

ABSTRACT
Herpes simplex virus 2 (HSV-2), the principal causative agent of recurrent genital herpes, is a highly prevalent viral infection worldwide. Limited information is available on the amount of genomic DNA variation between HSV-2 strains because only two genomes have been determined, the HG52 laboratory strain and the newly sequenced SD90e low-passage-number clinical isolate strain, each from a different geographical area. In this study, we report the nearly complete genome sequences of 34 HSV-2 low-passage-number and laboratory strains, 14 of which were collected in Uganda, 1 in South Africa, 11 in the United States, and 8 in Japan. Our analyses of these genomes demonstrated remarkable sequence conservation, regardless of geographic origin, with the maximum nucleotide divergence between strains being 0.4% across the genome. In contrast, prior studies indicated that HSV-1 genomes exhibit more sequence diversity, as well as geographical clustering. Additionally, unlike HSV-1, little viral recombination between HSV-2 strains could be substantiated. These results are interpreted in light of HSV-2 evolution, epidemiology, and pathogenesis. Finally, the newly generated sequences more closely resemble the low-passage-number SD90e than HG52, supporting the use of the former as the new reference genome of HSV-2.
IMPORTANCE Herpes simplex virus 2 (HSV-2) is a causative agent of genital and neonatal herpes. Therefore, knowledge of its DNA genome and genetic variability is central to preventing and treating genital herpes. However, only two full-length HSV-2 genomes have been reported. In this study, we sequenced 34 additional HSV-2 low-passage-number and laboratory viral genomes and initiated analysis of the genetic diversity of HSV-2 strains from around the world. The analysis of these genomes will facilitate research aimed at vaccine development, diagnosis, and the evaluation of clinical manifestations and transmission of HSV-2. This information will also contribute to our understanding of HSV evolution.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) and herpes simplex virus 2 (HSV-2) are two closely related human species of herpesviruses in the genus Simplexvirus of the family Herpesviridae (1). HSV-1 is mostly associated with orofacial infections, while HSV-2 is generally associated with genital herpes. Both viruses cause significant human disease, so knowledge of the structure of their DNA genomes and the extent of their genetic variation is very important. A high overall GC content and the presence of highly reiterated repeat regions in both noncoding and coding portions of the genome complicate sequence determination (2).
The HSV linear double-stranded DNA genomes consist of two covalent linked components, the long (L) and short (S) components, which invert relative to each other by intramolecular recombination (1). The L component consists of unique sequences (UL) bounded by inverted repeats (RL and RL′), and the S component consists of unique sequences (US) bounded by inverted repeats (RS and RS′) (3). The termini contain direct repeats of a sequence called the “a” sequence, and copies of this sequence are present in an inverted form, designated the a′ sequence, at the L-S junction (4). The genomic structure can therefore be diagrammed as an-RL-UL- RL′-a′m-RS′-US-RS-a (1). The inverted copies of the “a” sequences promote recombination between the termini and the internal repeats, resulting in the inversion of the L and S components. This results in four isomers of the viral genome, which are all packaged in virions (4). There are 84 recognized unique protein-coding open reading frames (ORFs) and several RNA transcripts that are not proven to encode proteins (1). They include the latency-associated transcripts (LATs) and several microRNAs. Five genes are located within the RL and RS sequences and are therefore diploid.
The complete genome of the HSV-1 laboratory strain 17 was determined in 1988 (5), and a large panel of HSV-1 genomes was recently sequenced (6, 7). Analysis of this large panel of HSV-1 genomes from several geographically distinct regions (6) has shown that despite high levels of sequence conservation, HSV-1 strains exhibit interstrain diversity, as well as geographic clustering (6). Furthermore, these whole-genome studies confirm that HSV-1 strains undergo recombination with high frequency across the entire genome (6).
The complete genome sequence of the HSV-2 HG52 laboratory strain was published in 1988, based on Sanger sequencing (8), and it has served as the reference genome for HSV-2. The original Sanger sequence of HSV-2 HG52 contains some errors, but these were corrected by Illumina sequencing (2) (GenBank accession number JN561323). The complete genome of the first low-passage-number HSV-2 isolate, SD90e, was published in 2014 (2). Currently, these are the only complete HSV-2 genomes that have been determined.
There is, however, some limited information about the evolution and diversity of HSV-2 genomes based on analysis of individual HSV-2 genes. Previous analysis of HSV-2 glycoprotein genes from 47 HSV-2 isolates from Europe and Africa has shown evidence of less genetic variation than HSV-1 and a high probability of recombination in HSV-2 (9). There is also evidence that the HSV-2 strains from the United States/Western Europe and Africa have diverged from each other (9) and have differences in immunological and pathogenic properties (10). Therefore, there has been a need to generate additional HSV-2 genome sequences for comparative purposes.
Based on the analysis of glycoprotein B (UL27) gene sequences, HSV-2 was originally reported to have diverged 6.6 million years ago from the closely related species HSV-1 (11), while analysis of 8 well-conserved genes led to a revised date of 8.4 to 8.5 million years (12). Analysis of the genome of a chimpanzee herpesvirus (ChHV) isolated in 2004 showed that HSV-2 was more closely related to ChHV than to HSV-1 (13, 14). Phylogenetic analysis suggested that HSV-2 might be the original human herpes simplex virus (14). However, using molecular dating, a recent study concluded that HSV-1 diverged from ChHV about 6 million years ago but that HSV-2 diverged from ChHV only 1.6 million years ago. The authors hypothesized that the latter occurred in a second, independent transmission to humans (15).
To facilitate comparative studies of HSV-2 evolution and pathogenesis, we present nearly full-length HSV-2 sequence data from 34 new strains, including low-passage-number isolates from diverse geographic locations throughout the world, and the initial comparative analysis of these genomes. These provide genomic information for the study of phenotypic differences, including antigenic diversity, among global isolates. This information will also assist in the development of therapeutic strategies, including accurate diagnostics, identification of naturally occurring drug resistance mutations, and vaccine design.
MATERIALS AND METHODS
Viruses.
The genomes of 34 HSV-2 strains were determined in this study. Fourteen of the viral isolates were obtained from individuals in the Rakai district in Uganda. These Ugandan isolates were cultured using cell monolayers of human foreskin fibroblasts (Hs27; Diagnostic Hybrids, Athens, OH). The cultures were monitored every 24 h for 4 days until a cytopathic effect (CPE) of at least 80% was reached, and the virus was harvested. These isolates underwent two additional passages in Vero cells prior to DNA isolation. Three viral isolates obtained at Johns Hopkins Hospital in Baltimore, MD, were cultured and identified using the ELVIS-HSV system (Diagnostic Hybrids, Athens, OH), which utilizes a genetically engineered baby hamster kidney cell line to indicate the presence of HSV. The isolates then underwent two additional passages in Vero cells prior to DNA preparation. Five samples from four subjects in Seattle, WA, were collected between 1996 and 2007. They were initially isolated in human diploid fibroblasts and then passaged twice in Vero cells prior to DNA preparation. The U.S. laboratory strain 333-R519 was propagated as described previously (16). The U.S. strain BethesdaP5 is a fresh human isolate that has been passaged only 4 times and only in human diploid fibroblasts (MRC5 cells). HSV-2 strain SD66 was isolated in Carletonville, South Africa, and propagated as described previously (10, 17). HSV-2 strain 89-390 was isolated in Boston, MA, and propagated as described previously (10, 18). The SD66 and 89-390 primary isolates were passaged 3 times on Vero cells to prepare stocks for these experiments. Eight HSV-2 strains obtained in a clinic in Tokyo, Japan, and provided by T. Kawana were isolated on R66 cells (19), passaged once in BJ-1 cells (human fibroblasts), and then passaged twice in Vero cells before viral DNA was isolated.
Preparation of viral DNA.
HSV-2 DNA from the Ugandan and Seattle strains was isolated from cytoplasmic and supernatant virions as described previously, with slight modifications (20). Viral DNAs from Vero cells infected with SD66 and 89-390 were prepared by double banding in NaI gradients, as described previously (2). Virion DNA was prepared from the Japanese isolates as described previously (21).
Genome sequencing and assembly.
Library construction and sequencing on the Illumina platform were performed at the Broad Institute as described previously (22). Consensus genome assembly was performed as described previously (2). Briefly, Illumina fragment pair data were first processed using ALLPATHS-LG (version R44182) to find overlaps between fragment pairs and to fill gaps where no overlap was present. This generated a set of sequencing fragments that consist of the complete sequence between two ends of a paired read set. These unpaired filled fragments were then analyzed using Roche's runMapping (version vMapAsmResearch-10/14/2011) program with default parameters and a reference genome, the original HSV-2 HG52 sequence. This reference consisted of unique segments (UL and US) and single copies of the repeat segments (RL and RS) of the HG52 genome flanked by a small amount of additional repetitive sequence at each terminus. The runMapping tool produced consensus sequences built from the placements of the filled fragment reads from each sample to the HSV-2 HG52 reference genome.
HSV-2 sequence alignments.
Alignments were generated to compare full-length HSV-2 sequence populations with FSA v1.15.7 using default parameters and the anchor-annealing technique for very long sequences (23). One alignment contained the 34 HSV-2 sequences generated in this study, along with four sequences from the GenBank database: the original Sanger sequence for HG52 (RefSeq; accession no. NC_001798.1), the updated Illumina sequence for HSV-2 strain HG52 (HG52 ILMN; accession no. JN561323), the HSV-2 SD90e sequence (2) (accession no. KF781518), and the ChHV genome sequence (accession no. NC_023677.1).
An additional HSV-2 alignment was generated containing the 34 newly sequenced genomes, the published SD90e genome, and the two genome sequences of the HG52 reference strains described above. Small repeat regions between and sometimes within HSV-2 coding domains and within the long and short terminal repeats characterize HSV-2. Therefore, to increase the quality of the alignments used for subsequent analyses, the full-length HSV-2 genome alignment was manually edited with MEGA5 software (24). This approach also allowed the localization of regions where sequence amplification was not efficient. Problematic regions were removed prior to phylogenetic analysis. This resulted in the exclusion of ∼3,000 bp out of a total of ∼152,000 bp, or approximately 2% of the genome sequence. Identity plots of this alignment were generated using Geneious version 6.0.5 (25).
