

ABSTRACT
Histone methyltransferase inhibitors (HMTis) and histone deacetylase inhibitors (HDACis) are reported to synergistically induce the expression of latent human immunodeficiency virus type 1 (HIV-1), but studies have largely been performed with cell lines. As specific and potent HMTis directed at EZH1 (enhancer of zeste 2 Polycomb repressive complex 2 subunit 1)/EZH2 are now in human testing, we wished to rigorously test such an inhibitor in a primary resting T-cell model of HIV latency. We found that GSK343, a potent and selective EZH2/EZH1 inhibitor, reduced trimethylation of histone 3 at lysine 27 (H3K27) of the HIV provirus in resting cells. Remarkably, this epigenetic change was not associated with increased proviral expression in latently infected resting cells. However, following the reduction in H3K27 at the HIV long terminal repeat (LTR), subsequent exposure to the HDACi suberoylanilide hydroxamic acid or vorinostat (VOR) resulted in increases in HIV gag RNA and HIV p24 antigen production that were up to 2.5-fold greater than those induced by VOR alone. Therefore, in primary resting CD4+ T cells, true mechanistic synergy in the reversal of HIV latency may be achieved by the combination of HMTis and HDACis. Although other cellular effects of EZH2 inhibition may contribute to the sensitization of the HIV LTR to subsequent exposure to VOR, and to increase viral antigen production, this synergistic effect is directly associated with H3K27 demethylation at nucleosome 1 (Nuc-1). Based upon our findings, the combination of HMTis and HDACis should be considered for testing in animal models or clinical trials.
IMPORTANCE Demethylation of H3K27 mediated by the histone methyltransferase inhibitor GSK343 in primary resting T cells is slow, occurring over 96 h, but by itself does not result in a significant upregulation of cell-associated HIV RNA expression or viral antigen production. However, following H3K27 demethylation, latent viral expression within infected primary resting CD4+ T cells is synergistically increased upon exposure to the histone deacetylase inhibitor vorinostat. Demethylation at H3K27 sensitizes the HIV promoter to the effects of an HDACi and provides a proof-of-concept for the testing of combination epigenetic approaches to disrupt latent HIV infection, a necessary step toward the eradication of HIV infection.
INTRODUCTION
Current antiretroviral therapy (ART) potently suppresses human immunodeficiency virus type 1 (HIV-1) replication; however, HIV infection persists (1, 2). The primary reservoir of persistent infection is a small pool of latently infected resting central memory CD4+ T cells (3–5). This reservoir must be targeted in any attempt to eradicate HIV-1 infection. Latent HIV infection is defined as dormant, nonreplicating virus that can be recovered following activation of the latently infected cell. However, a recent study found that a single exposure to latency-reversing agents, even a broad mitogenic signal, does not reverse latency universally throughout a population of infected cells, thus suggesting the need for serial and/or combination latency reversal strategies (6). As the toxicities of global T-cell activation may be clinically unacceptable (7), careful targeting of cellular pathways must be developed to effectively disrupt HIV latency within safe and tolerable clinical protocols.
The chromatin environment is critical for the establishment and maintenance of latent HIV infection (8–10). Epigenetic modifications such as histone methylation and deacetylation appear to play a role in maintaining HIV latency. Latent HIV-1 proviruses display a defined nucleosomal structure featuring a key nucleosome near the transcription start site (11, 12), with local occupancy of histone deacetylases (HDACs), deacetylated histones (13–15), and methylated histones (16–20). HDAC inhibitors (HDACis) have been extensively studied for their potential to reactivate latent virus in cultured-cell models of latency, ex vivo in resting CD4+ T cells from patients, and in vivo in patients with ART-suppressed viremia (21–32). However, in some model systems and assays, HDAC inhibition has been claimed to be insufficiently effective to reverse HIV latency in a clinically meaningful way, and the use of combination latency reversal strategies has been proposed (33–35). Currently, it is unclear which model system(s) will accurately predict the efficacy and safety of combination therapies in vivo.
PRC1 and PRC2, the Polycomb repressor group (PcG) protein complexes, and heterochromatin protein 1 (HP1) constitute the major histone methyltransferase (HMT) complexes that silence chromatin (36). Histone methylation at the lysine tails of histones H3 and H4 can have diverse effects, but generally, methylation of histones contributes to gene silencing, while demethylation allows gene expression. PcG complexes contribute to the conditional silencing at many cellular promoters (37). The trimethylation of histone H3 at lysine 27 is a silencing mark propagated by the histone methyltransferase EZH2 (enhancer of zeste 2 Polycomb repressive complex 2 subunit 2), which is a component of the multisubunit Polycomb repressive complex PRC2. EZH2 inhibition can reactivate the latent HIV long terminal repeat (LTR) in latent T-cell lines (18). The small-molecule inhibitor GSK343 is a highly specific EZH2/EZH1 inhibitor (38) and effectively treats B-cell lymphomas in murine models (39).
The use of a combination of drugs targeting different epigenetic targets, HMTs and HDACs, has been proposed as a strategy for latency reversal (35). We demonstrate for the first time in a primary resting T-cell model of HIV latency that pretreatment with the EZH2 inhibitor (EZH2i) GSK343 at levels sufficient to induce demethylation of H3K27 at the critical HIV LTR nucleosome 1 (Nuc-1) site is associated directly with sensitization of the HIV LTR to subsequent pharmacological exposures to the HDACi vorinostat (VOR) and leads to enhanced proviral expression and latency reversal beyond that achieved by VOR alone. This is the first demonstration of an effective, clinically feasible approach for the use of combination epigenetic therapies to more effectively disrupt latent HIV infection.
MATERIALS AND METHODS
Cell Lines, PBMCs, and tissue culture reagents.
The latently infected cell lines 2D10 and J-89 were maintained in RPMI medium with l-glutamine (Gibco, Life Technologies), 10% fetal bovine serum (FBS), penicillin (100 IU/ml), and streptomycin (100 μg/ml) in an incubator at 37°C with 5% CO2. Peripheral blood mononuclear cells (PBMCs) were isolated from the buffy coat of healthy donors by using Ficoll-Hypaque (GE Healthcare, USA) gradient centrifugation. After washing, the cells were plated in RPMI complete medium supplemented with human interleukin-2 (IL-2) (Peprotech, USA) at 10 U/ml and incubated in a humidified incubator at 37°C with 5% CO2. Written informed consent was obtained from all participants before study enrollment. Approval was obtained from the University of North Carolina (UNC) institutional biomedical review board and the Food and Drug Administration.
Reagents used for proviral activation.
GSK343 was a gift from P. Trojer (Constellation Pharmaceuticals, Cambridge, MA). UNC 1999 was a gift from J. Jian (Icahn School of Medicine at Mount Sinai, New York, NY). VOR was a gift from Merck, phytohemagglutinin (PHA) was obtained from Remel (Thermo Scientific, Waltham, MA), and JQ1 was a gift from J. Bradner (Dana-Farber Cancer Institute, Harvard University).
Establishment of a primary cell model of HIV latency.
Primary cell experiments were performed as described previously by Saleh et al. (40), with minor modifications. Experiments were repeated for confirmation at least three times, using primary cells from the same donor. Briefly, frozen PBMCs from a single healthy donor were thawed and kept overnight in RPMI medium with l-glutamine, 10% FBS, penicillin (100 IU/ml), streptomycin (100 μg/ml), and 10 U IL-2 in 5% CO2 at 37°C. They were then used to purify naive CD4+ T cells by using naive CD4+ T-cell isolation kit II (Miltenyi Biotech) according to the manufacturer's protocol. After purification, cells were maintained in medium supplemented with 50 nM CCL19 (chemokine [C-C motif] ligand 19) for 2 days. These cells were then washed and infected with NL4-3 virus (100 ng p24 viral equivalents per million cells) by spinoculation for 2 h at 300 × g at 37°C. After infection, cells were washed three times, resuspended in complete medium with 10 U IL-2, and cultured for 4 days.
