

ABSTRACT
The process of reverse transcription (RTN) in retroviruses is essential to the viral life cycle. This key process is catalyzed exclusively by the viral reverse transcriptase (RT) that copies the viral RNA into DNA by its DNA polymerase activity, while concomitantly removing the original RNA template by its RNase H activity. During RTN, the combination between DNA synthesis and RNA hydrolysis leads to strand transfers (or template switches) that are critical for the completion of RTN. The balance between these RT-driven activities was considered to be the sole reason for strand transfers. Nevertheless, we show here that a specific mutation in HIV-1 RT (L92P) that does not affect the DNA polymerase and RNase H activities abolishes strand transfer. There is also a good correlation between this complete loss of the RT's strand transfer to the loss of the DNA clamp activity of the RT, discovered recently by us. This finding indicates a mechanistic linkage between these two functions and that they are both direct and unique functions of the RT (apart from DNA synthesis and RNA degradation). Furthermore, when the RT's L92P mutant was introduced into an infectious HIV-1 clone, it lost viral replication, due to inefficient intracellular strand transfers during RTN, thus supporting the in vitro data. As far as we know, this is the first report on RT mutants that specifically and directly impair RT-associated strand transfers. Therefore, targeting residue Leu92 may be helpful in selectively blocking this RT activity and consequently HIV-1 infectivity and pathogenesis.
IMPORTANCE Reverse transcription in retroviruses is essential for the viral life cycle. This multistep process is catalyzed by viral reverse transcriptase, which copies the viral RNA into DNA by its DNA polymerase activity (while concomitantly removing the RNA template by its RNase H activity). The combination and balance between synthesis and hydrolysis lead to strand transfers that are critical for reverse transcription completion. We show here for the first time that a single mutation in HIV-1 reverse transcriptase (L92P) selectively abolishes strand transfers without affecting the enzyme's DNA polymerase and RNase H functions. When this mutation was introduced into an infectious HIV-1 clone, viral replication was lost due to an impaired intracellular strand transfer, thus supporting the in vitro data. Therefore, finding novel drugs that target HIV-1 reverse transcriptase Leu92 may be beneficial for developing new potent and selective inhibitors of retroviral reverse transcription that will obstruct HIV-1 infectivity.
INTRODUCTION
Reverse transcription (RTN) is a critical step in the life cycle of all retroviruses and the related long terminal repeat (LTR) retrotransposons. This complex multistep process is performed by a single enzyme, the retroviral reverse transcriptase (RT) that copies the viral plus strand RNA into integration competent double-stranded viral DNA (1–3). To perform RTN, RTs have two activities. These are the DNA polymerase activity, which copies both DNA and RNA, and hence is both a DNA-dependent and RNA-dependent DNA polymerase (DDDP and RDDP, respectively) activity and an RNase H activity, which, in conjunction with DNA synthesis, cleaves the RNA template in the generated RNA/DNA heteroduplexes (2, 4). During RTN, DNA synthesis produces both minus (−) and plus (+) DNA strands, whereas the RNase H removes the viral genomic (+)RNA template, as well as the tRNA-primer that is used to initiate minus-strand DNA synthesis. Throughout RTN, two strand transfer (ST) events take place. In both, the nascent DNA strand switches from the copied template to a second template that is further copied (1, 2, 5). In the first ST, designated (−)ST, the growing DNA strand (that was synthesized from the 5′-end of the viral RNA) is translocated onto the matching 3′-end of the RNA strand. In the second switch, designated (+)ST, the 3′-end of the (+)DNA strand, with the primer binding site (PBS) sequence, switches onto a complementary sequence in the already synthesized (−) DNA strand. Both template transfers depend on stable complementarities between the ends of the growing (donor) DNA and the acceptor RNA or DNA strands. Here, the matching sequences are relatively long. The terminal repeat (R) sequence, which promotes (−)ST, is 98 nucleotides (nt) long in human immunodeficiency virus type 1 (HIV-1) and 68 nt long in murine leukemia virus (MLV), while the PBS is usually 18 nt long in most retroviruses (including HIV-1 and MLV) (1).
New in vitro evidence, presented recently by us, show that RTs can perform also template switches with even a very short (1 to 2 nt) complementarity between the 3′ ends of the primer donor strand and the DNA or RNA template acceptor strands (6–8). These tiny duplexes are markedly stabilized thermodynamically by the RT that “clamps” together the duplex structures that are otherwise very unstable. The stabilization of this sequence microhomology efficiently promotes DNA synthesis, since the acceptor strand can be copied by RT in the presence of deoxynucleoside triphosphates (dNTPs) after the strand switch took place. With HIV and MLV RTs, this activity is accomplished when the functional template is adjoining a second DNA or RNA segment, annealed upstream to most of the primer (see Fig. 3C). This new clamp function is performed, though to variable extents, by all RTs tested but not by cellular DNA polymerases, suggesting that it is potentially essential to retroviral RTN (6). Interestingly, RTs, other than those of HIV and MLV, can also form a stable clamp with even a single nucleotide complementarity between the donor and acceptor strands, and these RTs can tolerate even short 1- to 2-nt gaps between the functional template and the adjacent strand (7).
FIG 3.

Clamp activity of the HIV-1 RT mutants. Equal DDDP activities of the indicated HIV-1 RT versions were assayed for the clamp assay, using the two methods described here (see Materials and Methods; see also panel C. (A) Clamp gel assay. S, 2- nt substrate; P, 59-nt products. (B) ELISA-based clamp method. Data are represented as means ± the standard deviations (SD). ***, P < 0.001 (as determined by paired two-tailed t test). (C) Nucleic acid clamp scheme. 1, Primer (P) annealed to template (T1) with 2-nt overhang in its 3′ end was incubated with a second template. This second template (T2) has 2 nt complementarily between their 3′ ends. Then, HIV-1 RT was added to the mixture along with all dNTPs. 2, HIV-1 RT clamps the 2 nt complementarily at the 3′ end of P and the 5′ end of T2. 3, T2 serves as the template for DNA elongation of the clamped primer. This scheme was prepared according to our previous publications (6, 7).
One explanation for the clamp function is that it can potentially allow the RT-associated DNA synthesis to bridge over nicks in the copied RNA or DNA templates that can be possibly encountered during RTN. In addition, some RTs have the capacity to perform template-independent DNA synthesis by their nontemplated addition (NTA) activity; thus, they can create de novo 3′-end overhangs of the primer strand (9–11). Therefore, when combined with their clamp activity, RTs can promote STs of the nascent DNA strands onto compatible acceptor strands. The combination between these RT functions depends on a variety of factors; namely, the extent of NTA activity, the stability of the terminal duplexes formed, the level of the RT's clamp activity, the bias of the dNTPs present, and the availability of proper acceptor strands (8).
To provide structural insights into the HIV-1 RT-DNA complex that allows a stable clamp, we have modeled such a three dimensional structure of this complex (6). This model suggested that several key HIV-1 RT residues could potentially participate in forming the RT-nucleic acids complex that allows a stable clamp structure. Consequently, further mutagenesis and structural studies are required to determine whether this model is experimentally valid and to test whether it is possible to molecularly dissect between DNA synthesis by itself and clamp activity of the RT. The results presented herein establish the involvement, to variable extents, of HIV-1 RT residues Glu89, Val90, Leu92, Lys154, and Pro157 in specifically affecting the RT's clamp activity. Remarkably, none of the major catalytic properties of all RT mutants generated (namely, the RDDP, the DDDP, the processivity of DNA synthesis, and the RNase H) were significantly affected.
