

ABSTRACT
Molluscum contagiosum virus (MCV) is unique in being the only known extant, human-adapted poxvirus, yet to date, it is very poorly characterized in terms of host-pathogen interactions. MCV causes persistent skin lesions filled with live virus, but these are generally immunologically silent, suggesting the presence of potent inhibitors of human antiviral immunity and inflammation. Fewer than five MCV immunomodulatory genes have been characterized in detail, but it is likely that many more remain to be discovered given the density of such sequences in all well-characterized poxviruses. Following virus infection, NF-κB activation occurs in response to both pattern recognition receptor (PRR) signaling and cellular activation by virus-elicited proinflammatory cytokines, such as tumor necrosis factor (TNF). As such, NF-κB activation is required for virus detection, antiviral signaling, inflammation, and clearance of viral infection. Hence, we screened a library of MCV genes for effects on TNF-stimulated NF-κB activation. This revealed MC132, a unique protein with no orthologs in other poxviral genomes, as a novel inhibitor of NF-κB. Interestingly, MC132 also inhibited PRR- and virus-activated NF-κB, since MC132 interacted with the NF-κB subunit p65 and caused p65 degradation. Unbiased affinity purification to identify host targets of MC132 revealed that MC132 acted by targeting NF-κB p65 for ubiquitin-dependent proteasomal degradation by recruiting p65 to a host Cullin-5/Elongin B/Elongin C complex. These data reveal a novel mechanism for poxviral inhibition of human innate immunity and further clarify how the human-adapted poxvirus MCV can so effectively evade antiviral immunity to persist in skin lesions.
IMPORTANCE How human cells detect and respond to viruses is incompletely understood, but great leaps in our understanding have been made by studying both the early innate immune response and the ways that viruses evade it. Poxviruses adapt to specific hosts over time by evolving elegantly precise inhibitors targeting the rate-limiting steps of immunity. These inhibitors reveal new features of the antiviral response while also offering potent new tools for approaching therapeutic intervention in autoimmunity. Molluscum contagiosum virus (MCV) is the only known extant poxvirus specifically adapted to human infection, yet it remains poorly characterized. In this study, we report the identification of the MCV protein MC132 as a potent inhibitor of NF-κB, an essential regulatory crux of innate immunity. Furthermore, identification of the mechanism of inhibition of NF-κB by MC132 reveals an elegant example of convergent evolution with human herpesviruses. This discovery greatly expands our understanding of how MCV so effectively evades human immunity.
INTRODUCTION
Host innate immune detection of virus infection employs pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), and cytosolic nucleic acid-sensing systems, which stimulate signal transduction cascades leading to the activation of NF-κB and IRF transcription factor families. Such transcription factors induce type I interferons (IFNs) and the proinflammatory cytokines tumor necrosis factor alpha (TNF-α) and interleukin-1 (IL-1) (1). IFNs and cytokines then stimulate pathways which limit viral spread and direct antiviral acquired immunity. In order to overcome these host defense mechanisms, viruses evolve inhibitors that target the rate-limiting steps in innate immune signaling. Thus, studying such inhibitors not only provides an understanding of viral pathogenesis but also reveals novel facets of the host innate signaling mechanisms which they target, providing new avenues for therapeutic intervention for disorders defined by aberrant innate immune responses and inflammation. This is particularly true of poxviruses, which have evolved small inhibitory proteins by integrating host sequences into their genomes which are then refined by mutation over long periods of virus-host evolution to better inhibit the continually diversifying immunity of their hosts (2). Although the discovery and characterization of such poxviral inhibitory proteins have been highly informative in defining host-virus interactions and uncovering novel aspects of innate immunity (3), most of these viral inhibitors have been described for non-human-adapted poxviruses, particularly for vaccinia virus (VACV).
The poxvirus molluscum contagiosum virus (MCV) is specifically adapted to human infection and has a genome predicted to encode 182 proteins, only 105 of which have orthologs in other orthopoxviruses (4). In contrast to the non-human-adapted VACV, which causes local inflammation in human skin lesions, MCV can inhabit human dermal lesions over long periods, with a minimal immune response and almost no inflammation. While this predicts that MCV has evolved unique, efficient inhibitors of human innate immunity, fewer than five MCV immunoregulators have been investigated in detail (5, 6). Hence, we screened 31 open reading frames (ORFs) of the MCV genome with no known function for inhibitors of known human antiviral innate immune signaling networks that culminate in the activation of NF-κB, a critical proinflammatory transcription factor. We then tested the effects of the MCV ORFs on TNF-stimulated NF-κB activation. This identified MC132, a unique protein with no orthologs in other poxviral genomes, as a novel inhibitor of NF-κB activation. Inhibition of NF-κB by MC132 was independent of the NF-κB activation pathways tested, as it was apparent for stimulation by proinflammatory cytokines, PRR ligands, DNA virus, or RNA virus. Further functional analysis, including unbiased affinity purification to identify host targets of MC132, showed that MC132 utilizes a host protein complex to degrade the p65 subunit of NF-κB in a fascinating example of convergent evolution in poxviruses and gammaherpesviruses. These discoveries greatly extend our understanding of how MCV, the only known extant human-adapted poxvirus, so efficiently evades human immunity.
MATERIALS AND METHODS
Cell culture and viruses.
Human embryonic kidney 293T (HEK293T) cells, HeLa cells, and COS-1 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% (vol/vol) fetal calf serum (FCS) and penicillin-streptomycin. Stable cells were selected with 300 μg Hygrogold (Invivogen), and expression of MC132 was induced with dilutions of sterile 10 mM cadmium chloride (Sigma). Sendai virus (ECACC), vesicular stomatitis virus (VSV; a gift from John C. Bell, Ottawa Hospital Research Institute), and modified vaccinia virus Ankara (MVA; a gift from Ingo Drexler, Düsseldorf University) were all used at a multiplicity of infection (MOI) of 10.
Plasmids and oligonucleotides.