Diversity and divergence calculations.
Diversity and divergence calculations were performed using MEGA5 software with all positions in alignments containing gaps and missing data eliminated. For the calculation of divergence between the 34 full-length HSV-2 genomes, a pairwise distance (p-distance) was calculated. Estimates of diversity within open reading frames were calculated using the Tamura-Nei molecular model (identified as the best-fitting model using the hierarchical test based on the Bayesian information criterion), and standard errors were calculated using a bootstrap procedure (1,000 replicates). Amino acid diversity was similarly calculated using the Poisson correction method. The ratio of nonsynonymous (dN) to synonymous (dS) substitutions for each site (dN/dS) was calculated by averaging over all sequence pairs using the Nei-Gojobori model. Divergence between the NCBI HSV-2 reference sequence HG52, the HG52 Illumina sequence, and the SD90e sequence and all other HSV-2 genomes was calculated using the Tamura-Nei substitution model. Additional analysis of the diversity and divergence of 7 HSV-2 ORF sequences available in GenBank (UL23, UL27, UL30, UL49, US4, US7, and US8) was performed as described above.
Construction of phylogenetic trees.
The randomized accelerated maximum-likelihood program (RAxML [26]) was run with 1,000 bootstrap replicates to construct phylogenies for the ChHV-1 and HSV-2 full-genome alignment and the HSV-2-only alignment. The single most likely tree from the 1,000 replicates is shown, along with the total percentage (0% to 100%) of bootstrap support for each branch. Bootstrap support values for branching of ChHV from the HSV-2 clade were robust (100%). Bootstrap support values within the HSV-2 clade were all below 10% and are not shown.
Analysis of recombination.
The recombination detection program (RDP) (27) was run on all full-length genome sequences representing all available genotypes and subtypes. Any sequences that produced consistently low P values among the RDP's multiple tests for recombination were subjected to further analysis. Simplot (28) was used to apply a boot-scanning approach to full-length sequences using the following parameters: 1,000-bp window, 1,000-bp step size, GapStrip:on, 100 repetitions, and F84 (maximum likelihood) T/t of 2.0. Highly related sequences were grouped to reduce phylogenetic noise during boot scanning. Groups were defined by phylogenetic analysis and a significant bootstrap of >90%. The groups included 13 genomes from Uganda (Uganda clade; strains M22987, D30613, F70764, M1119, L22861, H00066, A76191, J09622, J32715, G75809, A76832, D39650, and D39765), 4 genomes from Japan (Japan clade; strains JA2, JA3, JA6, and JA9), 2 genomes from the United States (US clade; 9335_2005_576 and 9335_2007_14), 2 genomes from Uganda and the United States (UG_US clade; K39924 and 44_619833), and 5 genomes from the United States and South Africa (US_ZA clade; 89_390, 44_419851, 333_9519, SD90e, and SD66). Recombination signal in Simplot was considered positive at a cutoff of 70% (29).
Nucleotide sequence accession numbers.
The sequences of the 34 HSV-2 isolates described were submitted to GenBank under the accession numbers given in Table 1.
TABLE 1.
Genomes, information on cell passage, and accession numbers
| Virus | Strain | Collection location | Collection yr | Primary clinical isolate | Passage history and notes (reference[s]) | Accession no. |
|---|---|---|---|---|---|---|
| HSV-2 | 8937_1999_3336 | Seattle, WA, USA | 2003 | Yes | 2 passages on Vero cells | KR135298 |
| HSV-2 | 10883_2001_13347 | Seattle, WA, USA | 2005 | Yes | 2 passages on Vero cells | KR135311 |
| HSV-2 | 9335_2005_576 | Seattle, WA, USA | 2009 | Yes | 2 passages on Vero cells | KR135312 |
| HSV-2 | 9335_2007_14 | Seattle, WA, USA | 2011 | Yes | 2 passages on Vero cells | KR135313 |
| HSV-2 | 7444_1996_25809 | Seattle, WA, USA | 1996 | Yes | 2 passages on Vero cells | KR135314 |
| HSV-2 | 89_390 | MA, USA | 1989 | Yes | 3 passages on Vero cells | KR135321 |
| HSV-2 | 44_619833 | MD, USA | 2007 | Yes | 2 passages on Vero cells | KR135308 |
| HSV-2 | 44_419851 | MD, USA | 2007 | Yes | 2 passages on Vero cells | KR135309 |
| HSV-2 | 44_319857 | MD, USA | 2007 | Yes | 2 passages on Vero cells | KR135310 |
| HSV-2 | BethesdaP5 | MD, USA | Unknown | Yes | 4 passages on human diploid fibroblasts (MRC5 cells) | KR135330 |
| HSV-2 | 333_R519 | TX, USA | Unknown | No | Plaque-purified version of HSV-2 strain 333; unknown no. of passages on Vero cells; virulent in animal models (16) | KR135331 |
| HSV-2 | HG52 | Scotland, UK | Prior to 1971 | No | Laboratory-adapted strain, attentuated for virulence (8, 10, 35) | NC_001798.1 |
| HSV-2 | HG52 ILMN | Scotland, UK | Prior to 1971 | No | Illumina sequence of strain HG52 | JN561323 |
| HSV-2 | M22987 | Rakai, Uganda | 2007 | Yes | 2 passages on Vero cells | KR135299 |
| HSV-2 | D30613 | Rakai, Uganda | 2007 | Yes | 2 passages on Vero cells | KR135300 |
| HSV-2 | F70764 | Rakai, Uganda | 2007 | Yes | 2 passages on Vero cells | KR135301 |
| HSV-2 | M1119 | Rakai, Uganda | 2007 | Yes | 2 passages on Vero cells | KR135302 |
| HSV-2 | L22861 | Rakai, Uganda | 2007 | Yes | 2 passages on Vero cells | KR135303 |
| HSV-2 | H00066 | Rakai, Uganda | 2007 | Yes | 2 passages on Vero cells | KR135304 |
| HSV-2 | K39924 | Rakai, Uganda | 2007 | Yes | 2 passages on Vero cells | KR135305 |
| HSV-2 | A76191 | Rakai, Uganda | 2007 | Yes | 2 passages on Vero cells | KR135306 |
| HSV-2 | J09622 | Rakai, Uganda | 2008 | Yes | 2 passages on Vero cells | KR135307 |
| HSV-2 | J32715 | Rakai, Uganda | 2007 | Yes | 2 passages on Vero cells | KR135315 |
| HSV-2 | G75809 | Rakai, Uganda | 2007 | Yes | 2 passages on Vero cells | KR135316 |
| HSV-2 | A76832 | Rakai, Uganda | 2007 | Yes | 2 passages on Vero cells | KR135317 |
| HSV-2 | D39650 | Rakai, Uganda | 2008 | Yes | 2 passages on Vero cells | KR135318 |
| HSV-2 | D39765 | Rakai, Uganda | 2008 | Yes | 2 passages on Vero cells | KR135319 |
| HSV-2 | SD90e | Carletonville, South Africa | 1994 | Yes | 3 passages on Vero cells (2, 10) | KF781518 |
| HSV-2 | SD66 | Carletonville, South Africa | 1994 | Yes | 3 passages on Vero cells | KR135320 |
| HSV-2 | JA1 | Japan | 2009 | Yes | 2 passages on Vero cells | KR135322 |
| HSV-2 | JA2 | Japan | 2010 | Yes | 2 passages on Vero cells | KR135323 |
| HSV-2 | JA3 | Japan | 2008 | Yes | 2 passages on Vero cells | KR135324 |
| HSV-2 | JA5 | Japan | 2009 | Yes | 2 passages on Vero cells | KR135325 |
| HSV-2 | JA6 | Japan | 2009 | Yes | 2 passages on Vero cells | KR135326 |
| HSV-2 | JA7 | Japan | 2010 | Yes | 2 passages on Vero cells | KR135327 |
| HSV-2 | JA8 | Japan | 2009 | Yes | 2 passages on Vero cells | KR135328 |
| HSV-2 | JA9 | Japan | 2010 | Yes | 2 passages on Vero cells | KR135329 |
| ChHV-1 | 105640 | USA | 2004 | Yes | Unknown no. of passages on Vero cells (14) | NC_023677.1 |
RESULTS
Genomic sequencing and assembly.
We performed high-throughput, paired-end Illumina sequencing of purified, randomly fragmented viral DNA with read lengths of 101 bp. Reference-assisted assembly of the genomes of 34 HSV-2 isolates gathered for this study (Table 1) generated contig sequence spanning the UL and US regions of the HSV-2 genome, as well as single copies of each of the long and short inverted-repeat regions (RL and RS). Average read coverage for these genomes ranged from 3,100- to 9,300-fold. The contigs were aligned and combined into single genomes using the HG52 reference genome (NC_001798) as a scaffold. As has been reported for other recent HSV-1 and HSV-2 genome sequences (2, 6), Illumina sequencing was unable to distinguish individual copies of the inverted-repeat regions and could not efficiently resolve all of the small repeat regions between HSV-2 coding domains and within the RL and RS terminal repeats that characterize HSV-2. Generation of a second copy of the RL and RS inverted repeats bounding their respective unique sequences was therefore accomplished by inverting a copy of each sequence in the final assemblies. The repeat structure of several of the regions flanking RL and RS resulted in low read depth in these regions and led to gaps in the genome assemblies. Because of the inability of automatic alignment algorithms to consistently handle small insertions and deletions in these problematic regions and to increase the quality of the alignments used for subsequent analyses, these regions (Fig. 1, red boxes, and data not shown) were removed from the assemblies, and trimmed versions of the genomes were used for alignment and phylogenetic analysis. As with previous HSV-2 genomes (2), numerous base substitutions and insertions/deletions (indels) were detected in the aligned sequences. No large indels were observed, however.
FIG 1.