We show here that the clamp activity was highly susceptible to mutations predominantly in residue 92 of HIV-1 RT. Interestingly, the ST activity of the tested mutants, and primarily the L92P mutant, was also severely impaired. This finding highlights the likely mechanistic linkage between the RT's clamp and ST activities. Such a result could be expected, because the clamp activity actually induces a “micro” ST with very short complementarities between the 3′ ends of the growing DNA strand and the acceptor template strand. This result also suggests that the ST process is not only an indirect outcome of a simple combination between DNA polymerase and RNase H activities but is rather a direct and unique activity of the RT. After these in vitro results, the L92P mutant of the RT that was introduced into an infectious HIV-1 clone led to a loss of all apparent viral replication in susceptible cells, since no ST products were observed in the virus-infected cells. As far as we know, this is the first report on mutants that specifically impair the ST activity of an RT. Therefore, targeting residue Leu92 of HIV-1 RT could prove advantageous is selectivity blocking the ST activity of the RT and consequently obstructing the infectivity of the virus and its pathogenesis.
MATERIALS AND METHODS
Cell lines.
Human embryonic kidney cells were transformed by sheared human adenovirus type 5 DNA (HEK293). Jurkat cells were an immortalized cell line of human T lymphocytes. MAGI-CCR5 are HeLa cells that express high levels of CD4 and contain a single integrated copy of a β-galactosidase gene that is under the control of a truncated HIV-1 LTR (12). MAGI-CCR5 cells are a clone of MAGI cells that express the human chemokine receptor, CCR5 (13). JLTRG-R5 cells are derived from Jurkat human T cells, which have been stably transfected with an LTR-green fluorescent protein (GFP) (14, 15).
Plasmids. (i) HIV-1 viral plasmid.
We used wild-type (WT) HIV-1 expressing plasmid (designated pSVC21) containing the complete infectious proviral DNA clone of the HXB2 HIV-1 strain that we had used in an earlier study of HIV-1 (16). From this plasmid, we also generated pSVC21 L92P, a single-mutation variant with the RT L92P mutation made by overlapping PCR.
(ii) Bacterial expression plasmid.
All RTs used in the present study were expressed in Escherichia coli. WT HIV-1 RT from the BH-10 isolate was expressed in bacteria using a plasmid (designated pHIV-1 RT), similar to that described earlier by us (17–19).
Antibodies.
We used several different antibodies. HIV-1SF2 p24 antibody was used to detect virus particles produced by HEK293 cell transfection. This reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Disease, National Institutes of Health (catalog no. 4250). Horseradish peroxidase-conjugated secondary anti-rabbit IgG antibodies were purchased from Abcam for Western blot analysis. Finally, anti-digoxigenin-POD(poly) Fab fragments from Roche were used to detect the clamp product in an enzyme-linked immunosorbent assay (ELISA) (6).
Site-directed mutagenesis of HIV-1 RT.
All RT mutants were derived from the BH10 isolate of WT HIV-1 RT, cloned by us (20). The specific RT residues were modified by overlapping PCR used for site-directed mutagenesis of the WT HIV-1 RT-expressing plasmid. All mutations were introduced in both p66 and p51 RT subunits. The presence of the introduced specific mutations was confirmed by sequencing the PCR-amplified DNA segment.
Expression and purification of the recombinant WT and mutant versions of HIV-1 RT.
All recombinant RTs were expressed in bacteria, with a six-histidine tag attached to the carboxyl terminus of the p66 subunit in the heterodimeric p66/p51 RT, and was purified as described previously (17–19).
Enzymatic activities of HIV-1 RT versions.
Basically, all assays, except for the RNase H assay, were based on the extension under the appropriate conditions of the end-labeled DNA primers that are part of the specific substrates used for each assay. The RNase H assay followed the hydrolysis of the 5′-32P-end-labeled RNA segment that is annealed to its cDNA. The assays were performed at their linear phases, after preliminary dose-response and time-dependent curves were determined.
(i) DDDP.
Each assay contained ∼0.4 pmol of the 5′-end-labeled primer DNA (E1 in Table 1), which was annealed to 0.1 pmol of a 54mer synthetic oligonucleotide DNA (E2) in the presence of 50 mM Tris-HCl (pH 8.0), 50 mM NaCl, 5 mM MgCl2, 2 mM dithiothreitol (DTT), 100 μg of acetylated bovine serum albumin (BSA)/ml, and 10 mM CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate} at a final volume of 12.5 μl, as described previously (6).
TABLE 1.
Synthetic oligonucleotides used in this studya
| Oligonucleotide | Assay | Length (nt) | Sequence (5′-3′) |
|---|---|---|---|
| E1 | Processivity + DDDP (primer) | 21 | TGAAAGACCCCCGCTGACGGG |
| E2 | Processivity + DDDP (template) | 54 | AATGAAAGACCCCACCTGTAGGTTGGATCCTTACCCGTCAGCGGGGGTCTTTCA |
| E3 | PAGE + ELISA clamp T1 | 19 | TGCTAGAGATTTTCCACAC |
| E4 | ELISA clamp T2 | 25 | Digoxigenin-TCCTAGAAAATATCCCCTCAGCCAC |
| E5 | ELISA clamp P | 21 | Biotin-GTGTGGAAAATCTCTAGCAGT |
| E6 | Clamp T2 | 40 | GCCGGCCCATGGTCTTCCTAGAAAATATCCCCTCAGCCAC |
| E7 | Clamp P | 21 | GTGTGGAAAATCTCTAGCAGT |
| E8 | ST-donor | 33 | GATCTGAGCCTGGGAGCTCTCTGGCTAACTAGG |
| E9 | ST-acceptor | 35 | GGTCTCTCTGGTTAGACCAGATCTGAGCCTGGGAG |
| E10 | RNase H (DNA) + RDDP primer | 20 | AGTTAGCCAGAGAGCTCCCA |
| E11 | RNase H (RNA) + ST + RDDP template | 33 | GAUCUGAGCCUGGGAGCUCUCUGGCUAACUAGG |
| R-U5 For | RT-qPCR | 21 | TCTGGCTAACTAGGGAACCCA |
| R-U5 Rev | RT-qPCR | 20 | CTGACTAAAAGGGTCTGAGG |
| U3-PPT For | RT-qPCR | 20 | CACAAGAGGAGGAGGAGGTG |
| U3-PPT Rev | RT-qPCR | 20 | TGGGAGTGAATTAGCCCTTC |
| Alu | Alu-Gag PCR | 25 | GCCTCCCAAACTGCTGGGATTACAG |
| Gag Rev | Alu-Gag PCR | 26 | GCACACAATAGAGGACTGCTATTGTA |
Oligonucleotides E1 to E11 were tested for their correct size, employing 7 M urea-PAGE. All primers used for RT-qPCR were tested for linearity by standard curve analysis and assayed in triplicates.
(ii) RDDP.
The extensions of DNA primers on RNA templates were performed as described above for DDDP but with an RNA template (designated E11 in Table 1) rather than a DNA template.
(iii) Processivity of DNA synthesis.
An assay to determine the processivity of DNA synthesis was performed as described in detail for various RTs (21–24).
(iv) RNase H.
This assay was performed as described earlier by us (17–19, 25). Each assay contained ∼0.1 pmol of the 5′-32P-end-labeled RNA transcript (E11), which was annealed to 0.4 pmol of a 20mer synthetic oligonucleotide DNA (E10 in Table 1). The RNase H reaction was initiated by adding an equal DDDP activity of the purified WT or mutant RTs, and the reaction mixtures were incubated at 37°C for 3 min.
(v) Nucleic acid clamp activity.