MC132 and MC014 were synthesized by Invitrogen and Genscript, respectively, and subcloned into the KpnI and NotI sites of pCEP and cadmium chloride-inducible pMEP4 plasmids (Invitrogen) with a C-terminal Flag tag (DYKDDDDK). The following forward (FP) and reverse (RP) primers were used for cloning of full-length and truncated MC132 into pCEP4: for pCEP4-MC132-Flag, GGGGTACCATGATGAACTTTTCCGACC (FP) and CCCAAGCTTACAACGCTGCAGAAGCAGG (RP); for pCEP4-T0, GGGGTACCATGGACCCGTGCTTGCTAGGT (FP); for pCEP4-T1, GGGGTACCATGTTGCGGCGGCTCGGTTGGC (FP); for pCEP4-T3, GGGAAGCTTGTGTGCCTGCTCGCGAAAG (RP); for pCEP4-T1.2, GGGGTACCATGACCCCCGAAGCAAAAGCG (FP); pCEP4-T3.2, GGGAAGCTTAGGCCAGGGTCCCGAGAGA (RP); and for pCEP4-T3.3, GGGAAGCTTCAAGTGCCGCGCGCAAAT (RP). pCEP4-MC132-HA was made by replacing the Flag sequence with a hemagglutinin (HA) tag. p65 truncations were made using the following primers: for full-length p65(1–551), p65F (GGGGTACCATGGACGAACTGTTCCCC) and p65R (CCCAAGCTTCTCTTCTATAGGAACTTG); for p65(1–147), p65F and 147R (CCCAAGCTTCTCTTCTATAGGAACTTG); for p65(148–300), 148F (GGGGTACCATGCAGCGTGGGGACTACGACC) and 300R (CCCAAGCTTCTCCTCAATCCGGTGACG); for p65(1–300), p65F and 300R; for p65(1–458), p65F and 458R (CCCAAGCTTTGTGCTGTTGCCAAGCAAGG); and for p65(148–458), 148F and 458R. Plasmids expressing Flag-IKKα, Flag-IKKβ, Flag-IKKε, Flag-TRAF2, and Flag-TRAF6 were from Tularik Inc. The sources of other expression plasmids were as follows: Flag-TRIF, S. Akira, Osaka University, Osaka, Japan; Myc-MyD88 and ISRE-luciferase, L. O'Neill, Trinity College Dublin, Dublin, Ireland; CD4-TLR9, A. Ozinsky, University of Washington, WA; CD4-TLR3, R. Medzhitov, Yale University, CT; Flag-MAVS and cGAMP synthase (cGAS), J. Chen, UT Southwestern Medical Centre; RasVHa, D. Cantrell, University of Dundee, Dundee, United Kingdom; Flag–p38 mitogen-activated protein kinase (MAPK), J. Saklatvala, Kennedy Institute of Rheumatology, London, United Kingdom; Elongin B and Elongin C, N. Stevenson, Trinity College Dublin, Dublin, Ireland; and HA-ubiquitin, A. Mansell, Monash University, Melbourne, Australia. The human STING coding region was amplified by PCR from full-length I.M.A.G.E. cDNA clones (IRATp970D0274D and IRAVp968F0688D; imaGenes) and cloned into the vector pCMV-myc (Clontech). The NF-κB–luciferase reporter gene was obtained from R. Hofmeister (Universitat Regensburg, Germany), and the pFR-luciferase reporter and pFA2-Elk1 were from Agilent. Cullin-5 small interfering RNA (siRNA) (5′-AGATTCCTGGCGTAAAAGCTT-3′) was from Invitrogen. Poly(dA:dT) was from Sigma-Aldrich. The sequences of the Elongin B PCR primers were as follows: ElonginB-FP, ATGGACGTGTTCCTCATGATC; and ElonginB-RP, TGCTGTTTGACTGGTGAAGC.
Real-time PCR.
RNAs from HEK293 cells grown in 12-well plates were extracted using an RNeasy kit (Qiagen) and converted to cDNAs by using a Quantitect RT kit (Qiagen). Elongin B mRNA was quantified by real-time PCR with the TaqMan gene expression assay Hs00277188_s1 and a β-actin endogenous control VIC-MGB probe (6-carboxyrhodamine–minor groove binder; Applied Biosystems). Experiments were performed in triplicate.
Abs.
Primary antibodies (Abs) used for immunoblotting were anti-β-actin (AC-74), anti-Myc (9E10), and anti-Flag (M2) from Sigma-Aldrich; anti-IκBα from R. Hay, University of Dundee, Dundee, United Kingdom; anti-phospho-p65 (Ser536; 93H1), anti-p65 (C-20), and anti-Cullin-5 from Santa Cruz; and anti-HA from Covance. The secondary Abs for immunoblotting were IRDye 680LT-conjugated anti-mouse, IRDye 800CW-conjugated anti-rabbit, and IRDye 680LT-conjugated anti-goat Abs (Li-Cor Biosciences). Secondary Abs for confocal microscopy were Alexa Fluor 647-conjugated anti-mouse or Alexa Fluor 488-conjugated anti-rabbit Abs (Invitrogen). An anti-rabbit isotype control antibody was obtained from Sigma.
ELISA.
Cell culture supernatants were assayed for expression of the IL-8 protein by use of an enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems) according to the manufacturer's instructions.
Immunoblotting.
Cells were seeded at 5 × 105 cells/well in 6-well dishes and transfected the next day with 3 μg total DNA by use of GeneJuice (Novagen). Twenty-four hours later, cells were lysed in 200 μl sample buffer (187.5 mM Tris [pH 6.8], 6% [wt/vol] SDS, 30% [vol/vol] glycerol, 0.3% [wt/vol] bromophenol blue, 150 mM dithiothreitol [DTT], and Benzonase), incubated on ice for 5 min, and then boiled for 5 min at 99°C. Twenty-microliter lysates were resolved by 10 to 20% SDS-PAGE, transferred to polyvinylidene difluoride membranes (Millipore), blocked for 1 h in 3% (wt/vol) bovine serum albumin (BSA) in phosphate-buffered saline (PBS), and probed overnight with a primary Ab (1:1,000 dilution in blocking solution). The next day, membranes were incubated with secondary Abs (1:10,000 dilution in blocking solution), and blots were visualized using an Odyssey imaging system (Li-Cor Biosciences).
Immunoprecipitation.
Cells were seeded at 4 × 106 cells/10-cm dish and transfected with 8 μg total plasmid the next day. Twenty-four hours later, cells were washed with ice-cold PBS and then scraped into lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 0.5% [vol/vol] NP-40, 30 mM NaF, 5 mM EDTA, 10% [vol/vol] glycerol, and 40 mM β-glycerophosphate containing inhibitors, i.e., 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride [PMSF], and 1% [vol/vol] aprotinin) and left on ice for 45 min. Anti-Flag beads (Sigma) were equilibrated with lysis buffer, and cleared lysates were mixed with beads and incubated for 2 h with rolling at 4°C. Beads were then washed three times with 1 ml lysis buffer, and immunoprecipitated material was eluted with 3× Flag peptide (Sigma) for 30 min with rolling at 4°C, after which it was separated and immunoblotted, with probing for the indicated proteins.
