

ABSTRACT
The four serotypes of dengue virus (DENV-1 to -4) represent the most prevalent mosquito-borne viral pathogens in humans. No clinically approved vaccine or antiviral is currently available for DENV. Here we report a spiropyrazolopyridone compound that potently inhibits DENV both in vitro and in vivo. The inhibitor was identified through screening of a 1.8-million-compound library by using a DENV-2 replicon assay. The compound selectively inhibits DENV-2 and -3 (50% effective concentration [EC50], 10 to 80 nM) but not DENV-1 and -4 (EC50, >20 μM). Resistance analysis showed that a mutation at amino acid 63 of DENV-2 NS4B (a nonenzymatic transmembrane protein and a component of the viral replication complex) could confer resistance to compound inhibition. Genetic studies demonstrate that variations at amino acid 63 of viral NS4B are responsible for the selective inhibition of DENV-2 and -3. Medicinal chemistry improved the physicochemical properties of the initial “hit” (compound 1), leading to compound 14a, which has good in vivo pharmacokinetics. Treatment of DENV-2-infected AG129 mice with compound 14a suppressed viremia, even when the treatment started after viral infection. The results have proven the concept that inhibitors of NS4B could potentially be developed for clinical treatment of DENV infection. Compound 14a represents a potential preclinical candidate for treatment of DENV-2- and -3-infected patients.
IMPORTANCE Dengue virus (DENV) threatens up to 2.5 billion people and is now spreading in many regions in the world where it was not previously endemic. While there are several promising vaccine candidates in clinical trials, approved vaccines or antivirals are not yet available. Here we describe the identification and characterization of a spiropyrazolopyridone as a novel inhibitor of DENV by targeting the viral NS4B protein. The compound potently inhibits two of the four serotypes of DENV (DENV-2 and -3) both in vitro and in vivo. Our results validate, for the first time, that NS4B inhibitors could potentially be developed for antiviral therapy for treatment of DENV infection in humans.
INTRODUCTION
Emerging infectious pathogens represent a major threat to public health. Vaccines and therapeutics are two key countermeasures against these pathogens. Many viruses of the genus Flavivirus within the family Flaviviridae are arthropod-borne human pathogens, among which the four serotypes of dengue virus (DENV) alone cause 390 million human infections each year (1). Several promising DENV vaccines are currently in clinical development (2). The most advanced vaccine (CYD-TDV) exhibited good efficacy against DENV-1, -3, and -4 but weak protection against DENV-2 (3–5). For antiviral development, four compounds have been tested in dengue clinical trials, including balapiravir (a nucleoside inhibitor) (6), celgosivir (a cellular α-glucosidase inhibitor) (7), chloroquine (a malaria drug with antiviral and immunomodulatory activities) (8), and prednisolone (a corticosteroid drug) (9). None of them showed any antiviral activity or clinical benefits in dengue patients. Notably, all these compounds were repurposed from existing drugs or compounds previously developed for other viruses. Bona fide inhibitors specifically designed for DENV have never advanced to clinical trials (10).
In this paper, we report the identification of a novel class of small-molecule anti-DENV agents, the spiropyrazolopyridones, using phenotypic screening. These inhibitors block DENV replication by targeting nonstructural protein 4B (NS4B), a nonenzymatic transmembrane protein functioning as an essential component of the viral replication complex. The lead candidate, compound 14a, is orally available and has good in vivo pharmacokinetic properties. Using a dengue mouse model, we show that compound 14a suppressed peak viremia on day 3 postinfection (p.i.), even when treatment started 2 days after viral infection. Our results have pharmacologically validated that inhibitors of NS4B could potentially be developed for clinical treatment of DENV infection.
MATERIALS AND METHODS
Cells, compounds, and antibodies.
A549 cells (human alveolar epithelial cells) were maintained in F-12 medium containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. BHK-21 cells (baby hamster kidney cells) were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% FBS and 1% penicillin-streptomycin. C6/36 mosquito cells were grown in RPMI 1640 medium containing 10% FBS and 1% penicillin-streptomycin. A549 cells containing a DENV-2 replicon were maintained in F-12 medium containing 10% FBS, 20 μg/ml puromycin, and 1% penicillin-streptomycin (11). Huh-7.5 cells containing a subgenomic replicon of hepatitis C virus (HCV) genotype 1b were licensed from Apath LLC (St. Louis, MO) (34) and were maintained in DMEM containing 10% FBS, 0.25 mg/ml Geneticin, and 1% penicillin-streptomycin. A549, BHK-21, DENV-2 replicon, and HCV replicon cell lines were incubated at 37°C. C6/36 cells were cultured at 28°C. All compounds were in-house synthesized. DENV-specific mouse monoclonal antibody 4G2 against the DENV envelope (E) protein was prepared from a hybridoma cell line purchased from the American Type Culture Collection (ATCC).
DENV-2 and HCV replicon assays.
A549 DENV-2 replicon cells were seeded at a density of 3,000 cells per well in a 384-well microplate. After incubation at 37°C with 5% CO2 overnight, the cells were treated with compounds. After 48 h of incubation, luciferase activities were measured by using the EndurRen live-cell substrate (Promega). Following luciferase activity measurement, the CellTiter-Glo reagent (Promega) was added to each well to determine the cytotoxicity of the compounds. For the HCV replicon assay, Huh-7.5 cells harboring the HCV replicon were seeded at a density of 20,000 cells per well in a 96-well microplate. At 48 h after compound treatment, the cells were assayed for luciferase activity by using a Bright-Glo luciferase assay (Promega). As a quality control, NITD-008, a nucleoside inhibitor of DENV and HCV (12), was included in our primary and secondary antiviral assays throughout the study.
Viral titer reduction assay.
The following viruses were used in the viral titer reduction assay: DENV-1, DENV-2, DENV-3, DENV-4, Japanese encephalitis virus (JEV), Powassan virus (POWV), Western equine encephalitis virus (WEEV), West Nile virus (WNV), yellow fever virus (YFV), and vesicular stomatitis virus (VSV). The sources of these viruses were reported previously (13). Approximately 2 × 104 cells (A549 or Vero cells) were seeded into each well of 96-well plates. At 24 h postseeding, A549 cells were infected with DENV (multiplicity of infection [MOI] of 0.5); Vero cells were infected with JEV, POWV, WNV, YFV, WEEV, or VSV (MOI of 0.1). The infected cells were immediately treated with serial dilutions of the compound. Because of the difference in replication kinetics among the different viruses, culture fluids were collected at different time points p.i.: culture fluids from JEV, POWV, WNV, WEEV, and YFV infections were collected at 42 h p.i.; those from DENV infections were collected at 48 h p.i; and those from VSV infections were collected at 16 h p.i. The viral titers in the collected samples were quantified by using a plaque assay.
Cytotoxicity assay.
Cell viability was measured by using Cell Counting Kit-8 (CCK-8) (Dojindo Molecular Technologies) according to the manufacturer's protocols. A549, Huh-7, or Vero cells were seeded at 5 × 103 cells per well in a 96-well plate. After 24 h of incubation at 37°C with 5% CO2, the cells were treated with 2-fold serial dilutions of the compound. At 48 h posttreatment, 4 μl of the CCK-8 solution was added to each well. After another 90 min of incubation at 37°C, the absorbance was measured at 450 nm by using a microplate reader (Tecan).
Transient-replicon assay.
The Renilla luciferase replicon of DENV-2 was in vitro transcribed using a T7 mMessage mMachine kit (Ambion, Austin, TX) from cDNA plasmids linearized with ClaI, as described previously (14). A549 cells were electroporated with 10 μg of replicon RNA by using a GenePulser Xcell system (Bio-Rad, Hercules, CA) and an established protocol (15). The transfected cells were seeded into a 12-well plate (3 × 105 cells per well), followed by treatment with the compound or the dimethyl sulfoxide (DMSO) control. At various time points posttransfection (p.t.), the cells were washed once with phosphate-buffered saline (PBS) and lysed in 200 μl 1× lysis buffer (Promega). The plates containing the lysis buffer were sealed with Parafilm and stored at −80°C. Once samples for all time points had been collected, 20 μl of cell lysates was transferred into a 96-well plate and assayed for luciferase signals with a Clarity luminescence microplate reader (BioTek).
Selection and sequencing of resistant virus.
Compound 1a-resistant virus was obtained by serial passaging of the DENV-2 NGC strain in A549 cells in the presence of increasing concentrations of the compound. Six selections for the compound-resistant virus were independently performed. Briefly, for each selection, A549 cell monolayers in a 12-well plate were inoculated at an MOI of 0.5 with wild-type (WT) virus or previously passaged virus and the compound. After incubation at 30°C for 1 h, the inoculum was removed, and fresh F-12 medium with 2% fetal calf serum (FCS) containing the compound was added to the wells. The cell cultures were incubated at 37°C for 3 days, after which the viruses from culture medium were quantified by reverse transcription-quantitative PCR (qRT-PCR) and used for the next round of infection. Resistance selection began at 27 nM compound (twice the 50% effective concentration [EC50]). After passaging of the cells for three rounds at the initial concentration (passage 1 [P1] to P3; 3 days per passage round), the compound concentration was increased to 80 nM (P4 to P8), 150 nM (P9 to P11), and 300 nM (P12 to P16). As a control, WT virus was passaged in the presence of 0.5% DMSO in parallel. The supernatant from 300 nM compound-selected virus (P16) was tested for compound sensitivity by using qRT-PCR. For viral whole-genome sequencing, viral RNA was extracted from P16 culture fluids by using the QIAamp viral RNA minikit (Qiagen) and amplified by using SuperScript One-Step reverse transcription (RT)-PCR with Platinum Taq (Invitrogen). The RT-PCR products were purified and subjected to DNA sequencing.
