

ABSTRACT
TorsinA is a membrane-tethered AAA+ ATPase implicated in nuclear envelope dynamics as well as the nuclear egress of herpes simplex virus 1 (HSV-1). The activity of TorsinA and the related ATPase TorsinB strictly depends on LAP1 and LULL1, type II transmembrane proteins that are integral parts of the Torsin/cofactor AAA ring, forming a composite, membrane-spanning assembly. Here, we use CRISPR/Cas9-mediated genome engineering to create single- and double knockout (KO) cell lines of TorA and TorB as well as their activators, LAP1 and LULL1, to investigate the effect on HSV-1 production. Consistent with LULL1 being the more potent Torsin activator, a LULL1 KO reduces HSV-1 growth by one order of magnitude, while the deletion of other components of the Torsin system in combination causes subtle defects. Notably, LULL1 deficiency leads to a 10-fold decrease in the number of viral genomes per host cell without affecting viral protein production, allowing us to tentatively assign LULL1 to an unexpected role that precedes HSV-1 nuclear egress.
IMPORTANCE In this study, we conduct the first comprehensive genetic and phenotypic analysis of the Torsin/cofactor system in the context of HSV-1 infection, establishing LULL1 as the most important component of the Torsin system with respect to viral production.
INTRODUCTION
Herpesviruses are enveloped, double-stranded DNA viruses that enter the host cell by fusing with the plasma membrane. Following the microtubule-dependent transport of the nucleocapsid to the nuclear pore complex, the linear herpesvirus genome is ejected into the nucleus of the host cell, where it is transcribed and replicated. Viral genome replication (1), transcription of viral genes, and assembly and packaging of new viral particles take place in designated replication compartments located at the periphery of the nucleus (2).
After the viral capsids are assembled and packaged, they must exit the nucleus to undergo further maturation in the cytoplasm. Herpesviruses undergo nuclear egress via a nuclear membrane budding mechanism in which the viral capsid first buds through the inner nuclear membrane (INM) to form an enveloped intermediate within the perinuclear space, which then fuses with the outer nuclear membrane (ONM) to release the deenveloped capsid into the cytosol (3, 4).
Several viral proteins are required for efficient nuclear egress. Viral kinase US3 and the virally manipulated host cell protein kinase C phosphorylate and locally disassemble the nuclear lamina, which represents a physical barrier between the viral capsid and the INM (5–7). Additionally, the soluble phosphoprotein UL31 and the type II inner nuclear membrane phosphoprotein UL34, which together constitute the viral nuclear egress complex, accumulate at the INM to facilitate capsid envelopment (8–10). Both UL31 and UL34 are essential for herpesvirus growth (11), and their coexpression without viral infection is sufficient to cause vesicle formation (12, 13). While the viral factors involved in nuclear egress are well characterized, much less is known about the importance of cellular factors in HSV-1 nuclear egress (14).
Torsins are a family of AAA+ ATPases (for ATPases associated with a variety of cellular activities) (15) that reside in the lumen of the endoplasmic reticulum (ER) and the contiguous perinuclear space (PNS). Deletion of a single glutamate residue near the C terminus of TorsinA (TorAΔE) is associated with the autosomal dominant movement disorder DYT1 dystonia (16). At a cellular level, this mutation causes budding of the nuclear membrane in neuronal cells (17), and this phenotype is recapitulated in nonneuronal cells when TorsinB (TorB), another member of the Torsin ATPase family, also is depleted (18), indicating that TorA and TorB have overlapping or redundant functions in nuclear envelope dynamics.
Although much remains to be learned about the precise cellular function of Torsin ATPases, recent evidence supports a role for Torsins in the transport of large ribonucleoprotein (RNP) particles via an alternative nuclear transport pathway that mechanistically resembles herpesvirus nuclear egress (19, 20). Both decreased Torsin expression and expression of a dominant-negative Torsin variant lead to an accumulation of RNP-containing vesicles in the perinuclear space, indicating that Torsin function is required for this alternative nuclear transport pathway (20). Consistent with these findings, overexpression of TorsinA WT decreases herpesvirus growth by ∼5-fold in PC6-3 cells (21). Taken together, these studies support a role for Torsin ATPases in nuclear membrane budding.
However, it is important to note that Torsins are highly unusual among AAA+ ATPases in that they are inactive in isolation and become active only upon the binding of stimulatory cofactors LAP1 (for lamina associated polypeptide 1) or LULL1 (for luminal domain like LAP1) (22, 23), type II transmembrane proteins residing in the nuclear envelope and ER, respectively (24, 25). The luminal domains of these proteins adopt AAA-like structures, enabling them to integrate as subunits into the Torsin ATPase ring in a heterohexameric assembly. By doing so, LAP1 and LULL1 contribute a key catalytic arginine residue into the nucleotide binding pocket of an adjacent Torsin subunit, complementing the fragmentary Torsin active site and enabling ATP hydrolysis to occur (26, 27). This activation mechanism and biological assembly is critical for generating the catalytically functional Torsin ATPase machine, highlighting the importance of LAP1 and LULL1 to Torsin cellular function. However, it currently is not known whether one or the other or both of the distinctively localizing Torsin ATPase activators is driving budding-based or alternative nuclear transport of large viral and cellular cargo.
In this study, we use the CRISPR/Cas9 genome editing system (28) to generate LAP1, LULL1, and TorA/TorB knockout HeLa cell lines and examine how modulating expression of these proteins affects HSV-1 growth. We find that a TorA/B double knockout (DKO) reduces viral production only marginally, but that knockout of LULL1 reduces HSV-1 production by more than one order of magnitude. Knockout of LAP1, the major Torsin regulator in the nuclear envelope, has no effect on viral production. Surprisingly, the defect in viral production in LULL1 knockout cells is not due to an accumulation of viral particles in the nucleus or the perinuclear space, as would be expected given the known role of Torsins in nuclear envelope budding in the context of RNP budding in Drosophila melanogaster (20). Instead, we find that the assembly and packaging of viral capsids in the nucleus is severely delayed in LULL1 knockout cells and that the viral genome copy number per cell is decreased by 10-fold, closely mirroring the observed decrease in viral production. These findings establish that LULL1 is required for herpesvirus maturation at a step prior to nuclear egress, leading us to propose that LULL1 serves additional functions independent of the previously established role of Torsin ATPases in nuclear membrane budding.
