms of bacterial type IV secretion machines. Philos Trans R Soc Lond B Biol Sci 367:1073–1087. doi: 10.1098/rstb.2011.0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cabezon E, Ripoll-Rozada J, Pena A, de la Cruz F, Arechaga I. 2015. Towards an integrated model of bacterial conjugation. FEMS Microbiol Rev 39:81–95. doi: 10.1111/1574-6976.12085. [DOI] [PubMed] [Google Scholar]
- 6.Cascales E, Christie PJ. 2003. The versatile bacterial type IV secretion systems. Nat Rev Microbiol 1:137–150. doi: 10.1038/nrmicro753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Voth DE, Broederdorf LJ, Graham JG. 2012. Bacterial type IV secretion systems: versatile virulence machines. Future Microbiol 7:241–257. doi: 10.2217/fmb.11.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alvarez-Martinez CE, Christie PJ. 2009. Biological diversity of prokaryotic type IV secretion systems. Microbiol Mol Biol Rev 73:775–808. doi: 10.1128/MMBR.00023-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trokter M, Felisberto-Rodriguez C, Christie PJ, Waksman G. 2014. Recent advances in structural and molecular biology of type IV secretion systems. Curr Opin Struct Biol 27:16–23. doi: 10.1016/j.sbi.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fronzes R, Schafer E, Wang L, Saibil HR, Orlova EV, Waksman G. 2009. Structure of a type IV secretion system core complex. Science 323:266–268. doi: 10.1126/science.1166101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chandran V, Fronzes R, Duquerroy S, Cronin N, Navaza J, Waksman G. 2009. Structure of the outer membrane complex of a type IV secretion system. Nature 462:1011–1015. doi: 10.1038/nature08588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fronzes R, Christie PJ, Waksman G. 2009. The structural biology of type IV secretion systems. Nat Rev Microbiol 7:703–714. doi: 10.1038/nrmicro2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Low HH, Gubellinin F, Rivera-Calzada A, Connery S, Dujeancourt A, Lu F, Fronzes R, Orlova EV, Waksman G. 2014. Structure of a type IV secretion system. Nature 508:550–553. doi: 10.1038/nature13081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Llosa M, Zunzunegui S, de la Cruz F. 2003. Conjugative coupling proteins interact with cognate and heterologous VirB10-like proteins while exhibiting specificity for cognate relaxosomes. Proc Natl Acad Sci U S A 100:10465–10470. doi: 10.1073/pnas.1830264100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cascales E, Atmakuri K, Sarkar MK, Christie PJ. 2013. DNA substrate-induced activation of the Agrobacterium VirB/VirD4 type IV secretion system. J Bacteriol 195:2691–2704. doi: 10.1128/JB.00114-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garza I, Christie PJ. 2013. A putative transmembrane leucine zipper of Agrobacterium VirB10 is essential for T-pilus biogenesis but not type IV secretion. J Bacteriol 195:3022–3034. doi: 10.1128/JB.00287-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Segura RL, Aguila-Arcos S, Ugarte-Uribe B, Vecino AJ, de la Cruz F, Goni FM, Alkorta I. 2013. The transmembrane domain of the T4SS coupling protein TrwB and its role in protein-protein interactions. Biochim Biophys Acta 1828:2015–2025. doi: 10.1016/j.bbamem.2013.05.022. [DOI] [PubMed] [Google Scholar]
- 18.Gomis-Ruth FX, Moncalian G, Perez-Luque R, Gonzalez A, Cabezon E, de la Cruz F, Coll M. 2001. The bacterial conjugation protein TrwB resembles ring helicases and F1-ATPase. Nature 409:637–641. doi: 10.1038/35054586. [DOI] [PubMed] [Google Scholar]
- 19.Hormaeche I, Alkorta I, Moro F, Valpuesta JM, Goni FM, De La Cruz F. 2002. Purification and properties of TrwB, a hexameric, ATP-binding integral membrane protein essential for R388 plasmid conjugation. J Biol Chem 277:46456–46462. doi: 10.1074/jbc.M207250200. [DOI] [PubMed] [Google Scholar]