On day 4, infected cells were either treated with 2 μM GSK343 or left untreated for a further 96 h (to day 8). On day 8, equal numbers of EZH2i-treated and untreated T cells were pulsed with 500 nM VOR for 24 h. As a positive comparative control, other cells were treated with 2 μg/ml PHA for 24 h, and at days 9 and 12 after infection, viral expression and release were assessed by PCR of cell-associated RNA and a p24 enzyme-linked immunosorbent assay (ELISA) of the culture supernatant. Latently infected cells were harvested for chromatin immunoprecipitation (ChIP) analysis on day 4 prior to GSK343 treatment and on day 8 after GSK343 treatment but prior to VOR pulsing. Supernatants were collected on days 0, 4, 8, 9, and 12 and stored at −80°C until p24 ELISAs (ABL Inc., Rockville, MD) were performed. The efficiency of infection varied from 3 to 10% in these experiments, and the cells were noncycling throughout the experiment, as observed by carboxyfluorescein succinimidyl ester (CFSE) labeling in the presence of 1 U/ml or 10 U/ml of IL-2 (data not shown).
ChIP.
A ChIP assay kit (catalog number 17-295; Millipore) was used, according to the manufacturer's protocol. To obtain sheared chromatin with DNA of 200 to 500 bp in length, extracted chromatin from 1 × 106 cells per ChIP was sonicated by using a Diagenode Bioruptor standard sonicator with a 1.5-ml tube holder at a medium setting for 24 min (30 s on and 15 s off). ChIP was performed as described above, with the following modifications. Sonicated cell lysates were centrifuged, and 7 to 20 μg of soluble chromatin was incubated with 4 to 5 μg antibody overnight. The following antibodies were used: anti-histone H3, anti-H3K27me3 (trimethylated histone 3 at lysine 27), anti-RNA polymerase II (Pol II), and anti-hemagglutinin (HA) (Abcam); anti-EZH2, anti-H3K27me2, anti-EED (embryonic ectoderm development gene), anti-RING1B, anti-BMI1, anti-H3K27ac, and anti-phospho-Ser2 Pol II (Active Motif); anti-H3K9me3 and anti-H3Ac (Millipore); and anti-HA, anti-rabbit, and anti-mouse immunoglobulin G (Cell Signaling). A quantitative real-time PCR assay of the products of ChIP was performed to verify the significance of changes in the occupancy of the specific proteins at the HIV-1 LTR with the use of Nuc-0, Nuc-1, Gag, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers, as reported previously (18). The sequences of primers are mentioned in Table S1 in the supplemental material. The percentage of input HIV LTR DNA recovered was determined by comparing the cycle threshold values of each reaction to a standard curve generated from input DNA. The fold change in occupancy of each protein at the LTR relative to the untreated control was calculated after subtracting background immunoprecipitation measured with nonspecific IgG or with an unrelated HA antibody.
Immunoblotting.
A total of 1.5 × 106 2D10 cells, J-89 cells, or PBMCs in 4 ml of RPMI medium were either untreated or treated with 0.25 μM, 0.5 μM, 1 μM, and 2 μM GSK343 for 96 h or kept with dimethyl sulfoxide (DMSO) as a vehicle control. Cells were washed twice with phosphate-buffered saline (PBS) and lysed in 200 μl of radioimmunoprecipitation assay (RIPA) buffer (20 mM Tris-HCl [pH 8.1], 1% Triton X-100, 2 mM EDTA, 500 mM NaCl) with freshly added 1% protease inhibitor cocktail and phosphatase inhibitor cocktail. Cells were incubated on ice for 10 min with intermittent vortexing. DNA was removed by centrifugation at 13,000 rpm for 10 min at 4°C. Protein was quantified by using the Bradford assay (41), and 20 to 25 μg of total protein was analyzed by SDS-PAGE. The following antibodies were used: primary anti-rabbit histone H3 and anti-rabbit H3K27me3 (Cell Signaling), anti-mouse alpha-tubulin (Abcam), and secondary anti-mouse-HRP (Sigma) and anti-rabbit-HRP (Santa Cruz Biotechnology). These antibodies were used at the specified dilutions, according to the manufacturers' instructions, with 5% milk or 5% bovine serum albumin (BSA) in 1× Tris-buffered saline (TBS) containing 0.1% Tween 20.
Cell-associated viral RNA (green fluorescent protein [GFP] mRNA or Gag RNA measurements).
2D10 cells, J-89 cells, or cells from the primary cell model were left untreated or treated with the selected drug concentrations mentioned below. Cells were washed, snap-frozen in an ethanol dry ice bath, and stored at −80°C until use. Cells were thawed on ice, and RNA was isolated by using the RNeasy RNA isolation kit (Qiagen, Valencia, CA) according to the manufacturer's protocol. DNA was removed from RNA extracts by DNase digestion (Promega, Madison, WI), and cDNA was synthesized by using the SuperScript III first-strand synthesis kit for reverse transcription-PCR (RT-PCR) (Invitrogen). Quantitative PCR was performed on cDNA with a Bio-Rad CFX96 or CFX384 system using QuantiTect Multiplex PCR Mastermix (Qiagen). The sequences of primers and probes are mentioned in Table S1 in the supplemental material. The relative mRNA expression level was calculated by using the 2−ΔΔCT method. The data shown are the means from at least three independent experiments, and the error bars represent the standard errors of the means.
MTT cell proliferation and viability assay.
To measure the proliferation and viability of 2D10 cells, J-89 cells, PBMCs, or primary resting T cells in the presence of compounds, cells were assayed by using the Vybrant 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) cell proliferation assay kit according to the manufacturer's instructions (Life Technologies). The percent viability over time was calculated relative to untreated samples.
shRNA constructs and infections.
Glycerol stocks carrying short hairpin RNA (shRNA) inserted into the pLKO.1 backbone (vector) (catalog number RHS4080; EZH2 clone TRCN0000040074) were obtained from Open Biosystems. Silenced 2D10 cell populations carrying HIV-1 proviruses encoding a d2EGFP (enhanced green fluorescent protein destabilized by residues 422 to 461 of mouse ornithine decarboxylase, giving an in vivo half-life of ∼2 h) marker (i.e., <5% of cells in the unstimulated cell population had detectable d2EGFP expression) were either superinfected with lentiviral vectors expressing either control or EZH2 shRNAs or transfected with either control scrambled shRNA or EZH2 shRNA constructs by using the Mirus Jurkat Trans-IT transfection reagent according to the manufacturer's instructions. One million 2D10 cells were infected with vesicular stomatitis virus glycoprotein (VSV-G)-pseudotyped lentiviral vectors expressing shRNAs. Cell viability and d2EGFP expression were assessed via fluorescence-activated cell sorting (FACS).
Statistical analysis.
Microsoft Excel or GraphPad Prism software was used to analyze the data. The Student paired t test was used for data analysis in pairs. In some experiments, one-way analysis of variance (ANOVA) was performed for multiple data comparisons. A P value of <0.05 was considered statistically significant.
RESULTS
PRC1 and PRC2 occupy the HIV promoter in 2D10 cells.
The Polycomb repressive complexes PRC1 and PRC2 act to compact chromatin but contain distinct components (37). The histone methyltransferase EZH2 is the catalytic component of the PRC2 silencing complex. We sought to document the presence of PRC2 in the 2D10 cell line. This clonal model of HIV latency is derived from Jurkat CD4+ T cells harboring a single copy of the integrated proviral genome with highly restricted expression, containing deletions in the HIV gag region and pol region and a mutation (H13L) in the tat gene encoding a short-lived green fluorescent protein (d2EGFP) reporter protein in place of the Nef gene (42). ChIP assays were performed on 2D10 cell extracts to measure the occupancy of key components of Polycomb repressive complexes at the HIV-1 LTR. EZH2 and trimethylated histone 3 at lysine 27 (H3K27me3) were found at the HIV Nuc-1 region, as previously shown in a related model (18), and occupancy of the PRC2 core component EED was observed. Furthermore, RING1B and BMI1, two core components of PRC1, were also detectable at the HIV-1 promoter (Fig. 1).
FIG 1.