MATERIALS AND METHODS
Cell culture and viral infection.
HeLa cells were cultured in 10% (vol/vol) fetal bovine serum in Dulbecco's modified Eagle medium (FBS-DMEM). Synchronized HSV-1 KOS infections were performed in 199V low-serum medium (29) at a multiplicity of infection (MOI) of 1.0 unless otherwise specified. Infections were incubated at 4°C for 90 min to allow viruses to adhere to the cell surface. The samples then were washed with phosphate-buffered saline (PBS) to remove unattached viruses and incubated in FBS-DMEM at 37°C for 90 min to allow for viral entry. The cells then were washed with acidic sodium citrate buffer (10 mM sodium citrate, pH 3.0, 4 mM KCl) to remove any remaining viruses from the cell surface to synchronize the infection. After the acidic citrate buffer wash, the cells were washed with PBS and incubated at 37°C in FBS-DMEM for the desired amount of time (30).
Antibodies and immunoblotting.
Immunoblotting was performed according to standard protocols in 5% (wt/vol) skim milk in Tris-buffered saline and 0.1% (vol/vol) Tween 20 (TBS-T) using Western Lightning plus ECL reagent (PerkinElmer). The antibodies used in this study were the following: rabbit polyclonal anti-LULL1 luminal domain (Covance custom antiserum) at 1:5,000, chicken polyclonal anti-LULL1 cytoplasmic domain (Covance custom antiserum) at 1:3,000, rabbit polyclonal anti-TorA (Covance custom antiserum) at 1:4,000, rabbit polyclonal anti-TorB (Covance custom antiserum) at 1:2,000, mouse monoclonal anti-β-actin (ab8226; Abcam) at 1:5,000, mouse monoclonal anti-ICP5 (ab6508; Abcam) at 1:2,000, mouse monoclonal anti-ICP8 (ab20194; Abcam) at 1:2,000, rabbit polyclonal anti-UL34 (Covance custom antiserum) at 1:5,000, and rabbit polyclonal anti-UL31 (Covance custom antiserum) at 1:5,000.
Generation of CRISPR/Cas9 knockout HeLa cell lines.
CRISPR guide sequences were identified using a published index of human exon guide RNA (gRNA) targets (28). The following guide sequences were used: LULL1 (single and double knockouts), 5′-GATTCTTGTCGTCCTGGTTGTGG-3′; LAP1B (single knockout), 5′-GAGAAAGCGCGTACTACCTTCGG-3′; LAP1B (knockout in LULL1 knockout background), 5′-GGGGTGTGTACGTCACCCCCAGG-3′; TorA, 5′-GATGTAGCCGGTGAGGACGC-3′; and TorB, 5′-GTAGGACAGGTAGCCGGTGA-3′. In each case the corresponding gRNA expression vector was constructed, incorporating the guide sequence, according to published protocols (28). The gRNA expression vector was cotransfected with a Cas9 nuclease expression construct into Flp-In TRex recombination-competent HeLa cells (Invitrogen) using X-tremeGENE 9 transfection reagent (Roche). The cells were cultured for 48 h and then split thinly into 10-cm dishes to facilitate the isolation of individual clonal HeLa cell colonies. Potential knockout colonies were screened both by genotyping PCR and by immunoblotting. The genotyping primers used were the following: LULL1 FW, 5′-CCACAGGCTTTGTTTCTG-3′; LULL1 RV, 5′-GGAATAGTAGCTATTCACAG-3′; LAP1 (single knockout) FW, 5′-GAAGAGTTCCGGTCCG-3′; LAP1 (single knockout) RV, 5′-CCTTCGCGTCTTCATTTC-3′; LAP1 (in LULL1 knockout background) FW, 5′-GATCGACTAAAGCTACGTC-3′; LAP1 (in LULL1 knockout background) RV, 5′-CCATTTTGAGGGGCGAG-3′; TorA FW, 5′-GCCTACCCTCCCGGCTAAG-3′; TorA RV, 5′-CATCAGCCTGGGACTGGC-3′; TorB FW, 5′-CGTGGGCCTAGCCATCGG-3′; and TorB RV, 5′-CCGTACCCGAAGCGTTGAG-3′.
Generation of LULL1 rescue cell lines.
LULL1 rescue constructs were generated via the insertion of a wild-type LULL1 cDNA into the site-specific recombination locus present in the LULL1 knockout Flp-In TRex HeLa cells that were generated as described above. LULL1 cDNA was integrated into the pcDNA5/FRT/TO recombination vector and cotransfected into LULL1 knockout cells with a 10-fold excess of Flp-recombinase expression construct. The cells then were cultured for 48 h and subjected to hygromycin B selection for 2 weeks, yielding a population of cells with genomically integrated LULL1 cDNA.
Viral plaque assay.
HSV-1 KOS (ATCC VR-1493) infection of HeLa cells was performed at an MOI of 1.0 as described above, and cells and cellular culture supernatant were harvested by scraping at the stated times. The samples were frozen at −80°C, thawed, and sonicated 3 times for 30 s each on a Branson 450 sonifier at 40% of maximum output level and constant duty cycle. Tenfold serial dilutions in 199V medium were made from these viral lysates, and the samples were incubated on confluent Vero cells for 2 h at 37°C. The virus then was aspirated and the cells were incubated in FBS-DMEM for an additional 2 h before the plates were overlaid with a 1:1 mixture of 1% (wt/vol) agarose in sterile water–FBS-DMEM. The overlaid samples were cultured for 4 days before the agarose/medium overlay was removed. The cells then were fixed with methanol and stained with crystal violet solution (2% [wt/vol] crystal violet in 40% [vol/vol] methanol) in order to quantify viral plaque formation.
Electron microscopy.
Cells were infected with HSV-1 KOS at an MOI of 1.0 as described above and cultured for either 12 or 18 h. The cells were rinsed with PBS and fixed for 1 h with 2.5% (vol/vol) glutaraldehyde–0.1 M sodium cacodylate buffer (pH 7.4). The cells then were prepared and stained with uranyl acetate as described previously (23). Electron microscopy was performed with the aid of the Yale Biological Electron Microscopy facility.
qPCR.