We used two systems to assay this activity. Both assays were described in detail earlier (6). The products of the first one were resolved by urea-PAGE. This clamp assay is shown schematically in Fig. 3C, and the assay conditions are similar to those performed with HIV-1 RT with two template substrates, designated in the scheme as T1 and T2 (E3+E6 in Table 1). The second method for assaying the clamp activity is an ELISA that was used previously and shown to be more sensitive and quantitative than the first method (6).
(vi) Strand transfer activity.
A 20mer DNA synthetic primer (E8), which is complementary to the E11 RNA oligonucleotide, was 5′ end labeled with [γ-32P]ATP using T4 polynucleotide kinase. The labeled primer was annealed to E11 (which is the primary RNA template) at a 4-to-1 molar ratio of template over primer in 10 mM Tris-HCl, 1 mM EDTA, and 40 mM KCl (pH 7.5). The 35-nt DNA oligonucleotide (E9), which is the acceptor DNA template, was mixed with the RNA-DNA hybrid. Each ST reaction assay contained 0.4 pmol of the 32P-end-labeled 20-nt synthetic oligonucleotide that was annealed to 1.6 pmol of the primary RNA template in the presence of 1.6 pmol of the acceptor DNA template. The ST reactions were initiated by adding the purified RT versions to each reaction mixture in the presence of 50 mM Tris-HCl, 50 mM KCl, 6 mM MgCl2, 1 mM DTT, 100 μg of acetylated BSA/ml, and 10 mM CHAPS (final pH of 8.0), supplemented by 80 μM concentrations of each dNTP in a final volume of 12.5 μl. The ST reaction mixtures were incubated at 37°C for 60 min and then terminated by adding stop solution, and the products were heat denatured and then separated by 7 M urea-PAGE.
Cell culture techniques.
Monolayer adherent MAGI-CCR5 and HEK293 cells were grown in Dulbecco modified Eagle medium (DMEM). The T-lymphocyte cell lines Jurkat and JLTRG-R5 were grown in RPMI medium. The cells were incubated at 37°C in a 5% CO2 atmosphere. All media were supplemented with 10% (vol/vol) fetal calf serum, 0.3 g of l-glutamine/liter, 100 U of penicillin/ml, and 100 U of streptomycin/ml.
DNA transfection and generation of WT and mutant HIV-1.
WT pSVC21 and the mutated HIV-1 L92P proviral plasmids were used to transfect HEK293 cells, employing the TurboFect cell transfection reagent (Fermentas), according to the manufacturer's instructions. At 1 day posttransfection, the medium was removed, and the cells were washed twice with PBS and supplemented with fresh medium. To check the virus titers, the transfected cells were cultured for 24, 48, or 72 h. At each time point, the medium was removed and filtered through a 0.45-μm-pore-size Millipore filter. The virus-containing medium was divided into aliquots and stored at −80°C. The number of virus particles in the medium was then analyzed by RT-quantitative PCR (RT-qPCR) using virus-specific U5 primers (26).
Multinucleate activation of galactosidase indicator (MAGI) protocol.
the protocol was previously described (16). Briefly, 104 MAGI-CCR5 cells were plated in a 96-well plate. One day later, the cells were infected at an MOI of 0.01 (i.e., with comparable numbers of WT HIV-1 and L92P HIV-1 particles, as determined by RT-qPCR). Before infection, the medium was removed from each well, and viral dilutions in a 50-μl total volume of complete DMEM in the presence of DEAE-dextran (20 μg/ml) were added to each well. After incubation for 2 h, 150 μl of DMEM was added to each well. At 2 days postinfection, the cells were stained, and positive infections were counted under light microscopy (in at least three wells per virus infection).
JLTRG-R5 protocol.
0A total of .3 × 106 JLTRG-R5 cells in 66 μl of phenol-free RPMI were spread in black 96-well plates (Greiner). Then, 50 μl of virus mix (HIV-1 WT, HIV-1 L92P, or mock infection in phenol-free RPMI medium) was used to infect three wells per virus. After 2 h, phenol-free RPMI medium was added to a final volume of 200 μl. The production of GFP by HIV-1-infected cells was measured daily for 28 consecutive days in a Synergy HT multidetection microplate reader, equipped with the following filter set: excitation, 485/20 nm; and emission, 528/20 nm. The results of the mock infection were deducted from the virus infection.
HIV-1 expressing stable lines.
HEK293 cells were cotransfected with HIV-1 plasmids (pSVC21 or pSVC21 L92P) and pBABE harboring puromycin resistance (27). Puromycin-resistant cells were obtained, and single cells were isolated, grown, and 6 weeks later tested for the production of HIV-1 WT or L92P viruses.
qPCR.
DNA was isolated from infected cells by using a Quick-gDNA miniprep kit (Zymo). HIV-1 R-U5 and U3-PPT segments were quantified by qPCR using specific primers, as described in Table 1. Universal ProbeLibrary 60 (Roche) primers were used to calculate the relative gDNA input.
RT-qPCR.
Viral RNA was isolated using a High-Pure viral RNA kit (Roche). The samples were treated with RNase-free DNase I (Fermentas) to eliminate any provirus-containing plasmid contaminations. Purified RNA was reverse transcribed using Transcriptor Universal cDNA Master (Roche). cDNA was amplified with FastStart SYBR green Master (Roche) on an Applied Biosystems StepOnePlus real-time PCR system.
RESULTS AND DISCUSSION
Mutating residues Glu89, Val90, Leu92, Lys154, Pro157, or Ala158 in HIV-1 RT.
Following the development of our initial three-dimensional (3D) model of the RT-nucleic acids clamp complex (6), which was based on the 1ROA HIV-1 RT crystal structure (28), we further narrowed the distance between the presumed RT clamp junction and the surrounding residues to only 7 Å (Fig. 1). By monitoring energy minimization (29), we found that HIV-1 RT residues 89 to 92, Lys154, Pro157, and Ala158 are expected to be selectively involved in the clamp activity of HIV-1 RT (see Fig. 1). Then, we have tested how these residues affect experimentally the RT's clamp activity. To do this, we generated recombinant RT versions in which each of the specific residues was mutated. The specific mutants generated were E89G, V90T, L92P, K154I, P157S, and A158T. The decision regarding which residue would replace each of the WT residues was made after determining the sequence homology to other RTs (as shown in reference 7). We have changed the HIV-1 RT residues to comparable residues found in these other RTs that have variable clamp activities; thus, the general aim was to identify residues that are specifically involved only in the clamp activity and not in other RT activities. This approach would allow “dissecting” between the clamp function and all other RT activities. Therefore, one of the suspected residues, Gln91, was not tested here, since it was already found that the Q91N HIV-1 RT mutation leads to a loss of >90% of the DNA polymerase activity (30). As far as we know, none of the other HIV-1 RT mutants investigated here had been studied before, except for the E89G mutant. This RT mutant was examined mainly due to its relative resistance to deoxynucleoside analog RT inhibitors and phosphonoformic acid (31). The E89G RT was also found to have a fidelity of DNA synthesis that is higher than that of WT RT and an increased processivity of DNA synthesis (see, for example, references 22, 32–34, and 35). In addition, virions that harbor this mutation were partially defective, producing HIV-1 titers that were ∼10-fold lower than those of the WT virus (36).
FIG 1.