Recombinant mutant virus construction.
A standard cloning procedure was used to generate recombinant viruses. To construct mutant cDNA plasmids of full-length DENV-2 NGC, we first engineered the NS4B V63 mutations into a shuttle vector by using a QuikChange III XL site-directed mutagenesis kit (Stratagene). The mutations in the shuttle vector were confirmed by DNA sequencing. The fragments containing the mutations were then engineered into the full-length cDNA clone (16) by using BspEI (a restriction enzyme site introduced at nucleotide position 6596 of the genome without changing the amino acid sequence) and NruI (nucleotide position 7738). All constructs were verified by DNA sequencing.
The pACYA177-DENV-1 (Western Pacific 74 strain) full-length cDNA clone was used to construct a shuttle vector and engineer the I64V mutation of NS4B. The shuttle vector was created by digestion of the pACYC-FLDENV-1 plasmid with restriction enzymes XhoI and ClaI, followed by ligation of the resulting 5,687-kb fragment (representing nucleotide positions 2607 to 8294 of the genome) into the predigested TOPO-TA vector (Life Technologies). The I64V mutation was first introduced into the shuttle vector by using the QuikChange III XL site-directed mutagenesis kit. The mutated DNA fragment from the shuttle vector was sequenced and then swapped into pACYC-FLDENV1 through the unique restriction enzymes XhoI and ClaI. The resulting I64V mutation was verified again by DNA sequencing.
Time-of-addition assay.
Approximately 2 × 105 A549 cells were seeded into each well of a 24-well plate. At 24 h postseeding, the cells were infected with DENV-2 (New Guinea C strain; MOI of 2.0) for 1 h at 4°C. Subsequently, the viral inocula were removed, and cells were washed three times with cold PBS to remove unabsorbed viruses. At 0, 2, 4, 6, 8, 10, 12, 14, 16, 18, or 20 h p.i., 10 μM compound was added to the infected cells. As negative controls, 0.9% DMSO was added to the infected cells. At 24 h p.i., culture fluids were collected, and viral titers were determined by a plaque assay (17).
In vitro transcription, RNA transfection, and immunofluorescence assay.
The full-length DENV-1 and DENV-2 cDNA plasmids were linearized with SacII and XbaI, respectively. Genome-length RNAs were in vitro transcribed by using the T7 mMessage mMachine kit (Ambion). RNA transfection and immunofluorescence assays (IFAs) were performed as previously described (15). Briefly, 10 μg RNAs was electroporated into BHK-21 cells. The transfected cells were monitored for viral E protein expression by IFA using mouse monoclonal antibody 4G2 (American Type Culture Collection) and anti-mouse immunoglobulin G conjugated with fluorescein isothiocyanate (FITC) as the primary and secondary antibodies, respectively. The culture medium was harvested on day 5 p.t. and stored at −80°C for subsequent plaque assays and viral genome sequencing.
Protein expression and purification.
We expressed and purified the N-terminal domain (NTD) (representing the first 125 amino acids) of DENV-2 NS4B. The cDNAs encoding the NS4B NTD were amplified by PCR and cloned into the NdeI and XhoI sites of pET-29b, resulting in plasmid pET29-NTD. The pET29-NTD plasmid encodes the NTD with a tag containing eight residues (LEHHHHHH) to aid in protein purification. The insertion of the tag into the plasmid was confirmed by DNA sequencing. To express the target protein, the plasmid was first transformed into Escherichia coli BL21(DE3) cells and grown on an LB plate that contained 30 μg/ml of kanamycin. One to three colonies on the plate were picked and incubated in 50 ml of LB medium. The culture grown overnight was transferred into 1 liter of LB medium with same antibiotic. When the optical density at 600 nm (OD600) reached 0.8, the protein was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and left in the shaker at 200 rpm overnight at 18°C. The E. coli cells were harvested by centrifugation at 8,000 × g for 10 min at 4°C. The NTD protein was purified in the presence of n-dodecyl β-d-maltoside (DDM) micelles as previously described (18).
Micro BioSpin column gel filtration assay.
Micro BioSpin 6 columns (Bio-Rad) capable of separating free ligands from bound ligands were used to measure the binding of 3H-labeled compound 14a to purified proteins. A binding mixture (20 μl) containing 20 mM sodium phosphate (pH 6.5), 0.1% 1-myristoyl-2-hydroxy-sn-glycero-3-[phospho-Rac-(1-glycerol)] (LMPG), 1 mM dithiothreitol (DTT), 150 mM NaCl, 5 to 50 μM NS4B, and 100 to 500 μM 3H-labeled compound 14a was incubated at room temperature for 10 min, adsorbed to a Micro BioSpin column, and centrifuged for 1 min. The eluent was collected and counted with a scintillation counter (PerkinElmer Life Sciences).
ISRE reporter assay.
HEK-293T cells were cotransfected with three plasmids by using X-treme 9 Gene reagent (Roche). The three plasmids included pISRE-Luc (firefly luciferase reporter under the control of the interferon [IFN]-stimulated response element [ISRE] [Clontech]), phRL-CMV (humanized Renilla luciferase under the control of the cytomegalovirus [CMV] promoter for normalizing the transfection efficiency [Promega]), and pXJ-2K-NS4B (expressing DENV NS4B) or the pXJ empty vector (control). At 24 h p.t., the cells were mocked treated or treated with 1,000 U of human IFN-β (Calbiochem) in the presence of 1 μM compound 1a or 0.5% DMSO. Cells were maintained in DMEM–10% FBS. At 24 h posttreatment, the cells were harvested, and luciferase activity was measured by using a dual-luciferase reporter assay system (Promega).
In vivo studies.
The Institutional Animal Care and Use Committee (IACUC) of the Novartis Institute for Tropical Diseases is registered with the Agri-Food and Veterinary Authority (AVA) of Singapore. All animal studies were approved by the IACUC. The pharmacokinetic (PK) profiles of compounds 1a and 14a (Table 1) (19) were determined in male Wistar rats (weighing 200 to 400 g) following intravenous (i.v.) and oral (p.o.) administration of 5 and 25 mg/kg of body weight, respectively. The PK profile of compound 14a was also determined in DENV-2-infected AG129 mice following p.o. administration of 100 mg/kg. The formulations used for i.v. and p.o. administrations were a solution in N-methylpyrrolidinone (NMP)-plasma (10:90, vol/vol) and a suspension in methylcellulose (0.5%, wt/vol) and Tween 80 (0.5%, vol/vol) in phosphate buffer (pH 6.8), respectively. Groups of three animals were used for each time point. Blood samples were collected at 0.17, 0.5, 1, 2, 4, 8, and 24 h after dosing (n = 3 per time point). Plasma concentrations of compounds 1a and 14a were measured by liquid chromatography-tandem mass spectrometry (LC-MS/MS) on an API 4000 mass spectrometer (AB Sciex, Framingham, MA, USA), using positive electrospray ionization and multiple-reaction monitoring (MRM) of m/z 415 → m/z 127 (compound 1a) or m/z 414 → m/z 144 (compound 14a). Chromatographic separation was performed on a Zorbax XDB-Phenyl column (4.6 by 75 mm, 3.5 μm; Agilent Technologies, Santa Clara, CA, USA), using a 5-min gradient elution with acetonitrile-water (containing 0.2% acetic acid) and a 1-ml/min flow rate. The lower limit of quantification achieved was 6 to 8 ng/ml. The PK parameters were calculated by a noncompartmental approach using WatsonLIMS software (Thermo Scientific).
TABLE 1.
In vitro pharmacokinetic and physicochemical properties of compounds 1a and 14a
| In vitro pharmacokinetic or physicochemical property | Value for compound |
|
|---|---|---|
| 1a | 14a | |
| LogP | 4.9 | 2.8 |
| Equilibrium solubility (mM) at pH 4.0/pH 6.8 | 0.025/0.011 | 0.729/0.504 |
| PAMPA, calculated fraction absorbed (%) | 68 | 79 |
| Plasma protein binding in mouse/rat (%) | >99/>99 | 98.6/96.4 |
| Microsomal stability, CLint in mouse/rat (μl · min−1 · mg−1) | 45/40 | 57/50 |
The in vivo efficacy of compound 14a was evaluated in a dengue viremia model (20). The model used 8- to 12-week-old AG129 mice (with knockout of IFN-α/β and IFN-γ receptors) purchased from B & K Universal and DENV-2 strain TSV01. The virus stock was propagated in C6/36 mosquito cells grown in RPMI 1640 medium, and 0.5 ml of 3 × 106 to 3 × 107 PFU/ml was injected intraperitoneally into the mice. The mice (n = 6 per group), either immediately or with a delay, as indicated, were dosed orally twice daily (BID) with compound 14a or the vehicle for 3 days. The formulation used was the same as the one described above for the PK studies. The mice were sacrificed on day 3 p.i. to collect blood samples for viral titer determinations using a plaque assay, as previously described (12). Statistical analysis was performed by a one-way analysis of variance (ANOVA) Tukey-Kramer test using Prism software (GraphPad).
