

ABSTRACT
Human herpes simplex viruses 1 and 2 (HSV-1 and HSV-2) are large-genome DNA viruses that establish a persistent infection in sensory neurons and commonly manifest with recurring oral or genital erosions that transmit virus. HSV encodes 12 predicted glycoproteins that serve various functions, including cellular attachment, entry, and egress. Glycoprotein G is currently the target of an antibody test to differentiate HSV-1 from HSV-2; however, this test has shown reduced capacity to differentiate HSV strains in East Africa. Until the recent availability of 26 full-length HSV-1 and 36 full-length HSV-2 sequences, minimal comparative information was available for these viruses. In this study, we use a variety of sequence analysis methods to compare all available sequence data for HSV-1 and HSV-2 glycoproteins, using viruses isolated in Europe, Asia, North America, the Republic of South Africa, and East Africa. We found numerous differences in diversity, nonsynonymous/synonymous substitution rates, and recombination rates between HSV-1 glycoproteins and their HSV-2 counterparts. Phylogenetic analysis revealed that while most global HSV-2 glycoprotein G sequences did not form clusters within or between continents, one clade (supported at 60.5%) contained 37% of the African sequences analyzed. Accordingly, sequences from this African subset contained unique amino acid signatures, not only in glycoprotein G, but also in glycoproteins I and E, which may account for the failure of sensitive antibody tests to distinguish HSV-1 from HSV-2 in some African individuals. Consensus sequences generated in the study can be used to improve diagnostic assays that differentiate HSV-1 from HSV-2 in global populations.
IMPORTANCE Human herpes simplex viruses 1 and 2 (HSV-1 and HSV-2) are large DNA viruses associated with recurring oral or genital erosions that transmit virus. Up to 12 HSV-1 and HSV-2 glycoproteins are involved in HSV cell entry or are required for viral spread in animals, albeit some are dispensable for replication in vitro. The recent availability of comparable numbers of full-length HSV-1 and HSV-2 sequences enabled comparative analysis of gene diversity of glycoproteins within and between HSV types. Overall, we found less glycoprotein sequence diversity within HSV-2 than within the HSV-1 strains studied, while at the same time, several HSV-2 glycoproteins were evolving under less selective pressure. Because HSV glycoproteins are the focus of antibody tests to detect and differentiate between infections with the two strains and are constituents of vaccines in clinical-stage development, these findings will aid in refining the targets for diagnostic tests and vaccines.
INTRODUCTION
Herpes simplex virus 1 and 2 (HSV-1 and HSV-2) are members of the subfamily Alphaherpesvirinae. They are large, enveloped DNA viruses that cause infections that cycle between a replication stage, where infectious virus particles are shed through mucocutaneous erosions, and a latent infection stage, where the virus persists in sensory neurons (1). While HSV-1 and HSV-2 induce antibody and T cell responses, they are insufficient to clear the virus. HSV-1 infection is primarily associated with oral lesions and is frequently diagnosed in children, while HSV-2 is primarily associated with genital lesions and is usually sexually transmitted (1). Coinfection of human immunodeficiency virus type 1 (HIV-1) with HSV-2 is common, with 50 to 90% of those infected with HIV-1 coinfected with HSV-2 (2). Infectious HIV-1 is shed in genital HSV-2 erosions, and the immune infiltrate in genital HSV-2 is enriched in CCR5+/CD4+ T cells that are targets for HIV-1 infection (3, 4). Therefore, HSV-2 lesions can serve as portals for both HIV-1 transmission and acquisition. Strategies for preventing HSV-2 infection provide a progressive approach for decreasing the HIV-1 epidemic (5).
HSV genomes span approximately 152,000 bp and include 77 unique open reading frames (ORFs) that encode proteins. The linear form of the genome contains two unique segments, the unique long (UL) and unique short (US) regions, flanked by inverted-repeat regions, RL and RS, respectively. The genome's high GC content and regions of low sequence complexity have limited the generation of finished full-genome sequences and evolutionary analysis among different isolates (6), as described in the accompanying paper by Newman et al. (7). While nucleic acid diversity among isolates across HSV-2 coding regions is low (0.1 to 0.6%) (8), small, variable-length repeats occur both between the ORFs and in a small number of ORFs, such as UL36, which encodes a large tegument protein, and RL2, which encodes ICP0. HSV recombination may complicate phylogenetic analysis of full-length genomes by presenting a conflicting signal (9); therefore, smaller coding regions have often been used to identify evolutionary relationships among genetic isolates of HSV collected in different geographic regions or to compare the two HSV species. Especially in the case of HSV-2, small data sets and a limited number of comparable regions have hindered extensive analyses.
The envelope of HSV is a lipid bilayer with up to 12 glycoproteins, based on evidence for covalent glycosylation of their protein products. Proteomic analysis of purified HSV-1 virions has detected at least 9 of these proteins (gL, gM, gH, gB, gC, gG, gD, gI, and gE) (10). Another protein (gN), encoded by UL49a, is also glycosylated in another alphaherpesvirus, pseudorabies virus (11), but not in HSV-1 (11, 12). It is included in this study because there is evidence that it localizes to the viral HSV-2 envelope and can also complex with gM (1). HSV glycoproteins are important during early interactions between the virus and the target cell, in virus egress and cell-to-cell spread, and in evasion strategies to avoid the immune response of the host (13); however, the function of each glycoprotein varies in relationship to the overall pathogenesis and immune evasion by the virus. Some glycoprotein genes are dispersed across the UL region; in contrast, others comprise the majority of the US region, where they are closely distributed, separated by small regions of length variation. Analysis across glycoproteins may help identify antigenic regions, support vaccine development, and provide better knowledge of the immune response as HSV evolves in global populations and in relationship to the HIV-1 epidemic. Previous studies of HSV-1 glycoprotein diversity have shown the presence of distinct clades and recombination (14–18); however, more recent studies suggest that no subgroups of HSV-1 exist (6, 19). Studies of four HSV-2 glycoproteins (gG, gI, gE, and gB) have suggested distinct HSV-2 clades with (20) and without (21) recombination.
Recently, 36 full-length HSV-2 genomes collected from four continents have been added to the NCBI sequence database, enabling a more robust analysis of HSV-2 glycoproteins (7, 22). Importantly, HSV glycoproteins, in particular gG, are the target of antibody detection assays used for the diagnosis of HSV-1 or HSV-2 infections (23); therefore, a comparison of HSV-1 and HSV-2 glycoproteins could show why these tests sometimes fail, especially in East Africa, where some serological tests have an assay specificity as low as 50.7% (24–28). We analyzed all putative glycoprotein genes in the UL and US regions, which included sequences derived from North America, Europe, Asia, the Republic of South Africa, and East Africa. The goal was to clarify how HSV glycoproteins have evolved in global populations and in comparison to each other. In addition, for the first time, an analysis of HSV-2 glycoproteins gL-2, gM-2, gH-2, gN-2, and gJ-2 was possible.
MATERIALS AND METHODS
Published HSV-1 and HSV-2 glycoprotein sequences and their location data (when available) were downloaded from the Virus Pathogen Resource (ViPR) (http://www.viprbrc.org/brc/home.spg?decorator=vipr) interface to NCBI GenBank (https://www.ncbi.nlm.nih.gov/GenBank/) (see Data Set S1 in the supplemental material).