Confocal microscopy.
Cells were seeded at 3 × 105 cells/ml on glass coverslips in 24-well plates and stimulated the next day as indicated. Cells were fixed for 12 min in 4% (wt/vol) paraformaldehyde and permeabilized for 15 min with 0.5% (vol/vol) Triton X-100 in PBS. Coverslips were blocked for 1 h in 5% (wt/vol) BSA-0.05% (vol/vol) Tween 20 in PBS and stained overnight with primary Abs (1:200 dilution in blocking solution). The following day, coverslips were incubated for 3 h with secondary Abs (1:500 dilution in blocking solution) and mounted in Mowiol 4-88 (Calbiochem) containing 1 mg/ml DAPI (4′,6-diamidino-2-phenylindole). Images were obtained with an Olympus FV1000 confocal microscope using a 360× oil-immersion objective.
Reporter gene assays.
For reporter gene assays, cells were seeded at 1 × 105 cells/ml in 96-well plates and transfected 16 h later, using GeneJuice transfection reagent (Novagen), with 80 ng reporter luciferase, 20 ng pGL3-renilla luciferase, and the indicated amounts of expression vectors and MCV ORFs, with the total amount of DNA adjusted to 230 ng with the empty vector pCMV-HA. Twenty-four hours after transfection, cells were either stimulated with cytokines, infected with virus for 24 h, or directly lysed in passive lysis buffer (Promega) and analyzed for luciferase activity. Firefly luciferase activity was normalized to renilla luciferase activity to control for transfection efficiency. For the Ras-driven Elk1 reporter assay, 1 ng of the pFA-Elk1 expression vector together with the pFR-luciferase reporter plasmid (80 ng) was employed, using a Ras-Ha expression vector for pathway activation.
Affinity purification and LC-MS/MS analysis.
Expression of Flag-tagged ORFs was induced by use of 1 μM CdCl2 in stably transfected HEK293T cells. After 24 h, cells were washed in 1× PBS, pelleted at 3,000 × g for 5 min at 4°C, and snap-frozen in liquid nitrogen. Cell pellets were thawed on ice and resuspended in 1 ml ice-cold TAP lysis buffer (50 mM Tris-HCl, pH 7.5, 4.3% glycerol, 0.2% NP-40, 1.5 mM MgCl2, 100 mM NaCl) supplemented with EDTA-free Complete protease inhibitor cocktail (Roche) and 250 U Benzonase (Sigma-Aldrich). After incubation on ice for 30 min, lysates were centrifuged at 12,000 × g for 5 min at 4°C. Cleared lysates were incubated with 40 μl anti-Flag M2 affinity gel (Sigma-Aldrich) for 1 h on a rotating wheel at 4°C. After incubation, the resin was washed in TAP lysis buffer (with the final 2 washes performed without NP-40) and resuspended in 40 μl 6 M guanidinium-HCl in 100 mM Tris, pH 8.5, supplemented with 10 mM tris(2-carboxyethyl)phosphine (TCEP) and 40 mM chloroacetamide. After 30 min of incubation at room temperature in the dark, lysates were diluted 1:10 with digestion buffer (10% acetonitrile, 25 mM Tris, pH 8.5) and incubated with 0.5 μg EndoLysC (Wako Chemicals) and 0.5 μg sequencing-grade modified trypsin (Promega) overnight on a rotating wheel at room temperature. After digestion, peptides were acidified with trifluoroacetic acid, desalted on reversed-phase C18 StageTips, and eluted before liquid chromatography-tandem mass spectrometry (LC-MS/MS) using buffer B (80% acetonitrile, 0.5% acetic acid). Eluted peptides were analyzed on a nanoflow EASY-nLC system coupled to an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific). Peptide separation was achieved on a C18 reversed-phase column (ReproSil-Pur C18-AQ; 1.9 μm by 200 mm by 0.075 mm; Dr.Maisch), using a 120-min linear gradient from 2 to 60% acetonitrile in 0.1% formic acid. The LTQ-Orbitrap XL MS was operated with a Top10 MS/MS spectrum acquisition method in the linear ion trap mode for each MS full scan. Raw files were processed with MaxQuant (version 1.4.1.4) and searched with the built-in Andromeda search engine against a human protein database (UniprotKB, release 2012_01) concatenated with a decoy of reversed sequences, using a label-free quantification (LFQ) algorithm as described previously (7). Carbamidomethylation was set as a fixed modification, while methionine oxidation and protein N-acetylation were included as variable modifications. The search was performed with an initial mass tolerance of 6 ppm for the precursor ion and 0.5 Da for the fragment ions. Search results were filtered in Perseus (version 1.4.1.8), with a false discovery rate (FDR) of 0.01. Prior to statistical analysis, known contaminants and reverse hits were removed. Proteins identified with at least 2 unique peptides and a minimum of 2 quantitation events in at least one experimental group were considered for analysis. LFQ protein intensity values were log transformed, and missing values were filled in by imputation, with random numbers drawn from a normal distribution. Significant interactors were determined using the two-sample t test with the Welch correction after 250 permutations, with the FDR threshold set to 0.01 and S0 empirically set to 1. Results were plotted using R (release version 2.15.3).
RNA interference.
Cells were seeded at 4 × 106 cells/10-cm dish and transfected with 2.5 nM Elongin B siRNA (AATGAACAAGCCGTGCAGTGA), 20 nM Cullin-5 siRNA (AGATTCCTGGCGTAAAAGCTT), or control siRNA, using Lipofectamine (Invitrogen). The next day, cells were transfected with a second dose of siRNA and simultaneously transfected with the indicated plasmids, and they were harvested the following day.
RESULTS
Identification of MC132 as a novel MCV inhibitor of TNF-α-stimulated NF-κB activation.