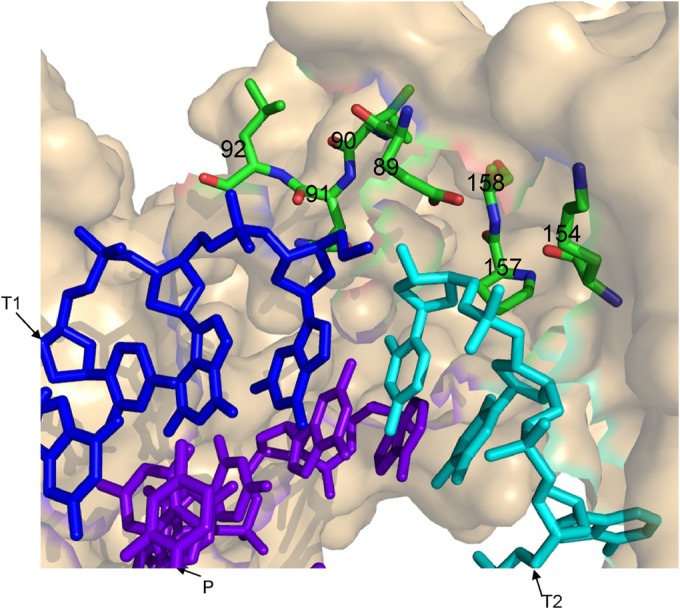
3D model for HIV-1 RT and clamp substrates. Using PyMol (PyMOL Molecular Graphics System, v1.4.1; Schrödinger, LLC), and based on 1ROA HIV-1 RT crystal structure (28), residues closer than 7 Å to the clamp site have been marked (i.e., residues 89 to 92, 154, 157, and 158 of the p66 RT subunit are highlighted and numbered). Blue, T1 (the first template); cyan, T2 (the second template); purple, P (the primer). Energy minimization was performed with the GROMACS 4.0.7 software suite, using the steepest descent algorithm (29).
DNA polymerase activities of all mutated RT versions.
All of the assays used to assess the clamp activity of RTs in fact measure a combination of both the initial capacity to clamp together the 2 nt complementary to the 3′ ends of the primer donor and acceptor strands and the resulting DNA polymerization that copies the acceptor strands (7, 9) (see also Fig. 3C). Therefore, all generated RT mutants were tested first to ensure that the DNA polymerase function (both DDDP and RDDP) was not impaired. The results exhibited in Fig. 2A and B show the extensions of the 5′-32P-end-labeled primers by copying the appropriate templates. All tested RT versions have similar apparent primer elongation capacities for each given substrate set, indicating that none of the mutants has lost a significant portion of its polymerase activity.
FIG 2.

DDDP, RDDP, and RNase H activities of HIV-1 RT mutants. The purified recombinant WT and mutant HIV-1 RT versions were tested. In the gels shown in panels A, B, and C, the order of all employed RT mutants is the same. (A) DDDP. (B) RDDP. Both assays were done by primer extensions on either DNA (DDDP) or RNA (RDDP) templates (see panel D). The mutant designations are indicated. Lanes C, control with no enzyme present. In panel A, “S” indicates the 21-nt substrate, and “P” indicates the 54-nt product of the primer extension. In panel B, “S” indicates the 20-nt substrate, and “P” indicates the 30-nt product of the primer extension. (C) The HIV-1 RT versions were assayed for their RNase H function, as described in Materials and Methods and in panel D. S, 33-nt labeled substrate. The reaction products are labeled as follows: C1, the 27-nt primary RNA product (that corresponds to the 17- to 18-nt cleavage according to the DNA 3′ end); C2, the secondary 18-nt product (corresponding to 8 to 9 nt from the DNA 3′ end). (D) Schematic descriptions of the performed assays. Upper panel, primer extensions (both RDDP and DDDP); lower panel, RNase H assay.
To test other RT functions that are related to the DNA-dependent activity, we assayed both the processivity of DNA synthesis of all RT versions generated and the strand displacement function (2). The processivity of a polymerase is directly proportional to the length of nascent polymeric products formed before the enzyme molecules dissociate from the product molecules and rebind other template–primer molecules. The extension of the 21-nt 5′-end-labeled primer to 54-nt products was tested in the absence or presence of an excess of a “quencher” DNA (namely, a trap of unlabeled activated DNA) (22, 37). Therefore, the synthesis obtained with the DNA trap reflects primer extensions due to single substrate-RT binding events. The results imply that all HIV-1 RT mutants have a processivity of DNA synthesis that is not substantially different from that of the WT RT (data not shown).
Another activity typical of RTs is the strand displacement, since DNA synthesis may require the strand displacement activity of RT to dislocate DNA strands that are annealed to the template strand and are located upstream to the elongated nascent DNA strand that is produced during DNA synthesis (2, 38). The DNA products generated by all tested HIV-1 RT versions in the strand displacement assay was found to be quite similar (data not shown).
RNase H activity of HIV-1 RT mutants.
After we confirmed that the polymerase activity of the mutants was not impaired (Fig. 2A and B), it was imperative also to assess whether the novel HIV-1 RT mutants retained their full RNase H function. Under the assay conditions used (see Materials and Methods), HIV-1 RT sequentially generates two products (17, 19, 39, 40). The primary cleavage product centers 17 to 18 nt from the 3′ end of the DNA strand in the RNA-DNA heteroduplex. The first product corresponds to the physical distance between the DNA polymerase and RNase H active sites of the RT. After making this cut, the RT molecule undergoes repositioning on the RNA-DNA heteroduplex and performs a secondary RNA cleavage at a position corresponding to a distance of 8 to 9 nt from the 3′ end of the DNA. The results presented in Fig. 2C show that all studied RTs mutants have an RNase H activity that is very similar to that of WT RT. This conclusion refers to both the extent of cleavage and the pattern of generated RNA cleavage products. In summary, none of the “basic” RT activities were impaired by the introduced single mutations, allowing us to test specifically the clamp and ST activity of all mutants.
Clamp activity of generated mutant HIV-1 RT variants.
To test the nucleic acid clamp activity of the different HIV-1 RT mutants, we used two methods (6, 7). The first one follows extensions of the 21-nt 5′-32P-end-labeled DNA primer that, after annealing to a cDNA strands, creates 3′-end 2-nt overhangs (the primary complex). These extensions are performed by copying the acceptor DNA strand, which is annealed or clamped at its 3′ end to 2-nt 3′ overhangs of the primer (for a schematic description, see Fig. 3C). A positive combined clamp/synthesis reaction is apparent from elongating primers to 59-nt products (as detected by urea-PAGE). This assay is semiquantitative. The second assay is an ELISA-based method (see Materials and Methods). This method was shown to be quantitative and is more sensitive than the first assay (6).
The results shown in Fig. 3A indicate that, relative to the clamp activity of WT HIV-1 RT, the A158T mutant was hardly affected, whereas the P157S, K154I, and V90T mutants lost more of their clamp activity. However, the E89G and L92P mutants did not possess any detectable activity. To further support these findings, we tested the clamp activity by also using an ELISA-based sensitive and quantitative technique (Fig. 3B). Here, there was relatively good agreement with the results of the previous method. Altogether, it is clear that only the L92P HIV-1 RT mutant has lost all its detectable activity, whereas the E89G, V90T, and K154I mutants still have some, albeit low, measurable residual activity (and the other two mutants were hardly affected).
Strand transfer activity of the HIV-1 RT mutants.
Basically, the ST assay that we used is composed of four consecutive steps (Fig. 4A). Step 1 was elongation of the 5′-end-labeled DNA primer by copying the RNA template (RDDP). In step 2, the RNA template was removed by the RNase H activity. Step 3 was the ST per se, namely, the switch to a second available template (a DNA strand with 3′-end sequence complementarity to the sequences added to the primer). Finally, in step 4, a further extension of the original DNA primer was performed by copying the second DNA template (DDDP). Hence, we used an assay system that involves these multistep reactions.
FIG 4.