- 20.Gomis-Ruth FX, de la Cruz F, Coll M. 2002. Structure and role of coupling proteins in conjugal DNA transfer. Res Microbiol 153:199–204. doi: 10.1016/S0923-2508(02)01313-X. [DOI] [PubMed] [Google Scholar]
- 21.Gomis-Ruth FX, Sola M, de la Cruz F, Coll M. 2004. Coupling factors in macromolecular type-IV secretion machineries. Curr Pharm Des 10:1551–1565. doi: 10.2174/1381612043384817. [DOI] [PubMed] [Google Scholar]
- 22.Cao Y, Hallet B, Sherratt DJ, Hayes F. 1997. Structure-function correlations in the XerD site-specific recombinase revealed by pentapeptide scanning mutagenesis. J Mol Biol 274:39–53. doi: 10.1006/jmbi.1997.1380. [DOI] [PubMed] [Google Scholar]
- 23.Ferreira H, Sherratt D, Arciszewska L. 2001. Switching catalytic activity in the XerCD site-specific recombination machine. J Mol Biol 312:45–57. doi: 10.1006/jmbi.2001.4940. [DOI] [PubMed] [Google Scholar]
- 24.Llosa M, Gomis-Ruth FX, Coll M, Cruz FDL. 2002. Bacterial conjugation: a two-step mechanism for DNA transport. Mol Microbiol 45:1–8. doi: 10.1046/j.1365-2958.2002.03014.x. [DOI] [PubMed] [Google Scholar]
- 25.Cascales E, Christie PJ. 2004. Definition of a bacterial type IV secretion pathway for a DNA substrate. Science 304:1170–1173. doi: 10.1126/science.1095211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Atmakuri K, Cascales E, Christie PJ. 2004. Energetic components VirD4, VirB11 and VirB4 mediate early DNA transfer reactions required for bacterial type IV secretion. Mol Microbiol 54:1199–1211. doi: 10.1111/j.1365-2958.2004.04345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schroder G, Krause S, Zechner EL, Traxler B, Yeo HJ, Lurz R, Waksman G, Lanka E. 2002. TraG-like proteins of DNA transfer systems and of the Helicobacter pylori type IV secretion system: inner membrane gate for exported substrates? J Bacteriol 184:2767–2779. doi: 10.1128/JB.184.10.2767-2779.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Christie PJ, Atmakuri K, Krishnamoorthy V, Jakubowski S, Cascales E. 2005. Biogenesis, architecture, and function of bacterial type IV secretion systems. Annu Rev Microbiol 59:451–485. doi: 10.1146/annurev.micro.58.030603.123630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Atmakuri K, Cascales E, Burton OT, Banta LM, Christie PJ. 2007. Agrobacterium ParA/MinD-like VirC1 spatially coordinates early conjugative DNA transfer reactions. EMBO J 26:2540–2551. doi: 10.1038/sj.emboj.7601696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y, Zhang X, Manias D, Yeo HJ, Dunny GM, Christie PJ. 2008. Enterococcus faecalis PcfC, a spatially localized substrate receptor for type IV secretion of the pCF10 transfer intermediate. J Bacteriol 190:3632–3645. doi: 10.1128/JB.01999-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yanofsky MF, Porter SG, Young C, Albright LM, Gordon MP, Nester EW. 1986. The virD operon of Agrobacterium tumefaciens encodes a site-specific endonuclease. Cell 47:471–477. doi: 10.1016/0092-8674(86)90604-5. [DOI] [PubMed] [Google Scholar]
- 32.Albright LM, Yanofsky MF, Leroux B, Ma DQ, Nester EW. 1987. Processing of the T-DNA of Agrobacterium tumefaciens generates border nicks and linear, single-stranded T-DNA. J Bacteriol 169:1046–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toro N, Datta A, Carmi OA, Young C, Prusti RK, Nester EW. 1989. The Agrobacterium tumefaciens virC1 gene product binds to overdrive, a T-DNA transfer enhancer. J Bacteriol 171:6845–6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo M, Jin S, Sun D, Hew CL, Pan SQ. 2007. Recruitment of conjugative DNA transfer substrate to Agrobacterium type IV secretion apparatus. Proc Natl Acad Sci U S A 104:20019–20024. doi: 10.1073/pnas.0701738104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Padavannil A, Jobichen