Automated alignments were generated using MEGA5 software (29) for each HSV species glycoprotein at the amino acid level and optimized by hand when necessary, after which the alignments were analyzed as nucleic acids, except where noted. Alignments containing both HSV-1 and HSV-2 sequences for each glycoprotein were also generated. To study recombination, concatenated alignments containing HSV-1 and HSV-2 glycoprotein sequences were generated from full-length HSV-1 (26 sequences) and HSV-2 (38 sequences) published sequences (6, 7, 22). The concatenated alignments contain glycoproteins in the order in which they reside in the UL (gL-gM-gH-gB-gC-gN-gK) and US (gG-gJ-gD-gI-gE) regions. Individual glycoprotein alignments were also used to assess recombination, as described below.
Assessments of overall evolutionary nucleotide diversity within an individual HSV-1 and HSV-2 glycoprotein alignment and divergence between HSV-1 and HSV-2 glycoprotein alignments (the average percent base substitutions per site over all sequence pairs) were conducted in MEGA5 (29) using the Tamura 3-parameter model (30). Standard error estimates were obtained by bootstrap (1,000 replicates). The best molecular model was identified using Model Selection in MEGA5. Nonsynonymous (dN) and synonymous (dS) substitutions per site were calculated using 1,000 bootstrap replicates and the Nei-Gojobori model (31). Amino acid divergence between HSV-1 and HSV-2 glycoprotein species was calculated in MEGA5 using the JTT correction. As described above, the molecular model was selected using Model Selection in MEGA5. Standard error estimates were obtained by bootstrap (1,000 replicates). All codon positions were included, and gapped positions were eliminated globally.
Several algorithms that use different statistical approaches were used to investigate recombination within individually aligned glycoprotein sequences and concatenated glycoprotein sequence alignments, as follows. The Recombination Detection Program (RDP) includes nine nonparametric recombination detection methods (32). The Genetic Algorithm Recombination Detection (GARD) program searches for evidence of segment-specific phylogenies; GARD searches the space of all possible locations in the alignment for breakpoints, inferring phylogenies for each putative nonrecombinant fragment, and assesses goodness of fit by the Akaike information criterion (33). Splitstree uses the pairwise homology test (PHI test) to statistically test for the presence of conflicting signal (recombination) in a set of aligned sequences with performance similar to that of coalescence-based methods; a P value of <0.05 is considered significant signal for recombination (34). In the analysis of each glycoprotein sequence, all genetic data available for each glycoprotein available in GenBank was used (see Data Set S1 in the supplemental material). In the analysis of concatenated alignments, only glycoprotein data from full-length sequences were used (6, 7). Simplot (35) was used to apply a boot-scanning approach to concatenated alignments with the following settings: window size, 1,000; step size, 50; strict consensus; F84 maximum-likelihood model of evolution; and 1,000 bootstrap replicates. A recombination signal in Simplot was considered positive at a cutoff of 70% (36). To corroborate the recombination signal in Simplot, neighbor-joining phylogenies (1,000 bootstrap replicates) were constructed using segments of the alignments that flanked the potential recombination breakpoint.
To investigate positive selection on a site-by-site basis, and because there is debate about whether counting methods or random-effects models are better approaches (37–39), we used an agreement-based inference that included the five methods available in the HyPhy analysis suite (40) (SLAC, FEL, iFEL, MEME, and FUBAR). A comparison of the SLAC and FEL methods is available in reference 41. iFEL is slightly different from FEL in that it investigates sitewise selection on internal branches of a phylogenetic tree to determine whether selection is occurring at the population level (42). MEME is a mixed-effects model of evolution that investigates episodic diversifying selection affecting individual codon positions (43). FUBAR uses a Bayesian approach for inferring selection (44). If at least three of these methods indicated positive selection at a particular coding position, positive selection was considered likely.
An amino acid phylogeny of HSV-1 gG was constructed using the JTT evolutionary model and 1,000 bootstrap replicate samples (45). This phylogeny was imported into Figtree software (http://www.molecularevolution.org) for graphic viewing of the results. The program Cluster Picker (46) was used to isolate clades in the tree meeting the criteria of >60% bootstrap significance and <0.5% diversity. Phylogenies for gI-2 and gE-2 were similarly constructed.
Signature pattern analysis was carried out for aligned HSV-2 gG, gI, and gE sequences using the program VESPA (47). The program detects positions for which the most common character in the query set differs from that in the background set. A background set of 15, 12, and 11 African sequences, which represented highly supported phylogenetic clades in gG, gI, and gE phylogenies, respectively, were isolated and compared to (i) other African sequences, (ii) all Western sequences (U.S. plus European), and (iii) all Asian sequences. Positions where two or more background sets were more than 70% different from one another were identified. Amino acid frequencies at distinguishing sites were calculated. The genetic distance within each query sequence population was also calculated. A gG, gI, and gE consensus sequence was generated for all HSV-1 sequences, the subset of HSV-1 African sequences that were phylogenetically distinct, and HSV-2 sequences using Geneious software (version 6.0.5). Consensus positions in the alignment were based on 95% identity. In cases where no consensus was identified, an “X” appears in the alignment. The three consensus sequences representing each glycoprotein were aligned, and amino acids were highlighted using a polarity color scheme (yellow, nonpolar; green, polar/uncharged; red, polar/acidic; blue, polar/basic) in Geneious software (version 6.0.5).
RESULTS
The numbers of sequences for each of the individual HSV-1 and HSV-2 glycoproteins were comparable among most sets of aligned glycoproteins, with two exceptions: for gB, there were three times more HSV-2 sequences, and for gJ, there were approximately three times more HSV-1 sequences (Table 1). The sequence populations for each glycoprotein were derived from viruses isolated in Europe, North America, Asia, East Africa, and South Africa. As expected, HSV-1 and HSV-2 glycoprotein genes were similar in length and aligned well, except for gG, for which the HSV-1 and HSV-2 genes were of different lengths and difficult to align, except at the 3′ ends of their coding sequences. The recently published 36 full-length HSV-2 genomes (7, 22) allowed a first-time evaluation of diversity in five glycoproteins (gL-2, gM-2, gN-2, gH-2, and gJ-2).
TABLE 1.