ST activity of HIV-1 RT mutants. (A) Schematic description of the ST assay. After the extension of the 5′-32P-end-labeled 20-nt DNA primer, which annealed to the 33-nt RNA template (step 1), the 30-nt products undergo the RT-directed RNase H cleavage to remove this RNA template; thus, the single-stranded (−) strong stop (SS) DNA is formed (step 2). Then, ST to the 35-nt acceptor DNA strand takes place (step 3). This is followed by further elongation of the nascent DNA to full-length 49-nt products (step 4). The products of this overall reaction were separated by urea-PAGE. (B) ST assay. Each reaction consists of equal DDDP activities of the HIV-1 RT mutants that were incubated for 30 min at 37°C. Reaction products were analyzed by urea-PAGE, followed by autoradiography. The strong-stop DNA transcript, that is, the products (30 nt) of the initial RDDP reactions, are marked as “P of RDDP.” The full-length (49-nt) DNA products, generated after ST had taken place, are marked as “P of ST.” S, 20-nt substrate.
Since the RDDP, RNase H, and DDDP activities of all mutant HIV-1 RT versions tested here had already been shown to be similar to those of the WT RT, the assay products are restricted by the only rate-limiting factor, namely, the ST activity. The results shown in Fig. 4B imply that the overall ST activity of HIV-1 RT was significantly affected by the introduced mutations, in a manner that generally matches the extent by which the clamp activity was modified. A quantification of the gel show that, while the mutants of residues 154, 157, and 158 were only slightly affected (∼58% residual activity for K154IA, ∼74% for A158T, and no effect on A158T), the other three mutants were severely affected (with residual activities of ∼8% for E89G, ∼17% for V90T, and an undetected activity for L92P). As expected, all of the mutants produced similar levels of single-stranded DNA products in step 1, since they have equal RDDP activities. Interestingly, the synthesis of full-length 49-nt final products of the STs was impaired by several mutants. Thus, the L92P and E89G mutants of HIV-1 RT lost all detectable ST activity. However, after using substantially higher enzyme amounts (up to 10-fold), the L92P mutant was always totally inactive, whereas the E89G mutant showed some residual activity (data not shown).
Taken together, these results strongly suggest that the clamp and ST activities of RT are mechanistically related. This finding is not entirely unexpected, since in both cases there is a switch from one template to a second one during DNA synthesis. Still, the clamp activity does not depend on the RNase H activity, whereas ST heavily depends on this RT function. This also explains why, as far as we know, all reported cases, in which the ST activity of RTs was found to be impaired were cases linked to a diminished RNase H function.
Serial mutagenesis of Leu92 in HIV-1 RT.
The attained data show that, out of all the mutated HIV-1 RT residues, mutating amino acid Leu92 to a Pro (a residue with a cyclic side chain) had the greatest impact on both the clamp and the ST activities of RT. However, it was not clear how changing the aliphatic Leu to a Pro could change the catalytic activities, since residue 92 faces away from the DNA (Fig. 1) and changing in silico Leu to other residues gave small movements of <0.1 Å (data not shown). To test this issue experimentally, we generated new Leu92 RT mutants, in which the Leu was modified to Ile (another aliphatic residue with a relatively similar side chain), Tyr (an aromatic residue), Lys (a basic residue), or Glu (an acidic residue). All RT Leu92 mutants were recombinantly expressed and purified in a manner similar to all of the HIV-1 RT versions described above.
“Basic” activities of Leu92 mutants of HIV-1 RT.
To confirm that none of the mutations of residue 92 affect the RDDP, DDDP, and RNase H and the processivity of DNA synthesis, the mutants were assayed for all of these activities. The results show that all mutant RTs retained nearly WT levels of these activities (data not shown).
Clamp activity of Leu92 mutants of HIV-1 RT.
The clamp activity was determined by the two assay methods described above (the PAGE-based primer extension [Fig. 5A] and the ELISA-based method [Fig. 5B]). The results show that all tested Leu92 mutants lost most of their clamp activity, indicating that position 92 in HIV-1 RT is critical for this specific activity. Still, the L92P mutant underwent the largest damage of activity (with no detectable activity even with the more sensitive ELISA-based technique [Fig. 5B]), indicating that the cyclic residue Pro, when present at position 92, has the largest impact.
FIG 5.

Clamp activity of HIV-1 RT Leu92 mutants. The HIV-1 RT versions were assayed for the clamp assay as described in Fig. 3. (A) Gel-based clamp assay. S, 21-nt substrate; P, 59-nt product. (B) ELISA-based clamp technique. The data represent the means ± the SD. ***, P < 0.001 (as determined by paired two-tailed t test).
ST activity of Leu92 mutants of HIV-1 RT.
The same HIV-1 Leu92 mutants were tested for their ST function (Fig. 6). It is apparent that the three mutants—L92P, L92K, and L92Y—do not possess any detectable activity, whereas L92E and L92I have apparently retained some activity (∼62% residual activity for L92E and ∼59% for L92I). These results indicate subtle differences between the clamp and ST sensitivities spectra as a function of the mutations introduced at residue 92 of HIV-1 RT. Nonetheless, both clamp and ST activities were fully lost in the L92P RT mutant.
FIG 6.
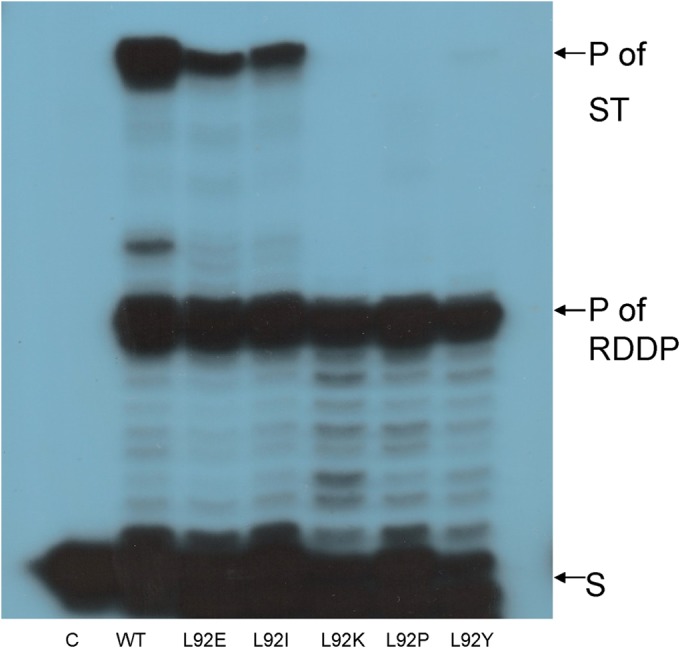
ST activity of WT HIV-1 RT of the Leu92 mutants. Each reaction consists of equal DDDP activities of the RT mutants that were incubated for 30 min at 37°C, as described in Materials and Methods. Reaction products were analyzed as marked in Fig. 4B.
The results presented here imply that the clamp and ST activities of the studied RT are linked, at least when the overall spectra of the affecting mutations are compared. This finding also indicates that there is a mechanistic link between the two activities that are related because in both cases the RT switches from copying one template (both RNA and DNA) to a second template. Another important deduction is that the mutants of HIV-1 RT show a distinct impairment of their ST activity. As far as we know, this is the first reported case in which a single mutation in an RT leads to a specific deficiency in only the ST function.
Introducing the RT L92P mutation into an infectious HIV-1 clone.