C, Qinghua Y, Seetharaman J, Velazquez-Campoy A, Yang L, Pan SQ, Sivaraman J. 2014. Dimerization of VirD2 binding protein is essential for Agrobacterium induced tumor formation in plants. PLoS Pathog 10:e1003948. doi: 10.1371/journal.ppat.1003948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Y, Staddon JH, Dunny GM. 2007. Specificity determinants of conjugative DNA processing in the Enterococcus faecalis plasmid pCF10 and the Lactococcus lactis plasmid pRS01. Mol Microbiol 63:1549–1564. doi: 10.1111/j.1365-2958.2007.05610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sciaky D, Montoya AL, Chilton MD. 1978. Fingerprints of Agrobacterium Ti plasmids. Plasmid 1:238–253. doi: 10.1016/0147-619X(78)90042-2. [DOI] [PubMed] [Google Scholar]
- 38.Zhu J, Oger PM, Schrammeijer B, Hooykaas PJ, Farrand SK, Winans SC. 2000. The bases of crown gall tumorigenesis. J Bacteriol 182:3885–3895. doi: 10.1128/JB.182.14.3885-3895.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Larebeke N, Genetello C, Schell J, Schilperoort RA, Hermans AK, Van Montagu M, Hernalsteens JP. 1975. Acquisition of tumour-inducing ability by non-oncogenic agrobacteria as a result of plasmid transfer. Nature 255:742–743. doi: 10.1038/255742a0. [DOI] [PubMed] [Google Scholar]
- 40.Ooms G, Hooykaas PJJ, Van Veen RJM, Van Beelen P, Regensburg-Tunik R, Schilperoort RA. 1982. Octopine Ti-plasmid deletion mutants of Agrobacterium tumefaciens with emphasis on the right side of the T-region. Plasmid 7:15–19. doi: 10.1016/0147-619X(82)90023-3. [DOI] [PubMed] [Google Scholar]
- 41.Fullner KJ. 1998. Role of Agrobacterium virB genes in transfer of T complexes and RSF1010. J Bacteriol 180:430–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou X-R, Christie PJ. 1999. Mutagenesis of Agrobacterium VirE2 single-stranded DNA-binding protein identifies regions required for self-association and interaction with VirE1 and a permissive site for hybrid protein construction. J Bacteriol 181:4342–4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dunny GM, Brown BL, Clewell DB. 1978. Induced cell aggregation and mating in Streptococcus faecalis: evidence for a bacterial sex pheromone. Proc Natl Acad Sci U S A 75:3479–3483. doi: 10.1073/pnas.75.7.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Staddon JH, Bryan EM, Manias DA, Chen Y, Dunny GM. 2006. Genetic characterization of the conjugative DNA processing system of enterococcal plasmid pCF10. Plasmid 56:102–111. doi: 10.1016/j.plasmid.2006.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jakubowski SJ, Krishnamoorthy V, Christie PJ. 2003. Agrobacterium tumefaciens VirB6 protein participates in formation of VirB7 and VirB9 complexes required for type IV secretion. J Bacteriol 185:2867–2878. doi: 10.1128/JB.185.9.2867-2878.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kovach ME, Phillips RW, Elzer PH, Roop RM II, Peterson KM. 1994. pBBR1MCS: a broad-host-range cloning vector. Biotechniques 16:800–802. [PubMed] [Google Scholar]
- 47.Bryan EM, Bae T, Kleerebezem M, Dunny GM. 2000. Improved vectors for nisin-controlled expression in gram-positive bacteria. Plasmid 44:183–190. doi: 10.1006/plas.2000.1484. [DOI] [PubMed] [Google Scholar]
- 48.Berger BR, Christie PJ. 1994. Genetic complementation analysis of the Agrobacterium tumefaciens virB operon: virB2 through virB11 are essential virulence genes. J Bacteriol 176:3646–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Otten L, De Greve H, Leemans J, Hain R, Hooykaas P, Schell J. 1984. Restoration of virulence of vir region mutants of Agrobacterium tumefaciens strain B6S3 by coinfection with normal and mutant Agrobacterium strains. Mol Gen Genet 195:159–163. doi: 10.1007/BF00332739. [DOI] [Google Scholar]