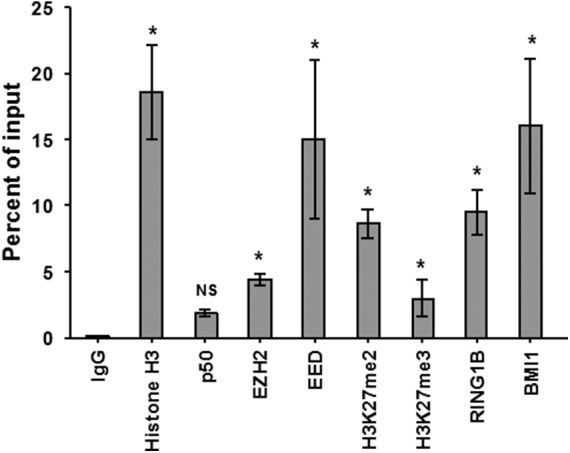
Components of Polycomb repressive complexes 1 and 2 occupy the HIV promoter in 2D10 cells. As expected, histone H3 occupancy was detected at the HIV LTR Nuc-1 region, but occupancy of the p50 homodimer of NF-κB was low or absent. Occupancy of the PRC2 components EZH2 and EED, occupancy of the PRC1 components RING1 and BMI1, H3K27me2, and H3K27me3 were detected; Results were compared with results for control IgG by using Student paired t tests. Error bars represent standard errors of the means (n = 3). *, P < 0.05; NS, not significant (P > 0.05).
The loss of the H3K27me3 mark induced by exposure to GSK343 has a negligible effect on proviral expression.
EZH2 is an HMT specifically involved in the di- and trimethylation of histone H3 at lysine 27. To assess the kinetic effects of the EZH2i GSK343 on the trimethylation of H3K27, 2D10 cells were treated with 0.25 to 2.0 μM GSK343 for up to 96 h. No overt toxic effects were observed, and viability, as measured by an MTT assay, was unaffected (see Fig. S2A in the supplemental material). Total cellular trimethylated H3K27 was reduced in a time- and dose-dependent manner, first discernible at 48 h (not shown) and maximally reduced at 96 h (Fig. 2A), without a change in the quantity of total histone H3. More specifically, in concert with this decrease, the measured occupancy of H3K27me3 at the HIV-1 LTR Nuc-1 site was not detectably reduced at 48 h (not shown) but was markedly reduced at 96 h (Fig. 2B). The effect was specific, as the H3K9me3 mark at the LTR remained unchanged (Fig. 2B). Furthermore, H3K27me2, EED, and EZH2 occupancy at the HIV LTR, determined by ChIP, and their total protein levels, detected by immunoblotting, were unchanged (data not shown). In parallel with H3K27me3 reduction, a modest increase in acetylated histone H3 and an appreciable increase in acetylation at the specific lysine H3K27 were also seen (Fig. 2C). Moreover, the cellular promoters for the heterochromatic gene SAT2 and the euchromatic gene MyoD also displayed reductions in the H3K27me3 mark, confirming a global cellular effect of GSK343 (Fig. 2D). To confirm that these epigenetic effects were not unique to the viral integrant found in 2D10 cells, the effects of GSK343 were tested in J-89 cells, a cell line containing full-length provirus with a gfp reporter gene followed by the env gene (43), where a similar specific loss of total cellular H3K27me3 was seen, as was reduced occupancy of this mark at the Nuc-1 region of the HIV LTR (see Fig. S1A and S1B in the supplemental material).
FIG 2.

The EZH2 inhibitor GSK343 reduces trimethylated H3K27 in 2D10 cells but has a negligible effect on proviral expression. (A) Western blot showing global reduction in H3K27me3 protein levels in whole-cell lysate after cells were treated with different concentrations of GSK343 for 96 h. The reported 50% inhibitory concentration of GSK343 in the cancer cell lines tested was <0.2 μM (38), so a range of concentrations of GSK343 of 0.25 to 2.0 μM, starting close to its 50% inhibitory concentration, was used to determine the drug effect. Histone H3 and α-tubulin are shown as loading controls. (V, vehicle control [0.02% DMSO]). (B) ChIP at the HIV LTR Nuc-1 promoter for H3K27me3 and H3K9me3. Hemagglutinin antibody (HA) is an unrelated isotype control. (C) Corresponding changes in total H3 acetylation and in acetyl H3K27. ChIP panels were normalized to H3 levels. The H3 level did not significantly change between the control and treated conditions. Results were compared with those for cells treated with the vehicle control DMSO by using Student paired t tests. Error bars represent standard errors of the means (n = 3). • or *, P < 0.05. (D) ChIP changes in H3K27me3 at the cellular promoters SAT2 (heterochromatic gene) and MyoD (euchromatic gene) in cells treated with 0.5 μM GSK343 for 96 h. *, P < 0.05, from 3 pairs of assays (determined by a Student paired t test). Results were compared to those of the respective DMSO controls (V, vehicle control). (E) Relative fold changes of cell-associated GFP RNA expression upon GSK343 exposure, normalized to GAPDH, as measured by quantitative RT-PCR. (E) HIV LTR-driven GFP expression in 2D10 cells, as measured by flow cytometry over 96 h. Treatment with VOR (500 nM) for 24 h served as the positive control. All the data shown represent means, and error bars represent standard deviations of the means, from three independent experiments.
Critically, despite the reduction in trimethylation at the HIV LTR, we observed negligible increases in LTR-driven RNA production and GFP expression levels following GSK343 treatment at concentrations of up to 2 μM for up to 96 h (Fig. 2E and F). This is of mechanistic interest, as in concert with reduced trimethylation, increased acetylation at the same residue was seen (Fig. 2D). It is therefore even more striking that no increase in LTR expression was seen, given the induction of expression seen in concert with histone acetylation when induced by HDAC inhibitors (Fig. 2F). A similar lack of an effect on LTR-driven gag RNA or GFP production was also seen in the J-89 model system (see Fig. S1C and S1D in the supplemental material). Moreover, another selective EZH2 inhibitor, UNC 1999 (44), had similar effects on proviral expression (data not shown).
EZH2 knockdown reduces H3K27me3 and PRC1 and PRC2 component occupancy without an increase in provirus expression.
To examine the impact of EZH2 inhibition by using an alternative approach, expression of EZH2 was inhibited by EZH2-specific shRNAs (Fig. 3A). EZH2 gene silencing was ca. 70% effective in 2D10 cells compared to the control scrambled shRNA. To directly address the role of EZH2 knockdown in HIV LTR expression, and to avoid secondary effects, we examined early time points (48 h) after knockdown but were unable to detect proviral induction in EZH2-silenced cells (Fig. 3B). However, we observed decreased occupancy of key components of both Polycomb repressive complexes, PRC1 and PRC2 (Fig. 3C and D). In contrast to EZH2 chemical inhibition, silencing of EZH2 reduced both the H3K27me2 and H3K27me3 marks at the LTR (Fig. 3C), but similar to the effects of GSK343, H3K27 demethylation did not by itself induce HIV LTR expression.
FIG 3.

EZH2 knockdown decreases H3K27me3 and PRC1 and PRC2 component occupancy in 2D10 cells without an increase in proviral expression. 2D10 cells were treated with either scrambled or EZH2 shRNAs for 48 h according to the Mirus Jurkat Trans-IT transfection reagent protocol. (A) Representative Western blot showing the effect of silencing of EZH2 on both EZH2 and H3K27me3. The relative quantification of each of the bands is indicated at the top of each blot. Histone H3 and α-tubulin are shown as loading controls. (B) Silencing of EZH2 does not lead to an increase in proviral gene expression, as measured by flow cytometry. (C) ChIP showing that the occupancy of the PRC2 components EZH2 and EED and H3K27 di- and trimethylation are reduced selectively at HIV LTR Nuc-1 by shRNA inhibition of EZH2. (D) ChIP showing that the RING1B and BMI1 components of PRC1 are also reduced at HIV LTR Nuc-1. H3 and H3K9me3 were used, as a control ChIP showed that silencing of EZH2 has no effect on the occupancies of total histone H3 or on the nonselective H3K9me3 mark. Sc, scrambled shRNA; Sh, EZH2 shRNA. ChIP was performed with the antibodies indicated at the top of each graph. Error bars represent standard errors of the means from three independent experiments. *, P < 0.05; NS, not significant (P > 0.05) (determined by the Student paired t test).