HSV-1 and -2 glycoprotein sequence population diversity within and divergence between strains
| Region | Glycoprotein | Gene | No. of sequences |
Alignment length (bp) |
% nucleic acid diversity (SE) within: |
% nucleic acid divergence (SE) between HSV-1 and -2 | % amino acid divergence (SE) between HSV-1 and -2 | |||
|---|---|---|---|---|---|---|---|---|---|---|
| HSV-1 | HSV-2 | HSV-1 | HSV-2 | HSV-1 | HSV-2 | |||||
| UL | gL | UL1 | 44 | 38 | 675 | 675 | 1.5 (0.3) | 0.01(0.0) | 32.3 (2.8) | 45.2 (5.0) |
| gM | UL10 | 39 | 38 | 1,422 | 1,401 | 0.8 (0.1) | 0.2 (0.1) | 19.8 (1.3) | 19.9 (2.2) | |
| gH | UL22 | 42 | 38 | 2,517 | 2,529 | 0.6 (0.1) | 0.3 (0.1) | 22.1 (1.0) | 24.1 (1.8) | |
| gB | UL27 | 57 | 156 | 2,739 | 2,721 | 0.6 (0.1) | 0.2 (0.0) | 11.8 (0.8) | 13.6 (1.2) | |
| gC | UL44 | 92 | 53 | 1,539 | 1,443 | 1.2 (0.1) | 0.4 (0.1) | 27.3 (1.6) | 38.9 (3.2) | |
| gN | UL49a | 37 | 41 | 276 | 264 | 0.5 (0.1) | 0.4 (0.2) | 37.8 (4.5) | 64.6 (12.8) | |
| gK | UL53 | 56 | 43 | 1,017 | 1,017 | 1.2 (0.1) | 0.5 (0.1) | 17.9 (1.3) | 17.2 (2.3) | |
| US | gG | US4 | 156 | 180 | 723 | 2,139 | 1.9 (0.3) | 0.4 (0.1) | 64.6 (4.5) | 91.8 (9.1) |
| gI | US7 | 75 | 85 | 1,317 | 1,137 | 1.3 (0.2) | 0.3 (0.1) | 31.2 (2.1) | 36.4 (3.7) | |
| gD | US6 | 61 | 87 | 1,185 | 1,182 | 0.7 (0.1) | 0.2 (0.0) | 21.6 (1.5) | 21.3 (2.5) | |
| gJ | US5 | 233 | 38 | 279 | 279 | 0.8 (0.2) | 0.2 (0.1) | 53.9 (0.7) | 103.9 (16.3) | |
| gE | US8 | 71 | 84 | 1,656 | 1,611 | 0.7 (0.1) | 0.2 (0.0) | 23.5 (1.3) | 29.5 (2.6) | |
Diversity within HSV glycoprotein genes at the nucleotide level ranged from 0.5 to 1.9% for each sequence population (Table 1). HSV-1 sequence alignments were ≥50% more diverse than HSV-2 sequences in all cases except in gN. The nonsynonymous (dN)-to-synonymous (dS) nucleotide substitution ratios were usually less than 0.5 in HSV-1 glycoprotein genes but were above 0.6 in seven HSV-2 glycoprotein genes (gL, gB, gC, gK, gG, gI, and gE), suggesting that different evolutionary constraints were acting on the two HSV populations (Fig. 1). gM, gH, gC, and gD had similar dN/dS ratios in the two HSV subtypes.
FIG 1.
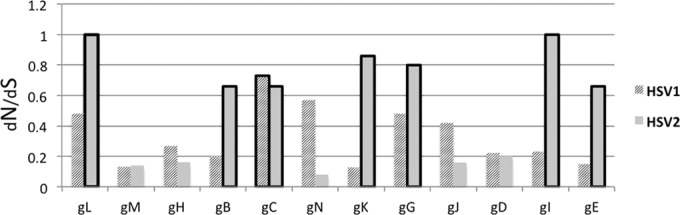
Ratios of nonsynonymous (dN) to synonymous (dS) substitutions. When dN/dS is greater than 0.6, the bar is outlined with a thick line.
Analysis of nucleotide divergence between HSV-1 and HSV-2 glycoprotein genes showed that three of the four HSV glycoprotein genes necessary for virus entry (gH, gB, and gD) were among the five most highly conserved between the two species; however, gL, which is also essential, was among the least conserved between species (Table 1). The most closely related glycoprotein between HSV-1 and HSV-2 was gB, at 11.8% divergence, followed by gK at 17.9%. In contrast, the most divergent glycoprotein genes were the gG gene at 64.6%, followed by the gJ gene at 53.9%. Amino acid divergence followed a similar pattern, except that in this case, the gJ gene was the most divergent glycoprotein gene between species (Table 1). Amino acid divergence between HSV-1 and HSV-2, calculated using the best-fitting molecular model (JTT), was sometimes much higher than corresponding nucleic acid divergence calculations. This is likely due to the nonlinear scaling for multiple substitutions with a complex model. Moreover, HSV genes have a high GC content, with about 80% G or C occurring at the 3rd codon position. This permits a biased codon usage for HSV, with an effective codon usage of approximately 40/61 different codons. These biases are expected to cause relatively low nucleotide diversity for a given degree of amino acid diversity.
A variety of computational approaches are classically used to detect genomic recombination events, and the strength of the positive signal for recombination varies between algorithms (9, 34, 48). These methods could be sensitive to sequence populations containing a very large amount of diversity that would result in phylogenetic noise; likewise, in a low-diversity data set, recombination would be impossible to detect using any computational approach (9). Using GARD, the PHI test in Splitstree, and nine other recombination algorithms in RDP, we detected moderate recombination in HSV-1 and infrequent recombination in HSV-2 glycoprotein alignments (Table 2). In most cases these alignments contained similar numbers of diverse global HSV isolates. The methods used were often inconsistent in their abilities to detect recombination, with the PHI test suggesting the most recombination events, in eight HSV-1 and HSV-2 glycoprotein alignments; GARD identified six recombination breakpoints within the aligned data, and RDP calculated recombination signal in only three sets of aligned data.
TABLE 2.
Recombination calculated within HSV1 and HSV2 glycoproteins
| Region | Product | Result |
||
|---|---|---|---|---|
| PHIa | GARDb | RDPc | ||
| UL | gL-1 | 0.00 | 306 | 1 |
| gL-2 | 1.00 | 0 | ||
| gM-1 | 0.00 | 0 | ||
| gM-2 | 0.01 | 0 | ||
| gH-1 | 0.01 | 0 | ||
| gH-2 | 0.15 | 762 | 0 | |
| gB-1 | 0.00 | 0 | ||
| gB-2 | 0.50 | 0 | ||
| gC-1 | 0.82 | 793 | 5 | |
| gC-2 | 0.37 | 576 | 0 | |
| gN-1 | 1.00 | 0 | ||
| gN-2 | 1.00 | 0 | ||
| gK-1 | 0.06 | 576 | 0 | |
| gK-2 | 0.44 | 1 | ||
| US | gG-1 | 0.57 | 0 | |
| gG-2 | 0.50 | 0 | ||
| gJ-1 | 0.00 | 0 | ||
| gJ-2 | 0.59 | 0 | ||
| gD-1 | 0.35 | 0 | ||
| gD-2 | 0.13 | 0 | ||
| gI-1 | 0.91 | 674 | 2 | |
| gI-2 | 1.00 | 0 | ||
| gE-1 | 0.00 | 0 | ||
| gE-2 | 0.05 | 0 | ||
The PHI test identifies recombination signal in a phylogenetic network; P values are listed (P values of <0.05 are considered positive for recombination).
GARD identifies putative breakpoints within aligned nucleic acid sequences; the locations of the calculated breakpoints in the nucleic acid alignments are listed.
RDP uses nine pairwise scanning approaches to detect recombination; the numbers of approaches that detected recombination are listed.
Concatenating glycoprotein sequence alignments and applying a boot-scanning approach could identify recombination within larger genetic segments of HSV. One would expect, given the physical distance between the locations of the glycoproteins in the full genome (especially in UL), clear breakpoints would occur near or at sites of concatenation. The same algorithms (GARD, PHI test, and RDP) applied to individual alignments were applied to the concatenated alignments. Again, HSV-1 exhibited more evidence of recombination than HSV-2 alignments, with a median value of 5 breakpoints for HSV-1 compared to 0 breakpoints for HSV-2 in UL and a median value of 3.5 breakpoints in HSV-1 compared to 0 breakpoints in HSV-2 in US. Boot-scanning (Simplot) analysis of concatenated sequence alignments suggested recombinant segments for both HSV-1 and HSV-2 in UL (Fig. 2 and 3), with recombination breakpoints appearing close to glycoprotein concatenation sites. To substantiate this result, phylogenies were generated using recombinant segments of the aligned data (Fig. 2B and 3B and C). Many highly supported branches were identified in each of the partial HSV-1 phylogenies, whereas very few highly supported branches were identified in the HSV-2 UL partial phylogenies, arguing that recombination in HSV-2 either is occurring less or is simply too difficult to detect using a computational approach. Also of note is that putative recombination was usually detected within continents and much less frequently between continents in both strains. Boot scanning was also performed for the HSV US concatenated sequence alignments. Again, recombination was detected in most HSV-1 sequences but could not be confirmed in any of the HSV-1 US sequences.