Overview of the features of the HSV-2 genome and sequence diversity in a genome alignment of 34 full genome sequences. The black bar at the top represents a consensus sequence drawn from the alignment of the genomes of 34 HSV-2 strains. The identity plot derived from this alignment, shown below, is colored as follows: green, 100% identity; green-brown, 30 to <100% identity; red, <30% identity. Below the identity plot, HSV-2 coding regions are shown, with arrows indicating the direction of the reading frame. The red boxes indicate regions of the multigenome alignment that either contained gaps or failed to align properly. These regions were deleted from the genome sequences in subsequent analyses.
Alignment and genomic diversity.
The generation of 34 additional nearly full-length HSV-2 genome sequences provided us with the opportunity to assess the relatedness of HSV-2 strains circulating in the United States, Africa, and Asia. Pairwise distance measurements of the 34 new genomes and the 2 previously reported genomes (Table 1) indicated that all the genomes were closely related to each other, as well as to HSV-2 HG52 and HSV-2 SD90e, with the maximum nucleotide divergence between strains being 0.4% across the genome (Table 2).
TABLE 2.
Estimates of evolutionary divergence between HSV-2 genome sequencesa
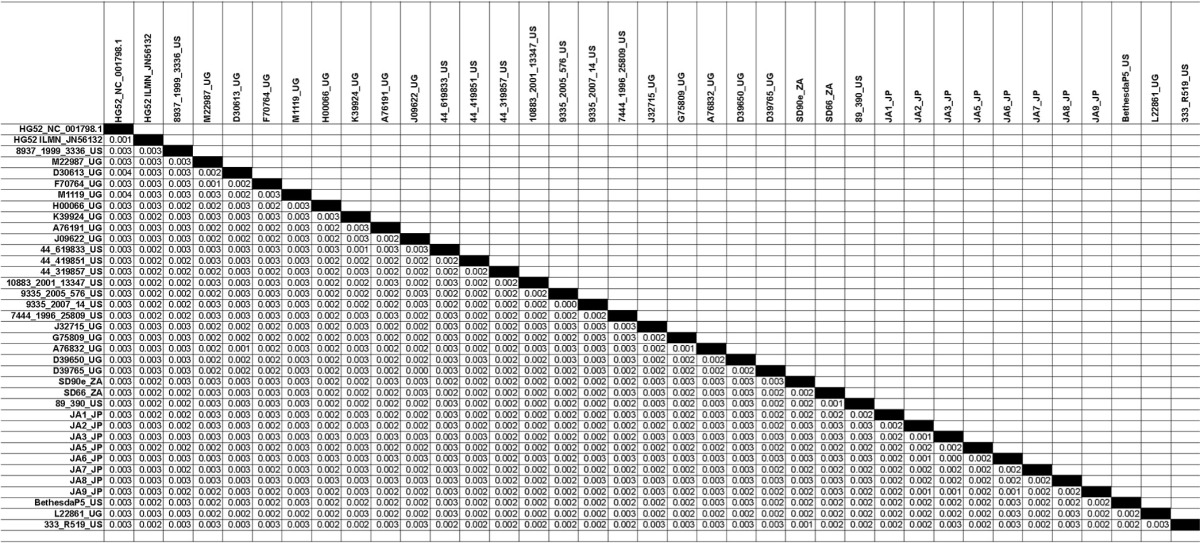
The numbers of base substitutions per site between sequences are shown. Analyses were conducted using the Maximum Composite Likelihood model (30). The analysis involved 37 nucleotide sequences. All positions containing gaps and missing data were eliminated. There were a total of 148,894 positions in the final data set. Evolutionary analyses were conducted in MEGA5 (24).
To compare these geographically diverse HSV-2 genomes, we first assessed the levels of DNA diversity of our sequenced strains, along with the two existing HG52 sequences, across the genome compared to the low-passage-number strain SD90e (Fig. 1). We noted that the genomes were largely conserved within the UL and US, with the highest variation observed in the intergenic regions. Regions with the highest levels of variation (>70%) were localized to known repetitive regions flanking the large internal and terminal repeat regions (Fig. 1). This clustering of variation could be attributed to the inherent variation in these regions, as well as to difficulties in sequencing and assembling these problematic regions with current deep-sequencing technologies.
Analysis of nucleotide and amino acid diversity among these HSV-2 genomes and within 70 UL and US ORFs confirmed high-level sequence conservation, with no pair of ORFs exceeding 0.5% diversity at the nucleotide level and 0.8% diversity at the amino acid level (Table 3). RL and RS ORFs were incomplete in a number of genomes, so they were not included in this analysis. Only one ORF exhibited nucleotide diversity levels over 0.4% (UL49), and ten ORFs showed amino acid diversity greater than 0.4% (UL20, UL26.5, UL27, UL39, UL44, UL49, UL53, US7, US4, and US11).
TABLE 3.
Nucleotide and amino acid diversity of HSV-2 strains in open reading frames
| ORF | Protein functiona | Orientationb | Length (bp) (nucleic acid reference sequence) | % Diversity (SE) |
dN/dS | No. of variable sites | |
|---|---|---|---|---|---|---|---|
| Nucleotide | Amino acid | ||||||
| UL1 | Glycoprotein L | F | 675 | 0.1 (0.0) | 0.2 (0.1) | 1.00 | 10 |
| UL2 | Uracil-DNA glycosylase | F | 1,005 | 0.2 (0.0) | 0.2 (0.1) | 0.25 | 20 |
| UL3 | Nuclear phosphoprotein | F | 702 | 0.1 (0.0) | 0.0 (0.0) | 0.00 | 7 |
| UL4 | Nuclear protein | R | 606 | 0.3 (0.2) | 0.3 (0.2) | 0.17 | 13 |
| UL5 | Component of DNA helicase-primase | R | 2,646 | 0.2 (0.0) | 0.1 (0.1) | 0.25 | 28 |
| UL6 | Minor capsid protein | F | 2,043 | 0.2 (0.0) | 0.3 (0.1) | 0.33 | 33 |
| UL7 | Virion egress protein | F | 891 | 0.4 (0.1) | 0.4 (0.2) | 0.20 | 16 |
| UL8 | Component of DNA helicase-primase | R | 2,259 | 0.3 (0.1) | 0.3 (0.1) | 0.40 | 47 |
| UL9 | Ori binding protein | R | 2,608 | 0.2 (0.0) | 0.1 (0.1) | 0.20 | 42 |
| UL10 | Glycoprotein M | F | 1,404 | 0.2 (0.1) | 0.2 (0.1) | 0.14 | 26 |
| UL11 | Myristylated tegument protein | R | 291 | 0.1 (0.1) | 0.3 (0.2) | 1.00 | 4 |
| UL12 | DNase | R | 1,863 | 0.2 (0.0) | 0.3 (0.1) | 0.33 | 31 |
| UL13 | Protein kinase; tegument protein | R | 1,496 | 0.2 (0.0) | 0.1 (0.0) | 0.00 | 19 |
| UL14 | Tegument protein | R | 660 | 0.0 (0.0) | 0.0 (0.0) | 0.00 | 4 |
| UL15 | Role in DNA packaging | F | 5,862 | 0.1 (0.0) | 0.2 (0.0) | 0.50 | 70 |
| UL16 | Proposed initiator CTG codon | R | 1,119 | 0.1 (0.1) | 0.2 (0.1) | 0.50 | 12 |
| UL17 | Tegument protein; DNA packaging | R | 2,118 | 0.2 (0.0) | 0.3 (0.1) | 0.67 | 29 |
| UL18 | Capsid protein | R | 957 | 0.1 (0.0) | 0.0 (0.0) | 0.00 | 6 |
| UL19 | Major capsid protein | R | 4,125 | 0.1 (0.0) | 0.1 (0.0) | 0.50 | 45 |
| UL20 | Virion membrane protein | R | 669 | 0.2 (0.1) | 0.6 (0.3) | 3.00 | 6 |
| UL21 | Tegument protein | F | 1,599 | 0.0 (0.0) | 0.0 (0.0) | 0.00 | 6 |
| UL22 | Glycoprotein H | R | 2,517 | 0.3 (0.1) | 0.3 (0.1) | 0.17 | 39 |
| UL23 | Thymidine kinase [2 possible poly(A) sites] | R | 1,131 | 0.2 (0.1) | 0.3 (0.1) | 1.00 | 16 |
| UL24 | Nuclear protein | F | 846 | 0.4 (0.1) | 0.4 (0.2) | 0.22 | 21 |
| UL25 | Virion protein | F | 1,758 | 0.1 (0.0) | 0.1 (0.0) | 0.00 | 20 |
| UL26 | Capsid maturation protease | F | 1,923 | 0.3 (0.1) | 0.4 (0.1) | 0.29 | 55 |
| UL26.5 | Capsid assembly protein | F | 990 | 0.4 (0.1) | 0.6 (0.2) | 0.38 | 39 |
| UL27 | Glycoprotein B | R | 2,718 | 0.2 (0.0) | 0.5 (0.1) | 0.67 | 45 |
| UL28 | Role in DNA packaging | R | 2,358 | 0.2 (0.0) | 0.2 (0.1) | 0.17 | 36 |
| UL29 | Single-stranded DNA binding protein | R | 3,609 | 0.2 (0.0) | 0.2 (0.1) | 0.14 | 61 |
| UL30 | DNA polymerase catalytic subunit | F | 3,723 | 0.1 (0.0) | 0.1 (0.0) | 0.50 | 32 |
| UL31 | Virion egress protein | R | 918 | 0.1 (0.0) | 0.0 (0.0) | 0.00 | 11 |
| UL32 | Role in DNA packaging | R | 1,797 | 0.2 (0.1) | 0.3 (0.1) | 0.17 | 29 |
| UL33 | Role in DNA packaging | F | 393 | 0.2 (0.1) | 0.1 (0.1) | 0.00 | 4 |
| UL34 | Membrane-associated phosphoprotein | F | 831 | 0.1 (0.1) | 0.2 (0.2) | 0.50 | 7 |