RESULTS
Identification of a spiropyrazolopyridone as a novel anti-DENV agent.
We used a dual-replicon approach (luciferase-reporting replicons of DENV-2 and HCV) (Fig. 1A) to identify inhibitors that selectively suppress DENV replication (Fig. 1B). “Hits” identified from the DENV-2 replicon were tested in the HCV replicon. This approach allowed us to identify compound 1, containing a spiropyrazolopyridone core, as a specific inhibitor of DENV-2 (Fig. 1C). Compound 1 inhibited the DENV-2 replicon (EC50, 14 nM) but not the HCV replicon (EC50, >5 μM); no cytotoxicity was detected at concentrations of up to 5 μM (Fig. 1D). Concentrations of >5 μM were not tested because of the low aqueous solubility of compound 1 (11 μM). As a positive control, NITD-008, a previously reported nucleoside inhibitor of both DENV and HCV (12), exhibited EC50s of 1.6 and 0.18 μM, respectively (Fig. 1D).
FIG 1.

(A) Schematic diagram of the Renilla luciferase (RLUC)-expressing DENV-2 replicon (New Guinea C strain) and the firefly luciferase (LUC)-expressing HCV replicon. The diagram is not drawn to scale. C22, N-terminal 22 amino acids of the DENV-2 capsid protein; E24, C-terminal 24 amino acids of the E protein; Pu, puromycin N-acetyltransferase gene; FMDV 2A, foot-and-mouth disease virus 2A protein; IRES, internal ribosomal entry site from encephalomyocarditis virus; Ub, ubiquitin; Neo, neomycin resistance gene; UTR, untranslated region. (B) Dual-replicon screening flow chart. Compound libraries were screened at 5 μM by using Renilla luciferase DENV-2 replicon cells. Compounds with >70% inhibition of Renilla luciferase activity in the primary screening were subjected to a dose-response test in Renilla luciferase DENV-2 replicon cells to derive EC50 and CC50 values. The compounds were also tested against the HCV firefly luciferase replicon. Compounds that selectively inhibited the DENV-2 replicon were further examined in a Renilla luciferase reporter cell line to rule out Renilla luciferase inhibitors. The inhibitors were further validated in a viral titer reduction assay using authentic DENV. (C) Chemical structure of compound 1 (racemate). (D) Antiviral activities of compound 1 (Cpd 1). NITD-008, a nucleoside inhibitor of DENV and HCV (12), was included as a positive control. DENV-2 Pu-Renilla luciferase replicon BHK-21 cells or HCV Neo-firefly luciferase replicon Huh-7 cells were cultured in medium containing 3-fold serial dilutions of compound 1 for 2 days at 37°C. ViviRen (Promega) and Britelite (PerkinElmer) were added to the DENV-2 and HCV replicons, respectively. The luciferase signal was detected with a Clarity 4.0 plate reader, followed by cell viability detection by the addition of CellTiter-Glo (Promega). The luciferase assay and the cell viability assay were performed according to the manufacturers' protocols. Antiviral activity and cell viability are presented as the left y axis and right y axis, respectively.
Since compound 1 is a racemate, we purified the R (compound 1a) and S (compound 1b) enantiomers (Fig. 2A) and tested their antiviral activities. As shown in Fig. 2B, the R enantiomer of compound 1a (EC50, 12 nM) was >83-fold more potent than the S enantiomer of compound 1b (EC50, >1,000 nM). Next, we examined the antiviral spectrum of compound 1a against a panel of flaviviruses (DENV-1 to -4, Japanese encephalitis virus [JEV], West Nile virus [WNV], yellow fever virus [YFV], and Powassan virus [POWV]) and nonflaviviruses (Western equine encephalitis virus [WEEV] and vesicular stomatitis virus [VSV]). Surprisingly, compound 1a inhibited only DENV-2 and -3 but not other viruses, including DENV-1 and -4 (Fig. 2C). To define the step of compound inhibition, we performed a transient-transfection assay using a DENV-2 luciferase replicon (Fig. 2D). Compound 1a did not inhibit viral RNA translation (as indicated by the luciferase signals at 2 to 9 h) but suppressed viral RNA synthesis or accumulation (as indicated by the luciferase signals at ≥24 h) (Fig. 2D). A time-of-addition experiment showed that compound 1a added at ≥6 h gradually lost its potency, suggesting that the compound exerts its inhibition at the viral RNA synthesis/accumulation step or at a late stage of the infection cycle after viral entry (Fig. 2E). Besides mammalian cells, we tested if the compound is active in mosquito C6/36 cells. As shown in Fig. 2F, compound 1a exhibited an EC50 of 9 nM in DENV-2-infected C6/36 cells, demonstrating that the compound is active in both mammalian and mosquito cells. In addition, we tested compound 1a in a panel of DENV-2 enzyme assays (including protease, nucleotriphosphatase [NTPase], methyltransferase, or polymerase, as described previously [21]); the compound did not inhibit any of these enzymatic activities (data not shown). Collectively, the results indicate that the R enantiomer of compound 1a specifically inhibits DENV-2 and -3 through blocking viral RNA synthesis or accumulation. However, the current data do not allow us to differentiate the mechanisms of compound-mediated suppression of viral RNA synthesis from those of compound-mediated enhancement of viral RNA degradation.
FIG 2.

In vitro antiviral profile of the spiropyrazolopyridone compounds. (A) Structures of spiropyrazolopyridone compound 1a (R), compound 1b (S), and compound 14a (R). (B) Antiviral activities of two enantiomers (compounds 1a and 1b) of compound 1 against DENV-2. A549 cells were infected with DENV-2 (New Guinea C strain) at an MOI of 0.5 in the presence of 2-fold serial dilutions of compounds. After incubation at 37°C for 48 h, cell culture fluids were harvested for a plaque assay. The data are plotted as logarithm (log10) values of average viral titers from triplicates versus the compound concentration. The error bars represent standard deviations (n = 3). (C) Antiviral spectra of compound 1a and its analog compound 14a. A549 cells were infected with DENV-1, -2, -3, or -4 (MOI of 0.5). Vero cells were infected with WNV, YFV, JEV, POW, WEEV, or VSV (MOI of 0.1). EC50s were calculated by nonlinear regression analysis using Prism software (GraphPad). See Materials and Methods for assay details. (D) Transient-replicon assay. A549 cells were transfected with 10 μg of DENV-2 luciferase replicon RNA. The transfected cells were immediately treated with different concentrations of compound 1a or 0.9% DMSO (as a negative control). At the indicated time points p.i., cells were assayed for luciferase signals (quantified as relative light units [RLU]). The log10 values of average luciferase signals and standard deviations are presented (n = 3). (E) Time-of-addition analysis. A549 cells were infected with DENV-2 (New Guinea C strain) at an MOI of 2 at 4°C for 1 h. After three washes with PBS to remove unbound viruses, cells were incubated at 37°C. At the indicated time points, compound 1a (1 μM) was added to the infected cells. As controls, the infected cells were treated with 0.5% DMSO. At 24 h p.i., the culture medium was collected, and viral titers were determined by a plaque assay. Average results and standard errors (n = 3) are presented. (F) Antiviral activity in mosquito C6/36 cells. C3/36 cells were infected with DENV-2 at an MOI of 1. Compound 1a was added at the indicated concentrations to cells immediately after infection. The culture supernatant was collected at 48 h p.i., and viral titers were quantified by a plaque assay.
Improvement of physicochemical properties of inhibitors.
Medicinal chemistry was used to improve the physicochemical properties of compound 1a, resulting in the analog compound 14a (Fig. 2A). Compared with compound 1a, compound 14a exhibited slightly decreased potency against DENV-2 and -3 and retained a similar antiviral spectrum (Fig. 2C). However, compound 14a showed improved aqueous solubility, from 11 μM (compound 1a) to 504 μM, and an improved logP value, from 4.9 (compound 1a) to 2.8 (Table 1). The detailed structure-activity relationship of medicinal chemistry was recently reported (19).
Spiropyrazolopyridone resistance maps to the viral NS4B protein.