In order to identify novel MCV inhibitors of human innate immunity, we analyzed the MCV genome for ORFs predicted to encode small, soluble proteins of unknown function and tested the effects of expression of 31 ORFs on inflammatory signaling. To do this, we stimulated HEK293T cells with TNF-α and measured the activation of an NF-κB-dependent reporter gene in the presence of transfected MCV ORF expression plasmids. This screen revealed MC132 as an inhibitor of TNF-activated NF-κB, in contrast to an unrelated MCV protein, MC014, which had no effect on this pathway (Fig. 1A). The effect of MC132 on NF-κB was not due to toxicity or nonspecific effects on reporter genes, since a constitutively active form of Ras (8), RasVHa, which activates Elk1 (using an Elk-1/GAL4 DNA-binding-domain fusion which monitors upstream activation of Elk-1 through a GAL4 promoter luciferase system), was unaffected by MC132 expression (Fig. 1D). Both MC132 and MC014 were expressed at similar levels (Fig. 1B), and MC132 exhibited cytoplasmic localization, whereas MC014 was localized to the nucleus (Fig. 1C).
FIG 1.

Identification of MC132 as an inhibitor of TNF-α-stimulated NF-κB activation. (A) HEK293T cells were seeded at 2 × 105 cells per ml, transfected with 80 ng NF-κB reporter gene, 40 ng TK renilla reporter gene, and 25 or 50 ng (indicated by wedge) empty vector (control) or pCEP4 plasmids expressing the indicated MCV ORFs, stimulated with 50 ng/ml TNF-α for 6 h, and then harvested and assayed for NF-κB reporter gene activity. Data are percentages of the stimulation activity for control cells and are means ± standard deviations (SD) for triplicate samples from a representative experiment (n = 3). (B) Extracts from the samples used for panel A were probed for expression of Flag-tagged viral proteins. (C) Localization of MC132 and MC014 in HEK293T cells. Cells were transfected with 3 μg pCEP4-Flag vector containing the indicated MCV ORFs. Cells were fixed 24 h later and stained with DAPI (blue) or for MCV protein expression (green). Representative images are shown (n = 4). (D) Elk1 activation by Ras in cells transfected with empty vector (control) or pCEP4 plasmids expressing MCV ORFs was measured by a reporter gene assay. Data are percentages of the stimulation activity for control cells for a representative experiment performed in triplicate (n = 3). (E to G) The same as panel A, except that HEK293T (E), COS-1 (F), or HeLa (G) cells were transfected with 50 ng TRAF2-Flag instead of TNF-α stimulation. (H) Cells stably expressing pMEP4-MC132 were seeded at 2 × 105 cells per ml, cultured with (+) or without (−) 1 μM CdCl2 to induce MC132 expression, and then, 24 h later, stimulated with 50 ng/ml TNF-α for 24 h. IL-8 production was then assayed by ELISA. Data are means ± SD for triplicate samples from a representative experiment (n = 3). *, P < 0.001 compared to the control.
We next examined the effect of MC132 on the activation of NF-κB by expression of TRAF2, a key regulatory component of the TNF receptor signaling pathway, and we also observed potent inhibition of this signal by MC132 (Fig. 1E). Inhibition of TRAF2-stimulated NF-κB by MC132 was independent of the cell type tested, since apart from HEK293T cells, inhibition was also observed in Cos-1 cells (Fig. 1F) and HeLa cells (Fig. 1G). We next established HEK293T cell lines with MC132 expression under the control of a metallothionein promoter, allowing expression to be induced by CdCl2. Using these cells, we demonstrated that NF-κB inhibition by MC132 translated into suppression of secretion of an important NF-κB-dependent cytokine, namely, IL-8 (Fig. 1H). Together, these results establish MC132 as a novel MCV NF-κB inhibitor.
MC132 inhibits NF-κB activation stimulated by IL-1 and multiple PRR viral detection pathways.
Since NF-κB inhibitors in other poxviruses often act downstream, at points of signaling convergence, in order to disable multiple detection pathways (3), we determined the effect of MC132 on NF-κB activation pathways other than the TNF pathway. Thus, we next examined the effect of MC132 on IL-1-activated NF-κB. MC132, but not MC014, potently inhibited activation of NF-κB by IL-1, as well as NF-κB activation by the IL-1 signaling components TRAF6 and MyD88 (Fig. 2A, C, and D), with similar levels of MCV protein expression (Fig. 2B). Since TLR9, like IL-1, also signals in a MyD88-dependent manner, we tested the effect of the MCV ORFs on TLR9 signaling and found that MC132 also inhibited TLR9-dependent NF-κB activation (Fig. 2E), while not affecting a TLR9-stimulated ISRE-dependent reporter gene which monitors IRF activation (Fig. 2F). Similarly, activation of NF-κB by TLR3, but not that of the ISRE reporter, was also inhibited (Fig. 2G and H), as was NF-κB stimulated by the TLR3 adapter protein TRIF (Fig. 2I). Both TLR9 and TLR3 have key roles in responding to poxviruses in vivo (9, 10), and both are characteristically upregulated in MCV-infected lesions, despite a lack of inflammation or viral clearance in such lesions under most circumstances (11). This is consistent with the notion that MC132 may prevent TLR3- and TLR9-driven responses in vivo.
FIG 2.