H3K27 demethylation of the HIV LTR induced by GSK343 increases the response of the proviral promoter to the HDAC inhibitor VOR.
HDACis and HMT inhibitors (HMTis) have been reported to synergistically enhance the expression of the latent HIV genome, although most of these studies were limited by the use of transformed cell line model systems and nonselective inhibitors (16, 18, 19, 45). As we found little effect on latent proviral expression following demethylation induced by exposure to GSK343 (Fig. 2), we then asked if the phenotype of the LTR had been altered, specifically if the HIV LTR was more sensitive to HDAC inhibition following H3K27 demethylation.
Following the reduction of H3K27me3 induced by 96 h of exposure to GSK343, we assessed the effect of VOR. We observed a dose-dependent proviral response following increasing exposures up to 500 nM VOR (Fig. 4A). Demethylation resulted in a >3-fold increase in LTR-driven GFP expression induced by VOR. However, GSK343 treatment for <72 h did not result in an increased response to subsequent VOR exposure (data not shown). Of note, although there was evidence of increased expression in the presence of VOR at each GSK343 concentration, there was not an augmented effect of VOR with exposure to the lowest concentration tested (250 nM). Furthermore, the effect of VOR was not enhanced by the simultaneous addition of GSK343 (data not shown). In addition, exposure to GSK343 for >96 h did not further increase the LTR response to subsequent VOR exposure (data not shown).
FIG 4.

H3K27 demethylation of the HIV LTR induced by GSK343 increases the response of the proviral promoter to the HDAC inhibitor VOR. (A) In 2D10 cells, following 96 h of preexposure to 0 to 2.0 μM GSK343, a dose-dependent increase in proviral activation following treatment with >350 nM VOR for 24 h is seen if cells are pretreated with >0.25 μM GSK343 (V, equivalent volume of vehicle [0.02% DMSO]; UT, untreated). Means and standard errors of the means were obtained from three independent experiments. *, P < 0.05 for samples treated with VOR and with VOR plus GSK343 (cells treated with 2.0 μM); •, P < 0.05 for samples treated with 2.0 μM GSK343 and combined drug treatment; NS, not significant (P > 0.05) (determined by a Student paired t test). (B) Enhanced proviral expression in cells treated with 2 μM GSK343 for 96 h (day 4), followed by 24 h of exposure to 500 nM VOR. The samples were assayed by flow cytometry on day 5. The inset shows a FACS overlay, using Flowjo X software, at day 5. Results were compared to those of samples treated with VOR only, and a Student paired t test was done for three pairs of assays. *, P < 0.05. (C) Similar effects were observed when GFP RNA was measured by real-time PCR. Significant increases following treatment with GSK343 and VOR compared to treatment with VOR alone were determined by using Student paired t tests. Error bars represent standard errors of the means (n = 3). *, P < 0.05. (D) Effects of inhibitor exposure, as described above for panel B, were determined by ChIP at the HIV LTR for H3Ac, H3K27me3, and Pol II and phospho-Pol II occupancy, using the respective antibodies at the positions and with the primers indicated. The line diagram at the bottom represents the 5′ HIV LTR and the relative nucleosome positions and primer positions. At Nuc-0, Nuc-1, and the downstream region, the chromatin marks H3K27me3 and H3Ac and the occupancy of the RNA Pol II and phosphorylated RNA Pol II (marker of processive Pol II) were measured. At Nuc-0, GSK343 treatment reduced H3K27me3 and VOR increased H3Ac levels, while the occupancy of Pol II and pPol II remained unchanged. At Nuc-1, which is close to the transcription start site, similar observations were made, but the Pol II occupancy was increased, while the pPol II occupancy was reduced by combined treatment with GSK343 and VOR. At the Gag gene site, in the absence of nearby nucleosomal histones, no effect on acetyl and methyl marks was observed, but pPol II occupancy increased more by treatment with the combination of GSK343 and VOR than by treatment with VOR alone, indicative of an augmentation of processive transcription. In panels B to D, error bars represent standard errors of the means from three independent experiments; for each chromatin mark/occupancy described and to compare the ChIP data obtained with the combined and individual drug treatments, we performed ANOVA of the 4 different treatments (DMSO, GSK343, VOR, and GSK343 plus VOR). All differences noted were significant; ⧫, P < 0.001. ChIP panels were normalized to H3 levels. The H3 level did not significantly change between the control and treated conditions.
However, exposure to GSK343 for 96 h is sufficient to sensitize the HIV LTR to a subsequent pulse with VOR. Using these optimized conditions, we confirmed that HIV RNA induction correlated with GFP induction (Fig. 4B and C). To further assess the effect of HMTi and HDACi on chromatin marks, RNA Pol II occupancy, and Pol II processivity (46), ChIP was performed (Fig. 4D). At both Nuc-1, the nucleosome adjacent to the transcriptional start site, and the upstream Nuc-0, treatment with GSK343 led to reduced H3K27me3 levels, and treatment with VOR alone led to increased H3Ac levels, which was further enhanced by combined treatment with GSK343. At Nuc-1, RNA Pol II occupancy increased after VOR exposure with or without GSK343. Consistent with increased HIV LTR transcription, the processive form of phosphorylated Pol II (pPol II) exhibited decreased occupancy at Nuc-1, while increased occupancy was observed (+611 Gag) after VOR exposure. This occupancy within the gene body was significantly enhanced following combined exposure to GSK343 and VOR. Of note, the level of H3K27me3 remained low under GSK343 treatment conditions even when VOR was added.
In order to test the hypothesis that GSK343 sensitization of the HIV LTR promoter is a general phenomenon, we tested JQ1, a small-molecule inhibitor of the bromodomain (BRD) and extra-C-terminal domain (BET) proteins that can alter epigenetic regulation. First, the minimum JQ1 exposure (50 nM) needed to activate HIV LTR-mediated GFP expression was established (Fig. 5A). As described above, a reduction of total trimethylated histone H3K27 was observed upon GSK343 treatment (Fig. 5B). When 2D10 cells were exposed to 2.0 μM GSK343 for 96 h and then treated with 50 nM JQ1 for a further 24 h, we observed enhanced proviral induction, as measured by GFP expression (Fig. 5C), and cell-associated HIV RNA (Fig. 5D) levels above those seen following exposure to only JQ1. To further assess the effect of the HMTi and the BET inhibitor (BETi) on chromatin, RNA Pol II occupancy, and Pol II processivity, ChIP was performed (Fig. 5E). At Nuc-0 and Nuc-1, GSK343 treatment significantly reduced H3K27me3 and modestly increased H3Ac in combination with JQ1. While the occupancy of Pol II and pPol II was unchanged at Nuc-0, their occupancy at Nuc-1 was increased by JQ1 alone or by combined exposure to GSK343 and JQ1. Again, the increase in LTR-mediated transcription was associated with increased pPol II occupancy at the downstream Gag region following JQ1 treatment, which was further enhanced upon combined drug treatment, indicative of processive transcription.
FIG 5.