To identify the target of the spiropyrazolopyridone inhibitor, we selected resistant virus by culturing DENV-2 with increasing concentrations of compound 1a (Fig. 3A). All viruses from six independent selections showed increased EC50s, from 12 nM to >1 μM (Fig. 3B), suggesting the emergence of resistant viruses. Complete genome sequencing of the resistant viruses revealed nucleotide changes at positions 7012 and/or 7013 of the genomic RNA, leading to a V63A, V63L, V63M, V63S, or V63T amino acid substitution in the NS4B protein (Fig. 3B). Specifically, selections I to IV contained mixed populations at amino acid position 63, whereas selections V and VI yielded identical V63M mutations. Based on the topology model of DENV NS4B (22), amino acid V63 is located in the second predicted transmembrane domain (pTMD2) in the endoplasmic reticulum (ER) lumen (Fig. 3C). To determine whether these mutations were responsible for resistance, we prepared a panel of recombinant DENV-2 strains containing individual NS4B mutations, including V63A, V63L, V63M, V63T, and V63S. Figure 3D shows the IFA results for the genome-length-RNA-transfected cells (monitoring viral E protein expression) (top), plaque morphology of the recombinant viruses (middle), and viral titers determined on day 5 p.t. (bottom). The results showed that none of the mutations significantly affected viral replication in cell culture, except that the V63S mutant virus exhibited smaller plaques (Fig. 3D). Resistance analysis (using a viral titer reduction assay) revealed that the V63A substitution conferred ∼10.6-fold resistance to compound 1a, while other substitutions conferred >83-fold resistance (Fig. 3E), demonstrating that the engineered mutations are responsible for resistance.
FIG 3.

Characterization of resistant viruses. (A) Scheme of selection and validation of DENV-2 that is resistant to compound 1a inhibition. WT DENV-2 was used for resistance selection. The virus was cultured in A549 cells with increasing concentrations of compound 1a. The EC50 for each selected isolate was measured at passage 16 (P16) by using qRT-PCR. The P16 viral RNA from each of the six independent selections was extracted for whole-genome sequencing. The extracted RNA was amplified by using SuperScript One-Step RT-PCR with Platinum Taq (Life Technologies). The RT-PCR products were sequenced. The identified mutations were introduced into a DENV-2 infectious cDNA clone to generate recombinant viruses. The resistance levels of recombinant viruses were evaluated by a viral titer reduction assay in A549 cells. (B) Resistance profile of the DENV-2 escape mutant. Mutations at position V63 of DENV-2 NS4B were consistently recovered from six independent resistant virus selections. DMSO (0.9%) was used as a negative control during resistance selection. Mixed mutations of V63 were obtained from selections I to IV. The sensitivities of the escape viruses to compound 1a (indicated by EC50s) were compared to that of the WT virus. Fold resistance was calculated as the EC50 for the resistant virus divided by the EC50 for the WT virus. (C) Membrane topology and sequence alignment of the DENV NS4B protein. The identified V63 mutation is located in predicted transmembrane domain 2 (pTMD2) of the NS4B topology (22). An amino acid sequence alignment of the pTMD2 regions (residues 60 to 83) of different flavivirus NS4B proteins is shown. The amino acid positions of NS4B are numbered according to DENV-2 numbering (GenBank accession number AY037116). The V63 mutation in DENV-2 NS4B is highlighted; the equivalent position in DENV-1 NS4B is I64, as indicated. (D) Characterization of DENV-2 NS4B V63 mutants. BHK-21 cells were transfected with 10 μg of WT or NS4B mutant genome-length RNA of DENV-2. The transfected cells were monitored for viral E protein expression by IFA at days 2 and 4 p.t. (Top) Anti-E monoclonal antibody 4G2 and Alexa Fluor 488 goat anti-mouse IgG were used as the primary and secondary antibodies, respectively. (Middle) Plaque morphology of recombinant WT and mutant viruses. (Bottom) Viral titers in the culture fluids on day 5 p.t. were quantitated by a plaque assay. (E) Summary of resistance of the recombinant DENV-2 V63 mutants. Six recombinant DENV-2 V63 mutants were prepared and tested against compound 1a. The EC50s and fold resistance values are shown.
Sequence alignment of the NS4B proteins among flaviviruses showed that V63 is absolutely conserved in DENV-2 and -3 but different in DENV-1 and -4 as well as in other flaviviruses (Fig. 3C). Remarkably, resistance residue 63L exists as the WT amino acid in the NS4B proteins of DENV-4, TBEV, and YFV; resistance residue 63T exists as the WT amino acid in the NS4B proteins of JEV and WNV. The variation at residue 63 of flavivirus NS4B could account for the selective inhibition of DENV-2 and -3 by compound 1a. To demonstrate this point, we prepared recombinant DENV-1 mutant containing an NS4B I64V mutation (equivalent to residue 63 of DENV-2 NS4B). Compared with WT DENV-1, the DENV-1 I64V mutant exhibited equivalent amounts of IFA-positive cells after transfection of the genome-length RNAs into BHK-21 cells (Fig. 4A, left). Cells transfected with WT and I64V mutant RNAs produced comparable viral titers on day 5 p.t. (Fig. 4A, right). Recombinant WT DENV-1 and the I64V mutant exhibited similar plaque morphologies (Fig. 4A, middle). These results indicate that the I64V mutation does not attenuate DENV-1 replication. Strikingly, the DENV-1 I64V mutant became sensitive to compound 1a inhibition, with an EC50 shift from >1 μM (WT DENV-1) to 140 nM (Fig. 4B). It should be noted that the EC50 of the DENV-1 I64V mutant (140 nM) is 11.6-fold higher than that of WT DENV-2 (12 nM), suggesting that residues outside I64 of DENV-1 NS4B also contribute to drug sensitivity. Nevertheless, these results demonstrate that single-amino-acid variation of NS4B plays an important role in the antiviral selectivity against the four serotypes of DENV.
FIG 4.
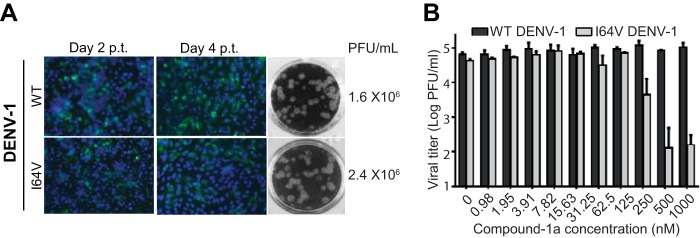
Characterization of the recombinant DENV-1 I64V mutant. (A) BHK-21 cells were transfected with 10 μg of WT or NS4B mutant genome-length RNA of DENV-1 (West Pacific strain). The transfected cells were monitored for viral E protein expression by IFA at days 2 and 4 posttransfection. (Left) Anti-E monoclonal antibody 4G2 and Alexa Fluor 488 goat anti-mouse IgG were used as the primary and secondary antibodies, respectively. (Middle) Plaque morphology of WT DENV-1 and the I64V mutant. (Right) Viral titers in the culture fluids at day 5 p.t. were determined by a plaque assay. (B) Conversion of DENV-1 to sensitivity to compound 1a inhibition by an I64V mutation. Recombinant DENV-1 containing the I64V mutation was generated. The effect of compound 1a on the DENV-1 I64V mutant was determined by a viral titer reduction assay in A549 cells. The logarithm (log10) values of average viral titers from triplicates are plotted against the compound 1a concentration. Average results and standard deviations (n = 3) are presented.
Cross-resistance analysis of distinct NS4B inhibitors.
We previously reported a different scaffold of the NS4B inhibitor NITD-618 (23). Resistance mutations for NITD-618 were mapped to NS4B residues P104L and A119T, both of which are located in the third transmembrane domain (TMD3) spanning residues 101 to 129 (Fig. 5A). To examine if the current inhibitors are cross-resistant to NITD-618, we tested the antiviral activities of compound 1a (Fig. 5B, top) and compound 14a (middle) in the DENV-2 replicon containing the P104L+A119T double mutation. Compound 1a showed comparable EC50s in the WT replicon (9 nM) and the P104L+A119T replicon (7 nM). Similarly, compound 14a showed equivalent EC50s in the WT replicon (43 nM) and the P104L+A119T replicon (39 nM). As expected, NITD-618 exhibited a higher EC50 in the P104L+A119T replicon (5 μM) than in the WT replicon (0.5 μM) (Fig. 5B, bottom). These results demonstrate that the current inhibitors are not cross-resistant to the previous NS4B inhibitor NITD-618.
FIG 5.