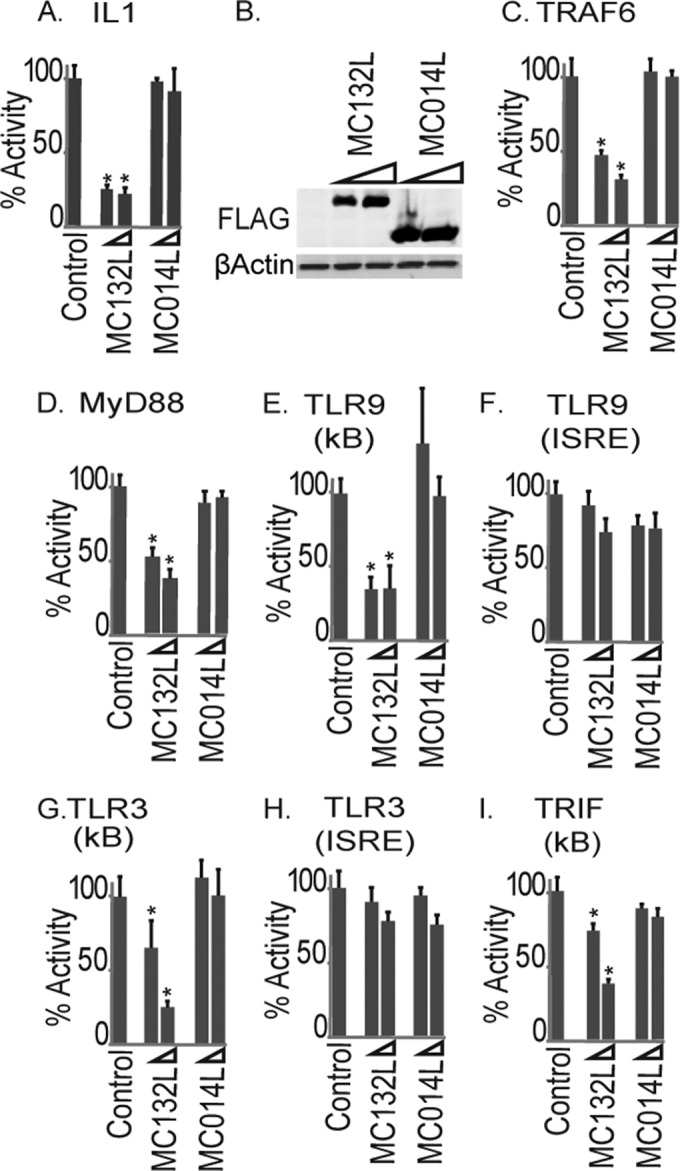
Inhibition of IL-1 and TLR activation of NF-κB by MC132. (A) HEK293T cells were seeded at 2 × 105 cells per ml, transfected with 80 ng NF-κB reporter gene, 40 ng TK renilla reporter gene, and 25 or 50 ng (indicated by wedge) empty vector (control) or pCEP4 plasmids expressing the indicated MCV ORFs, stimulated with 50 ng/ml IL-1β for 6 h, and then harvested and assayed for NF-κB reporter gene activity. Data are percentages of the stimulation activity for control cells and are means ± SD for triplicate samples from a representative experiment (n = 4). (B) Extracts from the samples for panel A were probed for expression of Flag-tagged viral proteins. (C to I) The same as panel A, except that instead of IL-1 stimulation, cells were transfected with 50 ng plasmid expressing TRAF6 (C), MyD88 (D), CD4-TLR9 (E and F), CD4-TLR3 (G and H), or TRIF (I) for 24 h. For panels F and H, cells were transfected with an ISRE-luciferase reporter in place of the NF-κB reporter. *, P < 0.001 compared to the control.
We next examined the effect of MC132 on the activation of innate signaling pathways during live poxvirus infection. As there are currently no experimental in vitro or in vivo models for MCV infection, we utilized a model comprising infection of HEK293T cells with modified vaccinia virus Ankara (MVA), an attenuated VACV strain that activates type I IFN gene induction in cell culture (Fig. 3C). Interestingly, while MC132 potently inhibited MVA-driven NF-κB activation (Fig. 3A), it had no effect on MVA-driven ISRE-luciferase activation (Fig. 3B). The human IFN-β gene promoter requires both NF-κB and IRF transcription factor transactivation during virus infection (12), and consistent with this, MC132 did inhibit induction of the MVA-stimulated IFN-β promoter (Fig. 3C).
FIG 3.
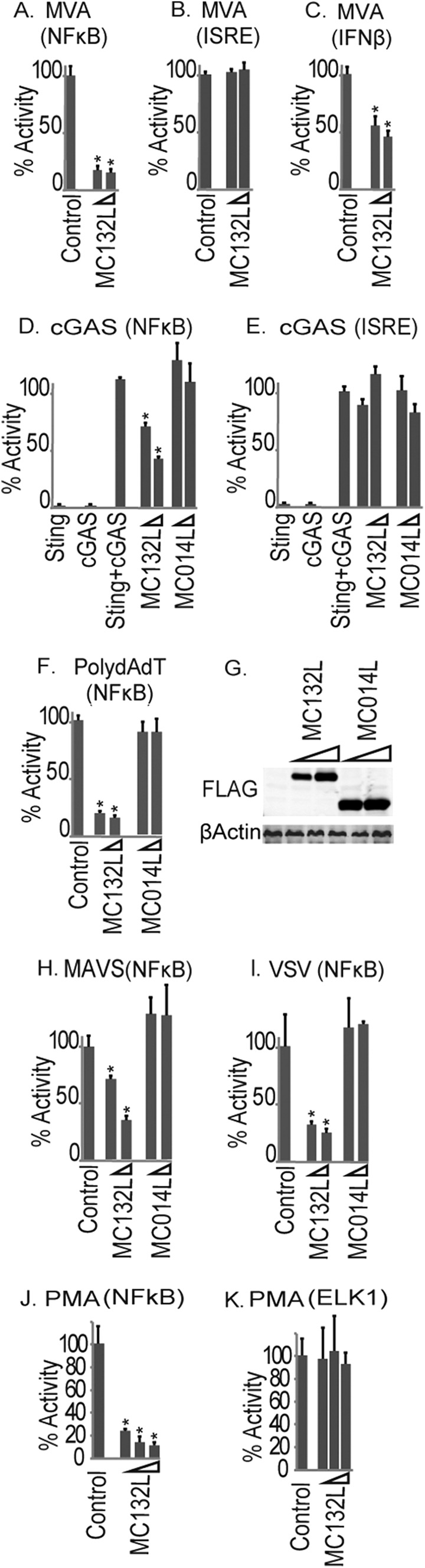
Inhibition of cytosolic DNA-sensing-pathway- and virus-stimulated NF-κB activation by MC132. HEK293T cells were seeded at 2 × 105 cells per ml and transfected with the indicated reporter genes and empty vector (control) or pCEP4 plasmids expressing MC132. Cells were then infected with MVA for 16 h, and NF-κB reporter activity (A), ISRE reporter activity (B), and IFN-β promoter reporter gene activity (C) were measured. Data are percentages of the stimulation activity for control cells and are means ± SD for triplicate samples from a representative experiment (n = 4). (D and E) The same as panel A, except that cells were transfected with cGAS- and STING-expressing plasmids (25 ng each), and 24 h later, NF-κB (D) or ISRE (E) reporter gene activity was measured. (F, H, and I) The same as panel A, except that cells were transfected with 500 ng/ml poly(dA:dT) (F) or 50 ng MAVS-expressing plasmid (H) or infected with VSV for 16 h (I). (G) Extracts from the samples used for panel F were probed for expression of Flag-tagged viral proteins. (J) The same as panel A, except that cells were transfected with 10 ng, 25 ng, and 50 ng of pCEP4-MC132flag and stimulated with 10 nM PMA for 6 h. (K) The same as panel J, except that Elk1 reporter gene activity was measured. *, P < 0.001 compared to the control.