H3K27 demethylation of the HIV LTR induced by GSK343 also increases the response of the proviral promoter to the BETi JQ1 in 2D10 cells. (A) Dose response of 2D10 cells to JQ1 at 10 to 1,000 nM, as measured by GFP production. (B) Representative Western blot showing global reduction in H3K27me3 in whole-cell lysates. Cells were assayed at day 0 (0 h) or were incubated for 5 days (120 h) with either the vehicle control DMSO or 2 μM GSK343 for 96 h. In selected cultures, 50 nM JQ1 was added for the final 24 h of culture. Histone H3 and α-tubulin were used as loading controls. (C) Enhanced proviral expression in cells treated with 2 μM GSK343 for 96 h (day 4), followed by 24 h of exposure to 50 nM JQ1. The Student paired t test was done for three pairs of assays. *, P < 0.05. (D) Similar effects were observed when GFP RNA was measured by real-time PCR. Relative fold values of JQ1 versus JQ1 and GSK343 were compared by using the Student paired t test. Error bars represent standard errors of the means (n = 3). *, P < 0.05. (E) The indicated treatment conditions were also evaluated by ChIP at the HIV LTR for H3Ac, H3K27me3, Pol II, and phospho-Pol II occupancy using the respective antibodies at the positions and with the primers indicated in Materials and Methods. The line diagram at the bottom represents the 5′ HIV LTR and the relative nucleosome positions and primer positions used for ChIP assays. At Nuc-0, Nuc-1, and the downstream region, the chromatin marks H3K27me3 and H3Ac and the occupancy of RNA Pol II and phospho-Ser RNA Pol II (marker of processive transcription or active Pol II) were measured. At Nuc-0, GSK343 treatment reduced H3K27me3 and modestly increased H3Ac when combined with JQ1, while the occupancy of Pol II and pPol II remained unchanged. At Nuc-1, which is close to the transcription start site, similar observations were made, but the Pol II and pPol II occupancy increased with combined treatments with GSK343 and JQ1. At the downstream Gag gene site, in the absence of nearby histones, no effect on acetyl and methyl marks was observed, but higher pPol II occupancy than that of Pol II was seen, indicative of processive transcription. Error bars represent standard errors of the means from three independent experiments. For each chromatin mark/occupancy described and to compare the ChIP data obtained with the combined and individual drug treatments, we performed ANOVA of the 4 different treatments (DMSO, GSK343, JQ1, and GSK343 plus JQ1). All differences noted were significant; ⧫, P < 0.001. ChIP panels were normalized to H3 levels. The H3 level did not significantly change between the control and treated conditions.
Of note, the recruitment of EZH2 and the level of H3K9me3 at the different HIV-1 regions (Nuc-0, Nuc-1, and Gag) were also determined by ChIP in 2D10 cells and were not significantly changed (not shown).
Occupancy of PRC components and activity of GSK343 at the HIV LTR in primary resting cells.
To extend these studies to more relevant primary cell models of latent HIV infection, we established HIV latency in primary CD4+ T cells using two previously reported protocols (40, 47). Components of PRC1 (RING1 and BMI1) and PRC2 (EZH2 and EED) and the silencing mark H3K27me3 were found at the HIV Nuc-1 region in both the Lewin CCL19 model (Fig. 6A) and the Bosque-Planelles model (not shown).
FIG 6.

Components of the Polycomb repressive complex occupy the HIV promoter in a latently infected primary T-cell model and validation of EZH2 inhibition by GSK343 in uninfected PBMCs. PRC1 (RING1B and BMI1) and PRC2 (EZH2 and EED) components occupy the HIV-1 promoter in latently infected primary T cells. (A) NL4-3-infected primary T cells at day 4 and day 8 postinfection (40), as described in the legend of Fig. 7A. ChIPs for total histone H3, H3K27me3, p50, H3K27me2, and mock IgG are also shown. Means and standard errors of the means from three independent experiments are displayed. For each chromatin occupancy described, the Student paired t test was done for 3 pairs of assays. *, P < 0.05 for mock IgG-treated versus untreated cells at day 4; •, P < 0.05 for mock IgG-treated versus untreated cells at day 8; NS, not significant (P > 0.05). (B) Representative blot showing a reduction in H3K27me3 in total uninfected PBMCs at 2 μM GSK343 for 96 h. Histone H3 and α-tubulin are shown as loading controls. (V, vehicle control [0.02% DMSO]). (C) ChIP changes in the samples described in panel B. Occupancies of H3K27me3 and control IgG were measured at the host gene promoters SAT2 (heterochromatic gene) and MyoD (euchromatic region gene). Results were compared with those for DMSO-treated cells by using Student paired t tests. Error bars represent standard errors of the means (n = 3). *, P < 0.05.
We then validated the effect of 2 μM GSK343 in primary uninfected PBMCs treated for 96 h and confirmed decreases in total H3K27me3 protein (Fig. 6B) and in promoter occupancy of H3K27me3 at the cellular MyoD and SAT2 promoters (Fig. 6C). Again, no effect of GSK343 on cell viability was observed (see Fig. S2B in the supplemental material). These findings validated the use of noncycling, primary cell models to study the combined effect of HDACi and HMTi on HIV latency.
HIV LTR demethylation mediated by an EZH2 inhibitor prior to exposure to the HDAC inhibitor VOR is associated with enhanced proviral expression in ex vivo cultures of resting CD4+ T cells.
We studied the combined effects of GSK343 and VOR using the model described previously by Saleh et al. (40), which was demonstrated to be responsive to VOR. The responses of this primary cell model to a variety of inducing stimuli also resemble those of resting CD4+ T cells obtained from HIV-infected patients (48). The design of this experiment, modified slightly from that described by Saleh et al. (40), is shown in Fig. 7A. Proviral quiescence was established 4 days after infection, as illustrated by the >10-fold reductions of viral p24 levels in culture supernatants and of cell-associated gag RNA levels (Fig. 7B and F).
FIG 7.

HIV LTR demethylation mediated by an EZH2 inhibitor prior to exposure to the HDAC inhibitor VOR is associated with enhanced proviral expression in ex vivo cultures of resting CD4+ T cells. (A) Schema for establishment of the primary cell model of HIV latency. Briefly, resting naive CD4+ cells were purified by negative selection using magnetically activated cell sorting (MACS) from the total PBMCs of the healthy donors. These resting cells were treated with 50 nM CCL19 in the presence of 10 U IL-2 for 2 days. The cells were infected with HIV-1 NL4-3 at day 0, and latency was established at day 4 postinfection. A total of 2 μM GSK343 was added at day 4 after latent infection was established. On day 8, GSK343-treated resting T cells were either exposed to 500 nM VOR or left untreated for a further 24 h. Also, day 8 untreated latently infected cells were treated with 2 μg/ml PHA (positive control) for 24 h. The effects were measured on days 9 and 12. Samples were washed on day 9 and kept in complete medium with 10 U IL-2/ml until the time of harvest at day 12. (B to E) Graphs showing viral p24 production in supernatants measured by a p24 ELISA at the times indicated. Means and standard errors of the means are from three independent experiments compared to the untreated control conditions; p24 values were normalized to 1 for the untreated control. UT, infected but untreated; PRE, before washing to remove the viral inoculum; POST, after washing to remove the viral inoculum; V, VOR; 343, GSK343. D0 refers to day 0 postinfection. The Student paired t test was done for paired samples as indicated, and values are from three experiments (n = 3). • or *, P < 0.05. (F) Enhanced viral production was also measured as cell-associated viral gag RNA fold changes compared to untreated infected samples for each time point group (days 0, 4, 8, 9, and 12) by using real-time PCR. The Student paired t test was done for the paired samples as indicated, and values from three experiments are shown (n = 3). • or *, P < 0.05. (G) ChIP assay of the cellular promoter MyoD and viral promoter HIV LTR Nuc-1 was performed for mock (IgG) and the H3K27me3, H3K9me3, and H3Ac chromatin marks for untreated and GSK343-treated samples on days 4 and 8. ChIP panels were normalized to H3 levels. The H3 level did not significantly change between the control and treated conditions. Results were compared with those for untreated day 4 chromatin by using Student paired t tests. Error bars represent standard errors of the means (n = 3). *, P < 0.05.
At day 4, selected cultures were then exposed to 2 μM GSK343 for 96 h. ChIP assays with day 8 cell extracts demonstrated a reduction of H3K27me3 at the HIV LTR Nuc-1 region and the host MyoD promoter in treated cultures (Fig. 7G). Again, this effect was specific, as H3K9me3 was unchanged (Fig. 7G) and, as in 2D10 cells, an increase in H3 acetylation was observed.