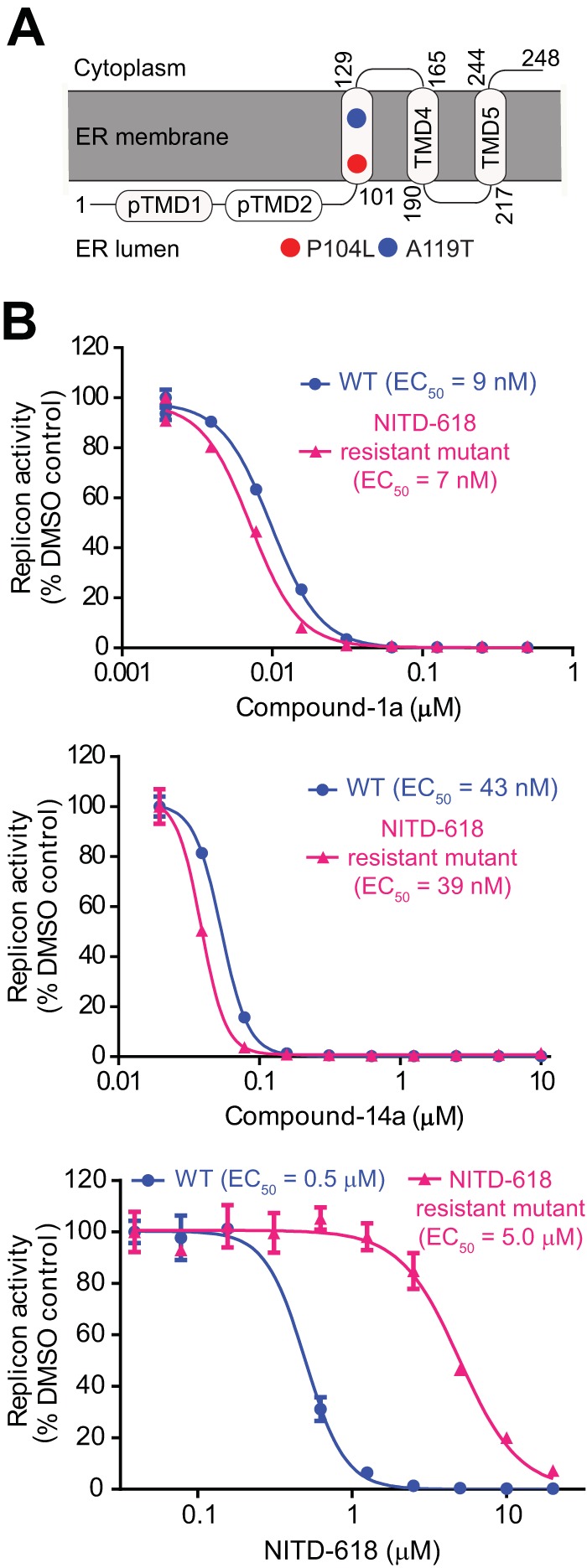
Cross-resistance analysis of distinct NS4B inhibitors. (A) Mapping of resistance mutations within NS4B. Amino acid changes (P104L and A119) associated with NITD-618 resistance are depicted in circles. (B) Analysis of cross-resistance between NITD-618 and spiropyrazolopyridone compounds. A549 cells containing the DENV-2 WT or P104L A119T mutant luciferase replicon were treated with compound 1a (top), compound 14a (middle), and NITD-618 (bottom) at the indicated concentrations for 48 h. The inhibition of viral replication was measured by the luciferase activity. EC50s are shown.
The spiropyrazolopyridone compound directly interacts with NS4B.
To demonstrate a direct compound-NS4B interaction, we prepared 3H-labeled compound 14a and two recombinant proteins (WT and resistant V63I mutant) representing the first 125 amino acids of DENV-2 NS4B (Fig. 6A). Based on the DENV-1 NS4B sequence (Fig. 3C), we selected the V63I mutation in DENV-2 NS4B for the compound-protein binding experiment. Gel filtration analysis showed that 3H-labeled compound 14a coeluted with the WT NS4B protein; in contrast, the resistant V63I mutant reduced the amount of 3H-labeled compound 14a coelution by 3-fold (Fig. 6B). As a control, 3H-labeled compound 14a did not coelute with recombinant NS4A, a different transmembrane protein of DENV-2 (Fig. 6B). These results suggest that compound 14a can directly bind to the DENV-2 NS4B protein. To confirm the gel filtration results, we attempted several biophysical methods (e.g., surface plasmon resonance and isothermal calorimetry); none of these methods proved to be feasible for measuring the compound-protein interaction (data not shown), possibly due to the challenging nature of the membrane protein.
FIG 6.
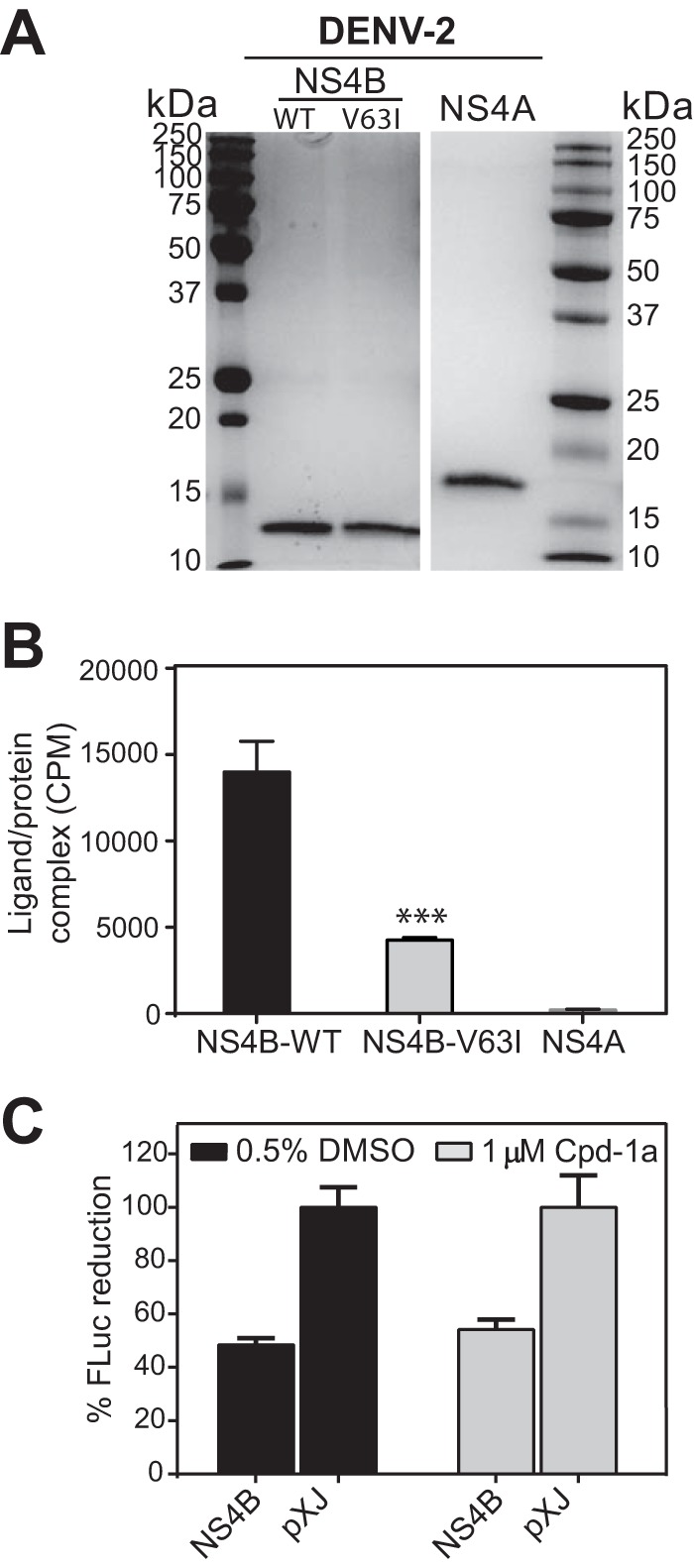
Binding of 3H-labeled compound 14a to recombinant NS4B. (A) SDS-PAGE analysis of recombinant DENV-2 NS4A and NS4B proteins. Full-length NS4A and the N-terminal 125 amino acids of DENV-2 NS4B were expressed and purified by using an E. coli expression system (18). The molecular masses (kilodaltons) of protein standards (marker) are indicated on the left and right. (B) Gel filtration analysis of binding of 3H-labeled compound 14a to the NS4B protein. 3H-labeled compound 14a was mixed with the indicated proteins and centrifuged through a Micro BioSpin 6 gel filtration column. Protein-bound 3H-labeled compound-14a was quantitated in the eluent by using a beta scintillation counter (PerkinElmer Life Sciences). NS4A served as a negative control. Analysis of each data point was carried out in triplicate. ***, P < 0.001 for comparison of binding of the compound to WT and V63I mutant NS4B proteins. (C) Effect of compound 1a on the induction of the ISRE-Luc reporter gene after treatment with IFN. A plasmid containing the coding region of DENV-2 2K-NS4B was cotransfected into HEK-293T cells together with the ISRE-Luc plasmid. At 24 h p.t., the cells were stimulated with 1,000 U of IFN-β in the presence or absence of 1 μM compound 1a. The cells were further incubated for 24 h, followed by quantification of luciferase expression. Results show the mean percentages of luciferase activity for each treatment (n = 3).
The compound does not inhibit DENV-2 through blocking of the NS4B-mediated antagonism of IFN signaling.
The pTMD1 and pTMD2 regions of DENV NS4B were reported to antagonize IFN signaling (23, 24). Since the resistance of spiropyrazolopyridone compounds was mapped to pTMD2 of NS4B, we examined the inhibitory effect of compound 1a on blocking the NS4B-mediated antagonism of IFN signaling. HEK-293T cells were cotransfected with a plasmid expressing DENV-2 2K-NS4B (the 2K fragment was included to ensure the correct topology of NS4B on the ER membrane) or an empty plasmid (as a control) together with another plasmid expressing firefly luciferase under the control of an ISRE promoter. A plasmid expressing Renilla luciferase was also cotransfected to normalize the transfection efficiency. At 24 h p.t., the cells were mock treated or treated with human IFN-β in the presence of either compound 1a or DMSO. Dual-luciferase activities were measured after incubation for another 24 h. As shown in Fig. 6C, the expression of NS4B reduced the ISRE promoter-driven firefly luciferase signal by 52% in the presence of DMSO, confirming the role of NS4B in inhibiting IFN signaling (23, 24). A similar reduction (46%) in the firefly luciferase activity was observed in cells that were treated with compound 1a, suggesting that the function of NS4B in antagonizing IFN signaling was not affected by the compound. These results allowed us to conclude that compound 1a exerts its anti-DENV activity via a mechanism other than antagonizing the anti-IFN function of NS4B.