Cells were infected with HSV-1 KOS at an MOI of 1.0 as described above and harvested by scraping at the stated times. Cell pellets were washed with PBS and lysed overnight at 55°C in DNA-isolation lysis buffer (10 mM Tris-HCl, pH 7.5, 10 mM EDTA, 0.5% [wt/vol] SDS, 10 mM NaCl, 200 μM proteinase K). Nucleic acids were isolated by phenol-chloroform extraction followed by ethanol precipitation. The resulting DNA/RNA pellets were resuspended in buffer EB (10 mM Tris-HCl, pH 8.5) and treated with RNase at 37°C for 1 h. Quantitative PCR (qPCR) then was performed on a Roche LightCycler 480 using the SYBR green real-time PCR master mix (Bio-Rad). Primers against the viral gene ICP27 were ICP27 FW (5′-CCGCCGGCCTGGATGTGACG-3′) and ICP27 RV (5′-CGTGGTGGCCGGGGTGGTGCTC-3′). Primers against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were GAPDH FW (5′-CTCCTCACAGTTGCCATGTA-3′) and GAPDH RV (5′-GTTGAGCACAGGGTACTTTATTG-3′). The input-adjusted fluorescence detection threshold crossing point or ΔCp value was calculated for each sample by subtracting the crossing point or Cp value obtained for GAPDH from the Cp value obtained for ICP27. All ΔCp values were then normalized to the corresponding 2-hpi timepoint, yielding a ΔΔCp or relative crossing point value for each timepoint for each sample. Biological and technical triplicates were performed simultaneously, and the data shown are representative of both biological and technical triplicate replications performed separately.
Statistical analysis.
Statistical analysis of the presence or absence of HSV-1 capsids in HSV-1-infected parental control, LULL1 knockout, and LULL1 rescue HeLa cells derived from analysis of large numbers of electron microscopy images was performed using a two-tailed Fisher exact test for statistical significance between contingencies with a small sample size.
RESULTS
Generation of LULL1 knockout and rescue cell lines.
LULL1 is not only the most potent Torsin activator, as judged by apparent affinity and catalytic efficacy (22, 23), it is also the most versatile Torsin cofactor in that it activates three out of four distinct members of the Torsin family of ATPases (22). Due to the established redundancy between Torsin ATPases (18), a LULL1 knockout cell line is expected to inhibit Torsin-specific activities far more profoundly than an individual Torsin deletion. In order to further investigate the functions of the Torsin/cofactor assembly in the context of viral infection, we generated a LULL1 KO HeLa cell line using the CRISPR/Cas9 genome editing system (28), creating a system in which Torsin ATPase activity is severely reduced without manipulating each Torsin ATPase individually.
LULL1 KO cells were generated in a Flp-In HeLa recombination-competent cell line, into which we can introduce a rescuing transgene via targeting at a defined locus (see below). After genome targeting, single-cell colonies were grown and screened via PCR using primers flanking the CRISPR target site to determine effective knockout, which is indicated by a change in the electrophoretic mobility of the PCR product (Fig. 1A). A PCR product of the expected size was observed for control cells, and we obtained one mutant clone that lacked a PCR product of the size indicative of a WT allele, suggesting that all alleles were successfully targeted in this case, presumably due to an internal deletion within the amplified region of the LULL1 locus (Fig. 1B). To ascertain that the genetic lesion resulted in the desired loss of LULL1 at the protein level, we prepared detergent extracts from WT cells and the mutant clone and subjected these to SDS-PAGE and immunoblotting (Fig. 1C). In order to rule out the presence of truncated LULL1 protein that could result from hypomorphic alleles, we used antibodies directed against both the N-terminal and the C-terminal domains of LULL1. Although a number of nonspecific (cross-reactive) species were detected with both antibodies in both WT cells and mutant cells, one band corresponding to the expected molecular mass of LULL1 was present in WT cells but lacking in the mutant LULL1 KO cell line (Fig. 1C). Since we did not detect truncation products that would be indicative of hypomorphic alleles, we conclude that the introduced genetic lesion(s) indeed result in the desired loss of the LULL1 protein.
FIG 1.

LULL1 knockout cells were generated using the CRISPR/Cas9 genome editing system. (A) Schematic diagram encompassing the TOR1AIP2 (LULL1) locus. Shown are the CRISPR guide RNA sequence used to create the knockout and the flanking genotyping PCR primers used to amplify the targeted region. Exons are black boxes, introns are black lines, and PCR primers are half arrows. (B) Genotyping PCR of the TOR1AIP2 (LULL1) locus using primers shown in panel A for parental and LULL1 knockout cells. GAPDH was used as a loading control. (C) Parental and LULL1 knockout cells were harvested and analyzed for the presence of LULL1 protein by SDS-PAGE followed by immunoblotting (IB) using antibodies raised against LULL1's C-terminal domain (left) and N-terminal domain (right). Arrows indicate LULL1. Blots were stripped and probed for β-actin as a control.
HSV-1 production is decreased in LULL1 knockout HeLa cells.
To investigate if LULL1 is implicated in HSV-1 growth, we used a single-step viral growth assay and directly compared WT and LULL1 KO cells. We found that viral production already was decreased in LULL1 KO cells at 9 hpi (Fig. 2A and B), and a more than 10-fold difference was observed at 24 hpi (Fig. 2A and B).
FIG 2.