Following the exposure of selected cultures to GSK343 and the validation of demethylation in treated cultures, cells were either cultured under standard conditions, with 500 nM VOR, or with PHA as a control to induce maximal proviral expression. Notably, 24 h of exposure to 500 nM VOR induced up to 2.5-fold more HIV p24 antigen production in cells that had been pretreated with GSK343 (Fig. 7B to E). This increase was evident at day 9 (Fig. 7D), following 24 h of exposure to VOR, and although VOR was removed after day 9, increased antigen production was even more apparent at day 12 (Fig. 7E). Consistent with the increased p24 release into the tissue culture supernatant, an average 2.5-fold induction of cell-associated HIV gag RNA was also measured in cultures treated with GSK343 and VOR (Fig. 7F). GSK343 and/or VOR exposures had no significant effect on cell viability, as measured by an MTT assay (see Fig. S2C in the supplemental material). In a parallel experiment, cells were exposed to a lower concentration of VOR (335 nM), and as expected, less p24 production was induced, although pretreatment with GSK343 still increased proviral induction (data not shown). Of note, the effect of VOR was not enhanced by the simultaneous addition of GSK343 (data not shown).
DISCUSSION
Strahl and Allis postulated that covalent histone modifications form a “histone code” read by effector proteins to bring about up- or downregulation of gene expression (49). The array of molecular mechanisms that first allow the establishment of latent, persistent proviral infection within resting CD4+ T cells and then later allow this transcriptionally quiescent state to be maintained for months or years in HIV-infected individuals is incompletely understood. However, it is clear that epigenetic modifications of the HIV LTR, most notably histone acetylation and methylation, contribute to the maintenance of the latent proviral state (2).
While DNA methylation does not appear to play a critical role in HIV silencing (50), acting by promoting histone methylation, Suv39H1 (17), G9a (19), and EZH2 (18) have been reported to participate in the maintenance of HIV latency. This activity of these enzymes was further validated by using pharmacological inhibitors such as BIX01294 (19), chaetocin (16, 45), and 3-deazaneplanocin A (DZNep) (18), alone or in combination with HDACis (16, 18, 19, 45, 51). However, these studies have serious limitations, as they were performed in either cycling, transformed T-cell line models of HIV latency (16, 18, 19, 45) or bulk PBMCs and resting CD4+ T cells isolated from HIV-positive (HIV+) aviremic patients (45), and the inhibitors used in these studies were either not highly selective or used at concentrations that allowed nonselective effects.
Following the demonstration that HDACis could disrupt HIV latency, the combined use of inhibitors of histone methylation and histone deacetylation to synergistically reactivate the expression of quiescent provirus within latently infected resting CD4+ T cells was considered (45). Although plausible, it remained unclear how the modulation of an epigenetic mark at the same histone residue would necessarily augment proviral expression beyond that achieved by maximal inhibition of deacetylation or methylation.
Therefore, before such a combination epigenetic approach, with the attendant risks of increased off-target toxic effects, is to be tested in animal models or human studies, evidence of the efficacy of combined therapy should be obtained with the most advanced and relevant experimental systems. Therefore, we attempted to address these limitations by (i) establishing exposure to an EZH2i that allows H3K27 demethylation but does not by itself result in HIV RNA expression; (ii) using the EZH2/EZH1 inhibitor GSK343, which is selective in primary cell systems (39); and (iii) performing studies using resting primary T-cell models of latency.
First, to validate the study of an EZH2 inhibitor, we established the presence of PRC2 and EZH2 itself in the 2D10 cell line. Although not unexpected, we also detected components of PRC1 occupying the HIV LTR in the model systems that we studied. Although further study of the activity and regulation of PRC1 at the HIV LTR is needed, taken together, the results of EZH2 inhibitor (unpublished data) and knockdown experiments suggest that PRC1 occupancy may be dependent on PRC2 occupancy and/or EZH2 activity.
To facilitate studies with the more demanding primary cell systems, we then established the appropriate exposure conditions to study the effect of the EZH2 inhibitor GSK343. We found that the action of GSK343 to remove the H3K27me3 mark at the critical HIV LTR Nuc-1 site and at two host gene loci was gradual, requiring 96 h of inhibitor exposure to markedly reduce the levels of trimethylation. Demethylation was selective, as there was no significant change in H3K9 trimethylation. While demethylation modestly increased the levels of acetylated H3 at the HIV Nuc-1 region at higher GSK343 concentrations over 96 h, it is critical to note that no significant increase in HIV LTR expression, as measured by both HIV RNA transcripts and the production of the GFP marker protein, was detected.
Small-molecule inhibitor studies were corroborated by experiments using shRNA inhibition of EZH2, similarly showing demethylation and reduced occupancy of components of both PRC2 and PRC1. Of note, previous work found that short hairpin RNA knockdown of EZH2 in 2D10 and E4 cell lines (18) resulted in the activation of LTR expression. The studies presented here utilized transient knockdown for only 48 h, measuring a more immediate effect of the loss of EZH2 activity.
We then tested the ability of GSK343, which did not significantly increase HIV LTR expression by itself, to sensitize the viral promoter to the inducing effect of the HDAC inhibitor VOR. The demonstration of the added value of such a combined effect is at the heart of the concept of combinations of “latency-reversing agents.” Of interest, minimal exposure to VOR (250 nM) had a modest effect (Fig. 4A) on LTR expression, which was unaffected by demethylation induced by GSK343. However, demethylation markedly sensitized the HIV LTR to exposures to higher concentrations of VOR, with a >3-fold increase of expression over that induced by VOR alone (8% to 26% GFP expression at 500 nM) (Fig. 4A). ChIP assays (Fig. 4D) documented that demethylation was induced by GSK343 at both Nuc-1, near the LTR transcriptional start site, and Nuc-0, in the enhancer region. Furthermore, the exposure to VOR resulted in acetylation at these sites, but acetylation was augmented if preceded by demethylation by GSK343. Of note, simultaneous exposure to VOR and GSK343 did not enhance the effect of VOR on LTR expression. While demethylation alone did not increase Pol II occupancy at Nuc-0, polymerase occupancy was increased by VOR, with or without demethylation. Pol II processivity, as marked by decreased occupancy of phosphorylated Pol II and increased occupancy in the downstream gene body, was increased by VOR and further augmented by demethylation.
A variety of agents are under study for their potential to effectively and safely reverse HIV latency. Among them, BET inhibitors have been suggested to have latency-reversing effects in some model systems, although they do not appear to allow the recovery of latent provirus from resting CD4+ T cells of aviremic patients as efficiently as HDACis (52–54). However, it is remarkable that demethylation of the HIV LTR sensitized the proviral promoter to the effects of JQ1 (Fig. 5), just as it did to the HDACi VOR.
Some previous studies of the activity of EZH2 at the HIV LTR have been complicated by the use of models utilizing cycling cells, notably transformed cell lines. The stability of epigenetic marks and the activities required to maintain them in such cells are likely to differ from those in primary, resting CD4+ T cells, the primary reservoir of latent HIV infection. Therefore, it was extremely important to extend these studies to ex vivo cultures of primary resting cells. We documented the occupancy of PRC2 and PRC1 components in two primary cell models of latent HIV infection (Fig. 6A) (not shown) and the kinetics of demethylation induced by GSK343 in primary cells (Fig. 6C and D). We then modified the primary cell model protocol described previously by Saleh et al. (40) to allow a period of pretreatment with GSK343, documenting demethylation at both the proviral promoter as well as a host gene, with modest increases in acetylation as well but without an increase in proviral gene expression or HIV p24 antigen production (Fig. 7). It was in this setting that we were able to observe a clear sensitization of the proviral promoter within resting, primary cells, mediated by demethylation induced by GSK343, to the subsequent HDACi effect of VOR. Importantly, this augmented disruption of HIV latency was achieved without inducing markers of cellular activation (data not shown).
In summary, it is widely believed that if treatments to eradicate HIV infection can be developed, they will involve a combination of interventions designed to disrupt latent infection and then allow infected cells to be targeted and cleared. HDAC inhibitors have shown some promise in the disruption of HIV latency in humans. However, in vitro evidence suggests that the effects of HDACis might be incomplete or insufficiently potent to allow complete ablation of persistent infection (33, 34). Combination approaches for the reversal of HIV latency have been demonstrated in vitro, but thus far, none appear safe enough to test in the clinic.
HMTis are entering clinical trials, and early reports suggest that such inhibitors may be safely administered with the significant caveat that, as was true with HDACis, current testing is ongoing in patients suffering from malignancies (55). Furthermore, as both HMTis and HDACis target cellular processes, their potential effects on immune activity must be examined, as eradication strategies will likely require unimpeded immune function to clear infected cells.