Pharmacology of spiropyrazolopyridones.
To select a compound for in vivo efficacy studies, we compared the pharmacokinetics of compound 1a and compound 14a in rats. Compound 14a showed a better pharmacological profile than did compound 1a. The detailed PK parameters were recently reported (19). The improved pharmacological parameters of compound 14a are most likely due to the improved compound solubility (Table 1). We therefore selected compound 14a for in vivo efficacy studies.
The in vivo efficacy of compound 14a was examined in a DENV-2/AG129 mouse (lacking IFN-α/β and IFN-γ receptors) model (20). AG129 mice were infected with DENV-2 and immediately treated with compound 14a via the oral route twice daily (BID) for 3 days. Peak viremia on day 3 p.i. was quantified by a plaque assay. Treatment with 5, 25, and 50 mg/kg of compound-14a BID reduced viremia by 1.7-, 10-, and 39-fold, respectively. Viremia was below the limit of detection (250 PFU/ml) in four out of six animals in the group that received 100 mg/kg BID (Fig. 7A). No disease or adverse events were observed in the infected animals. To correlate the observed antiviral activity with compound exposure, we measured the pharmacokinetics of compound 14a (100 mg/kg) in infected mice (Fig. 7B). Upon oral dosing, the plasma level of compound 14a increased steadily to a maximum concentration of drug in serum (Cmax) of 221 μM (equivalent to 91,450 ng/ml) at 0.25 h postdosing; the area under the concentration-time curve (AUC) from 0 to 24 h reached 517,728 ng · h/ml. At 24 h postdosing, the plasma concentration of compound 14a remained above the EC90 value (0.2 μM). Collectively, these results demonstrate that compound 14a has good in vivo pharmacokinetic properties, leading to in vivo antiviral efficacy.
FIG 7.
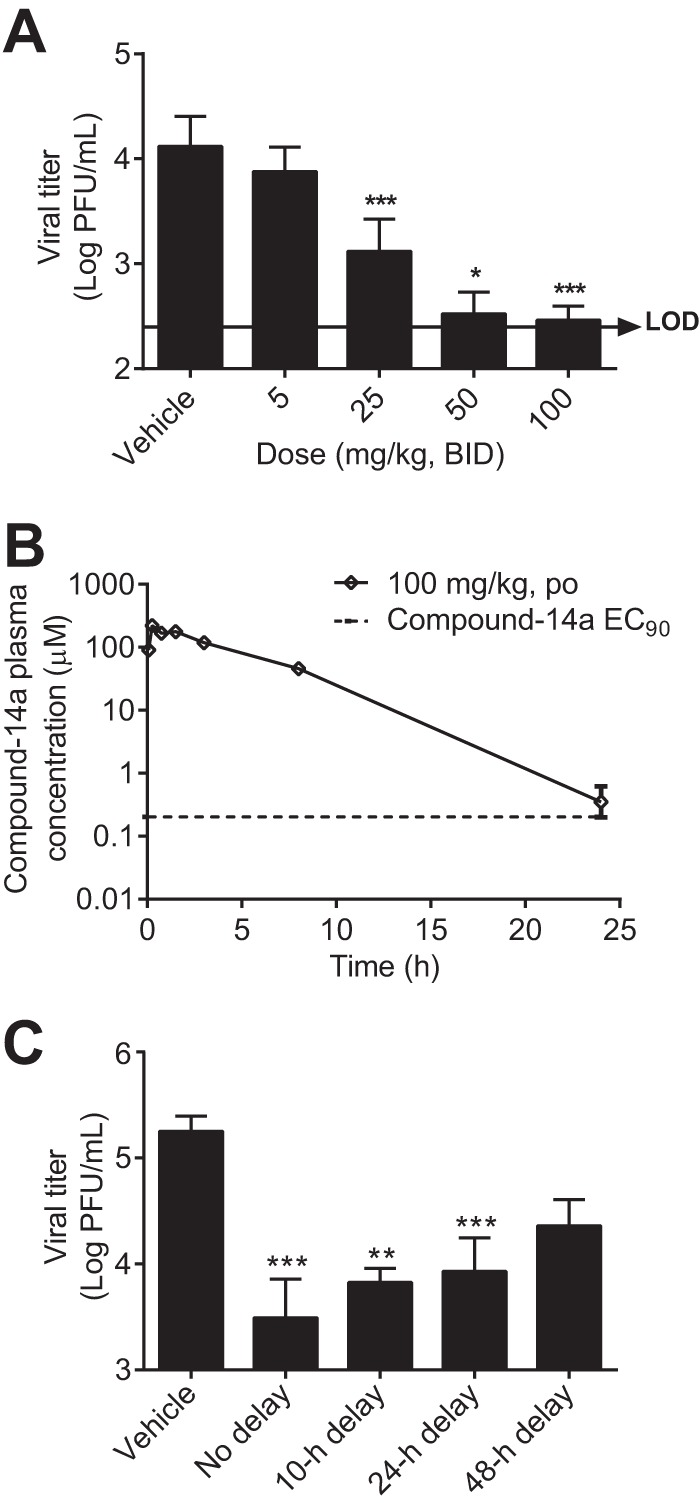
In vivo pharmacokinetics and efficacy of compound 14a. (A) AG129 mice (8 to 12 weeks old; 6 mice per treatment group) were infected with 1.5 × 106 PFU of DENV-2 (strain TSV01) on day 0. The mice were treated orally with 5, 25, 50, or 100 mg/kg compound 14a BID. The peak viremia on day 3 p.i. was quantified by a plaque assay. A one-way ANOVA Tukey-Kramer test indicates that, compared with the vehicle control, the reduction in viremia in all treatment groups, except for the 5-mg/kg group, is statistically significant. *, P < 0.05; **, P < 0.01; ***, P < 0.001. LOD, limit of detection (250 PFU/ml). Values below the limit of detection were set equal to 250 PFU/ml. The antiviral activities with treatment doses of 50 and 100 mg/kg were recently reported (19) (B) Plasma concentrations of compound 14a after oral administration to DENV-2-infected AG129 mice at a dosage of 100 mg/kg (n = 3 per time point). The EC90 (0.2 μM) of compound 14a is indicated by the dotted line. (C) Peak viremia of mice with a delayed start of treatment after infection with 1.5 × 107 PFU of DENV-2 (strain TSV01) (dosed at 100 mg/kg).
To simulate treatment in a clinical setting, we examined the efficacy of compound 14a (100 mg/kg BID) in a delayed treatment. Treatments started at 10, 24, or 48 h after infection; viremia was quantified at 72 h postinfection. Compared with immediate treatment, the viremia reduction level decreased from 57-fold to 27-fold (10-h delay), 21-fold (24-h delay), and 8-fold (48-h delay) (Fig. 7C). However, statistical analysis showed that viremia reduction in the treatment group with a 48-h delay was not significant. Overall, these results demonstrate that compound 14a is efficacious even when treatment starts postinfection.
In vitro safety pharmacological profiling of compound 14a.
Table 2 summarizes the in vitro safety profiling of compound 14a in various cellular receptors, ion channels, transporters, and metabolic enzymes. The compound did not show interference with these host proteins, with 50% inhibitory concentrations (IC50s) of >30 μM. These results suggest that compound 14a has a good in vitro safety profile.
DISCUSSION
We used a dual DENV-2 and HCV replicon approach to identify inhibitors that selectively suppress DENV replication. This approach allowed us to uncover the spiropyrazolopyridone scaffold as a potent inhibitor of DENV-2 and -3. Resistance analysis and direct ligand-protein binding indicate that DENV-2 NS4B is the target of the spiropyrazolopyridone inhibitors. Our results suggest that these inhibitors do not exert their antiviral activity via blocking of the NS4B-mediated evasion of innate immunity. Since NS4B functions as an essential component of the viral replication complex, the inhibitors could function by interfering with protein-protein interactions during viral RNA synthesis. In line with this hypothesis, NS4B was recently reported to oligomerize itself (18) as well as to interact with NS1 (24), NS3 (25, 26), and NS4A (27). Besides binding to viral proteins, NS4B is also expected to interact with host factors during viral replication. It is conceivable that small-molecule inhibitors blocking such interactions could lead to the suppression of viral replication. Further studies are needed to elucidate the exact mechanism of how these inhibitors block viral RNA synthesis.