| UL35 | Capsid protein | F | 339 | 0.1 (0.0) | 0.1 (0.1) | 0.00 | 4 |
| UL36 | Very large tegument protein | R | 9,412 | 0.2 (0.0) | 0.2 (0.0) | 0.25 | 160 |
| UL37 | Tegument protein | R | 3,345 | 0.1 (0.0) | 0.2 (0.1) | 0.50 | 43 |
| UL38 | Capsid protein | F | 1,401 | 0.4 (0.1) | 0.4 (0.1) | 0.22 | 36 |
| UL39 | Ribonucleotide reductase large subunit | F | 3,441 | 0.4 (0.1) | 0.5 (0.1) | 0.18 | 97 |
| UL40 | Ribonucleotide reductase small subunit | F | 1,014 | 0.1 (0.0) | 0.0 (0.0) | 0.00 | 11 |
| UL41 | Tegument protein; host shutoff factor | R | 1,479 | 0.1 (0.0) | 0.1 (0.1) | 0.00 | 14 |
| UL42 | DNA polymerase subunit | F | 1,413 | 0.2 (0.1) | 0.4 (0.1) | 1.00 | 18 |
| UL43 | Probable membrane protein | F | 1,245 | 0.3 (0.1) | 0.4 (0.1) | 0.40 | 22 |
| UL44 | Glycoprotein C | F | 1,443 | 0.4 (0.1) | 0.7 (0.2) | 0.80 | 24 |
| UL45 | Tegument/envelope protein | F | 519 | 0.1 (0.0) | 0.1 (0.1) | 1.00 | 5 |
| UL46 | Tegument protein | R | 2,169 | 0.3 (0.1) | 0.4 (0.1) | 0.33 | 40 |
| UL47 | Tegument protein | R | 2,091 | 0.1 (0.0) | 0.3 (0.1) | 1.00 | 26 |
| UL48 | Tegument protein; transactivator of immediate-early genes | R | 1,473 | 0.2 (0.1) | 0.1 (0.1) | 0.00 | 17 |
| UL49 | Tegument protein | R | 912 | 0.5 (0.1) | 0.8 (0.3) | 0.40 | 26 |
| UL49A | Probable virion membrane protein | R | 264 | 0.4 (0.2) | 0.2 (0.1) | 0.07 | 7 |
| UL50 | Deoxyuridine triphosphatase | F | 1,110 | 0.2 (0.1) | 0.3 (0.1) | 0.25 | 23 |
| UL51 | Tegument protein | R | 735 | 0.1 (0.1) | 0.0 (0.0) | 0.00 | 5 |
| UL52 | Component of DNA helicase-primase | F | 3,204 | 0.2 (0.0) | 0.3 (0.1) | 0.25 | 49 |
| UL53 | Glycoprotein K | F | 1,017 | 0.4 (0.1) | 0.7 (0.2) | 0.50 | 27 |
| UL54 | ICP27 immediate-early regulatory protein | F | 1,539 | 0.1 (0.0) | 0.2 (0.1) | 0.50 | 22 |
| UL55 | Nuclear protein | F | 561 | 0.1 (0.0) | 0.2 (0.1) | 0.50 | 7 |
| UL56 | Membrane protein | R | 708 | 0.2 (0.1) | 0.4 (0.2) | 0.50 | 13 |
| US1 | ICP4 immediate-early transactivator | F | 1,239 | 0.4 (0.1) | 0.4 (0.2) | 0.38 | 32 |
| US2 | Virion protein | F | 873 | 0.2 (0.1) | 0.4 (0.2) | 0.50 | 11 |
| US3 | Protein kinase | F | 1,443 | 0.2 (0.1) | 0.3 (0.1) | 0.25 | 22 |
| US4 | Glycoprotein G | F | 2,097 | 0.4 (0.1) | 0.7 (0.1) | 1.00 | 61 |
| US5 | Glycoprotein J | F | 276 | 0.2 (0.1) | 0.2 (0.1) | 0.17 | 9 |
| US6 | Glycoprotein D | F | 1,179 | 0.2 (0.0) | 0.3 (0.1) | 0.25 | 17 |
| US7 | Glycoprotein I | F | 1,116 | 0.3 (0.1) | 0.6 (0.2) | 0.75 | 24 |
| US8 | Glycoprotein E | F | 1,635 | 0.2 (0.0) | 0.4 (0.1) | 1.00 | 29 |
| US9 | Tegument protein | F | 267 | 0.4 (0.2) | 0.2 (0.1) | 0.08 | 6 |
| US10 | Virion protein | R | 906 | 0.3 (0.1) | 0.4 (0.2) | 0.40 | 16 |
| US11 | RNA binding protein | R | 453 | 0.2 (0.1) | 0.5 (0.3) | 1.00 | 7 |
| US12 | Immediate-early inhibitor of antigen presentation | R | 258 | 0.2 (0.1) | 0.0 (0.0) | 0.00 | 2 |
Reference 1.
F, forward; R, reverse.
We also examined the ratio of nonsynonymous to synonymous substitutions (dN/dS) to detect evidence of selection pressure within HSV-2 ORFs (Table 3). We found that while the majority of ORFs appeared to be under negative, purifying selection (dN/dS < 1), several ORFs (UL1, UL11, UL23, UL42, UL45, UL47, US4, US8, and US11) showed more evidence of neutral selection (dN/dS = 1) (Table 3). One ORF (UL20) appeared to show evidence of positive selection (dN/dS > 1), although the relatively small number of variable sites present in the ORF makes interpretation difficult.
Because nearly all of the 34 new genomes were from low-passage-number isolates, we compared the nucleotide and amino acid divergences of their ORFs from those of the HG52 laboratory strain (both the Sanger RefSeq [NC_001798.1] and corrected Illumina [JN561323] sequences) and from the SD90e low-passage-number clinical strain (KF781518) (Table 4). We found that pairwise divergence of these genomes from both HG52 sequences and SD90e ranged from 0 to 1.1% at the nucleotide level and 0 to 2.1% at the amino acid level (Table 4). Amino acid divergence between the HSV-2 strains was sometimes much higher than corresponding nucleic acid divergence calculations. This is likely due to the high GC content of HSV genes, with about 80% G or C occurring at the 3rd codon position. This permits a biased codon usage for HSV, with an effective codon usage of approximately 40 out of 61 different codons. These biases are expected to cause relatively low nucleotide diversity for a given degree of amino acid diversity.
TABLE 4.
Nucleotide and amino acid divergence of HSV-2 strains in open reading frames
| ORF | Protein functiona | % Nucleotide divergence from: |
% Amino acid divergence from: |
||||
|---|---|---|---|---|---|---|---|
| HG52 RefSeqb | HG52 ILMNc | SD90ed | HG52 RefSeqb | HG52 ILMNc | SD90ed | ||
| UL1 | Glycoprotein L | 0.1 (0.0) | 0.1 (0.0) | 0.1 (0.0) | 0.1 (0.0) | 0.1 (0.0) | 0.1 (0.0) |
| UL2 | Uracil-DNA glycosylase | 0.5 (0.2) | 0.5 (0.2) | 0.4 (0.2) | 0.5 (0.3) | 0.4 (0.3) | 0.1 (0.1) |
| UL3 | Nuclear phosphoprotein | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
| UL4 | Nuclear protein | 0.2 (0.1) | 0.2 (0.1) | 0.2 (0.1) | 0.2 (0.1) | 0.2 (0.1) | 0.2 (0.1) |
| UL5 | Component of DNA helicase-primase | 0.2 (0.1) | 0.2 (0.1) | 0.1 (0.0) | 0.1 (0.0) | 0.1 (0.0) | 0.1 (0.0) |
| UL6 | Minor capsid protein | 0.6 (0.2) | 0.2 (0.1) | 0.2 (0.1) | 0.6 (0.2) | 0.4 (0.2) | 0.2 (0.1) |
| UL7 | Virion egress protein | 0.3 (0.1) | 0.3 (0.1) | 0.3 (0.1) | 0.3 (0.2) | 0.3 (0.2) | 0.2 (0.1) |
| UL8 | Component of DNA helicase-primase | 0.5 (0.1) | 0.3 (0.1) | 0.3 (0.1) | 0.8 (0.2) | 0.5 (0.2) | 0.3 (0.1) |
| UL9 | Ori binding protein | 0.2 (0.1) | 0.1 (0.0) | 0.2 (0.1) | 0.2 (0.1) | 0.1 (0.1) | 0.1 (0.1) |
| UL10 | Glycoprotein M | 0.3 (0.1) | 0.2 (0.1) | 0.1 (0.0) | 0.3 (0.2) | 0.3 (0.2) | 0.1 (0.0) |
| UL11 | Myristylated tegument protein | 1.1 (0.5) | 1.0 (0.6) | 0.1 (0.0) | 2.1 (1.4) | 2.1 (1.5) | 0.2 (0.1) |
| UL12 | DNase | 0.3 (0.1) | 0.3 (0.1) | 0.1 (0.0) | 0.4 (0.3) | 0.3 (0.2) | 0.2 (0.1) |
| UL13 | Protein kinase; tegument protein | 0.3 (0.1) | 0.2 (0.1) | 0.1 (0.1) | 0.3 (0.2) | 0.2 (0.2) | 0.0 (0.0) |
| UL14 | Tegument protein | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