To further study the biological relevance of the loss of ST activity by the L92P mutant, this mutation was introduced into the RT-encoding gene of an infectious HIV-1 clone. The original pSVC21 plasmid that contains the full infectious cDNA of the HXB2 strain of HIV-1 (16), underwent site-directed mutagenesis. After transfecting HEK293 cells, the produced WT and L92P-mutated virions were quantified by RT-qPCR (Fig. 7A). The production of the properly cleaved capsid p24 protein was confirmed by Western analysis of the generated viral particles (Fig. 7B). It is apparent that the mutant HIV-1 and the WT virus produced relatively similar amounts of virions and that the virions of both viral strains possessed similar amounts of both cleaved p24 protein and noncleaved p55 Gag precursor. These similarities between the mutant and WT virus allowed further analyses of the infection capacity of the mutant.
FIG 7.
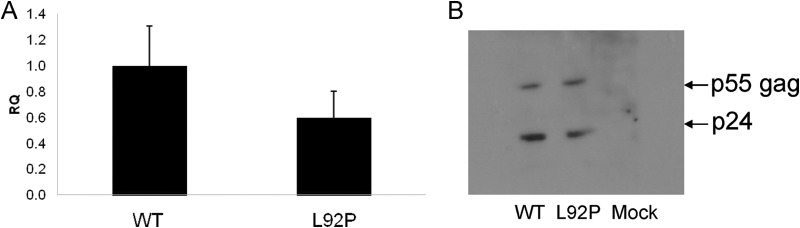
Virus production by transfecting HEK293 cells with plasmids harboring WT or L92P HIV-1 strains. (A) Two days after transfecting HEK293 cells, viruses from supernatants were collected and lysed, and the viral RNA was reverse transcribed. The presence of the viral RNA was then detected with virus-specific primers in qPCRs (see Materials and Methods). RQ, relative quantification. Error bars represent the means ± the SD. (B) Western blot analysis of p24 CA protein present in virus lysates. The anti-CA antibodies detect p24 and the CA polyprotein precursor, p55 gag. Mock, supernatant from HEK293 uninfected cells.
Infectivity of both the WT and the L92P mutant of HIV-1.
The transfection-produced virions were used to infect HIV-1-sensitive HeLa or Jurkat cells. Two assays were used to detect the extent of viral infectivity. The first was a MAGI assay conducted on HeLa cells that express the CD4 and CCR5 receptors. The second assay assessed the infectivity of human T cells, with the GFP gene under the control of a Tat-sensitive promoter. The results of the first assay, analyzed 48 h after infection, are shown in Fig. 8. It is apparent that only WT HIV-1 induced the appearance of cells with nuclei stained by the substrate specific to β-galactosidase (see Materials and Methods). Unlike the WT virus, the L92P mutant did not produce any detectable stained nuclei. In addition, most stained nuclei are clustered within multinucleated cells, a phenomenon resulting from the capacity of HIV-1 to form syncytia in these cells (12). Figure 8A shows the results seen in representative microscopic fields, whereas Fig. 8B summarizes the numbers of positive cells counted each in three wells obtained in three independent experiments. The counted stained cells were either singly nucleated or multiply nucleated (syncytium). Support for the lack of any detectable infectivity by the L92P mutant can be seen from the results of the second assay (Fig. 8C). Here, we assayed the activity of the GFP, induced by the viruses that infect JLTRG-R5 cells grown in suspension. This kinetic experiments show that the L92P mutant induced no GFP activity even 28 days after infection, whereas the WT virus induces a GFP appearance from day 8 postinfection. Collectively, these results indicate that the L92P mutant lost all of its detectable infectivity.
FIG 8.

Infectivity of HIV-1 WT and L92P virions. (A) Equal virion amounts of the WT HIV-1 and L92P viruses, as determined by RT-qPCR, were used to infect MAGI cells. At 2 days postinfection, the MAGI protocol was performed (see Materials and Methods), and three wells from each infection were inspected under light microscopy for positive blue cells. Scale bar, 50 μm. (B) The results of three independent MAGI experiments were quantified, showing the means ± the SD. Mock, infection with supernatants obtained from uninfected HEK293 cells. (C) JLTRG-R5 cells were infected with equal virion amounts of WT HIV-1 or the L92P mutant. Once a day, the level of GFP, produced in the infected cells, was quantified in a plate reader, as described in Materials and Methods. The numbers represent the mean results of three independent experiments ± the SD.
Integration of cDNA from WT and L92P mutant HIV-1 into the genome of HIV-1-infected cells.
At this stage, we tried to understand at which step viral replication was blocked in the L92P HIV-1 mutant. We infected the HIV-1 susceptible MAGI-CCR5 cells with equal HIV-1 virion numbers of the WT and the L92P mutant (as determined by RT-qPCR [see Materials and Methods]). At 1 day postinfection, the cellular genomic DNA was harvested and tested for the presence of integrated HIV-1 cDNA. First, we PCR amplified DNA segments that span the proviral and cellular DNA boundaries, by using a known Alu-specific repetitive human specific sequence (41) and an HIV-1-specific primer taken from the gag gene (26). The products were then amplified by nested qPCRs that were carried out with internal HIV-1-specific primers, derived from HIV-1 LTR (26). For further information regarding all of the primers, see Table 1. The results (Fig. 9A) imply that, unlike the infection with WT HIV-1, the L92P mutant did not lead to any cellular integration of viral cDNA. This result can easily explain why in the experimental data depicted in Fig. 8, the mutant was shown to lose its infectivity.
FIG 9.
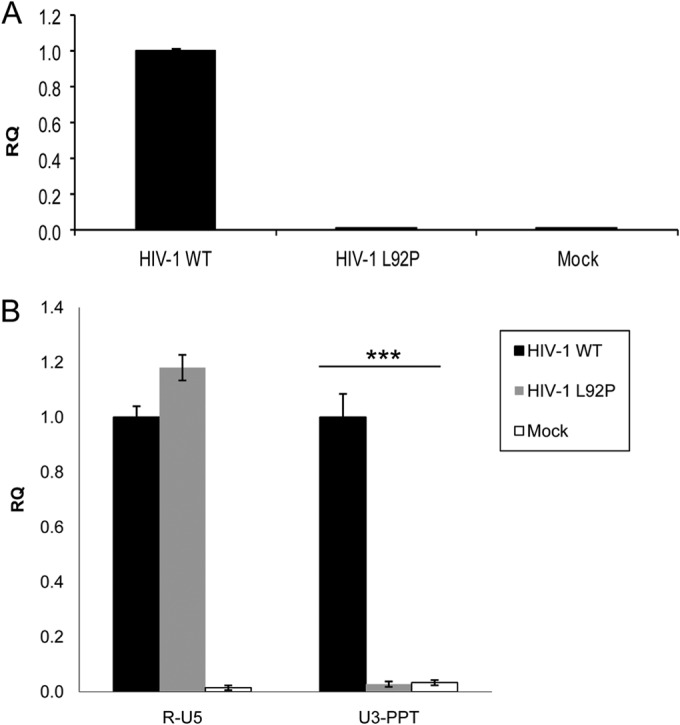
The RTN products generated 24 h postinfection in cells infected by the WT and L92P mutant HIV-1. (A) Integration quantification of HIV-1 by Alu-PCR, followed by nested viral qPCR. The details of this experiment are described in Materials and Methods. (B) HIV-1 RTN intermediates. Total DNA from HIV-1 WT, HIV-1 L92P, and mock-infected cells were tested for RTN products. R-U5, the first RTN product; U3-PPT, the first product following first ST. RQ, relative quantification. Error bars represent the means ± the SD of three experiments. ***, P < 0.001 (as determined by paired two-tailed t test).
Intracellular synthesis of viral cDNA by WT and L92P mutant HIV-1.