Our study has proven the concept that DENV NS4B is a valid antiviral target. Compared with previously reported flavivirus NS4B inhibitors (23, 28–30), the spiropyrazolopyridone compounds have superior in vitro potency and exhibited in vivo efficacy; none of the previous NS4B inhibitors showed any in vivo efficacy. In the AG129 mouse model, compound 14a showed potency when mice were treated with the inhibitor immediately after infection or when treatment was delayed by 48 h. Since peak viremia was measured at 72 h p.i. in the AG129 mouse model, the 48-h delay allowed only 24 h of compound treatment of the infected mice. These in vivo efficacy results underscore the potency of compound 14a. The resistance of spiropyrazolopyridone compounds (V63 mutation of DENV-2 NS4B) is also distinct from those of other NS4B inhibitors such as NITD-618 (P104L and A119T of NS4B) (23). Indeed, spiropyrazolopyridone compounds showed comparable antiviral activities in the WT and NS4B P104L+A119T mutant replicons (Fig. 5). More importantly, compound 14a exhibits good in vivo pharmacokinetics and physicochemical properties (Table 1). It also has a good in vitro safety pharmacology profile (Table 2). Collectively, the above-described profile warrants consideration of this compound as a bona fide DENV inhibitor for preclinical development.
TABLE 2.
In vitro safety pharmacology profiling of compound 14a
| Targeta | Assayb | Species | IC50 (μM) |
|---|---|---|---|
| Serotonin 5-HT 1A receptor | Functional | Human | |
| Serotonin 5-HT 2A receptor | Functional | Human | >30 |
| Serotonin 5-HT 2B receptor | Functional | Human | >30 |
| Serotonin 5-HT 2C receptor | Binding | Human | >30 |
| Serotonin 5-HT 3 channel | Binding | Human | >30 |
| Serotonin transporter | Binding | Human | >30 |
| Acetylcholinesterase | Binding | Human | >30 |
| Adenosine 1 receptor | Binding | Human | >30 |
| Adenosine 2a receptor | Binding | Human | >30 |
| Adenosine 3 receptor | Binding | Human | >30 |
| Adenosine transporter | Binding | Human | >30 |
| Adrenergic alpha 1A receptor | Functional | Human | >30 |
| Adrenergic alpha 2A receptor | Functional | Human | >30 |
| Androgen receptor | Functional | Rat | >30 |
| Angiotensin II AT1 receptor | Binding | Human | >30 |
| Adrenergic beta 1 receptor | Binding | Human | >30 |
| Adrenergic beta 2 receptor | Functional | Human | >30 |
| Bile salt export pump | Functional | Human | >30 |
| Cannabinoid 1 receptor | Functional | Human | >30 |
| Cholecystokinin A receptor | Binding | Human | >30 |
| Cyclooxygenase-1 | Binding | Human | >30 |
| Cyclooxygenase-2 | Binding | Human | >30 |
| Dopamine D1 receptor | Functional | Human | >30 |
| Dopamine D2 receptor | Binding | Human | >30 |
| Dopamine D3 receptor | Binding | Human | >30 |
| Dopamine transporter | Binding | Human | 14 |
| Estrogen alpha receptor | Functional | Human | >30 |
| Endothelin A receptor | Binding | Human | >30 |
| GABA-A receptor | Functional | Human | >30 |
| Ghrelin receptor | Binding | Human | >30 |
| Glucocorticoid receptor | Functional | Human | >30 |
| Histamine H1 receptor | Binding | Human | >30 |
| Histamine H3 receptor | Binding | Human | >30 |
| Epidermal growth factor receptor | Binding | Human | >30 |
| v-erb-b2 erythroblastic leukemia viral oncogene homolog 2, transcript variant 1 | Binding | Human | >30 |
| Kinase insert domain receptor | Binding | Human | >30 |
| Muscarinic M1 receptor | Binding | Human | >30 |
| Muscarinic M2 receptor | Functional | Human | >30 |
| Muscarinic M3 receptor | Binding | Human | >30 |
| Monoamine oxidase A | Binding | Human | >30 |
| Melanocortin 3 receptor | Binding | Human | >30 |
| Motilin receptor | Binding | Human | >30 |
| Norepinephrine transporter | Binding | Human | >30 |
| Nicotinic receptor nonrecombinant central | Binding | Human | >30 |
| Opiate delta receptor | Binding | Human | >30 |
| Opiate mu receptor | Binding | Human | >30 |
| Phosphodiesterase 3 | Binding | Human | >30 |
| Phosphodiesterase 4D | Binding | Human | >30 |
| PPAR gamma receptor | Functional | Human | >30 |
| Progesterone receptor | Functional | Human | >30 |
| Pregnane X receptor | Functional | Human | >30 |
| Vasopressin 1a receptor | Binding | Human | >30 |
| Cathepsin D | Enzymatic | Human | >30 |
| Caspase 3 | Enzymatic | Human | >30 |
| Thrombin | Enzymatic | Human | >30 |
| MMP8 | Enzymatic | Human | >30 |
PPAR, peroxisome proliferator-activated receptor; MMP8, matrix metalloproteinase-8.
The functional assays were performed by using purified membrane fractions bearing the receptors of interest and radioligand. The binding assays were performed by using radioligand on mostly G-protein-coupled receptors and some transporters, ion channels, and enzymes. The enzymatic assays were performed by using a peptide substrate carrying the fluorophore PT14.
The major weakness of compound 14a is the lack of efficacy against DENV-1 and -4. Medicinal chemistry is needed to improve its panserotype activity. Achieving panserotype activity will be challenging, since the amino acid variation among the four DENV serotypes is ∼30 to 35% (31). The same challenge was encountered when developing pangenotype inhibitors of HCV (32). The potent antiviral activity of compound 14a against DENV-2 and -3 complements the weak efficacy against DENV-2 of the current frontrunner vaccine CYD-TDV (3–5). Most importantly, the current compound has provided an opportunity to answer two paramount questions for dengue therapeutics development: can an antiviral drug reduce viremia in dengue patients (infected with DENV-2 or -3), and, if so, will viremia reduction prevent patients from developing severe hemorrhagic fever and shock? Since compound 14a is not a prodrug and directly inhibits viral NS4B, it is expected to eliminate the complications of nucleoside inhibitors (which require host kinases to convert them to their triphosphate forms before being antiviral active), as evidenced by the efficacy failure of balapiravir in dengue patients (6, 33).
ACKNOWLEDGMENTS
We thank colleagues at the Novartis Institute for Tropical Diseases (NITD) for helpful discussions and technical support during the course of this study.
This project was partially supported by funding from the TCR flagship STOP Dengue program of the National Medical Research Council in Singapore to the NITD.
REFERENCES
- 1.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI. 2013. The global distribution and burden of dengue. Nature 496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fink K, Shi PY. 2014. Live attenuated vaccine: the first clinically approved dengue vaccine? Expert Rev Vaccines 13:185–188. doi: 10.1586/14760584.2014.870888. [DOI] [PubMed] [Google Scholar]
- 3.Sabchareon A, Wallace D, Sirivichayakul C, Limkittikul K, Chanthavanich P, Suvannadabba S, Jiwariyavej V, Dulyachai W, Pengsaa K, Wartel TA, Moureau A, Saville M, Bouckenooghe A, Viviani S, Tornieporth NG, Lang J. 2012. Protective efficacy of the recombinant, live-attenuated, CYD tetravalent dengue vaccine in Thai schoolchildren: a randomised, controlled phase 2b trial. Lancet 380:1559–1567. doi: 10.1016/S0140-6736(12)61428-7. [DOI] [PubMed] [Google Scholar]
- 4.Capeding MR, Tran NH, Hadinegoro SR, Ismail HI, Chotpitayasunondh T, Chua MN, Luong CQ, Rusmil K, Wirawan DN, Nallusamy R, Pitisuttithum P, Thisyakorn U, Yoon IK, van der Vliet D, Langevin E, Laot T, Hutagalung Y, Frago C, Boaz M, Wartel TA, Tornieporth NG, Saville M, Bouckenooghe A, CYD14 Study Group. 2014. Clinical efficacy and safety of a novel tetravalent dengue vaccine in healthy children in Asia: a phase 3, randomised, observer-masked, placebo-controlled trial. Lancet 384:1358–1365. doi: 10.1016/S0140-6736(14)61060-6. [DOI] [PubMed] [Google Scholar]
- 5.Villar L, Dayan GH, Arredondo-Garcia JL, Rivera DM, Cunha R, Deseda C, Reynales H, Costa MS, Morales-Ramirez JO, Carrasquilla G, Rey LC, Dietze R, Luz K, Rivas E, Miranda Montoya MC, Cortes Supelano M, Zambrano B, Langevin E, Boaz M, Tornieporth N, Saville M, Noriega F, CYD15 Study Group. 2015. Efficacy of a tetravalent dengue vaccine in children in Latin America. N Engl J Med 372:113–123. doi: 10.1056/NEJMoa1411037. [DOI] [PubMed] [Google Scholar]