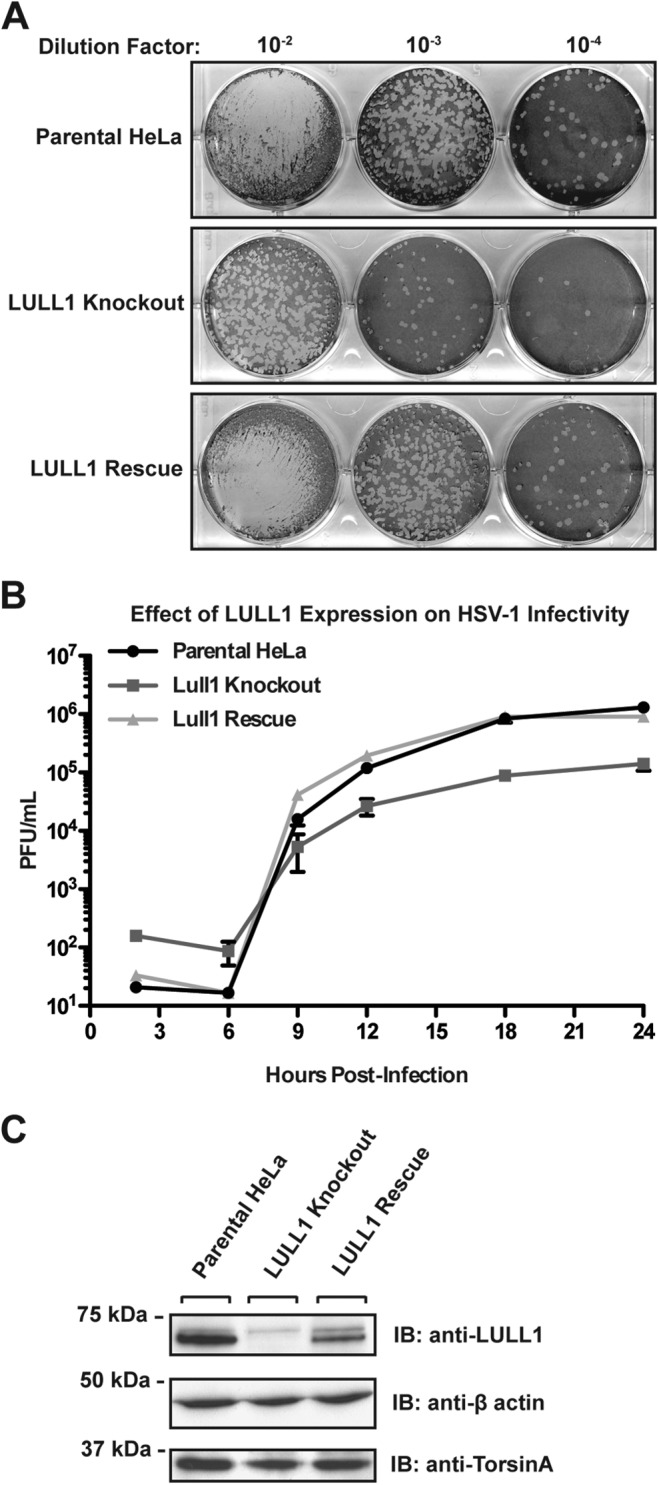
LULL1 knockout inhibits HSV-1 production by 10-fold. (A) HSV-1 viral plaque assay. Parental control, LULL1 KO, and LULL1 rescue cells were infected with HSV-1 KOS strain at an MOI of 1.0. Viral particles were harvested from the whole-cell culture at 24 hpi and reinfected on Vero cells in a serial dilution. Cells were stained with crystal violet for quantification of viral plaques. (B) Single-step growth assay of HSV-1 KOS strain on parental control, LULL1 KO, and LULL1 rescue cells at an MOI of 1.0. Plaque-forming units per milliliter were quantified from parental control (circle), LULL1 KO (square), and LULL1 rescue (triangle) cell lines at various time points postinfection. Each point represents the mean from three experiments with standard deviations shown. (D) Detergent extracts were prepared from parental, LULL1 KO, and LULL1 rescue cell lines, resolved by SDS-PAGE, and subjected to immunoblotting using the indicated antibodies.
To validate this phenotype and to eliminate the possibility of false-positive results due to off-target effects that may be caused by the CRISPR/Cas9 system, a LULL1 rescue cell line was generated in the LULL1 knockout HeLa cell background. LULL1 rescue was performed by site-specific recombination of wild-type LULL1 cDNA into the genomic Flp-In recombination locus. LULL1 expression levels in this rescue cell line are somewhat less than, but within severalfold of, the expression observed in parental HeLa cells, as judged by immunoblotting of extracts obtained from the rescue cell line compared to control cells (Fig. 2C). Importantly, the defect in viral infectivity resulting from LULL1 deletion is fully restored (Fig. 2A and B), indicating that the HSV-1 growth defect is indeed attributable to LULL1.
Expression of viral proteins is unchanged in LULL1 knockout cells.
In order to narrow down the specific stage(s) of the viral life cycle that could be affected by the LULL1 deletion, we monitored viral protein production over the course of viral infection. To this end, we probed HeLa cell lysates isolated from HSV-1-infected parental, LULL1 knockout, and LULL1 rescue cells harvested at various stages of infection with antibodies against several key proteins of the HSV-1 lytic cycle. The viral proteins analyzed include ICP8 (UL29), a viral DNA binding protein required for viral genome replication (31), ICP5 (UL19), the major HSV-1 capsid protein (32), and UL31 and UL34, which together constitute the viral nuclear egress complex (8). In analyzing the expression of these viral proteins over the course of a 24-h infection, we are able to assess the production of proteins expressed at various stages of HSV-1 replication, as ICP8 is an early protein, UL31 is a late protein, and ICP5 and UL34 are leaky late or early-late proteins (1). Analysis of these particular proteins also allows us to track the production of proteins involved in three major steps of viral replication: viral genome replication, viral capsid production, and nuclear egress.
Surprisingly, we found that despite the greater than 10-fold decrease in virus production observed in LULL1 KO cells compared to that of control cells, both the timing and overall levels of viral protein production were comparable (Fig. 3). In all three cell lines, ICP8, UL31, and UL34 expression were detected first at 9 to 12 hpi, while expression of significant levels of ICP5 was not observed until after 12 h. Production of all four proteins increased over time relative to the cellular protein beta-actin, with comparable expression levels between parental, LULL1 KO, and LULL1 rescue cells throughout the course of infection (Fig. 3).
FIG 3.