- 50.Li F, Alvarez-Martinez C, Chen Y, Choi KJ, Yeo HJ, Christie PJ. 2012. Enterococcus faecalis PrgJ, a VirB4-like ATPase, mediates pCF10 conjugative transfer through substrate binding. J Bacteriol 194:4041–4051. doi: 10.1128/JB.00648-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 52.Gelvin SB. 2012. Traversing the cell: Agrobacterium T-DNA's journey to the host genome. Front Plant Sci 3:52. doi: 10.3389/fpls.2012.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Simone M, McCullen CA, Stahl LE, Binns AN. 2001. The carboxy-terminus of VirE2 from Agrobacterium tumefaciens is required for its transport to host cells by the virB-encoded type IV transport system. Mol Microbiol 41:1283–1293. doi: 10.1046/j.1365-2958.2001.02582.x. [DOI] [PubMed] [Google Scholar]
- 54.Vergunst AC, van Lier MC, den Dulk-Ras A, Grosse Stuve TA, Ouwehand A, Hooykaas PJ. 2005. Positive charge is an important feature of the C-terminal transport signal of the VirB/D4-translocated proteins of Agrobacterium. Proc Natl Acad Sci U S A 102:832–837. doi: 10.1073/pnas.0406241102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Kregten M, Lindhout BI, Hooykaas PJ, van der Zaal BJ. 2009. Agrobacterium-mediated T-DNA transfer and integration by minimal VirD2 consisting of the relaxase domain and a type IV secretion system translocation signal. MPMI 22:1356–1365. doi: 10.1094/MPMI-22-11-1356. [DOI] [PubMed] [Google Scholar]
- 56.Lee MH, Kosuk N, Bailey J, Traxler B, Manoil C. 1999. Analysis of F factor TraD membrane topology by use of gene fusions and trypsin-sensitive insertions. J Bacteriol 181:6108–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Paz HD, Larrea D, Zunzunegui S, Dehio C, de la Cruz F, Llosa M. 2010. Functional dissection of the conjugative coupling protein TrwB. J Bacteriol 192:2655–2669. doi: 10.1128/JB.01692-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tato I, Zunzunegui S, de la Cruz F, Cabezon E. 2005. TrwB, the coupling protein involved in DNA transport during bacterial conjugation, is a DNA dependent ATPase. Proc Natl Acad Sci U S A 102:8156–8161. doi: 10.1073/pnas.0503402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matilla I, Alfonso C, Rivas G, Bolt EL, de la Cruz F, Cabezon E. 2010. The conjugative DNA translocase TrwB is a structure-specific DNA-binding protein. J Biol Chem 285:17537–17544. doi: 10.1074/jbc.M109.084137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cascales E, Christie PJ. 2004. Agrobacterium VirB10, an ATP energy sensor required for type IV secretion. Proc Natl Acad Sci U S A 101:17228–17233. doi: 10.1073/pnas.0405843101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de la Cruz F, Frost LS, Meyer RJ, Zechner EL. 2010. Conjugative DNA metabolism in Gram-negative bacteria. FEMS Microbiol Rev 34:18–40. doi: 10.1111/j.1574-6976.2009.00195.x. [DOI] [PubMed] [Google Scholar]
- 62.Hirt H, Manias DA, Bryan EM, Klein JR, Marklund JK, Staddon JH, Paustian ML, Kapur V, Dunny GM. 2005. Characterization of the pheromone response of the Enterococcus faecalis conjugative plasmid pCF10: complete sequence and comparative analysis of the transcriptional and phenotypic responses of pCF10-containing cells to pheromone induction. J Bacteriol 187:1044–1054. doi: 10.1128/JB.187.3.1044-1054.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nagai H, Cambronne ED, Kagan JC, Amor JC, Kahn RA, Roy CR. 2005. A C-terminal translocation signal required for Dot/Icm-dependent delivery of the Legionella RalF protein to host cells. Proc Natl Acad Sci U S A 102:826–831. doi: 10.1073/pnas.0406239101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Atmakuri K, Ding Z, Christie PJ. 2003. VirE2, a type IV secretion substrate, interacts with the VirD4 transfer protein at cell poles of Agrobacterium tumefaciens. Mol Microbiol 49:1699–1713. doi: 10.1046/j.1365-2958.2003.03669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hormaeche I, Iloro I, Arrondo JL, Goni FM, de la Cruz F, Alkorta I. 2004. Role of the transmembrane domain in the stability of TrwB, an integral protein involved in bacterial conjugation. J Biol Chem 279:10955–10961. doi: 10.1074/jbc.M310422200. [DOI] [PubMed] [Google Scholar]