FIG 2.

Boot-scanning and phylogenetic analyses for evidence of recombination within concatenated HSV-1 UL glycoprotein sequences. (A) HSV-1 strain 17 was used as the query sequence against other HSV-1 UL concatenated glycoprotein sequences using a boot-scanning approach. The x axis in the boot scan reflects the position in the aligned set of sequences, and the y axis shows the percentage of permuted trees in which an individual HSV-1 strain clustered with the query. A recombination cutoff value of 70% is indicated by the dashed line. Regions of the scan associated with each glycoprotein segment are boxed, with the glycoprotein name above the plot. Three putative recombination events occurred and are numbered. The arrows indicate positions where sequence concatenation may be linked to the recombination signal. Directly below the boot-scanning plot, numbered black lines indicate regions of the alignment used to substantiate the recombination signal in the phylogenies shown in panel C. (B) Concatenated glycoprotein phylogeny with taxa colored by region (green, Europe; red, Africa; orange, Asia). The tree was used as a guide to group some sequences in the boot-scanning analysis plot shown in panel A. Boot-scanning groups, indicated by colored cones subtending the sequence names, were defined by bootstrap significance of >90% and included three African groups (1 to 3), one European group, one Asian group, and one mixed group containing sequences from Asia and Europe. (C) Three midpoint-rooted phylogenies were generated and are shown in cladogram format. The phylogenies were generated with segments of the alignment as noted in panel A. The lengths of the segments used to create the phylogeny are shown at the bottom. Portions of the tree containing highly supported clades, similar to the phylogeny containing the full concatenated sequence (panel B), are indicated. The strain containing the most closely related recombinant segment is indicated next to the phylogeny with the query sequence (strain 17). The asterisks indicate bootstrap values of 70% or higher.
FIG 3.

Boot-scanning and phylogenetic analyses for evidence of recombination within concatenated HSV-2 UL glycoprotein sequences. (A) HSV-2 Uganda strain G19090_UG was used as the query sequence against other HSV-2 UL concatenated glycoprotein sequences using a boot-scanning approach. The x axis reflects the position in the aligned set of sequences, and the y axis shows the percentage of permuted trees in which an individual HSV-1 strain clustered with the query. A recombination cutoff value of 70% is indicated by the dashed line. Regions of the scan associated with each glycoprotein segment are boxed, with the glycoprotein name above the plot. Two putative recombination events occurred and are numbered. The arrows indicate positions where sequence concatenation may be linked to the recombination signal. Directly below the boot-scanning plot, numbered black lines indicate regions of the alignment used to substantiate recombination signal in the phylogenies shown in panel C. (B) Concatenated glycoprotein phylogeny (far left) with taxa colored by region (green, Europe; blue, United States; red, Africa; orange, Asia). The tree was used as a guide to group some sequences in the boot-scanning analysis plot shown in panel A. Boot-scanning groups, indicated by colored cones subtending the sequence names, were defined by bootstrap significance of >90% and included two African groups (1 and 2), one European group, one Asian group, one U.S. group, one South African group, and two mixed groups containing sequences from (i) Uganda and the United States and (ii) South Africa and the United States. (C) Two midpoint-rooted phylogenies were generated and are shown in cladogram format. These phylogenies were generated with segments of the alignment as noted in panel A. The lengths of the segments used to create the phylogeny are shown at the bottom. The strain containing the most closely related recombinant segment is indicated next to the phylogeny with the query sequence (G19090_UG). The asterisks indicate bootstrap values of 70% or higher.
Positive selection occurs in coding regions when evolution selects for advantageous amino acid residue changes. Of the methods used for detecting amino acids under positive selection, MEME and FUBAR frequently identified many positively selected sites compared to the older methods (FEL, iFEL, and SLAC) (Table 3). Both of these algorithms are considered superior to the other methods used because they reflect selection under a wider range of scenarios that may vary over a larger number of site classes (43, 44). Glycoprotein C, which plays a significant role in attachment to the cell surface and the adsorption of virus to cells, contained the most positively selected amino acids in HSV-1 (Table 3). Combined with high diversity (1.2 ± 0.1) and a high dN/dS ratio, this indicates that HSV-1 gC is perhaps more flexible than other glycoproteins while simultaneously fixing potentially beneficial mutations. In HSV-2, gG contained the most positively selected sites and a corresponding high dN/dS ratio. Again, this indicates a region that is randomly accumulating mutations while fixing those that are advantageous.
TABLE 3.
Positively selected amino acid positions for HSV-1 and HSV-2 starting from the first codon in the reference sequence using five methods
| Region | Product | Amino acid position(s) |
|||||
|---|---|---|---|---|---|---|---|
| FEL (0.1)a | SLAC (0.1)b | iFEL (0.1)a | MEME (0.1)a | FUBAR (0.9)c | Agreementd | ||
| UL | gL-1 | 212 | 212 | 212 | 157, 171, 212 | 212 | |
| gL-2 | 37 | ||||||
| gM-1 | 203 | 203 | 203 | 203 | 203 | 203 | |
| gM-2 | 13, 243 | ||||||
| gH-1 | 284 | 284 | 284 | 284, 369 | 284, 369, 370 | 284 | |
| gH-2 | 516, 634 | 516 | 516 | 634 | 112, 154, 516, 634 | 516, 634 | |
| gB-1 | 59 | 549 | |||||
| gB-2 | 7, 336, 851 | 336, 402, 851 | 7, 336, 851 | 7, 43, 336, 488, 851 | 7, 42, 55, 336, 402, 851 | 7, 336, 851 | |
| gC-1 | 23, 289, 306, 366 | 23, 289, 306, 366 | 23, 75, 132,242, 262, 289, 309, 349 | 16, 23, 71, 80, 132, 242, 245, 248, 262, 264, 289, 305, 306, 383, 393, 414, 424 | 16, 23, 132, 262, 289, 300, 306, 366, 424 | 23, 289, 306, 366 | |
| gC-2 | 53 | 344, 428 | 194 | 53, 194, 195 | 53, 169, 214 | 53 | |
| gN-1 | |||||||
| gN-2 | |||||||
| gK-1 | 224 | 224, 309 | 40, 224, 226, 309, 325 | 224 | 224 | ||
| gK-2 | 247 | 115, 247 | |||||
| US | gG-1 | 3, 167, 225, 238 | 167 | 97, 166, 167, 207 | 3, 167, 207, 214, 238 | 3, 97, 167, 238 | 3, 167, 238 |
| gG-2 | 204, 212, 291, 338, 426 | 204, 338, 426 | 204, 212, 291, 338, 400, 426, 569 | 136, 204, 212, 291, 331, 338, 400, 426, 469, 562, 563, 604 | 35, 136, 204, 212, 291, 328, 338, 349, 408, 426, 445, 512, 592 | 204, 212, 291, 338, 426 | |
| gJ-1 | 60 | 25 | 54, 60, 91 | ||||
| gJ-2 | 30, 86 | ||||||
| gD-1 | 50 | 84, 369, 374 | |||||
| gD-2 | 353 | ||||||
| gI-1 | 189, 297 | 18, 179, 189, 297 | 18, 179, 189, 221, 222 | 189 | |||
| gI-2 | 246 | 215, 246 | 120, 259 | ||||
| gE-1 | 188 | 259 | 120, 259 | ||||
| gE-2 | 194 | 43, 51, 410 | |||||
The number in parentheses is the cutoff P value for the single degree of freedom likelihood ratio test to classify a site as positively selected.