| UL15 | Role in DNA packaging | 0.1 (0.0) | 0.1 (0.0) | 0.1 (0.0) | 0.2 (0.1) | 0.2 (0.1) | 0.2 (0.1) |
| UL16 | Proposed initiator CTG codon | 0.1 (0.1) | 0.1 (0.0) | 0.1 (0.0) | 0.1 (0.0) | 0.1 (0.0) | 0.1 (0.0) |
| UL17 | Tegument protein; DNA packaging | 0.3 (0.1) | 0.2 (0.1) | 0.2 (0.0) | 0.3 (0.1) | 0.2 (0.1) | 0.2 (0.1) |
| UL18 | Capsid protein | 0.1 (0.1) | 0.1 (0.1) | 0.1 (0.1) | 0.2 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
| UL19 | Major capsid protein | 0.3 (0.1) | 0.1 (0.0) | 0.2 (0.0) | 0.5 (0.1) | 0.2 (0.1) | 0.1 (0.0) |
| UL20 | Virion membrane protein | 0.3 (0.2) | 0.3 (0.2) | 0.1 (0.1) | 0.4 (0.2) | 0.4 (0.2) | 0.4 (0.2) |
| UL21 | Tegument protein | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
| UL22 | Glycoprotein H | 0.3 (0.1) | 0.3 (0.1) | 0.3 (0.1) | 0.3 (0.1) | 0.3 (0.1) | 0.2 (0.1) |
| UL23 | Thymidine kinase [2 possible poly(A) sites] | 0.2 (0.1) | 0.2 (0.1) | 0.2 (0.1) | 0.5 (0.3) | 0.5 (0.3) | 0.2 (0.1) |
| UL24 | Nuclear protein | 0.2 (0.1) | 0.2 (0.1) | 0.2 (0.1) | 0.2 (0.1) | 0.2 (0.1) | 0.2 (0.1) |
| UL25 | Virion protein | 0.2 (0.1) | 0.1 (0.1) | 0.0 (0.0) | 0.3 (0.2) | 0.2 (0.2) | 0.0 (0.0) |
| UL26 | Capsid maturation protease | 0.3 (0.1) | 0.3 (0.1) | 0.2 (0.1) | 0.4 (0.2) | 0.4 (0.2) | 0.2 (0.1) |
| UL26.5 | Capsid assembly protein | 0.4 (0.2) | 0.4 (0.2) | 0.3 (0.1) | 0.8 (0.4) | 0.8 (0.4) | 0.3 (0.1) |
| UL27 | Glycoprotein B | 0.2 (0.1) | 0.2 (0.0) | 0.2 (0.1) | 0.4 (0.1) | 0.4 (0.1) | 0.5 (0.2) |
| UL28 | Role in DNA packaging | 0.2 (0.1) | 0.2 (0.1) | 0.1 (0.0) | 0.2 (0.1) | 0.2 (0.1) | 0.1 (0.0) |
| UL29 | Single-stranded DNA binding protein | 0.3 (0.1) | 0.3 (0.1) | 0.2 (0.1) | 0.2 (0.1) | 0.2 (0.1) | 0.2 (0.1) |
| UL30 | DNA polymerase catalytic subunit | 0.1 (0.1) | 0.1 (0.0) | 0.1 (0.0) | 0.3 (0.1) | 0.3 (0.1) | 0.2 (0.1) |
| UL31 | Virion egress protein | 0.1 (0.1) | 0.1 (0.1) | 0.1 (0.1) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
| UL32 | Role in DNA packaging | 0.3 (0.1) | 0.2 (0.1) | 0.2 (0.1) | 0.4 (0.2) | 0.3 (0.1) | 0.2 (0.1) |
| UL33 | Role in DNA packaging | 0.2 (0.2) | 0.2 (0.1) | 0.2 (0.2) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
| UL34 | Membrane-associated phosphoprotein | 0.1 (0.0) | 0.2 (0.1) | 0.1 (0.1) | 0.1 (0.1) | 0.5 (0.4) | 0.3 (0.2) |
| UL35 | Capsid protein | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
| UL36 | Very large tegument protein | 0.2 (0.0) | 0.2 (0.0) | 0.2 (0.0) | 0.3 (0.1) | 0.2 (0.1) | 0.2 (0.0) |
| UL37 | Tegument protein | 0.2 (0.1) | 0.1 (0.0) | 0.1 (0.0) | 0.3 (0.1) | 0.2 (0.1) | 0.2 (0.1) |
| UL38 | Capsid protein | 0.7 (0.1) | 0.3 (0.1) | 0.4 (0.1) | 1.0 (0.3) | 0.4 (0.2) | 0.6 (0.3) |
| UL39 | Ribonucleotide reductase large subunit | 0.4 (0.1) | 0.8 (0.1) | 0.4 (0.1) | 0.7 (0.2) | 0.7 (0.2) | 0.4 (0.1) |
| UL40 | Ribonucleotide reductase small subunit | 0.1 (0.0) | 0.1 (0.0) | 0.1 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
| UL41 | Tegument protein; host shutoff factor | 0.1 (0.0) | 0.1 (0.0) | 0.3 (0.1) | 0.0 (0.0) | 0.0 (0.0) | 0.2 (0.2) |
| UL42 | DNA polymerase subunit | 0.3 (0.1) | 0.3 (0.1) | 0.2 (0.1) | 0.5 (0.3) | 0.5 (0.2) | 0.5 (0.2) |
| UL43 | Probable membrane protein | 0.2 (0.1) | 0.2 (0.1) | 0.2 (0.0) | 0.4 (0.2) | 0.4 (0.2) | 0.2 (0.1) |
| UL44 | Glycoprotein C | 0.4 (0.1) | 0.3 (0.1) | 0.4 (0.1) | 0.5 (0.2) | 0.5 (0.2) | 0.6 (0.2) |
| UL45 | Tegument/envelope protein | 0.1 (0.1) | 0.0 (0.0) | 0.0 (0.0) | 0.4 (0.3) | 0.0 (0.0) | 0.0 (0.0) |
| UL46 | Tegument protein | 0.6 (0.1) | 0.4 (0.1) | 0.5 (0.1) | 0.7 (0.2) | 0.5 (0.2) | 0.5 (0.2) |
| UL47 | Tegument protein | 0.1 (0.0) | 0.1 (0.0) | 0.1 (0.0) | 0.2 (0.1) | 0.2 (0.1) | 0.2 (0.1) |
| UL48 | Tegument protein; transactivator of immediate-early genes | 0.1 (0.1) | 0.1 (0.0) | 0.2 (0.1) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
| UL49 | Tegument protein | 0.5 (0.1) | 0.7 (0.2) | 0.8 (0.2) | 0.7 (0.3) | 0.7 (0.3) | 1.1 (0.4) |
| UL49A | Probable virion membrane protein | 0.5 (0.3) | 0.4 (0.3) | 1.0 (0.6) | 0.1 (0.1) | 0.1 (0.1) | 1.2 (1.1) |
| UL50 | Deoxyuridine triphosphatase | 0.2 (0.1) | 0.2 (0.1) | 0.2 (0.1) | 0.3 (0.2) | 0.3 (0.2) | 0.3 (0.2) |
| UL51 | Tegument protein | 0.1 (0.1) | 0.1 (0.1) | 0.1 (0.1) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
| UL52 | Component of DNA helicase-primase | 0.3 (0.1) | 0.2 (0.1) | 0.2 (0.1) | 0.4 (0.2) | 0.4 (0.1) | 0.3 (0.1) |
| UL53 | Glycoprotein K | 0.4 (0.1) | 0.4 (0.1) | 0.4 (0.2) | 0.7 (0.3) | 0.5 (0.2) | 0.7 (0.3) |
| UL54 | ICP27 immediate-early regulatory protein | 0.1 (0.0) | 0.1 (0.0) | 0.1 (0.1) | 0.2 (0.1) | 0.1 (0.0) | 0.1 (0.0) |
| UL55 | Nuclear protein | 0.1 (0.1) | 0.1 (0.0) | 0.1 (0.0) | 0.1 (0.0) | 0.1 (0.1) | 0.1 (0.1) |
| UL56 | Membrane protein | 0.1 (0.1) | 0.1 (0.0) | 0.5 (0.2) | 0.2 (0.1) | 0.2 (0.1) | 1.0 (0.6) |
| US1 | ICP4 immediate-early transactivator | 0.7 (0.2) | 0.5 (0.2) | 0.3 (0.1) | 1.0 (0.4) | 0.7 (0.3) | 0.3 (0.1) |
| US2 | Virion protein | 0.3 (0.1) | 0.3 (0.1) | 0.3 (0.1) | 0.6 (0.4) | 0.6 (0.3) | 0.6 (0.4) |
| US3 | Protein kinase | 0.2 (0.1) | 0.1 (0.0) | 0.2 (0.1) | 0.4 (0.2) | 0.2 (0.1) | 0.3 (0.2) |
| US4 | Glycoprotein G | 0.4 (0.1) | 0.4 (0.1) | 0.4 (0.1) | 0.7 (0.2) | 0.6 (0.2) | 0.7 (0.2) |
| US5 | Glycoprotein J | 0.1 (0.0) | 0.1 (0.1) | 0.1 (0.1) | 0.1 (0.1) | 0.1 (0.1) | 0.1 (0.1) |
| US6 | Glycoprotein D | 0.2 (0.0) | 0.1 (0.0) | 0.2 (0.1) | 0.6 (0.2) | 0.1 (0.0) | 0.1 (0.0) |
| US7 | Glycoprotein I | 0.4 (0.1) | 0.3 (0.1) | 0.3 (0.1) | 0.7 (0.3) | 0.7 (0.3) | 0.5 (0.3) |
| US8 | Glycoprotein E | 0.2 (0.1) | 0.2 (0.1) | 0.2 (0.1) | 0.6 (0.2) | 0.4 (0.2) | 0.3 (0.2) |
| US9 | Tegument protein | 0.3 (0.2) | 0.3 (0.2) | 0.3 (0.2) | 0.1 (0.1) | 0.1 (0.1) | 0.1 (0.1) |
| US10 | Virion protein | 0.3 (0.1) | 0.3 (0.1) | 0.2 (0.1) | 0.6 (0.1) | 0.6 (0.3) | 0.5 (0.3) |
| US11 | RNA binding protein | 0.1 (0.1) | 0.1 (0.1) | 0.1 (0.1) | 0.3 (0.2) | 0.3 (0.2) | 0.3 (0.2) |
| US12 | Immediate-early inhibitor of antigen presentation | 0.1 (0.1) | 0.1 (0.1) | 0.4 (0.3) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
| Avg divergence | 0.26 (0.02) | 0.22 (0.02) | 0.21 (0.02) | 0.36 (0.04) | 0.30 (0.04) | 0.25 (0.03) | |
Reference 1.
HG52 RefSeq accession no. NC_001798.1.
HG52 ILMN accession no. JN561323.
SD90e accession no. KF781518.