- 66.Hormaeche I, Segura RL, Vecino AJ, Goni FM, de la Cruz F, Alkorta I. 2006. The transmembrane domain provides nucleotide binding specificity to the bacterial conjugation protein TrwB. FEBS Lett 580:3075–3082. doi: 10.1016/j.febslet.2006.04.059. [DOI] [PubMed] [Google Scholar]
- 67.Segura RL, Aguila-Arcos S, Ugarte-Uribe B, Vecino AJ, de la Cruz F, Goni FM, Alkorta I. 2014. Subcellular location of the coupling protein TrwB and the role of its transmembrane domain. Biochim Biophys Acta 1838:223–230. doi: 10.1016/j.bbamem.2013.08.016. [DOI] [PubMed] [Google Scholar]
- 68.Wong JJ, Lu J, Edwards RA, Frost LS, Glover JN. 2011. Structural basis of cooperative DNA recognition by the plasmid conjugation factor, TraM. Nucleic Acids Res 39:6775–6788. doi: 10.1093/nar/gkr296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wong JJ, Lu J, Glover JN. 2012. Relaxosome function and conjugation regulation in F-like plasmids: a structural biology perspective. Mol Microbiol 85:602–617. doi: 10.1111/j.1365-2958.2012.08131.x. [DOI] [PubMed] [Google Scholar]
- 70.Parker C, Meyer RJ. 2007. The R1162 relaxase/primase contains two type IV transport signals that require the small plasmid protein MobB. Mol Microbiol 66:252–261. doi: 10.1111/j.1365-2958.2007.05925.x. [DOI] [PubMed] [Google Scholar]
- 71.Lang S, Gruber K, Mihajlovic S, Arnold R, Gruber CJ, Steinlechner S, Jehl MA, Rattei T, Frohlich KU, Zechner EL. 2010. Molecular recognition determinants for type IV secretion of diverse families of conjugative relaxases. Mol Microbiol 78:1539–1555. doi: 10.1111/j.1365-2958.2010.07423.x. [DOI] [PubMed] [Google Scholar]
- 72.Alperi A, Larrea D, Fernandez-Gonzalez E, Dehio C, Zechner EL, Llosa M. 2013. A translocation motif in relaxase TrwC specifically affects recruitment by its conjugative type IV secretion system. J Bacteriol 195:4999–5006. doi: 10.1128/JB.00367-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Redzej A, Ilangovan A, Lang S, Gruber CJ, Topf M, Zangger K, Zechner EL, Waksman G. 2013. Structure of a translocation signal domain mediating conjugative transfer by type IV secretion systems. Mol Microbiol 89:324–333. doi: 10.1111/mmi.12275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rashkova S, Spudich GM, Christie PJ. 1997. Characterization of membrane and protein interaction determinants of the Agrobacterium tumefaciens VirB11 ATPase. J Bacteriol 179:583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burgess RR, Arthur TM, Pietz BC. 2000. Mapping protein-protein interaction domains using ordered fragment ladder far-western analysis of hexahistidine-tagged fusion proteins. Methods Enzymol 328:141–157. doi: 10.1016/S0076-6879(00)28396-1. [DOI] [PubMed] [Google Scholar]
- 76.Stahl LE, Jacobs A, Binns AN. 1998. The conjugal intermediate of plasmid RSF1010 inhibits Agrobacterium tumefaciens virulence and VirB-dependent export of VirE2. J Bacteriol 180:3933–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhou XR, Christie PJ. 1997. Suppression of mutant phenotypes of the Agrobacterium tumefaciens VirB11 ATPase by overproduction of VirB proteins. J Bacteriol 179:5835–5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kristich CJ, Manias DA, Dunny GM. 2005. Development of a method for markerless genetic exchange in Enterococcus faecalis and its use in construction of a srtA mutant. Appl Environ Microbiol 71:5837–5849. doi: 10.1128/AEM.71.10.5837-5849.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]