- 6.Nguyen NM, Tran CN, Phung LK, Duong KT, Huynh HA, Farrar J, Nguyen QT, Tran HT, Nguyen CV, Merson L, Hoang LT, Hibberd ML, Aw PP, Wilm A, Nagarajan N, Nguyen DT, Pham MP, Nguyen TT, Javanbakht H, Klumpp K, Hammond J, Petric R, Wolbers M, Nguyen CT, Simmons CP. 2013. A randomized, double-blind placebo controlled trial of balapiravir, a polymerase inhibitor, in adult dengue patients. J Infect Dis 207:1442–1450. doi: 10.1093/infdis/jis470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Low JG, Sung C, Wijaya L, Wei Y, Rathore AP, Watanabe S, Tan BH, Toh L, Chua LT, Hou Y, Chow A, Howe S, Chan WK, Tan KH, Chung JS, Cherng BP, Lye DC, Tambayah PA, Ng LC, Connolly J, Hibberd ML, Leo YS, Cheung YB, Ooi EE, Vasudevan SG. 2014. Efficacy and safety of celgosivir in patients with dengue fever (CELADEN): a phase 1b, randomised, double-blind, placebo-controlled, proof-of-concept trial. Lancet Infect Dis 14:706–715. doi: 10.1016/S1473-3099(14)70730-3. [DOI] [PubMed] [Google Scholar]
- 8.Tricou V, Minh NN, Van TP, Lee SJ, Farrar J, Wills B, Tran HT, Simmons CP. 2010. A randomized controlled trial of chloroquine for the treatment of dengue in Vietnamese adults. PLoS Negl Trop Dis 4:e785. doi: 10.1371/journal.pntd.0000785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tam DT, Ngoc TV, Tien NT, Kieu NT, Thuy TT, Thanh LT, Tam CT, Truong NT, Dung NT, Qui PT, Hien TT, Farrar JJ, Simmons CP, Wolbers M, Wills BA. 2012. Effects of short-course oral corticosteroid therapy in early dengue infection in Vietnamese patients: a randomized, placebo-controlled trial. Clin Infect Dis 55:1216–1224. doi: 10.1093/cid/cis655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim SP, Wang QY, Noble CG, Chen YL, Dong H, Zou B, Yokokawa F, Nilar S, Smith P, Beer D, Lescar J, Shi PY. 2013. Ten years of dengue drug discovery: progress and prospects. Antiviral Res 100:500–519. doi: 10.1016/j.antiviral.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 11.Ng CY, Gu F, Phong WY, Chen YL, Lim SP, Davidson A, Vasudevan SG. 2007. Construction and characterization of a stable subgenomic dengue virus type 2 replicon system for antiviral compound and siRNA testing. Antiviral Res 76:222–231. doi: 10.1016/j.antiviral.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 12.Yin Z, Chen YL, Schul W, Wang QY, Gu F, Duraiswamy J, Kondreddi RR, Niyomrattanakit P, Lakshminarayana SB, Goh A, Xu HY, Liu W, Liu B, Lim JY, Ng CY, Qing M, Lim CC, Yip A, Wang G, Chan WL, Tan HP, Lin K, Zhang B, Zou G, Bernard KA, Garrett C, Beltz K, Dong M, Weaver M, He H, Pichota A, Dartois V, Keller TH, Shi PY. 2009. An adenosine nucleoside inhibitor of dengue virus. Proc Natl Acad Sci U S A 106:20435–20439. doi: 10.1073/pnas.0907010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qing M, Zou G, Wang QY, Xu HY, Dong H, Yuan Z, Shi PY. 2010. Characterization of dengue virus resistance to brequinar in cell culture. Antimicrob Agents Chemother 54:3686–3695. doi: 10.1128/AAC.00561-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zou G, Chen YL, Dong H, Lim CC, Yap LJ, Yau YH, Shochat SG, Lescar J, Shi PY. 2011. Functional analysis of two cavities in flavivirus NS5 polymerase. J Biol Chem 286:14362–14372. doi: 10.1074/jbc.M110.214189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi PY, Tilgner M, Lo MK, Kent KA, Bernard KA. 2002. Infectious cDNA clone of the epidemic West Nile virus from New York City. J Virol 76:5847–5856. doi: 10.1128/JVI.76.12.5847-5856.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zou G, Xu HY, Qing M, Wang QY, Shi PY. 2011. Development and characterization of a stable luciferase dengue virus for high-throughput screening. Antiviral Res 91:11–19. doi: 10.1016/j.antiviral.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 17.Puig-Basagoiti F, Tilgner M, Forshey BM, Philpott SM, Espina NG, Wentworth DE, Goebel SJ, Masters PS, Falgout B, Ren P, Ferguson DM, Shi PY. 2006. Triaryl pyrazoline compound inhibits flavivirus RNA replication. Antimicrob Agents Chemother 50:1320–1329. doi: 10.1128/AAC.50.4.1320-1329.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zou J, Xie X, Lee LT, Chandrasekaran TR, Reynaud A, Yap L, Wang QY, Dong H, Kang C, Yuan Z, Lescar J, Shi PY. 2014. Dimerization of flavivirus NS4B protein. J Virol 88:3379–3391. doi: 10.1128/JVI.02782-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zou B, Chan WL, Ding M, Leong SY, Nilar S, Seah PG, Liu W, Karuna R, Blasco F, Yip A, Chao A, Susila A, Dong H, Wang QY, Xu HY, Chan K, Wan KF, Gu F, Diagana TT, Wagner T, Dix I, Shi P-Y, Smith PW. 2015. Lead optimization of spiropyrazolopyridones: a new and potent class of dengue virus inhibitors. ACS Med Chem Lett 6:344–348. doi: 10.1021/ml500521r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schul W, Liu W, Xu HY, Flamand M, Vasudevan SG. 2007. A dengue fever viremia model in mice shows reduction in viral replication and suppression of the inflammatory response after treatment with antiviral drugs. J Infect Dis 195:665–674. doi: 10.1086/511310. [DOI] [PubMed] [Google Scholar]
- 21.Noble CG, Chen YL, Dong H, Gu F, Lim SP, Schul W, Wang QY, Shi PY. 2010. Strategies for development of dengue virus inhibitors. Antiviral Res 85:450–462. doi: 10.1016/j.antiviral.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 22.Miller S, Sparacio S, Bartenschlager R. 2006. Subcellular localization and membrane topology of the dengue virus type 2 non-structural protein 4B. J Biol Chem 281:8854–8863. doi: 10.1074/jbc.M512697200. [DOI] [PubMed] [Google Scholar]
- 23.Xie X, Wang QY, Xu HY, Qing M, Kramer L, Yuan Z, Shi PY. 2011. Inhibition of dengue virus by targeting viral NS4B protein. J Virol 85:11183–11195. doi: 10.1128/JVI.05468-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Youn S, Li T, McCune BT, Edeling MA, Fremont DH, Cristea IM, Diamond MS. 2012. Evidence for a genetic and physical interaction between nonstructural proteins NS1 and NS4B that modulates replication of West Nile virus. J Virol 86:7360–7371. doi: 10.1128/JVI.00157-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Umareddy I, Chao A, Sampath A, Gu F, Vasudevan SG. 2006. Dengue virus NS4B interacts with NS3 and dissociates it from single-stranded RNA. J Gen Virol 87:2605–2614. doi: 10.1099/vir.0.81844-0. [DOI] [PubMed] [Google Scholar]
- 26.Zou J, Lee LT, Wang QY, Xie XP, Lu SY, Yau YH, Yuan ZM, Shochat SG, Kang CB, Lescar J, Shi PY. 2015. Mapping the interactions between the NS4B and NS3 proteins of dengue virus. J Virol 89:3471–3483. doi: 10.1128/JVI.03454-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou J, Xie X, Wang QY, Dong H, Lee MY, Kang C, Yuan Z, Shi PY. 2015. Characterization of dengue virus NS4A and NS4B protein interaction. J Virol 89:3455–3470. doi: 10.1128/JVI.03453-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patkar CG, Larsen M, Owston M, Smith JL, Kuhn RJ. 2009. Identification of inhibitors of yellow fever virus replication using a replicon-based high-throughput assay. Antimicrob Agents Chemother 53:4103–4114. doi: 10.1128/AAC.00074-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Cleef KW, Overheul GJ, Thomassen MC, Kaptein SJ, Davidson AD, Jacobs M, Neyts J, van Kuppeveld FJ, van Rij RP. 2013. Identification of a new dengue virus inhibitor that targets the viral NS4B protein and restricts genomic RNA replication. Antiviral Res 99:165–171. doi: 10.1016/j.antiviral.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 30.Xie X, Zou J, Wang QY, Shi PY. 2015. Targeting dengue virus NS4B protein for drug discovery. Antiviral Res 118:39–45. doi: 10.1016/j.antiviral.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 31.Green S, Rothman A. 2006. Immunopathological mechanisms in dengue and dengue hemorrhagic fever. Curr Opin Infect Dis 19:429–436. doi: 10.1097/01.qco.0000244047.31135.fa. [DOI] [PubMed] [Google Scholar]
- 32.Sofia MJ. 2014. Beyond sofosbuvir: what opportunity exists for a better nucleoside/nucleotide to treat hepatitis C? Antiviral Res 107:119–124. doi: 10.1016/j.antiviral.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 33.Chen YL, Abdul Ghafar N, Karuna R, Fu Y, Lim SP, Schul W, Gu F, Herve M, Yokohama F, Wang G, Cerny D, Fink K, Blasco F, Shi PY. 2014. Activation of peripheral blood mononuclear cells by dengue virus infection depotentiates balapiravir. J Virol 88:1740–1747. doi: 10.1128/JVI.02841-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blight KJ, Kolykhalov AA, Rice CM. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972–1974. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]