We next examined the effect of MC132 on other pathways known to be involved in primary sensing of poxvirus infection, such as the cGAMP synthase (cGAS) cytosolic DNA sensor, which operates via STING (13, 14). Coexpression of cGAS and STING in HEK293T cells, which reconstitutes cGAS signaling (15), led to NF-κB and IRF activation. Consistent with the MVA data, MC132 potently inhibited cGAS-induced NF-κB activation but not cGAS-induced ISRE induction (Fig. 3D and E). In HEK293T cells, which lack TLR3, TLR9, and cGAS sensing pathways, it is likely that MVA activates NF-κB through the RNA polymerase III (Pol III) response (3). Pol III transcribes AT-rich DNA into an RNA ligand that activates the RIG-I–MAVS sensing system and has been implicated in sensing intracellular poxviral DNA (16). Thus, we stimulated cells expressing similar levels of MCV proteins (Fig. 3G) with poly(dA:dT) to activate Pol III-dependent signaling to NF-κB, and we found that MC132 potently inhibited both RIG-I- and MAVS-driven NF-κB activation (Fig. 3F and H).
To determine if MC132 could act as a general inhibitor of virus-induced NF-κB activation, we examined its effect on RNA virus-activated NF-κB by using vesicular stomatitis virus (VSV), and again, we observed a potent inhibition of this signal by MC132 (Fig. 3I). Since VSV infection is chiefly detected through RIG-I activation in these cells (17), these data are consistent with what was seen for the Pol III response, which operates via RIG-I (Fig. 3F). As an additional confirmation of the specificity of MC132 inhibition of the NF-κB pathway, we examined the effect of MC132 on phorbol myristate acetate (PMA)-stimulated activation of signaling pathways. While MC132 caused a potent, dose-dependent inhibition of PMA-activated NF-κB, it had no effect on PMA-activated Elk1 (Fig. 3J and K).
The data thus far suggested that the novel MCV inhibitor MC132 was targeting NF-κB activation at a common convergence point in the network of signal transduction pathways that would be activated during viral infection, namely, the IKK complex (Fig. 4A). The IKK complex consists of two kinases, IKKα and -β, which are required for NF-κB activation and directly regulate the transcription factor through phosphorylation of both IκΒ, driving its degradation, and the p65 subunit, with phosphorylation at key sites required for transactivation (3). We examined the effect of MC132 on NF-κB activation by IKKs by determining the expression of IKKα and IKKβ. In each case, we observed a potent inhibition of IKK-stimulated NF-κB activation (Fig. 4B and C) with similar levels of MCV protein expression (Fig. 4D), suggesting that MC132 was exerting its effect further downstream.
FIG 4.
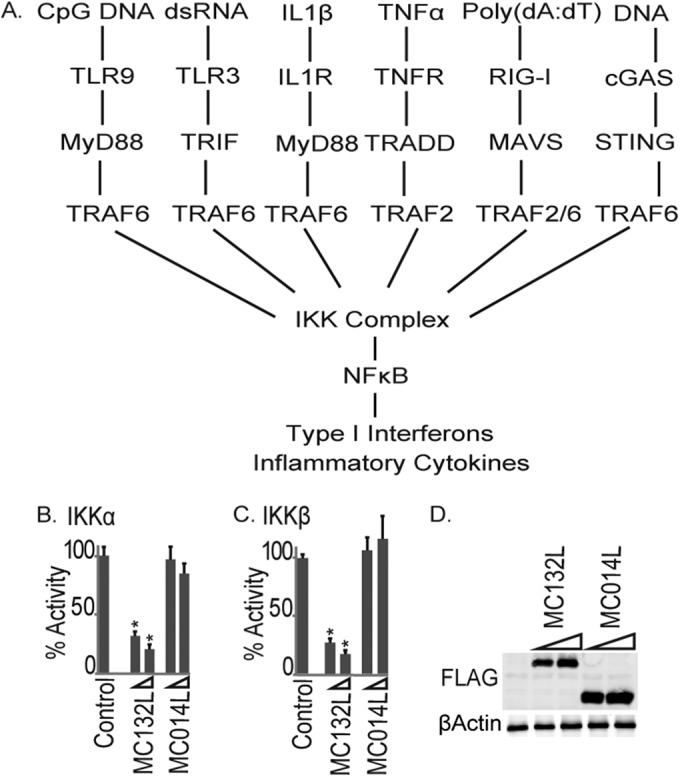
MC132 inhibits NF-κB activation by IKKs, a common convergence point in the network of signal transduction pathways activated during viral infection. (A) Schematic showing multiple signal transduction pathways to NF-κB expected to be activated during a poxviral infection, all of which were shown to be sensitive to MC132 inhibition (Fig. 1 to 3). (B and C) HEK293T cells were seeded at 2 × 105 cells per ml, transfected with 25 or 50 ng (indicated by wedge) empty vector (control) or pCEP4 plasmids expressing the indicated MCV ORFs, together with 50 ng IKKα (B) or IKKβ (C), and then harvested and assayed for NF-κB reporter gene activity 24 h later. Data are percentages of the stimulation activity for control cells and are means ± SD for triplicate samples from a representative experiment (n = 3). *, P < 0.001 compared to the control. (D) Extracts from the samples used for panel C were probed for expression of Flag-tagged viral proteins.
MC132 targets the NF-κB subunit p65.
We next investigated the precise mechanism by which MC132 inhibits NF-κB downstream of IKK activation by examining direct NF-κB activation, as measured by detection of phosphorylated p65. To do this, we stimulated the MC132-expressing CdCl2-induced stable cell line described earlier (Fig. 1H) with TNF. This showed a profound inhibition of p65 phosphorylation in MC132-expressing cells compared to pMEP4 empty vector control cells (Fig. 5A, top panel). Inhibition of p65 phosphorylation in the presence of MC132 expression was associated with a notable decrease in the protein expression levels of p65 (Fig. 5A, second panel). Thus, we next investigated whether MC132-stimulated decreased levels of p65 protein expression might be the primary mechanism through which MC132 exerts its effect on NF-κB. Using the MC132-expressing cells, different concentrations of CdCl2 allowed titrated expression of the MC132 protein, and this demonstrated a direct correlation between increased expression of MC132 and reduced expression of p65 compared to that in control cells stably carrying the empty vector (Fig. 5B). Consistent with this, confocal microscopy showed that cells transiently transfected with the plasmid expressing MC132 displayed decreased staining of p65 compared to nontransfected bystander cells (Fig. 5C). In order to determine whether an MC132-mediated reduction of p65 expression explained the observed inhibition of NF-κB activity, the ability of ectopic p65 expression to overcome MC132 inhibition of NF-κB was examined. While MC132 was still able to inhibit p65-induced NF-κB activation when p65 was expressed at low levels, this inhibition was overcome when more of the plasmid encoding p65 was transfected into cells (Fig. 5D).