In general, we saw that HG52 ILMN was more closely related to the other 34 genomes than HG52 RefSeq when comparing either individual ORFs or the average divergence for all ORFs (Table 4). This was presumably a result of the sequencing errors in the original RefSeq sequence. When we compared the divergence of ORF amino acid sequence from SD90e and HG52 ILMN, we observed that 25 of the ORFs in the 34 new sequences were more closely related to SD90e than to HG52 ILMN, while 10 ORFs were closer to HG52 ILMN. Most ORFs were only slightly more divergent from one strain or the other, but four were noticeably different. UL49 and UL49A were strikingly diverged from SD90e (1.1 and 1.2%, respectively), while two ORFs, UL11 and US1, were significantly diverged from HG52 ILMN. The origin of the divergence in these strains is not immediately obvious. Furthermore, the average divergence for all ORFs was greater for HG52 ILMN than for SD90e; therefore, the 34 new genomes are more closely related to SD90e than to HG52 ILMN.
To determine if the levels of diversity and divergence seen in the ORFs of these 34 HSV-2 genomes were reflected in a larger data set, we calculated the levels of diversity in the GenBank sequences available for 7 ORFs (Table 5), as well as the levels of divergence from the HG52 RefSeq, HG52 ILMN, and SD90e strains. We observed that the nucleotide and amino acid diversities in the larger, independent sequence sets were similar to what we had observed for the 34 full-length HSV-2 genomes examined. While larger numbers of sequences for ORFs that were most divergent from HG52 (UL11 and US1) were not available in GenBank, the levels of amino acid divergence of the available ORFs from the two HG52 sequences were similar to those seen for our 34 new sequences. An additional 122 sequences were available for one of the ORFs that displayed >1% amino acid divergence from SD90e (UL49). Again, we found that divergence of these UL49 GenBank sequences was greater for SD90e than for HG52 ILMN (Table 5).
TABLE 5.
Diversity and divergence of GenBank sequences for select HSV-2 open reading frames
| ORF | Protein | GenBank |
New sequences |
||||||
|---|---|---|---|---|---|---|---|---|---|
| No. of sequences | Parametera | % Diversity/divergencea (SE) |
No. of sequences | Parametera | % Diversity/divergencea (SE) |
||||
| Nucleotide | Amino acid | Nucleotide | Amino acid | ||||||
| UL23 | Thymidine kinase | 185 | Diversity | 0.2 (0.1) | 0.4 (0.2) | 34 | Diversity | 0.2 (0.1) | 0.3 (0.1) |
| Divergence from HG52 RefSeq | 0.1 (0.1) | 0.3 (0.2) | Divergence from HG52 RefSeq | 0.2 (0.1) | 0.5 (0.3) | ||||
| Divergence from HG52 ILMN | 0.1 (0.1) | 0.3 (0.2) | Divergence from HG52 ILMN | 0.2 (0.1) | 0.5 (0.3) | ||||
| Divergence from SD90e | 0.2 (0.1) | 0.3 (0.1) | Divergence from SD90e | 0.2 (0.1) | 0.2 (0.1) | ||||
| UL27 | Virion membrane glycoprotein B | 108 | Diversity | 0.2 (0.0) | 0.4 (0.1) | 34 | Diversity | 0.2 (0.0) | 0.5 (0.1) |
| Divergence from HG52 RefSeq | 0.2 (0.1) | 0.4 (0.1) | Divergence from HG52 RefSeq | 0.2 (0.1) | 0.4 (0.1) | ||||
| Divergence from HG52 ILMN | 0.2 (0.0) | 0.3 (0.1) | Divergence from HG52 ILMN | 0.2 (0.0) | 0.4 (0.1) | ||||
| Divergence from SD90e | 0.2 (0.1) | 0.4 (0.1) | Divergence from SD90e | 0.2 (0.1) | 0.5 (0.2) | ||||
| UL30 | DNA polymerase catalytic subunit | 54 | Diversity | 0.1 (0.0) | 0.2 (0.1) | 34 | Diversity | 0.1 (0.0) | 0.1 (0.0) |
| Divergence from HG52 RefSeq | 0.1 (0.0) | 0.3 (0.1) | Divergence from HG52 RefSeq | 0.1 (0.1) | 0.3 (0.1) | ||||
| Divergence from HG52 ILMN | 0.1 (0.0) | 0.3 (0.1) | Divergence from HG52 ILMN | 0.1 (0.0) | 0.3 (0.1) | ||||
| Divergence from SD90e | 0.1 (0.0) | 0.2 (0.1) | Divergence from SD90e | 0.1 (0.0) | 0.2 (0.1) | ||||
| UL49 | Tegument protein | 122 | Diversity | 0.8 (0.2) | 0.9 (0.3) | 34 | Diversity | 0.5 (0.1) | 0.8 (0.3) |
| Divergence from HG52 RefSeq | 0.6 (0.2) | 0.7 (0.3) | Divergence from HG52 RefSeq | 0.5 (0.1) | 0.7 (0.3) | ||||
| Divergence from HG52 ILMN | 0.6 (0.2) | 0.7 (0.3) | Divergence from HG52 ILMN | 0.7 (0.2) | 0.7 (0.3) | ||||
| Divergence from SD90e | 0.9 (0.2) | 1.5 (0.6) | Divergence from SD90e | 0.8 (0.2) | 1.1 (0.4) | ||||
| US4 | Virion membrane glycoprotein G | 141 | Diversity | 0.4 (0.1) | 0.7 (0.1) | 34 | Diversity | 0.4 (0.1) | 0.7 (0.1) |
| Divergence from HG52 RefSeq | 0.4 (0.1) | 0.7 (0.2) | Divergence from HG52 RefSeq | 0.4 (0.1) | 0.7 (0.2) | ||||
| Divergence from HG52 ILMN | 0.4 (0.1) | 0.7 (0.2) | Divergence from HG52 ILMN | 0.4 (0.1) | 0.6 (0.2) | ||||
| Divergence from SD90e | 0.5 (0.1) | 0.8 (0.2) | Divergence from SD90e | 0.4 (0.1) | 0.7 (0.2) | ||||
| US7 | Virion membrane glycoprotein I | 49 | Diversity | 0.3 (0.1) | 0.6 (0.3) | 34 | Diversity | 0.3 (0.1) | 0.6 (0.2) |
| Divergence from HG52 RefSeq | 0.3 (0.1) | 0.6 (0.3) | Divergence from HG52 RefSeq | 0.4 (0.1) | 0.7 (0.3) | ||||
| Divergence from HG52 ILMN | 0.3 (0.1) | 0.6 (0.3) | Divergence from HG52 ILMN | 0.3 (0.1) | 0.7 (0.3) | ||||
| Divergence from SD90e | 0.3 (0.1) | 0.6 (0.3) | Divergence from SD90e | 0.3 (0.1) | 0.5 (0.3) | ||||
| US8 | Virion membrane glycoprotein E | 50 | Diversity | 0.2 (0.0) | 0.4 (0.1) | 34 | Diversity | 0.2 (0.0) | 0.4 (0.1) |
| Divergence from HG52 RefSeq | 0.2 (0.1) | 0.6 (0.3) | Divergence from HG52 RefSeq | 0.2 (0.1) | 0.6 (0.2) | ||||
| Divergence from HG52 ILMN | 0.1 (0.0) | 0.2 (0.1) | Divergence from HG52 ILMN | 0.2 (0.1) | 0.4 (0.2) | ||||
| Divergence from SD90e | 0.1 (0.1) | 0.3 (0.2) | Divergence from SD90e | 0.2 (0.1) | 0.3 (0.2) | ||||
GenBank accession numbers: HG52 RefSeq, NC_001798.1; HG52 ILMN, JN561323; SD90e, KF781518.
Analysis of HSV-2 recombination.
To determine if HSV-2 genomes display the extensive recombination reported for HSV-1 sequences (6, 30, 31), we employed boot-scanning and phylogenetic analyses of full-length HSV-2 strain alignments. First, to confirm that we could detect recombination, we performed our analysis on HSV-1 and were able to detect recombination crossover events over large segments (2,500 to 8,500 bp) at levels comparable to those previously seen (references 6, 30, and 31 and results not shown). In contrast, our analysis of recombination in HSV-2 showed only five major crossover events, with detectable recombination seen only over small segments of the aligned sequences (700 to 1,170 bp) (Fig. 2). To confirm the recombination signals observed in the HSV-2 boot scans, we performed phylogenetic analysis of these five small regions between recombination breakpoints. These analyses showed few highly supported branches and could confirm potential recombination over 1,170 bp and 700 bp between HG52 and two U.S. strains, BethesdaP5 and 8937_1999_3336, respectively (data not shown). The weak signal for recombination in HSV-2 suggested either that recombination does not occur in HSV-2 genomes as frequently as is seen in HSV-1 or that the high level of sequence similarity among HSV-2 genome sequences makes lateral gene transfer difficult to detect. Additional analyses using a variety of methods to confirm the lack of recombination in these HSV-2 strains are described in more detail in the accompanying paper by Lamers et al. (32).
FIG 2.
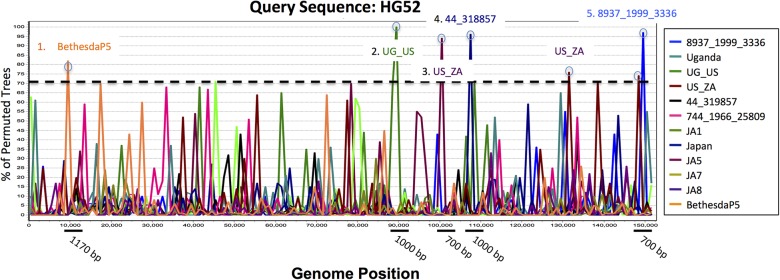
Boot-scanning analysis for evidence of recombination within HSV-2 strains. Shown is a boot-scanning plot of the HSV-2 reference sequence HG52 (accession no. NC_001798.1) versus all other HSV-2 full genome sequences. The x axis reflects the position in the aligned set of sequences, and the y axis shows the percentage of permuted trees in which an individual HSV-2 strain clusters with the query. A recombination cutoff value of 70% is indicated by the dashed line. Positive signals for recombination with HG52 are indicated by a small circle at the peak recombination score and with the name of the strain that most closely resembles the query strain. Directly below the boot-scanning plot, the black lines indicate the recombinant breakpoint regions and their lengths.