The development of effective latency-reversing agents and reservoir depletion strategies is challenging given the extraordinary health benefits of ART. The demonstration that EZH2 inhibition can sensitize HIV hidden in the persistent reservoir of infection within primary, resting CD4+ T cells to the latency-reversing effects of an HDAC inhibitor provides a proof-of-principle for this type of combination antilatency therapy. Such combinations should be considered for further study in animal model systems or, if safe and practical, in clinical studies.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health grant U19 AI096113 to D.M.M., National Institutes of Health grants RR024383 to the UNC TRaCS Institute and AI50410 to the staff of the UNC Center for AIDS Research, an NCI center core support grant (P30CA016086), and an equipment grant from the James B. Pendleton Charitable Trust.
We thank S. Choudhary, B. Allard, J. Kirchherr, K. Sholtis, and S. Gupta for technical support; J. Kuruc and A. Crooks for clinical sample coordination; Y. Park and the staff of the UNC Blood Bank; L. Bixby, E. Trudeau, and the UNC Flow Cytometry Core Facility, UNC-CH Genome Analysis Facility; J. Jin for the gift of UNC 1999; and P. Trojer for the gift of GSK343. Finally, we are grateful for the contributions of the volunteers who have participated in these studies.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00572-15.
REFERENCES
- 1.Archin NM, Sung JM, Garrido C, Soriano-Sarabia N, Margolis DM. 2014. Eradicating HIV-1 infection: seeking to clear a persistent pathogen. Nat Rev Microbiol 12:750–764. doi: 10.1038/nrmicro3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siliciano RF, Greene WC. 2011. HIV latency. Cold Spring Harb Perspect Med 1:a007096. doi: 10.1101/cshperspect.a007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, Lloyd AL, Nowak MA, Fauci AS. 1997. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A 94:13193–13197. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF. 1997. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 5.Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, Richman DD. 1997. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278:1291–1295. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- 6.Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, Lai J, Blankson JN, Siliciano JD, Siliciano RF. 2013. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 155:540–551. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prins JM, Jurriaans S, van Praag RM, Blaak H, van Rij R, Schellekens PT, ten Berge IJ, Yong SL, Fox CH, Roos MT, de Wolf F, Goudsmit J, Schuitemaker H, Lange JM. 1999. Immuno-activation with anti-CD3 and recombinant human IL-2 in HIV-1-infected patients on potent antiretroviral therapy. AIDS 13:2405–2410. doi: 10.1097/00002030-199912030-00012. [DOI] [PubMed] [Google Scholar]
- 8.Hakre S, Chavez L, Shirakawa K, Verdin E. 2011. Epigenetic regulation of HIV latency. Curr Opin HIV AIDS 6:19–24. doi: 10.1097/COH.0b013e3283412384. [DOI] [PubMed] [Google Scholar]
- 9.Mbonye U, Karn J. 2011. Control of HIV latency by epigenetic and non-epigenetic mechanisms. Curr HIV Res 9:554–567. doi: 10.2174/157016211798998736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tripathy MK, Abbas W, Herbein G. 2011. Epigenetic regulation of HIV-1 transcription. Epigenomics 3:487–502. doi: 10.2217/epi.11.61. [DOI] [PubMed] [Google Scholar]
- 11.Van Lint C, Emiliani S, Ott M, Verdin E. 1996. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J 15:1112–1120. [PMC free article] [PubMed] [Google Scholar]
- 12.Verdin E, Paras P Jr, Van Lint C. 1993. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J 12:3249–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coull JJ, Romerio F, Sun JM, Volker JL, Galvin KM, Davie JR, Shi Y, Hansen U, Margolis DM. 2000. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J Virol 74:6790–6799. doi: 10.1128/JVI.74.15.6790-6799.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keedy KS, Archin NM, Gates AT, Espeseth A, Hazuda DJ, Margolis DM. 2009. A limited group of class I histone deacetylases acts to repress human immunodeficiency virus type 1 expression. J Virol 83:4749–4756. doi: 10.1128/JVI.02585-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC. 2006. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J 25:139–149. doi: 10.1038/sj.emboj.7600900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernhard W, Barreto K, Saunders A, Dahabieh MS, Johnson P, Sadowski I. 2011. The Suv39H1 methyltransferase inhibitor chaetocin causes induction of integrated HIV-1 without producing a T cell response. FEBS Lett 585:3549–3554. doi: 10.1016/j.febslet.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 17.du Chene I, Basyuk E, Lin YL, Triboulet R, Knezevich A, Chable-Bessia C, Mettling C, Baillat V, Reynes J, Corbeau P, Bertrand E, Marcello A, Emiliani S, Kiernan R, Benkirane M. 2007. Suv39H1 and HP1gamma are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J 26:424–435. doi: 10.1038/sj.emboj.7601517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friedman J, Cho WK, Chu CK, Keedy KS, Archin NM, Margolis DM, Karn J. 2011. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J Virol 85:9078–9089. doi: 10.1128/JVI.00836-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imai K, Togami H, Okamoto T. 2010. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J Biol Chem 285:16538–16545. doi: 10.1074/jbc.M110.103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marban C, Suzanne S, Dequiedt F, de Walque S, Redel L, Van Lint C, Aunis D, Rohr O. 2007. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J 26:412–423. doi: 10.1038/sj.emboj.7601516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Archin NM, Espeseth A, Parker D, Cheema M, Hazuda D, Margolis DM. 2009. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res Hum Retroviruses 25:207–212. doi: 10.1089/aid.2008.0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Archin NM, Keedy KS, Espeseth A, Dang H, Hazuda DJ, Margolis DM. 2009. Expression of latent human immunodeficiency type 1 is induced by novel and selective histone deacetylase inhibitors. AIDS 23:1799–1806. doi: 10.1097/QAD.0b013e32832ec1dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC, Richman DD, Hudgens MG, Bosch RJ, Coffin JM, Eron JJ, Hazuda DJ, Margolis DM. 2012. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 487:482–485. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Contreras X, Schweneker M, Chen CS, McCune JM, Deeks SG, Martin J, Peterlin BM. 2009. Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J Biol Chem 284:6782–6789. doi: 10.1074/jbc.M807898200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edelstein LC, Micheva-Viteva S, Phelan BD, Dougherty JP. 2009. Short communication: activation of latent HIV type 1 gene expression by suberoylanilide hydroxamic acid (SAHA), an HDAC inhibitor approved for use to treat cutaneous T cell lymphoma. AIDS Res Hum Retroviruses 25:883–887. doi: 10.1089/aid.2008.0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elliott JH, Wightman F, Solomon A, Ghneim K, Ahlers J, Cameron MJ, Smith MZ, Spelman T, McMahon J, Velayudham P, Brown G, Roney J, Watson J, Prince MH, Hoy JF, Chomont N, Fromentin R, Procopio FA, Zeidan J, Palmer S, Odevall L, Johnstone RW, Martin BP, Sinclair E, Deeks SG, Hazuda DJ, Cameron PU, Sekaly RP, Lewin SR. 2014. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog 10:e1004473. doi: 10.1371/journal.ppat.1004473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matalon S, Palmer BE, Nold MF, Furlan A, Kassu A, Fossati G, Mascagni P, Dinarello CA. 2010. The histone deacetylase inhibitor ITF2357 decreases surface CXCR4 and CCR5 expression on CD4(+) T-cells and monocytes and is superior to valproic acid for latent HIV-1 expression in vitro. J Acquir Immune Defic Syndr 54:1–9. doi: 10.1097/QAI.0b013e3181d3dca3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rasmussen TA, Tolstrup M, Brinkmann CR, Olesen R, Erikstrup C, Solomon A, Winckelmann A, Palmer S, Dinarello C, Buzon M. 2014. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV 1:e13–e21. doi: 10.1016/S2352-3018(14)70014-1. [DOI] [PubMed] [Google Scholar]