The number in parentheses is the cutoff P value for the two-tailed extended binomial test to classify a site as positively selected.
The number in parentheses is a cutoff value for the Bayes factor for the event of positive selection at a given site, sel 2.
At least three methods indicated positive selection at the coding position(s) listed; boldface indicates agreement among all selection methods.
Because serology tests used to discriminate HSV-1 and HSV-2 infections are frequently inaccurate in East African populations (25–28), we further examined HSV-2 gG amino acid sequences using phylogenetic methods. The analysis demonstrated that most global strains were dispersed around the tree, with few clusters with bootstrap support greater than 50%; however, there was one large (14 sequences), low-diversity clade (<0.1%) with moderate bootstrap support (>60%) (Fig. 4A). The viruses represented in this clade were similarly identified in phylogenies of glycoproteins gI-2 and gE-2 (Fig. 4B and C). This group of African sequences comprised 37% of the total East African sequences in GenBank and was derived from subjects in Tanzania and Uganda.
FIG 4.

Maximum-likelihood phylogenetic analysis of HSV-2 gG, gI, and gE amino acid sequences. Individual sequences are colored by global region. The asterisks indicate clades identified with Cluster Picker with a bootstrap value of >60% and within-clade diversity of <0.5%.
To understand the changes in amino acids that resulted in the phylogenetic grouping of the African subset, a signature pattern analysis was performed that compared the sequences to alignments with the other African, the Western-derived, and the Asian sequences. A unique amino acid signature emerged for the HSV-2 gG, gI, and gE glycoproteins in the East African subset (Table 4). Four positions in gG-2 varied significantly from those in the other groups. gG-2 position 291 was always a neutral glycine in the African subset and primarily a negatively charged glutamic acid in the other groups. At gG-2 position 338, a lysine was identified in 98% of the African subset, whereas arginine was present in most other sequences. Significant polymorphisms in positions gG-2 372 and 542 were not departures in charge but could represent slight differences in hydrophobicity or polarity. Position 542 is within a known immunodominant region of gG (49). Additional signature amino acid polymorphisms occurred in glycoproteins gI-2 and gE-2 (Table 4). Alignments containing consensus amino acid sequences from HSV-1, HSV-2, and the African subset for gG, gI, and gE contain numerous regions with enough sequence identity for the development of new enzyme-linked immunosorbent assays (ELISAs) that would improve the diagnostic serology for HSV-1 and HSV-2 in global populations (Fig. 5 to 7).
TABLE 4.
U.S. HSV-2 glycoprotein signatures for a subset of African sequences
| Alignment | % amino acid distance in gG (SE) | Amino acid(s) at positiona: |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| gG-2 |
gI-2 |
gE-2, 134 | |||||||
| 291 | 338 | 372 | 546 | 10 | 159 | 230 | |||
| African subset (background; 14 sequences) | 0.1 (0.1) | G100 | L93/I7 | V86/A14 | T100 | V64 | L100 | L79/P21 | R79/L21 |
| Other African (query; 27 sequences) | 0.5 (0.1) | E82/G18 | R78/L22 | A100 | A100 | A100 | R63/L37 | S52/P44/L2 | L85/R15 |
| Western (query; 118 sequences) | 0.6 (0.1) | E95/G5 | R90/L10 | A100 | A100 | A100 | R71/L29 | P84/S16 | L90/R10 |
| Asia (query; 8 sequences) | 0.6 (0.2) | E87/G13 | R100 | A100 | A100 | A100 | L88/R22 | P100 | L75/R25 |
FIG 5.

Amino acid alignment of gG consensus sequences from HSV-2, the HSV-2 African subset identified in Fig. 2, and HSV-1. The amino acids are colored according to a polarity scale (yellow, nonpolar; green, polar/uncharged; red, polar/acidic; blue, polar/basic). The dashes represent gaps inserted to maximize the alignment. The boxes indicate differences from HSV-1 and HSV-2 sequences. The red circles indicate differences between the majority of HSV-2 and the African subgroup.
FIG 7.

Amino acid alignment of gE consensus sequences from HSV-2, the HSV-2 African subset identified in Fig. 2, and HSV-1. The amino acids are colored according to a polarity scale (yellow, nonpolar; green, polar/uncharged; red, polar/acidic; blue, polar/basic). The dashes represent gaps inserted to maximize the alignment. The boxes indicate differences from HSV-1 and HSV-2 sequences. The red circle indicates a difference between the majority of HSV-2 and the African subgroup.
FIG 6.

Amino acid alignment of gI consensus sequences from HSV-2, the HSV-2 African subset identified in Fig. 2, and HSV-1. The amino acids are colored according to a polarity scale (yellow, nonpolar; green, polar/uncharged; red, polar/acidic; blue, polar/basic). The dashes represent gaps inserted to maximize the alignment. The boxes indicate differences from HSV-1 and HSV-2 sequences. The red circles indicate differences between the majority of HSV-2 and the African subgroup.
DISCUSSION
Until the recent availability of 36 full-length HSV-2 sequences (7, 22) to add to a panel of 26 full-length HSV-1 genomic sequences (6), our ability to analyze the diversity between HSV-1 and HSV-2 has been limited. The genetic distance between HSV-1 and HSV-2 glycoprotein nucleotide and amino acid sequence populations can be quite large (11 to 64% and 13.5 to 83%, respectively). Despite the finding that HSV-1 glycoproteins are significantly more diverse than those of HSV-2 (P > 0.001), nonsynonymous substitutions were more frequently observed in HSV-2 glycoproteins. Overall, this implies that while HSV-2 is accumulating both synonymous and nonsynonymous changes, the change is occurring at such a low level that there is limited biological effect.
Nucleotide and amino acid diversity revealed a number of contrasting results between HSV-1 and HSV-2 strain glycoproteins. For example, this study was the first to examine a large number of sequences of gL-2, gM-2, gN-2, gH-2, and gJ-2 and to compare them to their HSV-1 counterparts. Nucleotide diversity in gL-2, a protein that chaperones gH through the Golgi apparatus to the plasma membrane (1), was almost nonexistent, in contrast to considerably high diversity in gL-1. Nucleotide diversity levels in gM, gH, and gN were similar in HSV-2 and HSV-1 strains. Calculations of gJ diversity showed a difference between the two species; however, in both species, gJ maintained the same length and showed a highly conserved region from amino acid positions 48 to 75, which may be important in the glycoprotein's function of blocking apoptosis (1). The large genetic diversity between the gG proteins of the two species is due to the fact that they share homologous segments only at the 3′ end of the gene, and because of this divergence, the viral protein product has historically been used to differentiate HSV-1 and HSV-2 antibody responses (1). Glycoprotein B has diverged least between HSV-1 and HSV-2, which may be related to the essential nature of gB for viral entry via fusion of the envelope with the plasma membrane (1). Glycoprotein K, which appears to be associated with neurovirulence, is the second most conserved glycoprotein between the two virus types. The fact that it is conserved more than glycoprotein H is particularly interesting, because gH is thought to be essential for virus entry and virus-induced cell fusion (1). This is important, because it provide evidence that gK has important in vivo functions that are not essential for viral replication in cell culture. While the results presented here are not intended to give an exhaustive review of diversity in all glycoproteins, they provide evidence that will be useful for additional studies, such as protein modeling and elucidating how glycoprotein variation affects structure-function relationships among HSV species.