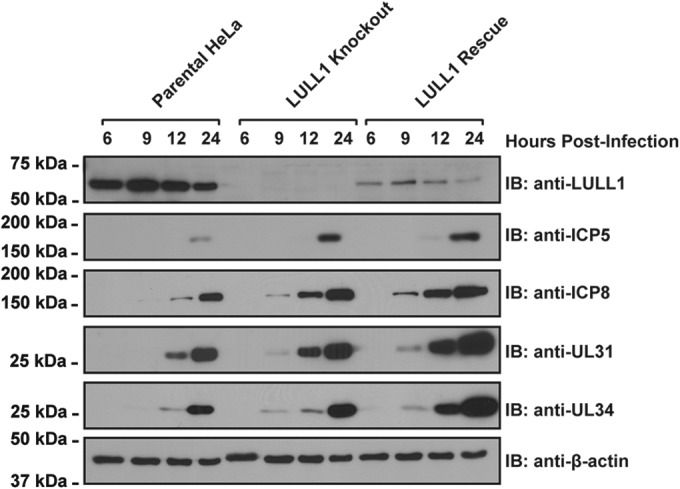
Viral protein production and kinetics are similar between HSV-1-infected parental, LULL1 knockout, and LULL1 rescue HeLa cells. Parental control, LULL1 KO, and LULL1 rescue cells were infected with HSV-1 KOS strain at a multiplicity of infection (MOI) of 10. Cells were harvested at 6, 9, 12, and 24 hpi and lysed in 1% (wt/vol) SDS. Detergent extracts were resolved by SDS-PAGE and subjected to immunoblotting using the indicated antisera.
These results indicate that the decrease in viral infectivity observed in LULL1 KO cells is not due to a general defect in viral protein production. Furthermore, this finding argues against a possible delay or failure of viral entry into the host cell, or a major defect in the transcription/translation of viral genes. Therefore, virus production likely is inhibited at a stage of viral maturation that occurs after viral protein production.
Electron microscopy reveals that HSV-1 capsid assembly and maturation is compromised in LULL1 knockout cells.
As a first step toward understanding if viral capsid assembly, DNA packaging, or nuclear egress of HSV-1 are perturbed in LULL1 KO cells, we used electron microscopy to observe viral particles in HSV-1-infected parental, LULL1 KO, and LULL1 rescue HeLa cells at 12 and 18 hpi. As expected, assembled HSV-1 nucleocapsids were observed at 12 hpi in parental control cells (Fig. 4A, arrow). A subset of these capsids are packaged with viral DNA and can be seen in the perinuclear space, indicating that they were undergoing nuclear egress at the time of fixation (Fig. 4A, arrowhead). In LULL1 KO cells, however, very few assembled nucleocapsids were observed at 12 hpi (Fig. 4B). Instead, the nuclei of infected cells contained electron-dense particles that may represent defective or immature viral capsids (Fig. 4B, arrow). In LULL1 rescue cells, as in parental control cells, assembled HSV-1 nucleocapsids (Fig. 4C, arrow) were observed at 12 hpi, and a subset of those capsids could be observed undergoing nuclear egress, as judged by their location in the perinuclear space (Fig. 4C, arrowhead). Quantification of these observations (Fig. 4G) with an n of 30 showed that while more than half of HSV-1-infected parental control and LULL1 rescue cells (53% and 57%, respectively) contained assembled nucleocapsids at 12 hpi, only 13% of infected LULL1 knockout cells had assembled nucleocapsids. The vast majority of infected LULL1 knockout cells (87%) contained only electron-dense noncapsid particles, as seen in Fig. 4B at 12 hpi. The difference between the number of infected parental control and infected LULL1 knockout HeLa cells containing viral capsids at 12 hpi was statistically significant (P = 0.0022). Similarly, the difference between the number of infected LULL1 knockout cells and LULL1 rescue cells containing viral capsids at 12 hpi was statistically significant (P = 0.0009). The difference between the number of infected parental control cells and LULL1 rescue cells containing viral capsids was not statistically significant, indicating that the reintroduction of the LULL1 gene fully rescues the observed phenotype.
FIG 4.

HSV-1 capsid assembly is compromised in LULL1 knockout cells. Parental control, LULL1 KO, and LULL1 rescue cells were infected with HSV-1 KOS strain at a multiplicity of infection (MOI) of 1.0. (A) Electron microscopy (EM) of HSV-1-infected parental HeLa cells at 12 hpi. Both assembled nucleocapsids (arrow) and packaged viral capsids undergoing nuclear egress (arrowhead) can be seen in HSV-1-infected parental HeLa cells at 12 hpi. (B) EM image of HSV-1-infected LULL1 knockout cells at 12 hpi. Partially formed capsids/scaffolding proteins indicative of early stages of HSV-1 infection (arrow), very few assembled capsids, and no nuclear egress, as judged by the presence of assembled and packaged viral particles in the perinuclear space, were seen in LULL1 KO cells at 12 hpi. (C) EM image of HSV-1-infected LULL1 rescue cells at 12 hpi. Both assembled nucleocapsids (arrow) and packaged viral capsids undergoing nuclear egress (arrowhead) can be seen in HSV-1-infected LULL1 rescue HeLa cells at 12 hpi. (D) As described for panel A, but at 18 hpi. Viral particles can be seen undergoing secondary envelopment in the cytoplasm of HSV-1-infected parental HeLa cells at 18 hpi. (E) As described for panel B, but at 18 hpi. Assembled capsids can be seen in the nuclei of HSV-1-infected LULL1 knockout cells at 18 hpi, but most capsids have not undergone nuclear egress and/or secondary envelopment at 18 hpi. (F) As described for panel C, but at 18 hpi. Viral particles can be seen undergoing secondary envelopment in the cytoplasm of HSV-1-infected LULL1 rescue cells at 18 hpi. (G) Quantification of the effect of LULL1 knockout on HSV-1 capsid formation at 12 hpi (n = 30). Fifty-three percent (16/30) of HSV-1-infected parental HeLa cells and 57% (17/30) of infected LULL1 rescue cells have assembled nucleocapsids (either empty or packaged) present at 12 hpi, while only 13% (4/30) of HSV-1-infected LULL1 knockout cells have assembled nucleocapsids at that time. Eighty-seven percent (26/30) of infected LULL1 knockout cells have only partially assembled capsids and/or scaffolding protein, as seen in panel B at 12 hpi. (H) Quantification of the effect of LULL1 knockout on HSV-1 capsid formation at 18 hpi (n = 30). Eighty-three percent (25/30) of HSV-1-infected parental HeLa cells and 93% (28/30) of infected LULL1 rescue HeLa cells have assembled capsids at 18 hpi, while assembled capsids (either empty or packaged) are seen in only 67% (20/30) of infected LULL1 knockout cells at that time.