- 29.Reuse S, Calao M, Kabeya K, Guiguen A, Gatot JS, Quivy V, Vanhulle C, Lamine A, Vaira D, Demonte D, Martinelli V, Veithen E, Cherrier T, Avettand V, Poutrel S, Piette J, de Launoit Y, Moutschen M, Burny A, Rouzioux C, De Wit S, Herbein G, Rohr O, Collette Y, Lambotte O, Clumeck N, Van Lint C. 2009. Synergistic activation of HIV-1 expression by deacetylase inhibitors and prostratin: implications for treatment of latent infection. PLoS One 4:e6093. doi: 10.1371/journal.pone.0006093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wei DG, Chiang V, Fyne E, Balakrishnan M, Barnes T, Graupe M, Hesselgesser J, Irrinki A, Murry JP, Stepan G, Stray KM, Tsai A, Yu H, Spindler J, Kearney M, Spina CA, McMahon D, Lalezari J, Sloan D, Mellors J, Geleziunas R, Cihlar T. 2014. Histone deacetylase inhibitor romidepsin induces HIV expression in CD4 T cells from patients on suppressive antiretroviral therapy at concentrations achieved by clinical dosing. PLoS Pathog 10:e1004071. doi: 10.1371/journal.ppat.1004071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ying H, Zhang Y, Lin S, Han Y, Zhu HZ. 2010. Histone deacetylase inhibitor Scriptaid reactivates latent HIV-1 promoter by inducing histone modification in in vitro latency cell lines. Int J Mol Med 26:265–272. [DOI] [PubMed] [Google Scholar]
- 32.Ylisastigui L, Archin NM, Lehrman G, Bosch RJ, Margolis DM. 2004. Coaxing HIV-1 from resting CD4 T cells: histone deacetylase inhibition allows latent viral expression. AIDS 18:1101–1108. doi: 10.1097/00002030-200405210-00003. [DOI] [PubMed] [Google Scholar]
- 33.Bullen CK, Laird GM, Durand CM, Siliciano JD, Siliciano RF. 2014. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat Med 20:425–429. doi: 10.1038/nm.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cillo AR, Sobolewski MD, Bosch RJ, Fyne E, Piatak M Jr, Coffin JM, Mellors JW. 2014. Quantification of HIV-1 latency reversal in resting CD4+ T cells from patients on suppressive antiretroviral therapy. Proc Natl Acad Sci U S A 111:7078–7083. doi: 10.1073/pnas.1402873111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Colin L, Van Lint C. 2009. Molecular control of HIV-1 postintegration latency: implications for the development of new therapeutic strategies. Retrovirology 6:111. doi: 10.1186/1742-4690-6-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beisel C, Paro R. 2011. Silencing chromatin: comparing modes and mechanisms. Nat Rev Genet 12:123–135. doi: 10.1038/nrg2932. [DOI] [PubMed] [Google Scholar]
- 37.Margueron R, Reinberg D. 2011. The Polycomb complex PRC2 and its mark in life. Nature 469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Verma SK, Tian X, LaFrance LV, Duquenne C, Suarez DP, Newlander KA, Romeril SP, Burgess JL, Grant SW, Brackley JA, Graves AP, Scherzer DA, Shu A, Thompson C, Ott HM, Aller GS, Machutta CA, Diaz E, Jiang Y, Johnson NW, Knight SD, Kruger RG, McCabe MT, Dhanak D, Tummino PJ, Creasy CL, Miller WH. 2012. Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med Chem Lett 3:1091–1096. doi: 10.1021/ml3003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, Martinez-Garcia E, Zhang H, Zheng Y, Verma SK, McCabe MT, Ott HM, Van Aller GS, Kruger RG, Liu Y, McHugh CF, Scott DW, Chung YR, Kelleher N, Shaknovich R, Creasy CL, Gascoyne RD, Wong KK, Cerchietti L, Levine RL, Abdel-Wahab O, Licht JD, Elemento O, Melnick AM. 2013. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23:677–692. doi: 10.1016/j.ccr.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saleh S, Solomon A, Wightman F, Xhilaga M, Cameron PU, Lewin SR. 2007. CCR7 ligands CCL19 and CCL21 increase permissiveness of resting memory CD4+ T cells to HIV-1 infection: a novel model of HIV-1 latency. Blood 110:4161–4164. doi: 10.1182/blood-2007-06-097907. [DOI] [PubMed] [Google Scholar]
- 41.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 42.Pearson R, Kim YK, Hokello J, Lassen K, Friedman J, Tyagi M, Karn J. 2008. Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J Virol 82:12291–12303. doi: 10.1128/JVI.01383-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kutsch O, Benveniste EN, Shaw GM, Levy DN. 2002. Direct and quantitative single-cell analysis of human immunodeficiency virus type 1 reactivation from latency. J Virol 76:8776–8786. doi: 10.1128/JVI.76.17.8776-8786.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, Macnevin CJ, Liu F, Gao C, Huang XP, Kuznetsova E, Rougie M, Jiang A, Pattenden SG, Norris JL, James LI, Roth BL, Brown PJ, Frye SV, Arrowsmith CH, Hahn KM, Wang GG, Vedadi M, Jin J. 2013. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem Biol 8:1324–1334. doi: 10.1021/cb400133j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bouchat S, Gatot JS, Kabeya K, Cardona C, Colin L, Herbein G, De Wit S, Clumeck N, Lambotte O, Rouzioux C, Rohr O, Van Lint C. 2012. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4(+) T cells from HIV-1-infected HAART-treated patients. AIDS 26:1473–1482. doi: 10.1097/QAD.0b013e32835535f5. [DOI] [PubMed] [Google Scholar]
- 46.Kim YK, Bourgeois CF, Pearson R, Tyagi M, West MJ, Wong J, Wu SY, Chiang CM, Karn J. 2006. Recruitment of TFIIH to the HIV LTR is a rate-limiting step in the emergence of HIV from latency. EMBO J 25:3596–3604. doi: 10.1038/sj.emboj.7601248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bosque A, Planelles V. 2011. Studies of HIV-1 latency in an ex vivo model that uses primary central memory T cells. Methods 53:54–61. doi: 10.1016/j.ymeth.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spina CA, Anderson J, Archin NM, Bosque A, Chan J, Famiglietti M, Greene WC, Kashuba A, Lewin SR, Margolis DM, Mau M, Ruelas D, Saleh S, Shirakawa K, Siliciano RF, Singhania A, Soto PC, Terry VH, Verdin E, Woelk C, Wooden S, Xing S, Planelles V. 2013. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog 9:e1003834. doi: 10.1371/journal.ppat.1003834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Strahl BD, Allis CD. 2000. The language of covalent histone modifications. Nature 403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 50.Blazkova J, Murray D, Justement JS, Funk EK, Nelson AK, Moir S, Chun TW, Fauci AS. 2012. Paucity of HIV DNA methylation in latently infected, resting CD4+ T cells from infected individuals receiving antiretroviral therapy. J Virol 86:5390–5392. doi: 10.1128/JVI.00040-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsuda Y, Kobayashi-Ishihara M, Fujikawa D, Ishida T, Watanabe T, Yamagishi M. 2015. Epigenetic heterogeneity in HIV-1 latency establishment. Sci Rep 5:7701. doi: 10.1038/srep07701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Banerjee C, Archin N, Michaels D, Belkina AC, Denis GV, Bradner J, Sebastiani P, Margolis DM, Montano M. 2012. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J Leukoc Biol 92:1147–1154. doi: 10.1189/jlb.0312165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bartholomeeusen K, Xiang Y, Fujinaga K, Peterlin BM. 2012. Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein. J Biol Chem 287:36609–36616. doi: 10.1074/jbc.M112.410746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boehm D, Calvanese V, Dar RD, Xing S, Schroeder S, Martins L, Aull K, Li PC, Planelles V, Bradner JE, Zhou MM, Siliciano RF, Weinberger L, Verdin E, Ott M. 2013. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 12:452–462. doi: 10.4161/cc.23309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ribrag V, Soria JC, Reyderman L, Chen R, Salazar P, Kumar N, Kuznetsov G, Keilhack H, Ottesen LH, Italiano A. 2014. Phase 1 first-in-human study of the enhancer of zeste-homolog 2 (EZH2) histone methyl transferase inhibitor E7438 as a single agent in patients with advanced solid tumors or B cell lymphoma. Eur J Cancer 50(Suppl 6):197. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.