FIG 5.
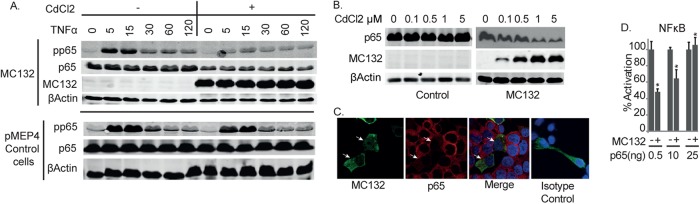
MC132 expression causes a depletion of p65 protein expression. (A) Effect of inducible MC132 expression on TNF-α-stimulated NF-κB activation. HEK293T cells stably transfected with pMEP4 and pMEP4-MC132 were seeded at 6 × 105 cells per well in 6-well dishes and treated with (+) or without (−) 1 μM CdCl2 to induce MCV protein expression. Twenty-four hours later, cells were stimulated with 50 ng/ml TNF-α for the indicated times, and cell lysates were immunoblotted with the indicated antibodies. Representative blots are shown (n = 3). (B) HEK293T cells stably containing pMEP4-MC132 or control cells containing pMEP4 were seeded at 6 × 105 cells per well in 6-well dishes for 24 h with the indicated concentrations of CdCl2 to induce MC132 expression, and cell lysates were immunoblotted for p65 and MC132. Representative blots are shown (n = 3). (C) HEK293T cells were seeded at 6 × 105 cells per well in 6-well dishes, transfected with 3 μg pCEP4-MC132, fixed 24 h later, and stained with DAPI (blue), for MC132-Flag (green) and endogenous p65 (red), and with an anti-rabbit isotype control. Representative images are shown (n = 3). MC132-positive cells are indicated with white arrows. (D) HEK293T cells were seeded at 2 × 105 cells per ml and transfected with the indicated amounts of p65 expression vector, with or without 50 ng of pCEP4-MC132. Cells were harvested 24 h later and assayed for NF-κB reporter gene activity. Data are presented as percentages of reporter activation by increasing amounts of p65 and display the effect of 50 ng of MC132 on this activation, with data given as means ± SD for triplicate samples from a representative experiment (n = 3). *, P < 0.001 compared to the control.
Further analysis of the relationship between MC132 and p65 showed that the proteins strongly coimmunoprecipitated (Fig. 6A), and construction of a series of truncation mutants of p65 demonstrated that MC132 interacted only with any p65 truncation containing the N-terminal half of the DNA-binding Rel homology domain (RHD), indicating that MC132 interacts with p65 through this region (Fig. 6B). To determine the region of MC132 responsible for binding p65, we also constructed a series of truncation mutants of MC132, gradually removing distinct portions from the N and C termini of the protein and testing the expression of the mutants (Fig. 6C, upper panel). Even minimal truncations of MC132, where as few as 6 amino acids at the N terminus or 18 amino acids at the C terminus were absent, abrogated its ability to bind p65 (Fig. 6C, lower panels). Importantly, there was a direct correlation between the lack of binding of MC132 truncation mutants to p65 (Fig. 6C, lower panels) and a lack of inhibition of NF-κB (Fig. 6D). These data suggest that the entire MC132 protein is required to inhibit p65 function through direct binding to the p65 RHD, thereafter somehow stimulating p65 degradation.
FIG 6.
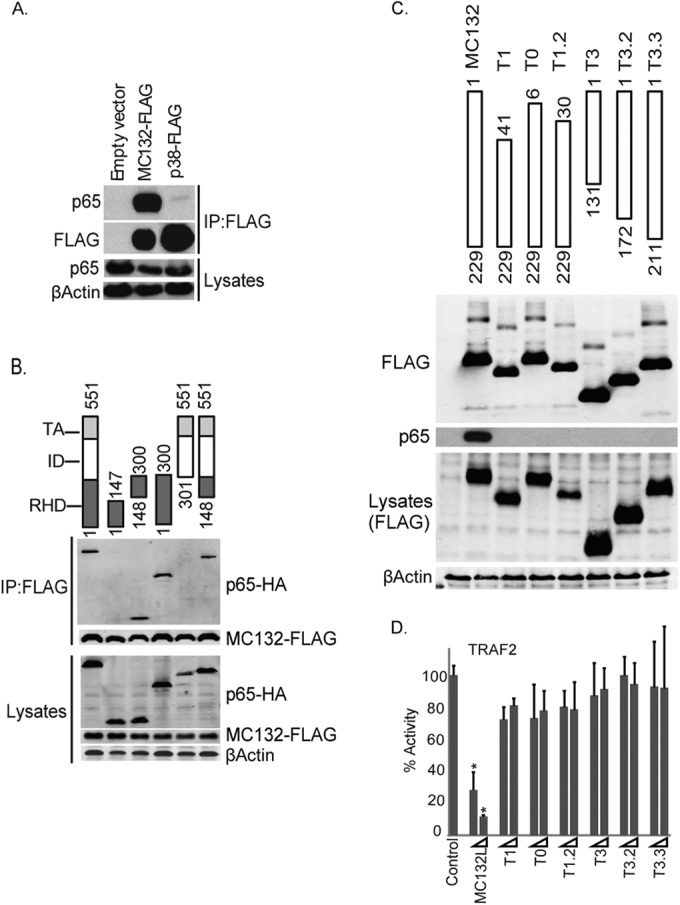
MC132 interacts with p65 via the RHD of p65. (A) MC132 and p65 interact. HEK293T cells were seeded at 4 × 106 cells per 10-cm plate and transfected with 8 μg pCEP4-MC132 or a plasmid expressing control p38-Flag, and 24 h later, the cells were lysed and immunoprecipitated (IP) with anti-Flag beads. Immunoblots were probed with the indicated antibodies. Representative blots are shown (n = 4). (B) MC132 interacts with the p65 RHD. The schematic shows full-length p65(1–551) and the different truncations tested. HEK293T cells were seeded at 4 × 106 cells per 10-cm plate and transfected with 4 μg pCEP4-MC132 together with 4 μg vector encoding full-length p65-HA or the indicated p65 truncation mutants. Cells were lysed the following day, immunoprecipitated with anti-Flag beads, and probed for Flag-tagged MC132 or p65-HA. Representative blots are shown (n = 4). (C) Truncation of MC132 abolishes interaction with p65 and inhibition of NF-κB. HEK293T cells were seeded at 4 × 106 cells per ml and transfected the following day with 8 μg pCEP4-MC132 or the MC132 truncation mutants indicated in the schematic. Cells were lysed the following day, immunoprecipitated with anti-Flag beads, and probed along with lysate controls for Flag-tagged proteins and endogenous p65. Representative blots are shown (n = 4). (D) The effect of MC132 truncations on TRAF2-stimulated NF-κB activation was measured by a reporter gene assay as described in the legend to Fig. 1. Data are percentages of the stimulation activity for control cells and are means ± SD for triplicate samples from a representative experiment (n = 4). *, P < 0.001 compared to the control.