At 18 hpi in HSV-1 infected parental HeLa cells, the majority of nucleocapsids have completed nuclear egress and can be seen undergoing secondary envelopment in the cytosol (Fig. 4D, arrowhead). On the other hand, although assembled nucleocapsids were observed in some infected LULL1 knockout cells at 18 hpi (Fig. 4E, arrow), less DNA packaging, nuclear egress, and secondary envelopment were observed in infected LULL1 knockout cells than in infected parental control cells at 18 hpi. However, this defect in capsid assembly and maturation is restored in infected LULL1 rescue cells, where the majority of viral capsids can be seen undergoing secondary envelopment in the cytosol, as in infected parental control cells at 18 hpi (Fig. 4F, arrowhead). Quantification of the effect of LULL1 knockout on HSV-1 capsid assembly at 18 hpi (Fig. 4H) with an n of 30 showed that assembled capsids (either packaged or unpackaged) were observed in 83% (25/30) of infected parental control cells and 93% (28/30) of infected LULL1 rescue HeLa cells, while assembled capsids were observed in only 67% (20/30) of infected LULL1 knockout cells at that time.
Taken together with the previous observation that viral protein production is not affected in LULL1 knockout cells (Fig. 3), these data suggest that the defect in HSV-1 production observed in the absence of LULL1 involves a defect or delay in the assembly and packaging of viral capsids in the nucleus. Furthermore, the fact that the observed capsid assembly defect was restored to wild-type conditions at both 12 and 18 hpi in LULL1 rescue cells indicates that the defect in question is in fact due to the absence of LULL1 and not to an off-target effect of CRISPR/Cas9 genome editing. These results are unexpected in that we observe a delay or defect in capsid assembly rather than the accumulation of fully assembled and packaged nucleocapsids in the nucleus and/or perinuclear space, as would be expected given that Torsin ATPases have been implicated in nuclear membrane budding (20, 21). However, we certainly cannot exclude the possibility that Torsin ATPases play a role in viral nuclear egress or more generally in alternative nuclear transport, since it is possible that there is an additional defect in nuclear egress that escapes our detection due to the earlier capsid assembly defect seen here. Moreover, it is possible that the virus has evolved mechanisms that bypass the requirement for Torsin ATPases observed in D. melanogaster (20). Regardless, our data are consistent with the interpretation that LULL1 deficiency causes a defect in HSV-1 production at a stage after viral entry and before nuclear egress.
HSV-1 genome copy number is decreased by one order of magnitude in LULL1 knockout cells.
Because we observed that viral particle assembly but not the production of viral proteins is delayed in LULL1 knockout cells compared to that in control cells, we next used qPCR to determine whether knockout of LULL1 is associated with a defect in viral genome replication. To this end, DNA samples were harvested from infected parental, LULL1 KO, and LULL1 rescue HeLa cells over the course of a 24-h viral infection and subjected to qPCR. By performing duplicate qPCR reads from each DNA sample using primers against both the viral gene ICP27 and the cellular housekeeping gene GAPDH, we were able to arrive at a ΔΔCp value that directly reflects the number of viral genomes per cell present in the sample.
At both 18 and 24 hpi, we found that there is an approximately 10-fold or 1-log decrease in the number of viral genomes per cell in the LULL1 KO cells compared to the level in control cells (Fig. 5). Furthermore, viral genome copy number is largely restored in the LULL1 rescue cell line, indicating that this phenotype indeed is due to the absence of LULL1. This 10-fold decrease in the number of viral genomes detected in infected LULL1 KO cells closely reflects the 10-fold decrease in viral infectivity observed in the single-step growth assay at both 18 and 24 hpi.
FIG 5.
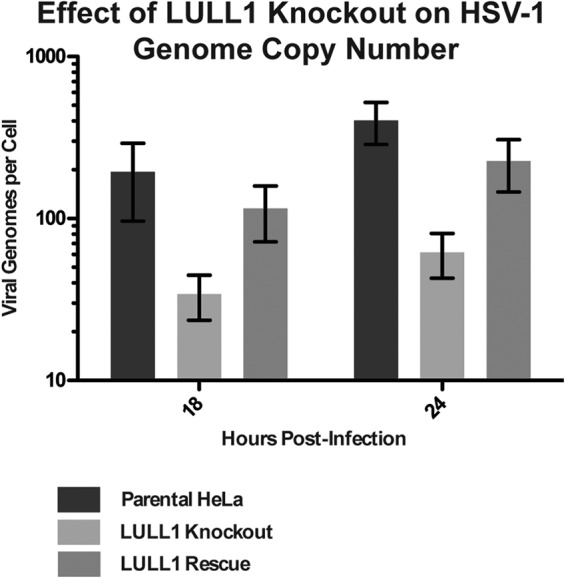
HSV-1 genome copy number is reduced by 10-fold in LULL1 knockout cells. DNA samples from HSV-1-infected parental, LULL1 knockout, and LULL1 rescue HeLa cells were analyzed by quantitative PCR (qPCR) using primers against both the viral gene ICP27 and the cellular housekeeping gene GAPDH. ΔCp values were calculated by subtracting the GAPDH Cp values from the corresponding ICP27 ΔCp values in order to obtain a ΔCp value that directly reflects the number of viral genomes per cell. The resulting ΔCp values were normalized to the 2-hpi time point, and these ΔΔCp values were plotted for each of the three cell lines at both 18 and 24 hpi.
LAP1 is dispensable for HSV-1 growth, while TorsinA and TorsinB depletion decreases HSV-1 production only marginally.
Finally, in order to determine if the viral production defect observed in LULL1 KO cells is LULL1 specific or if Torsin machinery in general is important for HSV-1 production, we generated LAP1 knockout, LAP1/LULL1 double knockout, and TorA/TorB double knockout HeLa cell lines. For all targeted genes, we obtained clonal cell lines in which we did not detect the signature PCR product corresponding to the WT allele (Fig. 6A). Correspondingly, we were unable to detect the protein products of the targeted alleles in the desired cell lines (Fig. 6B), indicating that all knockouts were successful.
FIG 6.