Because of the potentially confounding effect on evolution and important biological implications, viral recombination is essential to consider when studying HSV-1 and HSV-2 diversity. A high degree of recombination has been suggested in the literature for both HSV-1 (6, 14, 16) and HSV-2 (20) strains. While we were able to identify recombination using numerous algorithms in more than 50% of the HSV-1 glycoproteins, differing from previous reports (20), recombination in HSV-2 was much more difficult to detect. This is likely due to the decreased sequence divergence in HSV-2 sequence populations compared to HSV-1. Even if recombination was occurring at a high rate in HSV-2, the limited glycoprotein diversity results in difficult-to-detect recombination events. Analysis of recombination within full-length HSV-2 genomes in the accompanying paper by Newman et al. (7) also documents much less detectable recombination occurring in HSV-2 than previously reported in HSV-1 (6). The lower HSV-2 recombination rate could be related to the fact that there has been less time for HSV-2 to recombine because it evolved after HSV-1 (50), or it may be the result of the lower seroprevalence of HSV-2 worldwide, so that people are less likely be coinfected by two strains.
Positively selected amino acid residues were frequently identified in HSV glycoproteins, with the highest number of positively selected sites occurring in gC-1 and gG-2. This finding, along with the detection of five or more positively selected sites in gG-2 and a high number of nonsynonymous substitutions, was an indication that perhaps nucleotide variation and the resulting antigenic variation were impacting the sensitivity or specificity of the Western blot assay in East Africa, where there is a significant difference between ELISA and Western blot results (25–28). This may be due in part to the U.S. strain of HSV-2 used to provide the antigens in the Western blot and extends to the antigen used in commercial ELISAs. Serologic differences in performance with African sera may be partially based on the finding that the gG-2 gene differed the most between African and Western strains of HSV-2. Further testing of HSV amino acid sites under positive selection may show how these sites affect glycoprotein structure. Phylogenetic analysis indicated that a subset of East African sequences that were collected from Uganda (7) and Tanzania (20) frequently segregated together in a low-diversity clade, especially in the US glycoproteins. Signature pattern analysis revealed that these sequences possess a unique signature in gG-2 that includes one position in an antigenic region bound by HSV-2-reactive antibodies (49). An amino acid signature in this East African subset was also identified in gI-2 and gE-2. gI and gE form a complex with each other, and this complex binds to the Fc domain of IgG (1); therefore, unique signature patterns across these US glycoproteins could be due to coevolution of amino acids based on necessary structural or physical functions. While this group of African sequences does not represent a true unique clade of HSV-2, the study demonstrates that virus evolving over many years in localized regions could perhaps generate enough unique signatures to impact serological testing. Future studies will explore whether other HSV-2 glycoproteins or different HSV-1 gG consensus sequences are more suited for antibody detection assays.
Several vaccines to prevent or treat HSV-2 infection have recently been tested in clinical trials, and additional vaccines are being tested (51–53). Each of these vaccines has a different composition, ranging from a single truncated gD-2 in the case of the GlaxoSmithKline vaccine (51) to a nearly complete HSV-2 in the case of the replication-incompetent strain with mutations in UL5 and UL29 (54). The implications of variation between HSV-1 and HSV-2 and between HSV species vary for these different vaccine formats. The truncated gD-2 protein vaccine (51) would not be expected to differ in efficacy with geographic region, as gD-2 sequences show very little variation. This vaccine showed better efficacy against HSV-1 than against HSV-2 in HSV-seronegative women, with the efficacy correlating with anti-gD2 antibody levels (55). This cross-protection is not completely unexpected, as gD-2 and gD-1 are highly similar in amino acid sequence. The absence of variability within gD-1 sequences bodes well for this candidate should it be advanced for HSV-1 protection. gB-1, also required for cell entry, has a similar favorable absence of diversity within and between strains, while our analyses show that gH and gL, which are also the targets of neutralizing antibodies, exhibit greater diversity. In contrast, HSV-2 therapeutic vaccines designed to elicit antibodies and T cells (56) are typically multivalent and may be less prone to sequence variation effects (57). Amino acid alignments containing consensus sequences from HSV-1, HSV-2, and the African subset further elucidate the differences between glycoprotein strains and will be used to generate new ELISAs that can better discriminate HSV-1 from HSV-2 antibodies in human sera.
Summary.
In this study, several analyses indicated that HSV-2 isolates are less diverse and perhaps evolving under different evolutionary pressures than those of HSV-1. This finding is likely due to the different evolutionary histories of the two strains (50, 58, 59). In the most recent analysis of the origins of the herpesvirus, HSV-1 was estimated to have emerged approximately 6.6 million years ago due to codivergence of chimpanzees and humans, whereas HSV-2 arose later, the latter hypothesized to be a cross-species transmission from chimpanzee to humanoids about 1.6 million years ago (50). Several factors may contribute to the higher worldwide seroprevalence and diversity of HSV-1 than of HSV-2, such as the time difference between the origins of the two species (60, 61) and more frequent exchange of oral secretions than genital secretions. In cell culture, HSV-1 also provides higher yields than HSV-2, so there may be more copies of HSV-1 shedding (1), which could impact viral diversity and the yield of recombinant viruses during a cellular coinfection event. Viruses evolving in different cellular reservoirs may also have impacted the difference in evolution between the two species. One final hypothesis is that HSV-1 may have reached a saturation point where positive selection is acting to control diversity that is not yet observable or essential for viral success in some HSV-2 genes. The influence of HSV-2 infection on the HIV-1 epidemic emphasizes the importance of studies that provide in-depth genetic analysis of HSV genes, especially the glycoproteins, because they permit further exploration of the evolutionary history of these viruses and because of their importance in designing serological tests and in vaccine development.
Supplementary Material
ACKNOWLEDGMENTS
This project has been funded in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract no. HHSN272200900018C to the Broad Institute's Genomic Sequencing Center for Infectious Diseases; grant AI057552 to D. M. Knipe; and grants AI094019 and AI030731 to D. M. Koelle. This work was also supported by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01302-15.