Phylogenetic analysis.
Alignment and subsequent phylogenetic analysis of the newly sequenced HSV-2 genomes with existing HSV-2 sequences using the ChHV genome sequence as an outgroup allowed us to determine the relationship between these geographically distinct HSV-2 strains. As expected, there was distinct clustering of HSV-2 sequences away from ChHV sequences in the whole-genome phylogeny (Fig. 3). The dendrogram also showed close relationship among all HSV-2 sequences regardless of geographic origin. This was in contrast to HSV-1 genome sequences, which exhibit robust geographical clustering (6). While clinical HSV-2 strains isolated in Uganda and Japan tended to cluster together, there was very low bootstrap support for this clustering, indicating a lack of strong phylogenetic evidence for grouping of these strains. Similarly, the grouping of HSV-2 strains isolated from the United States with strains from South Africa also had low bootstrap support.
FIG 3.

Phylogenetic relationships of HSV-2 genome sequences to that of chimpanzee herpesvirus. Shown is a maximum-likelihood tree of 34 nearly full-length HSV-2 genome nucleotide sequences generated as part of this study and the publicly available HSV-2 sequences for HG52 (accession no. NC_001798.1 and JN561323) and SD90e (accession no. KF781518), along with ChHV sequence from strain 105640 (accession no. NC_023677). Problematic regions from the multiple-sequence alignment were removed from all sequences. The tree is rooted using the ChHV sequence, and all horizontal branch lengths are drawn to a scale of nucleotide substitutions per site. Bootstrap resampling (1,000 replications) was performed. Bootstrap support values for each node, other than that separating ChHV from HSV-2 sequences, were <10% and are not shown on the tree.
To further explore the relationship between geographically diverse HSV-2 sequences, we generated a dendrogram of full-length and nearly full-length HSV-2 sequences (Fig. 4). The complete genome tree recapitulated the clustering of Uganda and Japan sequences into separate branches and again showed loose association of U.S. and South Africa sequences. Similar results were observed with analyses of UL or US regions alone (results not shown). However, in all phylogenetic analyses, there was strong support (>65% bootstrap value in multiple genomic regions) for the relatedness of U.S. sequence 44_619833 and Uganda sequence K39924_UG.
FIG 4.

Phylogenetic relationships of HSV-2 genome sequences. Shown is a maximum-likelihood tree of 34 nearly full-length HSV-2 genome nucleotide sequences generated as part of this study and the publicly available HSV-2 sequences for HG52 (accession no. NC_001798.1 and JN561323) and SD90e (accession no. KF781518). Problematic regions from the multiple-sequence alignment were removed from all sequences. The tree is unrooted, and all horizontal branch lengths are drawn to a scale of nucleotide substitutions per site. Bootstrap resampling (1,000 replications) support values are shown at the nodes.
DISCUSSION
The availability of a large number of nearly complete genome sequences from low-passage-number clinical isolates of HSV-2 allowed us to explore the sequence evolution and diversity of low-passage-number virus strains isolated from Asia, Africa, and the United States to infer viral evolution and potential viral determinants of pathogenicity and disease outcome. Here, we report the sequencing and assembly of 34 additional strains, 33 low-passage-number clinical strains and 1 laboratory strain, isolated in the United States, Uganda, South Africa, and Japan. Illumina sequencing of these samples generated high-quality, nearly complete genome assemblies of the unique regions of the genome. However, as has been previously reported for both HSV-1 and HSV-2 (2, 6), accurate sequences of both copies of the terminal and internal large repeat regions (RL and RS) and of intergenic repetitive regions proved difficult. Further sequencing of the terminal and internal repeat regions with additional methods, such as single-molecule, long-read sequencing technology, as has been done for another Herpesviridae family member, pseudorabies virus (PRV), may allow single-base resolution of these difficult regions of the genome (33).
Sequence diversity of HSV-2 isolates.
These newly sequenced HSV-2 strains showed remarkable sequence conservation, regardless of geographic origin. The level of HSV-2 diversity for the 34 full-length genomes was less than reported previously for HSV-1 (6), and similar levels of diversity were observed for 7 specific HSV-2 genes in the larger GenBank database. However, as in previous studies, problematic areas in or near repeat regions of the genome were excluded from our analyses due to technical difficulties in sequencing and assembling genome repeats. Because these regions may represent locations of real biological variability (34), it is likely that future advances in genome sequencing and assembly technology could accurately fill in these missing regions, and these improvements could highlight additional diversity within these genomes.
Although the HSV-2 sequences were generally highly conserved, certain ORFs showed higher diversity. UL49 was more diverse than the other ORFs (0.8% at the amino acid level). In addition, certain ORFs were divergent in specific strains. For example, UL49 and UL49A showed increased divergence from SD90e, while UL11 and US1 showed increased divergence from HG52. The origin of the divergence in these ORFs remains to be defined.
The lower level of diversity in HSV-2 than in HSV-1 has implications for viral evolution. The decreased diversity seen in HSV-2 is consistent with its diverging more recently than HSV-1 from ChHV or another herpesvirus progenitor (15) but could also be the result of a greater bottleneck during genital transmission than in oral transmission or the lower prevalence of HSV-2. While all of the strains sequenced here were passaged in cell culture prior to preparation of viral DNA for sequencing, passage numbers were kept low to minimize the potential accumulation of single nucleotide polymorphisms (SNPs) during cell culture. Previous sequence comparison of an HSV-2 low-passage-number viral genome with a derivative that had undergone plaque purification revealed high levels of sequence conservation and minimal changes to the viral genome (2); therefore, we do not anticipate high numbers of cell-culture-associated SNPs in these genomes.
An understanding of viral diversity is also important for vaccine design. The high level of diversity in human immunodeficiency virus type 1 (HIV-1) is one of the factors that have limited the development of an effective vaccine. Therefore, the limited genetic diversity of these 34 HSV-2 strains bodes well for the potential of an HSV-2 vaccine to contain sufficient antigens to protect against these strains from around the world. Identification of the key protective antigens will be necessary before this question can be answered adequately.
Phylogenetic analysis of the full genome sequences of these HSV-2 strains, as well as of unique regions of the genome, showed a lack of robust support for geographic clustering. Norberg et al. previously reported evidence for two genogroups, one from isolates from Tanzania and one from isolates from Tanzania and Scandinavia (9). This was based on removing isolates that showed “conflicting phylogenetic signals” from the analysis. The differences between this study and our finding may be due to the difference between whole-genome analysis and individual gene analysis or to the removal of recombination from the genes being analyzed in the Norberg et al. analysis. Further analysis of the genes that show the greatest diversity is needed to determine whether they represent distinct clades in HSV-2.
Recombination.
We found less evidence of recombination in HSV-2 genomes than in HSV-1, although the low sequence diversity may limit the ability to detect recombination in HSV-2. Norberg et al. (9) had reported significant recombination in HSV-2 through analysis of three glycoprotein genes. It is conceivable that recombination is less for HSV-2 than HSV-1, but cell culture studies show equal frequencies of recombination between HSV-2 mutants versus HSV-1 mutants (C. Zhou and D. M. Knipe, unpublished results). Thus, this is not likely to be the explanation for the apparent low level of recombination evidenced in the HSV-2 strains. More likely, the low level of recombination is due to the low level of genetic diversity, making recombination less detectable in the HSV-2 genomes.
HSV-2 reference genome.
The HSV-2 HG52 genome has served as the reference genome because, until recently, it was the only sequence available. However, HG52 is very attenuated in animals relative to other HSV-2 strains (10, 35). Upon sequencing of the SD90e low-passage-number clinical isolate (GenBank accession no. KF781518), which shows pathogenicity in mice similar to that of other HSV-2 strains (10), Colgrove et al. proposed that the SD90e genome should serve as a new HSV-2 reference genome (2). In this study, we found that, on average, SD90e is closer to the new group of HSV-2 genomes than even the revised HG52 sequence at both the whole-genome and individual ORF levels. Therefore, the results from this study further support the proposal that SD90e serve as the HSV-2 reference genome sequence.
Taken together, the low level of sequence diversity, low rate of recombination, and relative lack of geographic clustering of HSV-2 strains are in contrast to what has been reported for geographically diverse HSV-1 strains. Several studies report that HSV-1 genomes display high levels of DNA diversity, as well as extensive recombination (6, 30, 31). Furthermore, analysis of genetic distances among HSV-1 strains isolated from Asia, Africa, North America, and Europe shows strong sequence clustering of strains based on geographic location. Possible explanations for the differences in genome diversity between HSV-1 and HSV-2 could be (i) that HSV-2 entered the human population later than HSV-1 and has not borne the cumulative selection pressures that HSV-1 has endured or (ii) that differences between HSV-1 and HSV-2 infection rates and age at the time of infection could lead to fewer opportunities for divergence and recombination in HSV-2. Recent analysis of the evolutionary origins of HSV-1 and HSV-2 supports the idea that HSV-2 entered the human lineage through divergence from ChHV only around 1.6 million years ago, while HSV-1 diverged from ChHV about 6 million years ago (15). However, the increased worldwide prevalence of HSV-1 compared to HSV-2 (36) and subsequent interaction with host selective pressures could also account for the increased sequence diversity seen in HSV-1. The reduced sequence diversity and genome recombination that we see in HSV-2 clinical isolates is consistent with either of these hypotheses, and further work is necessary to discriminate between these and other hypotheses.
The nearly complete genome sequences of geographically diverse HSV-2 low-passage-number isolates reported here permits assessment of the genetic diversity of HSV-2 strains/isolates in circulation and will facilitate study of the relationship of this diversity to pathogenicity and epidemiology. Metagenomic analysis with the relevant reference genomic sequences could assist research aimed at diagnosis and the evaluation of clinical manifestations and transmission of HSV-2. An understanding of HSV-2 genetic variation may also contribute to deciphering aspects of disease transmission and pathogenesis. For example, variation in T-cell or B-cell epitopes would extend the concept of immune selection from RNA to DNA viruses. HSV-2 proteins interact with and are restricted by host proteins at many points. As human genome data accumulate, viral sequence variation from geographically distinct specimens will be important. HSV-2 has likely traveled with humans during migrations over the millennia (15), and definition of clades and tag SNPs will allow analysis of how populations of a sexually transmitted, persistent latent pathogen covary among and between isolated and cosmopolitan human populations. Examination of the biological and clinical implications of specific SNPs is under way. Additional mining of these genome sequences could yield insights into the sequence determinants of HSV-2 pathogenicity and can serve as a tool in the design of future therapies and vaccines.