HSV-1 production is only moderately affected by LAP1, TorA, and TorB knockout. (A) LAP1/LULL1 double knockout, TorA/TorB double knockout, and LAP1 knockout cell lines were generated using the CRISPR/Cas9 genome editing system. (Left) Genotyping PCR of the CRISPR-targeted LAP1 and LULL1 gene loci in LAP1/LULL1 double knockout HeLa cells. (Middle) Genotyping PCR of the CRISPR-targeted TorA and TorB gene loci in TorA and TorB double knockout HeLa cells. (Right) Genotyping PCR of the targeted LAP1 locus in a LAP1 single HeLa cell knockout. PCR using primers against GAPDH was used as a loading control in all three cases. (B) Immunoblots against LULL1, LAP1, TorA, and TorB demonstrating knockout of the stated protein in each of the cell lines used for panel A, with β-actin as a loading control. (C) HSV-1 KOS production at 24 hpi was quantified by viral plaque assay in parental, LULL1 knockout, LAP1 knockout, LAP1/LULL1 double knockout, and TorA/B double knockout HeLa cells. Plaque-forming units per milliliter represent the means from three experiments with standard deviations shown.
We next assessed HSV-1 production in these cell lines by a single-step growth assay, using a 24-hpi endpoint measurement. We found that LAP1 KO alone, unlike LULL1 KO, has at best a minor effect on virus production (Fig. 6C). Importantly, there is no additional decrease in HSV-1 production in LAP1/LULL1 double KO cells compared to LULL1 single KO cells, indicating that these two proteins do not function redundantly to promote HSV-1 growth. Of note, we observe only a 2-fold decrease in virus production in TorA/TorB double KO cells, indicating that the decrease in virus production observed in LULL1 KO cells is not solely attributable to a lack of TorA/TorB activity in the absence of LULL1. The fact that LULL1, but not LAP1, affects viral production indicates that these Torsin cofactors have distinct and independent cellular roles. Taken together, these data establish a new role for LULL1 as a host cell factor required for efficient HSV-1 production.
DISCUSSION
In this study, we find that HSV-1 production is decreased by one order of magnitude in LULL1 KO HeLa cells, which establishes LULL1 as the most important component of the Torsin system with respect to viral production. This defect is fully rescued when LULL1 expression is restored (Fig. 2), unambiguously demonstrating that this defect in HSV-1 growth is strictly due to LULL1 expression. Expression of the viral proteins ICP5, ICP8, UL31, and UL34 is unaffected in LULL1 KO cells (Fig. 3), leading us to conclude that the observed phenotype is not due to a generic defect in viral entry, transcription of viral genes, or production of viral proteins. The earliest defect in the lytic cycle that we can detect is a reduction of the viral genome copy number to an extent that closely matches the overall decrease in virus production observed in LULL1 KO cells (Fig. 5).
These data indicate that either the replication or the maintenance/stability of the HSV-1 genome is affected by deletion of LULL1. Additionally, the fact that both viral genome copy number and virus production are decreased by approximately 10-fold in LULL1 KO cells compared to control cells suggests that the defect in virus production is in fact caused primarily by a defect in viral genome replication or maintenance. This result is unexpected, as the process of viral genome replication occurs prior to nuclear egress (4), the step of viral reproduction predicted to be affected by a defect in the Torsin nuclear membrane budding machinery by analogy to RNP budding (20). Whether the observed HSV-1 capsid assembly defect (Fig. 4) is merely a consequence of the absence of viral genomes or the decrease in viral genome count is due to a defect in the coupling of viral genome replication, processing and capsid maturation remain to be seen. It also is possible that both the viral genome production/maintenance and viral capsid assembly defects are caused by an unknown factor upstream of these two events. However, from our data we can deduce that knockout of LULL1 intersects the HSV-1 life cycle at a stage after viral entry, transcription of viral mRNA, and the production of viral proteins but prior to primary envelopment and nuclear egress. The mechanistic underpinnings of how LULL1, being a type II transmembrane protein primarily residing in the ER (25), can affect processes in the nuclear compartment have yet to be elucidated. At this point, we cannot exclude the possibility that LULL1 exerts its effect indirectly. For example, it is possible that the synthesis or nuclear trafficking of a specific HSV-1 or host cell protein implicated in viral DNA replication/processing or capsid biogenesis is affected in LULL1 knockout cells. In this context, it is noteworthy that LULL1 previously has been implicated in transport events from the ER to the nuclear envelope (33), and more recently, the Torsin system was shown to be implicated in the biogenesis of the nuclear pore complex in Caenorhabditis elegans (34). These outstanding mechanistic questions notwithstanding, our study firmly establishes the importance of the Torsin regulator LULL1 for the lytic growth of HSV-1. Additional studies will be required to address the precise underlying mechanism. Given these findings, however, it appears reasonable to speculate that LULL1 depletion leads to a reduction in genome replication, maintenance, or processing, which is either causative or at least correlated with the observed delay/defect in capsid assembly in the nucleus.
ACKNOWLEDGMENTS
We thank George Miller, Richard Park, and members of the Schlieker laboratory for critical readings of the manuscript and Morven Graham for her assistance with electron microscopy.
This work was funded by the Charles Hood Foundation, Ellison Medical Foundation (AG-NS-0662-10), and NIH (DP2 OD008624-01 and 5T32GM007223-39).