To understand why no detectable integrated cDNA was produced by the L92P mutant, we have gone back through the life cycle of the retrovirus to determine whether cDNA, or a segments of it, was synthesized after infection. In order to eliminate plasmid background contaminations from the original transfections, we generated stable HIV-1-expressing HEK293 cells. We then infected MAGI-CCR5 cells with these viruses, isolated the total cellular DNA, and specifically amplified by qPCR the viral R-U5 region (corresponding to the accumulated minus strong-stop DNA) or the product after the first ST (U3-PPT). The data presented in Fig. 9B show that both cDNA segments were amplified in WT virus-infected cells, whereas in the case of the L92P mutant, only the R-U5 product was detected, and no significant U3-PPT levels (above mock-infected cell levels) were observed. In agreement with the in vitro data presented above, this in vivo information implies that, although DNA synthesis was hardly affected by the L92P mutation (as evident from the high level of the minus strong-stop R-U5 DNA), the first ST was completely blocked. Therefore, it is reasonable to conclude that the ST activity of the RT L92P mutant was severely damaged both in vitro and in vivo.
In summary, the results presented here emphasize a likely mechanistic linkage between HIV-1 RT's clamp and ST activities. Such a deduction is not unexpected, because the clamp activity actually induces a “micro” ST with very short complementaries between the 3′ end of the growing DNA strand and the 5′ end of the acceptor template strand. Following these in vitro results, the L92P mutant of the RT that was introduced into an infectious HIV-1 clone led to a total loss of any apparent viral replication in susceptible cells. This finding confirms the pivotal role of HIV-1 RT residue 92 in the viral life cycle. As far as we know, this is the first report on HIV-1 RT mutants that specifically impair the ST activity of an RT.
Taken together, Leu92 is critical for the replication of HIV-1, probably due to its cardinal importance in the ST function. Thus far, the ST function of RTs has been regarded as an indirect outcome that results from a simple combination between both DNA polymerase and RNase H activities. We show here that ST results also from a direct and unique activity of the RT in which Leu92 has a major role. L92 is located in the 3D structure of HIV-1 RT, close to the so called “template grip,” and is situated between the β strands (β5A and β5B) in the palm subdomain of the p66 subunit of the RT (1, 42). An important support for the conclusion regarding the critical role of Leu92 stems from the analyses of the currently available data of the sequences of numerous naturally occurring HIV-1 isolates, or sequences HIV-1 segments, obtained from NCBI database (ncbi.nlm.nih.gov). The impressive finding is that out of 12,534 HIV-1 RT sequences, 12,526 (99.94%) have a Leu92 and only 5 have a Met92 (0.04%) and 3 have an Iso92 (0.02%), and not even one isolate has a Pro92 (data not shown). Interestingly, the amino acids sequences that surround Leu92 in the numerous HIV-1 isolates analyzed are also conserved. In an inspection of the RT's sequence (residues 84 to 100), TQDFWEVQLGIPHPAGL, spanning Leu92 (underlined), all 17 residues are conserved in >98% of the virus isolates (data not shown). This information supports the importance of this whole RT segment to viral fitness. We could not find any structural clues to explain mechanistically the uniqueness of the RT-driven ST activity. Therefore, further structural studies are required to elucidate how HIV-1 RT Leu92 is specifically and critically involved in the ST activity and why a proline at this site has the highest deleterious effect. Once this new structural information has been obtained, it is possible that targeting residue 92 of HIV-1 RT could be beneficial for selectively blocking the unique ST activity of the RT and, consequently, the infectivity and pathogenicity of the virus.
ACKNOWLEDGMENTS
We are very thankful to the National Institutes of Health AIDS Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Disease, for the generous gifts of the MAGI-CCR5 and JLTRG-R5 cells and the anti-HIV-1 p24 antibody.
This research was supported in part by a grant from the Israel Science Foundation (grant 411/07).
REFERENCES
- 1.Coffin JM, Hughes SH, Varmus HE. 1997. Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [PubMed] [Google Scholar]
- 2.Herschhorn A, Hizi A. 2010. Retroviral reverse transcriptases. Cell Mol Life Sci 67:2717–2747. doi: 10.1007/s00018-010-0346-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Menendez-Arias L, Berkhout B. 2008. Retroviral reverse transcription. Virus Res 134:1–250. doi: 10.1016/j.virusres.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 4.Schultz SJ, Champoux JJ. 2008. RNase H activity: structure, specificity, and function in reverse transcription. Virus Res 134:86–103. doi: 10.1016/j.virusres.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basu VP, Song M, Gao L, Rigby ST, Hanson MN, Bambara RA. 2008. Strand transfer events during HIV-1 reverse transcription. Virus Res 134:19–38. doi: 10.1016/j.virusres.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 6.Oz-Gleenberg I, Herschhorn A, Hizi A. 2011. Reverse transcriptases can clamp together nucleic acids strands with two complementary bases at their 3′ termini for initiating DNA synthesis. Nucleic Acids Res 39:1042–1053. doi: 10.1093/nar/gkq786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oz-Gleenberg I, Herzig E, Voronin N, Hizi A. 2012. Substrate variations that affect the nucleic acid clamp activity of reverse transcriptases. FEBS J 279:1894–1903. doi: 10.1111/j.1742-4658.2012.08570.x. [DOI] [PubMed] [Google Scholar]
- 8.Oz-Gleenberg I, Hizi A. 2011. Strand selections resulting from the combined template-independent DNA synthesis and clamp activities of HIV-1 reverse transcriptase. Biochem Biophys Res Commun 408:482–488. doi: 10.1016/j.bbrc.2011.04.063. [DOI] [PubMed] [Google Scholar]
- 9.Oz-Gleenberg I, Herzig E, Hizi A. 2012. The template-independent DNA synthesis activity associated with the reverse transcriptase of the LTR retrotransposon Tf1. FEBS J 279:142–153. doi: 10.1111/j.1742-4658.2011.08406.x. [DOI] [PubMed] [Google Scholar]
- 10.Golinelli MP, Hughes SH. 2002. Nontemplated base addition by HIV-1 RT can induce nonspecific strand transfer in vitro. Virology 294:122–134. doi: 10.1006/viro.2001.1322. [DOI] [PubMed] [Google Scholar]
- 11.Golinelli MP, Hughes SH. 2002. Nontemplated nucleotide addition by HIV-1 reverse transcriptase. Biochemistry 41:5894–5906. doi: 10.1021/bi0160415. [DOI] [PubMed] [Google Scholar]
- 12.Kimpton J, Emerman M. 1992. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated β-galactosidase gene. J Virol 66:2232–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. 1996. Identification of a major coreceptor for primary isolates of HIV-1. Nature 381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 14.Kutsch O, Levy DN, Bates PJ, Decker J, Kosloff BR, Shaw GM, Priebe W, Benveniste EN. 2004. Bis-anthracycline antibiotics inhibit human immunodeficiency virus type 1 transcription. Antimicrob Agents Chemother 48:1652–1663. doi: 10.1128/AAC.48.5.1652-1663.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ochsenbauer-Jambor C, Jones J, Heil M, Zammit KP, Kutsch O. 2006. T-cell line for HIV drug screening using EGFP as a quantitative marker of HIV-1 replication. Biotechniques 40:91–100. doi: 10.2144/000112072. [DOI] [PubMed] [Google Scholar]