REFERENCES
- 1.Whitley R, Roizman B, Knipe D, Knipe D, Howley P (ed). 2013. Fields virology. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Tobian AA, Charvat B, Ssempijja V, Kigozi G, Serwadda D, Makumbi F, Iga B, Laeyendecker O, Riedesel M, Oliver A, Chen MZ, Reynolds SJ, Wawer MJ, Gray RH, Quinn TC. 2009. Factors associated with the prevalence and incidence of herpes simplex virus type 2 infection among men in Rakai, Uganda. J Infect Dis 199:945–949. doi: 10.1086/597074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu J, Hladik F, Woodward A, Klock A, Peng T, Johnston C, Remington M, Magaret A, Koelle DM, Wald A, Corey L. 2009. Persistence of HIV-1 receptor-positive cells after HSV-2 reactivation is a potential mechanism for increased HIV-1 acquisition. Nat Med 15:886–892. doi: 10.1038/nm.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schacker T, Ryncarz AJ, Goddard J, Diem K, Shaugnessy M, Corey L. 1998. Frequent recovery of HIV-1 from genital herpes simplex virus lesions in HIV-1-infected men. JAMA 280:61–66. doi: 10.1001/jama.280.1.61. [DOI] [PubMed] [Google Scholar]
- 5.Abu-Raddad LJ, Magaret AS, Celum C, Wald A, Longini IM Jr, Self SG, Corey L. 2008. Genital herpes has played a more important role than any other sexually transmitted infection in driving HIV prevalence in Africa. PLoS One 3:e2230. doi: 10.1371/journal.pone.0002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szpara ML, Gatherer D, Ochoa A, Greenbaum B, Dolan A, Bowden RJ, Enquist LW, Legendre M, Davison AJ. 2014. Evolution and diversity in human herpes simplex virus genomes. J Virol 88:1209–1227. doi: 10.1128/JVI.01987-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newman RM, Lamers SL, Weiner B, Ray SC, Colgrove RC, Diaz F, Jing L, Wang K, Saif S, Young S, Henn M, Laeyendecker O, Tobian AAR, Cohen JI, Koelle DM, Quinn TC, Knipe DM. 2015. Genome sequencing and analysis of geographically diverse clinical isolates of herpes simplex virus 2. J Virol 89:8219–8232. doi: 10.1128/JVI.01303-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dolan A, Jamieson FE, Cunningham C, Barnett BC, McGeoch DJ. 1998. The genome sequence of herpes simplex virus type 2. J Virol 72:2010–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salemi M, Vandamme A-M, Lemey P. 2009. The phylogenetic handbook: a practical approach to phylogenetic analysis and hypothesis testing, 2nd ed Cambridge University Press, Cambridge, United Kingdom. [Google Scholar]
- 10.Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol 82:8605–8618. doi: 10.1128/JVI.00904-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jons A, Granzow H, Kuchling R, Mettenleiter TC. 1996. The UL49.5 gene of pseudorabies virus codes for an O-glycosylated structural protein of the viral envelope. J Virol 70:1237–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koyano S, Mar EC, Stamey FR, Inoue N. 2003. Glycoproteins M and N of human herpesvirus 8 form a complex and inhibit cell fusion. J Gen Virol 84:1485–1491. doi: 10.1099/vir.0.18941-0. [DOI] [PubMed] [Google Scholar]
- 13.Haarr L, Skulstad S. 1994. The herpes simplex virus type 1 particle: structure and molecular functions. APMIS 102:321–346. doi: 10.1111/j.1699-0463.1994.tb04882.x. [DOI] [PubMed] [Google Scholar]
- 14.Norberg P, Bergstrom T, Rekabdar E, Lindh M, Liljeqvist JA. 2004. Phylogenetic analysis of clinical herpes simplex virus type 1 isolates identified three genetic groups and recombinant viruses. J Virol 78:10755–10764. doi: 10.1128/JVI.78.19.10755-10764.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolb AW, Adams M, Cabot EL, Craven M, Brandt CR. 2011. Multiplex sequencing of seven ocular herpes simplex virus type-1 genomes: phylogeny, sequence variability, and SNP distribution. Invest Ophthalmol Vis Sci 52:9061–9073. doi: 10.1167/iovs.11-7812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bowden R, Sakaoka H, Donnelly P, Ward R. 2004. High recombination rate in herpes simplex virus type 1 natural populations suggests significant co-infection. Infect Genet Evol 4:115–123. doi: 10.1016/j.meegid.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 17.Liljeqvist JA, Tunback P, Norberg P. 2009. Asymptomatically shed recombinant herpes simplex virus type 1 strains detected in saliva. J Gen Virol 90:559–566. doi: 10.1099/vir.0.007070-0. [DOI] [PubMed] [Google Scholar]
- 18.Rose L, Crowley B. 2013. Molecular characterization of clinical isolates of herpes simplex virus type 1 collected in a tertiary-care hospital in Dublin, Ireland. J Med Virol 85:839–844. doi: 10.1002/jmv.23541. [DOI] [PubMed] [Google Scholar]
- 19.Harishankar A, Jambulingam M, Gowrishankar R, Venkatachalam A, Vetrivel U, Ravichandran S, Yesupadam SM, Madhavan HN. 2012. Phylogenetic comparison of exonic US4, US7 and UL44 regions of clinical herpes simplex virus type 1 isolates showed lack of association between their anatomic sites of infection and genotypic/sub genotypic classification. Virol J 9:65. doi: 10.1186/1743-422X-9-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Norberg P, Kasubi MJ, Haarr L, Bergstrom T, Liljeqvist JA. 2007. Divergence and recombination of clinical herpes simplex virus type 2 isolates. J Virol 81:13158–13167. doi: 10.1128/JVI.01310-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmidt-Chanasit J, Bialonski A, Heinemann P, Ulrich RG, Gunther S, Rabenau HF, Doerr HW. 2010. A 12-year molecular survey of clinical herpes simplex virus type 2 isolates demonstrates the circulation of clade A and B strains in Germany. J Clin Virol 48:208–211. doi: 10.1016/j.jcv.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 22.Colgrove R, Diaz F, Newman R, Saif S, Shea T, Young S, Henn M, Knipe DM. 2014. Genomic sequences of a low passage herpes simplex virus 2 clinical isolate and its plaque-purified derivative strain. Virology 450-451:140–145. doi: 10.1016/j.virol.2013.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Summerton J, Riedesel M, Laeyendecker O, Gaydos C, Maldeis NE, Hardick A, Morrow RA, Quinn TC. 2007. Effect of sexually transmitted disease (STD) coinfections on performance of three commercially available immunosorbent assays used for detection of herpes simplex virus type 2-specific antibody in men attending Baltimore, Maryland, STD clinics. Clin Vaccine Immunol 14:1545–1549. doi: 10.1128/CVI.00120-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gamiel JL, Tobian AA, Laeyendecker OB, Reynolds SJ, Morrow RA, Serwadda D, Gray RH, Quinn TC. 2008. Improved performance of enzyme-linked immunosorbent assays and the effect of human immunodeficiency virus coinfection on the serologic detection of herpes simplex virus type 2 in Rakai, Uganda. Clin Vaccine Immunol 15:888–890. doi: 10.1128/CVI.00453-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hogrefe W, Su X, Song J, Ashley R, Kong L. 2002. Detection of herpes simplex virus type 2-specific immunoglobulin G antibodies in African sera by using recombinant gG2, Western blotting, and gG2 inhibition. J Clin Microbiol 40:3635–3640. doi: 10.1128/JCM.40.10.3635-3640.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith JS, Bailey RC, Westreich DJ, Maclean I, Agot K, Ndinya-Achola JO, Hogrefe W, Morrow RA, Moses S. 2009. Herpes simplex virus type 2 antibody detection performance in Kisumu, Kenya, using the Herpeselect ELISA, Kalon ELISA, Western blot and inhibition testing. Sex Transm Infect 85:92–96. doi: 10.1136/sti.2008.031815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Dyck E, Buve A, Weiss HA, Glynn JR, Brown DW, De Deken B, Parry J, Hayes RJ. 2004. Performance of commercially available enzyme immunoassays for detection of antibodies against herpes simplex virus type 2 in African populations. J Clin Microbiol 42:2961–2965. doi: 10.1128/JCM.42.7.2961-2965.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laeyendecker O, Henson C, Gray RH, Nguyen RH, Horne BJ, Wawer MJ, Serwadda D, Kiwanuka N, Morrow RA, Hogrefe W, Quinn TC. 2004. Performance of a commercial, type-specific enzyme-linked immunosorbent assay for detection of herpes simplex virus type 2-specific antibodies in Ugandans. J Clin Microbiol 42:1794–1796. doi: 10.1128/JCM.42.4.1794-1796.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 30.Tamura K. 1992. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol Biol Evol 9:678–687. [DOI] [PubMed] [Google Scholar]