REFERENCES
- 1.Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed). 2007. Fields virology, 5th ed Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Weller SK, Coen DM. 2012. Herpes simplex viruses: mechanisms of DNA replication. Cold Spring Harb Perspect Biol 4:a013011. doi: 10.1101/cshperspect.a013011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mettenleiter TC. 2002. Herpesvirus assembly and egress. J Virol 76:1537–1547. doi: 10.1128/JVI.76.4.1537-1547.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 5.Mou F, Wills E, Baines JD. 2009. Phosphorylation of the U(L)31 protein of herpes simplex virus 1 by the U(S)3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J Virol 83:5181–5191. doi: 10.1128/JVI.00090-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muranyi W, Haas J, Wagner M, Krohne G, Koszinowski UH. 2002. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 297:854–857. doi: 10.1126/science.1071506. [DOI] [PubMed] [Google Scholar]
- 7.Park R, Baines JD. 2006. Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J Virol 80:494–504. doi: 10.1128/JVI.80.1.494-504.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ. 2001. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J Virol 75:8803–8817. doi: 10.1128/JVI.75.18.8803-8817.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J Virol 76:8939–8952. doi: 10.1128/JVI.76.17.8939-8952.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simpson-Holley M, Baines J, Roller R, Knipe DM. 2004. Herpes simplex virus 1 U(L)31 and U(L)34 gene products promote the late maturation of viral replication compartments to the nuclear periphery. J Virol 78:5591–5600. doi: 10.1128/JVI.78.11.5591-5600.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang YE, Van Sant C, Krug PW, Sears AE, Roizman B. 1997. The null mutant of the U(L)31 gene of herpes simplex virus 1: construction and phenotype in infected cells. J Virol 71:8307–8315. http://jvi.asm.org/content/71/11/8307.long. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klupp BG, Granzow H, Fuchs W, Keil GM, Finke S, Mettenleiter TC. 2007. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc Natl Acad Sci U S A 104:7241–7246. doi: 10.1073/pnas.0701757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bigalke JM, Heuser T, Nicastro D, Heldwein EE. 2014. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat Commun 5:4131. doi: 10.1038/ncomms5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rose A, Schlieker C. 2012. Alternative nuclear transport for cellular protein quality control. Trends Cell Biol 22:509–514. doi: 10.1016/j.tcb.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neuwald AF, Aravind L, Spouge JL, Koonin EV. 1999. AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res 9:27–43. http://genome.cshlp.org/content/9/1/27.full. [PubMed] [Google Scholar]
- 16.Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, Fahn S, Risch NJ, Buckler AJ, Gusella JF, Breakefield XO. 1997. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet 17:40–48. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- 17.Goodchild RE, Kim CE, Dauer WT. 2005. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron 48:923–932. doi: 10.1016/j.neuron.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 18.Kim CE, Perez A, Perkins G, Ellisman MH, Dauer WT. 2010. A molecular mechanism underlying the neural-specific defect in torsinA mutant mice. Proc Natl Acad Sci U S A 107:9861–9866. doi: 10.1073/pnas.0912877107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Speese SD, Ashley J, Jokhi V, Nunnari J, Barria R, Li Y, Ataman B, Koon A, Chang YT, Li Q, Moore MJ, Budnik V. 2012. Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic Wnt signaling. Cell 149:832–846. doi: 10.1016/j.cell.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jokhi V, Ashley J, Nunnari J, Noma A, Ito N, Wakabayashi-Ito N, Moore MJ, Budnik V. 2013. Torsin mediates primary envelopment of large ribonucleoprotein granules at the nuclear envelope. Cell Rep 3:988–995. doi: 10.1016/j.celrep.2013.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maric M, Shao J, Ryan RJ, Wong CS, Gonzalez-Alegre P, Roller RJ. 2011. A functional role for TorsinA in herpes simplex virus 1 nuclear egress. J Virol 85:9667–9679. doi: 10.1128/JVI.05314-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao C, Brown RS, Chase AR, Eisele MR, Schlieker C. 2013. Regulation of Torsin ATPases by LAP1 and LULL1. Proc Natl Acad Sci U S A 110:E1545–E1554. doi: 10.1073/pnas.1300676110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rose AE, Zhao C, Turner EM, Steyer AM, Schlieker C. 2014. Arresting a Torsin ATPase reshapes the endoplasmic reticulum. J Biol Chem 289:552–564. doi: 10.1074/jbc.M113.515791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foisner R, Gerace L. 1993. Integral membrane proteins of the nuclear envelope interact with lamins and chromosomes, and binding is modulated by mitotic phosphorylation. Cell 73:1267–1279. doi: 10.1016/0092-8674(93)90355-T. [DOI] [PubMed] [Google Scholar]
- 25.Goodchild RE, Dauer WT. 2005. The AAA+ protein torsinA interacts with a conserved domain present in LAP1 and a novel ER protein. J Cell Biol 168:855–862. doi: 10.1083/jcb.200411026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown RS, Zhao C, Chase AR, Wang J, Schlieker C. 2014. The mechanism of Torsin ATPase activation. Proc Natl Acad Sci U S A 111:E4822–E4831. doi: 10.1073/pnas.1415271111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sosa BA, Demircioglu FE, Chen JZ, Ingram J, Ploegh H, Schwartz TU. 2014. How lamina-associated polypeptide 1 (LAP1) activates Torsin. eLife 3:e03239. doi: 10.7554/eLife.03239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blaho JA, Morton ER, Yedowitz JC. 2005. Herpes simplex virus: propagation, quantification, and storage. Curr Protoc Microbiol Chapter 14:Unit 14E 1. doi: 10.1002/9780471729259.mc14e01s00. [DOI] [PubMed] [Google Scholar]
- 30.Roller RJ, Zhou Y, Schnetzer R, Ferguson J, DeSalvo D. 2000. Herpes simplex virus type 1 U(L)34 gene product is required for viral envelopment. J Virol 74:117–129. doi: 10.1128/JVI.74.1.117-129.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conley AJ, Knipe DM, Jones PC, Roizman B. 1981. Molecular genetics of herpes simplex virus. VII. Characterization of a temperature-sensitive mutant produced by in vitro mutagenesis and defective in DNA synthesis and accumulation of gamma polypeptides. J Virol 37:191–206. http://jvi.asm.org/content/37/1/191.long. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gibson W, Roizman B. 1972. Proteins specified by herpes simplex virus. 8. Characterization and composition of multiple capsid forms of subtypes 1 and 2. J Virol 10:1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vander Heyden AB, Naismith TV, Snapp EL, Hodzic D, Hanson PI. 2009. LULL1 retargets TorsinA to the nuclear envelope revealing an activity that is impaired by the DYT1 dystonia mutation. Mol Biol Cell 20:2661–2672. doi: 10.1091/mbc.E09-01-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Gompel MJW, Nguyen KCQ, Hall DH, Dauer WT, Rose LS. 2015. A novel function for the Caenorhabditis elegans torsin OOC-5 in nucleoporin localization and nuclear import. Mol Biol Cell 26:1752–1763. doi: 10.1091/mbc.E14-07-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]