- 16.Rosenbluh J, Hayouka Z, Loya S, Levin A, Armon-Omer A, Britan E, Hizi A, Kotler M, Friedler A, Loyter A. 2007. Interaction between HIV-1 Rev and integrase proteins: a basis for the development of anti-HIV peptides. J Biol Chem 282:15743–15753. doi: 10.1074/jbc.M609864200. [DOI] [PubMed] [Google Scholar]
- 17.Sevilya Z, Loya S, Adir N, Hizi A. 2003. The ribonuclease H activity of the reverse transcriptases of human immunodeficiency viruses type 1 and type 2 is modulated by residue 294 of the small subunit. Nucleic Acids Res 31:1481–1487. doi: 10.1093/nar/gkg235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sevilya Z, Loya S, Duvshani A, Adir N, Hizi A. 2003. Mutagenesis of cysteine 280 of the reverse transcriptase of human immunodeficiency virus type-1: the effects on the ribonuclease H activity. J Mol Biol 327:19–30. doi: 10.1016/S0022-2836(03)00052-4. [DOI] [PubMed] [Google Scholar]
- 19.Sevilya Z, Loya S, Hughes SH, Hizi A. 2001. The ribonuclease H activity of the reverse transcriptases of human immunodeficiency viruses type 1 and type 2 is affected by the thumb subdomain of the small protein subunits. J Mol Biol 311:957–971. doi: 10.1006/jmbi.2001.4904. [DOI] [PubMed] [Google Scholar]
- 20.Hizi A, McGill C, Hughes SH. 1988. Expression of soluble, enzymatically active, human immunodeficiency virus reverse transcriptase in Escherichia coli and analysis of mutants. Proc Natl Acad Sci U S A 85:1218–1222. doi: 10.1073/pnas.85.4.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Avidan O, Bochner R, Hizi A. 2006. The catalytic properties of the recombinant reverse transcriptase of bovine immunodeficiency virus. Virology 351:42–57. doi: 10.1016/j.virol.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 22.Avidan O, Hizi A. 1998. The processivity of DNA synthesis exhibited by drug-resistant variants of human immunodeficiency virus type-1 reverse transcriptase. Nucleic Acids Res 26:1713–1717. doi: 10.1093/nar/26.7.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Avidan O, Loya S, Tonjes RR, Sevilya Z, Hizi A. 2003. Expression and characterization of a recombinant novel reverse transcriptase of a porcine endogenous retrovirus. Virology 307:341–357. doi: 10.1016/S0042-6822(02)00131-9. [DOI] [PubMed] [Google Scholar]
- 24.Kirshenboim N, Hayouka Z, Friedler A, Hizi A. 2007. Expression and characterization of a novel reverse transcriptase of the LTR retrotransposon Tf1. Virology 366:263–276. doi: 10.1016/j.virol.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 25.Bochner R, Duvshani A, Adir N, Hizi A. 2008. Mutagenesis of Gln294 of the reverse transcriptase of human immunodeficiency virus type-2 and its effects on the ribonuclease H activity. FEBS Lett 582:2799–2805. doi: 10.1016/j.febslet.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 26.Julias JG, Ferris AL, Boyer PL, Hughes SH. 2001. Replication of phenotypically mixed human immunodeficiency virus type 1 virions containing catalytically active and catalytically inactive reverse transcriptase. J Virol 75:6537–6546. doi: 10.1128/JVI.75.14.6537-6546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morgenstern JP, Land H. 1990. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res 18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peletskaya EN, Kogon AA, Tuske S, Arnold E, Hughes SH. 2004. Nonnucleoside inhibitor binding affects the interactions of the fingers subdomain of human immunodeficiency virus type 1 reverse transcriptase with DNA. J Virol 78:3387–3397. doi: 10.1128/JVI.78.7.3387-3397.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. 2005. GROMACS: fast, flexible, and free. J Comput Chem 26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 30.Pandey N, Mishra CA, Manvar D, Upadhyay AK, Talele TT, Comollo TW, Kaushik-Basu N, Pandey VN. 2011. The glutamine side chain at position 91 on the beta5a-beta5b loop of human immunodeficiency virus type 1 reverse transcriptase is required for stabilizing the dNTP binding pocket. Biochemistry 50:8067–8077. doi: 10.1021/bi200815e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prasad VR, Lowy I, de los Santos T, Chiang L, Goff SP. 1991. Isolation and characterization of a dideoxyguanosine triphosphate-resistant mutant of human immunodeficiency virus reverse transcriptase. Proc Natl Acad Sci U S A 88:11363–11367. doi: 10.1073/pnas.88.24.11363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drosopoulos WC, Prasad VR. 1996. Increased polymerase fidelity of E89G, a nucleoside analog-resistant variant of human immunodeficiency virus type 1 reverse transcriptase. J Virol 70:4834–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quan Y, Inouye P, Liang C, Rong L, Gotte M, Wainberg MA. 1998. Dominance of the E89G substitution in HIV-1 reverse transcriptase in regard to increased polymerase processivity and patterns of pausing. J Biol Chem 273:21918–21925. doi: 10.1074/jbc.273.34.21918. [DOI] [PubMed] [Google Scholar]
- 34.Rezende LF, Curr K, Ueno T, Mitsuya H, Prasad VR. 1998. The impact of multidideoxynucleoside resistance-conferring mutations in human immunodeficiency virus type 1 reverse transcriptase on polymerase fidelity and error specificity. J Virol 72:2890–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rubinek T, Bakhanashvili M, Taube R, Avidan O, Hizi A. 1997. The fidelity of 3′ misinsertion and mispair extension during DNA synthesis exhibited by two drug-resistant mutants of the reverse transcriptase of human immunodeficiency virus type 1 with Leu74→Val and Glu89→Gly. Eur J Biochem 247:238–247. doi: 10.1111/j.1432-1033.1997.00238.x. [DOI] [PubMed] [Google Scholar]
- 36.Nikolenko GN, Svarovskaia ES, Delviks KA, Pathak VK. 2004. Antiretroviral drug resistance mutations in human immunodeficiency virus type 1 reverse transcriptase increase template-switching frequency. J Virol 78:8761–8770. doi: 10.1128/JVI.78.16.8761-8770.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Avidan O, Meer ME, Oz I, Hizi A. 2002. The processivity and fidelity of DNA synthesis exhibited by the reverse transcriptase of bovine leukemia virus. Eur J Biochem 269:859–867. doi: 10.1046/j.0014-2956.2001.02719.x. [DOI] [PubMed] [Google Scholar]
- 38.Whiting SH, Champoux JJ. 1998. Properties of strand displacement synthesis by Moloney murine leukemia virus reverse transcriptase: mechanistic implications. J Mol Biol 278:559–577. doi: 10.1006/jmbi.1998.1720. [DOI] [PubMed] [Google Scholar]
- 39.Gao HQ, Sarafianos SG, Arnold E, Hughes SH. 1999. Similarities and differences in the RNase H activities of human immunodeficiency virus type 1 reverse transcriptase and Moloney murine leukemia virus reverse transcriptase. J Mol Biol 294:1097–1113. doi: 10.1006/jmbi.1999.3325. [DOI] [PubMed] [Google Scholar]
- 40.Wisniewski M, Balakrishnan M, Palaniappan C, Fay PJ, Bambara RA. 2000. Unique progressive cleavage mechanism of HIV reverse transcriptase RNase H. Proc Natl Acad Sci U S A 97:11978–11983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Agosto LM, Yu JJ, Dai J, Kaletsky R, Monie D, O'Doherty U. 2007. HIV-1 integrates into resting CD4+ T cells even at low inoculums as demonstrated with an improved assay for HIV-1 integration. Virology 368:60–72. doi: 10.1016/j.virol.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding J, Hughes SH, Arnold E. 1997. Protein-Nucleic acid interactions and DNA conformation in a complex of human immunodeficiency virus type 1 reverse transcriptase with a double-stranded DNA template-primer. Biopolymers 44:125–138. doi:. [DOI] [PubMed] [Google Scholar]