- 31.Nei M, Gojobori T. 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3:418–426. [DOI] [PubMed] [Google Scholar]
- 32.Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. 2010. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26:2462–2463. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SD. 2006. GARD: a genetic algorithm for recombination detection. Bioinformatics 22:3096–3098. doi: 10.1093/bioinformatics/btl474. [DOI] [PubMed] [Google Scholar]
- 34.Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267. [DOI] [PubMed] [Google Scholar]
- 35.Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salminen MO, Carr JK, Burke DS, McCutchan FE. 1995. Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Res Hum Retroviruses 11:1423–1425. doi: 10.1089/aid.1995.11.1423. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki Y, Nei M. 2001. Reliabilities of parsimony-based and likelihood-based methods for detecting positive selection at single amino acid sites. Mol Biol Evol 18:2179–2185. doi: 10.1093/oxfordjournals.molbev.a003764. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki Y, Nei M. 2002. Simulation study of the reliability and robustness of the statistical methods for detecting positive selection at single amino acid sites. Mol Biol Evol 19:1865–1869. doi: 10.1093/oxfordjournals.molbev.a004010. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki Y, Nei M. 2004. False-positive selection identified by ML-based methods: examples from the Sig1 gene of the diatom Thalassiosira weissflogii and the tax gene of a human T-cell lymphotropic virus. Mol Biol Evol 21:914–921. doi: 10.1093/molbev/msh098. [DOI] [PubMed] [Google Scholar]
- 40.Pond SL, Frost SD, Muse SV. 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- 41.Kosakovsky Pond SL, Frost SD. 2005. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22:1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- 42.Pond SL, Frost SD, Grossman Z, Gravenor MB, Richman DD, Brown AJ. 2006. Adaptation to different human populations by HIV-1 revealed by codon-based analyses. PLoS Comput Biol 2:e62. doi: 10.1371/journal.pcbi.0020062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL. 2012. Detecting individual sites subject to episodic diversifying selection. PLoS Genet 8:e1002764. doi: 10.1371/journal.pgen.1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL, Scheffler K. 2013. FUBAR: a fast, unconstrained Bayesian approximation for inferring selection. Mol Biol Evol 30:1196–1205. doi: 10.1093/molbev/mst030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zuckerkandl E, Pauling L. 1965. Evolutionary divergence and convergence in proteins. Academic Press, New York, NY. [Google Scholar]
- 46.Ragonnet-Cronin M, Hodcroft E, Hue S, Fearnhill E, Delpech V, Brown AJ, Lycett S. 2013. Automated analysis of phylogenetic clusters. BMC Bioinformatics 14:317. doi: 10.1186/1471-2105-14-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Korber B, Myers G. 1992. Signature pattern analysis: a method for assessing viral sequence relatedness. AIDS Res Hum Retroviruses 8:1549–1560. doi: 10.1089/aid.1992.8.1549. [DOI] [PubMed] [Google Scholar]
- 48.Martin D, Rybicki E. 2000. RDP: detection of recombination amongst aligned sequences. Bioinformatics 16:562–563. doi: 10.1093/bioinformatics/16.6.562. [DOI] [PubMed] [Google Scholar]
- 49.Grabowska A, Jameson C, Laing P, Jeansson S, Sjogren-Jansson E, Taylor J, Cunningham A, Irving WL. 1999. Identification of type-specific domains within glycoprotein G of herpes simplex virus type 2 (HSV-2) recognized by the majority of patients infected with HSV-2, but not by those infected with HSV-1. J Gen Virol 80:1789–1798. [DOI] [PubMed] [Google Scholar]
- 50.Wertheim JO, Smith MD, Smith DM, Scheffler K, Kosakovsky Pond SL. 2014. Evolutionary origins of human herpes simplex viruses 1 and 2. Mol Biol Evol 31:2356–2364. doi: 10.1093/molbev/msu185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Belshe RB, Leone PA, Bernstein DI, Wald A, Levin MJ, Stapleton JT, Gorfinkel I, Morrow RL, Ewell MG, Stokes-Riner A, Dubin G, Heineman TC, Schulte JM, Deal CD. 2012. Efficacy results of a trial of a herpes simplex vaccine. N Engl J Med 366:34–43. doi: 10.1056/NEJMoa1103151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dutton JL, Li B, Woo WP, Marshak JO, Xu Y, Huang ML, Dong L, Frazer IH, Koelle DM. 2013. A novel DNA vaccine technology conveying protection against a lethal herpes simplex viral challenge in mice. PLoS One 8:e76407. doi: 10.1371/journal.pone.0076407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jones CA, Cunningham AL. 2003. Development of prophylactic vaccines for genital and neonatal herpes. Expert Rev Vaccines 2:541–549. doi: 10.1586/14760584.2.4.541. [DOI] [PubMed] [Google Scholar]
- 54.Hoshino Y, Pesnicak L, Dowdell KC, Burbelo PD, Knipe DM, Straus SE, Cohen JI. 2009. Protection from herpes simplex virus (HSV)-2 infection with replication-defective HSV-2 or glycoprotein D2 vaccines in HSV-1-seropositive and HSV-1-seronegative guinea pigs. J Infect Dis 200:1088–1095. doi: 10.1086/605645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Belshe RB, Heineman TC, Bernstein DI, Bellamy AR, Ewell M, van der Most R, Deal CD. 2014. Correlate of immune protection against HSV-1 genital disease in vaccinated women. J Infect Dis 209:828–836. doi: 10.1093/infdis/jit651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Long D, Skoberne M, Gierahn TM, Larson S, Price JA, Clemens V, Baccari AE, Cohane KP, Garvie D, Siber GR, Flechtner JB. 2014. Identification of novel virus-specific antigens by CD4(+) and CD8(+) T cells from asymptomatic HSV-2 seropositive and seronegative donors. Virology 464-465:296–311. doi: 10.1016/j.virol.2014.07.018. [DOI] [PubMed] [Google Scholar]
- 57.Cairns TM, Huang ZY, Whitbeck JC, Ponce de Leon M, Lou H, Wald A, Krummenacher C, Eisenberg RJ, Cohen GH. 2014. Dissection of the antibody response against herpes simplex virus glycoproteins in naturally infected humans. J Virol 88:12612–12622. doi: 10.1128/JVI.01930-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McGeoch DJ, Cook S, Dolan A, Jamieson FE, Telford EA. 1995. Molecular phylogeny and evolutionary timescale for the family of mammalian herpesviruses. J Mol Biol 247:443–458. doi: 10.1006/jmbi.1995.0152. [DOI] [PubMed] [Google Scholar]
- 59.McGeoch DJ, Cook S. 1994. Molecular phylogeny of the Alphaherpesvirinae subfamily and a proposed evolutionary timescale. J Mol Biol 238:9–22. doi: 10.1006/jmbi.1994.1264. [DOI] [PubMed] [Google Scholar]
- 60.Fleming DT, McQuillan GM, Johnson RE, Nahmias AJ, Aral SO, Lee FK, St Louis ME. 1997. Herpes simplex virus type 2 in the United States, 1976 to 1994. N Engl J Med 337:1105–1111. doi: 10.1056/NEJM199710163371601. [DOI] [PubMed] [Google Scholar]
- 61.Nahmias AJ, Lee FK, Beckman-Nahmias S. 1990. Sero-epidemiological and -sociological patterns of herpes simplex virus infection in the world. Scand J Infect Dis Suppl 69:19–36. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.