ACKNOWLEDGMENTS
This project has been funded in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract no. HHSN272200900018C to the Broad Institute's Genomic Sequencing Center for Infectious Diseases; grant AI057552 to D. M. Knipe; and grant AI030731 to D. M. Koelle. This work was also supported by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases.
We thank Tatsuo Suzutani from the Fukushima Medical University School of Medicine and Takashi Kawana from Teikyo University in Japan for supplying the Japanese HSV-2 isolates.
REFERENCES
- 1.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Colgrove R, Diaz F, Newman R, Saif S, Shea T, Young S, Henn M, Knipe DM. 2014. Genomic sequences of a low passage herpes simplex virus 2 clinical isolate and its plaque-purified derivative strain. Virology 450-451:140–145. doi: 10.1016/j.virol.2013.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roizman B, Jacob RJ, Knipe DM, Morse LS, Ruyechan WT. 1979. On the structure, functional equivalence, and replication of the four arrangements of herpes simplex virus DNA. Cold Spring Harbor Symp Quant Biol 43:809–826. doi: 10.1101/SQB.1979.043.01.088. [DOI] [PubMed] [Google Scholar]
- 4.Hayward GS, Jacob RJ, Wadsworth SC, Roizman B. 1975. Anatomy of herpes simplex virus DNA: evidence for four populations of molecules that differ in the relative orientations of their long and short components. Proc Natl Acad Sci U S A 72:4243–4247. doi: 10.1073/pnas.72.11.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McGeoch DJ, Moss HW, McNab D, Frame MC. 1987. DNA sequence and genetic content of the HindIII l region in the short unique component of the herpes simplex virus type 2 genome: identification of the gene encoding glycoprotein G, and evolutionary comparisons. J Gen Virol 68:19–38. doi: 10.1099/0022-1317-68-1-19. [DOI] [PubMed] [Google Scholar]
- 6.Szpara ML, Gatherer D, Ochoa A, Greenbaum B, Dolan A, Bowden RJ, Enquist LW, Legendre M, Davison AJ. 2014. Evolution and diversity in human herpes simplex virus genomes. J Virol 88:1209–1227. doi: 10.1128/JVI.01987-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szpara ML, Parsons L, Enquist LW. 2010. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J Virol 84:5303–5313. doi: 10.1128/JVI.00312-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dolan A, Jamieson FE, Cunnigham C, Barnett BC, McGeogh DJ. 1998. The genome sequence of herpes simplex virus type 2. J Virol 72:2010–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Norberg P, Kasubi MJ, Haarr L, Bergstrom T, Liljeqvist JA. 2007. Divergence and recombination of clinical herpes simplex virus type 2 isolates. J Virol 81:13158–13167. doi: 10.1128/JVI.01310-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dudek TE, Torres-Lopez E, Crumpacker C, Knipe DM. 2011. Evidence for differences in immunologic and pathogenesis properties of herpes simplex virus 2 strains from the United States and South Africa. J Infect Dis 203:1434–1441. doi: 10.1093/infdis/jir047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGeoch DJ, Cook S. 1994. Molecular phylogeny of the Alphaherpesvirinae subfamily and a proposed evolutionary timescale. J Mol Biol 238:9–22. doi: 10.1006/jmbi.1994.1264. [DOI] [PubMed] [Google Scholar]
- 12.McGeoch DJ, Cook S, Dolan A, Jamieson FE, Telford EA. 1995. Molecular phylogeny and evolutionary timescale for the family of mammalian herpesviruses. J Mol Biol 247:443–458. doi: 10.1006/jmbi.1995.0152. [DOI] [PubMed] [Google Scholar]
- 13.Luebcke E, Dubovi E, Black D, Ohsawa K, Eberle R. 2006. Isolation and characterization of a chimpanzee alphaherpesvirus. J Gen Virol 87:11–19. doi: 10.1099/vir.0.81606-0. [DOI] [PubMed] [Google Scholar]
- 14.Severini A, Tyler SD, Peters GA, Black D, Eberle R. 2013. Genome sequence of a chimpanzee herpesvirus and its relation to other primate alphaherpesviruses. Arch Virol 158:1825–1828. doi: 10.1007/s00705-013-1666-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wertheim JO, Smith MD, Smith DM, Scheffler K, Kosakovsky Pond SL. 2014. Evolutionary origins of human herpes simplex viruses 1 and 2. Mol Biol Evol 31:2356–2364. doi: 10.1093/molbev/msu185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K, Kappel JD, Canders C, Davila WF, Sayre D, Chavez M, Pesnicak L, Cohen JI. 2012. A herpes simplex virus 2 glycoprotein D mutant generated by bacterial artificial chromosome mutagenesis is severely impaired for infecting neuronal cells and infects only Vero cells expressing exogenous HVEM. J Virol 86:12891–12902. doi: 10.1128/JVI.01055-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lai W, Chen CY, Morse SA, Htun Y, Fehler HG, Liu H, Ballard RC. 2003. Increasing relative prevalence of HSV-2 infection among men with genital ulcers from a mining community in South Africa. Sex Transm Infect 79:202–207. doi: 10.1136/sti.79.3.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chatis PA, Crumpacker CS. 1991. Analysis of the thymidine kinase gene from clinically isolated acyclovir-resistant herpes simplex viruses. Virology 180:793–797. doi: 10.1016/0042-6822(91)90093-Q. [DOI] [PubMed] [Google Scholar]
- 19.Taguchi F, Toba M, Tada A. 1979. Establishment of a permanent cell line (HEL-R66) from human embryonic lung cells with high susceptibility to viruses. Brief report. Arch Virol 60:347–351. doi: 10.1007/BF01317506. [DOI] [PubMed] [Google Scholar]
- 20.Koelle DM, Chen HB, Gavin MA, Wald A, Kwok WW, Corey L. 2001. CD8 CTL from genital herpes simplex lesions: recognition of viral tegument and immediate early proteins and lysis of infected cutaneous cells. J Immunol 166:4049–4058. doi: 10.4049/jimmunol.166.6.4049. [DOI] [PubMed] [Google Scholar]
- 21.Denniston KJ, Madden MJ, Enquist LW, Vande Woude G. 1981. Characterization of coliphage lambda hybrids carrying DNA fragments from herpes simplex virus type 1 defective interfering particles. Gene 15:365–378. doi: 10.1016/0378-1119(81)90180-3. [DOI] [PubMed] [Google Scholar]
- 22.Grad YH, Lipsitch M, Feldgarden M, Arachchi HM, Cerqueira GC, Fitzgerald M, Godfrey P, Haas BJ, Murphy CI, Russ C, Sykes S, Walker BJ, Wortman JR, Young S, Zeng Q, Abouelleil A, Bochicchio J, Chauvin S, Desmet T, Gujja S, McCowan C, Montmayeur A, Steelman S, Frimodt-Moller J, Petersen AM, Struve C, Krogfelt KA, Bingen E, Weill FX, Lander ES, Nusbaum C, Birren BW, Hung DT, Hanage WP. 2012. Genomic epidemiology of the Escherichia coli O104:H4 outbreaks in Europe, 2011. Proc Natl Acad Sci U S A 109:3065–3070. doi: 10.1073/pnas.1121491109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bradley RK, Roberts A, Smoot M, Juvekar S, Do J, Dewey C, Holmes I, Pachter L. 2009. Fast statistical alignment. PLoS Comput Biol 5:e1000392. doi: 10.1371/journal.pcbi.1000392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 27.Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. 2010. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26:2462–2463. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salminen MO, Carr JK, Burke DS, McCutchan FE. 1995. Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Res Hum Retroviruses 11:1423–1425. doi: 10.1089/aid.1995.11.1423. [DOI] [PubMed] [Google Scholar]
- 30.Norberg P, Tyler S, Severini A, Whitley R, Liljeqvist JA, Bergstrom T. 2011. A genome-wide comparative evolutionary analysis of herpes simplex virus type 1 and varicella zoster virus. PLoS One 6:e22527. doi: 10.1371/journal.pone.0022527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bowden R, Sakaoka H, Donnelly P, Ward R. 2004. High recombination rate in herpes simplex virus type 1 natural populations suggests significant co-infection. Infect Genet Evol 4:115–123. doi: 10.1016/j.meegid.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 32.Lamers SL, Newman R, Laeyendecker O, Tobian AAR, Colgrove RC, Ray SC, Koelle DM, Cohen J, Knipe DM, Quinn TC. 2015. Global diversity within and between human herpesvirus 1 and 2 glycoproteins. J Virol 89:8206–8218. doi: 10.1128/JVI.01302-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tombacz D, Sharon D, Olah P, Csabai Z, Snyder M, Boldogkoi Z. 2014. Strain Kaplan of pseudorabies virus genome sequenced by PacBio single-molecule real-time sequencing technology. Genome Announc 2:e00628-14. doi: 10.1128/genomeA.00628-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tognon M, Cassai E, Rotola A, Roizman B. 1983. The heterogenous regions in herpes simplex virus 1 DNA. Microbiologica 6:191–198. [PubMed] [Google Scholar]
- 35.Mitchell WJ, Deshmane SL, Dolan A, McGeoch DJ, Fraser NW. 1990. Characterization of herpes simplex virus type 2 transcription during latent infection of mouse trigeminal ganglia. J Virol 64:5342–5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith JS, Robinson NJ. 2002. Age-specific prevalence of infection with herpes simplex virus types 2 and 1: a global review. J Infect Dis 186(Suppl 1):S3–S28. doi: 10.1086/343739. [DOI] [PubMed] [Google Scholar]