MC132 recruits the Elongin B/Elongin C/Cullin-5 ubiquitin ligase complex to NF-κB p65 to mediate p65 degradation.
In order to reveal the mechanism whereby MC132 can mediate p65 degradation, we explored which cellular proteins, apart from p65, MC132 might interact with by performing unbiased affinity purification combined with mass spectrometry (AP-MS), using the MC132-expressing inducible stable cell line (Fig. 5A and B). Analysis of MC132 interaction partners by AP-MS showed that four proteins were significantly enriched in the MC132 immunoprecipitates: TCEB2 (Elongin C), TCEB2 (Elongin B), CUL5 (Cullin-5), and PP1A (peptidyl-prolyl cis-trans isomerase A) (Fig. 7A). Peptides corresponding to p65 were detected in MC132 immunoprecipitates but not significantly enriched (data not shown), likely due to the stimulated degradation of p65 in the MC132-induced line. Note that Cullin-5 and Elongin B/C have previously been demonstrated to facilitate ubiquitin-mediated p65 degradation by gammaherpesvirus proteins with no sequence similarity to MC132, i.e., latent nuclear protein-1 (LANA-1) in Kaposi's sarcoma-associated herpesvirus (KSHV) (18) and the ORF73 protein in murid herpesvirus 4 (MuHV-4) (19). Thus, we examined the ability of MC132 to recruit p65 to the Elongin B/Elongin C/Cullin-5 E3 ligase complex to effect p65 degradation. Coimmunoprecipitation of ectopically expressed Elongin B showed that MC132 could associate with Elongin B in the presence or absence of ectopically expressed Elongin C (Fig. 7B, lanes 6 and 8). Note that p65 associated with Elongin B only when MC132 was present, not when it was absent (Fig. 7B, compare lane 9 to lane 10), suggesting that MC132 recruits p65 to the Elongin complex. Thus, we assayed the ability of MC132 to cause ubiquitination of endogenous p65 (in the presence of the proteasome inhibitor MG132 to prevent degradation of ubiquitin-tagged p65). Interestingly, p65 was ubiquitinated in the presence of full-length MC132 but not in the presence of the T3 MC132 truncation (Fig. 7C), which we previously observed to be defective in both p65 binding and NF-κB inhibition (Fig. 6C and D). This suggested that MC132 recruitment of p65 to the Elongin B/C complex might target p65 for ubiquitination and subsequent degradation via Cullin-5 E3 ligase activity. To test this, we employed RNA interference to target Elongin B. When Elongin B expression was reduced by the use of siRNA, as determined by real-time PCR (Fig. 7D), the ability of MC132 to cause p65 ubiquitination was significantly compromised (Fig. 7E, compare lane 6 to lane 3). Additionally, siRNA knockdown of Elongin B also marginally but reproducibly enhanced TRAF6-driven NF-κB activity (Fig. 7F, compare bar 3 and bar 1) and completely reversed the inhibition caused by MC132 (compare bar 4 and bar 6). We also depleted Cullin-5 by RNA interference (Fig. 7G) and observed that the ability of MC132 to cause p65 ubiquitination was partially compromised (Fig. 7H, compare lane 4 to lane 2), suggesting that Cullin-5 may play a less essential role in MC132-driven p65 degradation than that of Elongin B, possibly due to redundancy between Cullin family members. Overall, the data suggest that MCV utilizes MC132 to capture p65 for ubiquitin-mediated degradation, leading to effective suppression of NF-κB activity (Fig. 8).
FIG 7.

MC132 recruits p65 to a Cullin-5/Elongin B/Elongin C complex to target p65 for ubiquitin-mediated degradation. (A) Volcano plot of MC132-interacting proteins identified from MC132-expressing HEK293T cells by AP-MS. (B) MC132 targets p65 to the Elongin B/C complex. HEK293T cells were seeded at 4 × 106 cells per 10-cm plate and transfected with 2 μg expression vector(s) for MC132-HA, Elongin B-Flag, and/or Elongin c-Myc, as indicated. Twenty-four hours later, cells were lysed, immunoprecipitated with anti-Flag beads, and then immunoblotted with the indicated antibodies. Representative blots are shown (n = 3). (C) MC132 induces polyubiquitination of p65. HEK293T cells were seeded at 4 × 106 cells per 10-cm plate, transfected with 4 μg expression vector(s) for ubiquitin-HA, MC132-Flag, and/or T3-Flag, as indicated, and treated with 20 μM MG132 for 4 h prior to harvest. Twenty-four hours later, cells were lysed, immunoprecipitated with anti-Flag beads, and then immunoblotted with the indicated antibodies. Ubi-p65, polyubiquitinated p65. A representative blot is shown (n = 3). (D) HEK293T cells were seeded at 2 × 105 cells per ml in 24-well dishes and transfected with 2.5 nM Elongin B siRNA at 24 and 48 h, and then cDNAs from cells were assayed for levels of Elongin B normalized to β-actin by real-time PCR. (E) HEK293T cells were seeded at 4 × 106 cells per 10-cm plate, transfected with 2.5 nM Elongin B or control siRNA for 24 h and again at 48 h, and transfected with the indicated constructs prior to MG132 treatment for 4 h and harvest. Total p65 was immunoprecipitated, and immunoblots were probed with the indicated antibodies. (F) The effect of Elongin B siRNA on TRAF6-stimulated NF-κB activation and MC132 inhibition was measured by a reporter gene assay as described in the legend to Fig. 1. Data are percentages of the stimulation activity for control cells and are means ± SD for triplicate samples from a representative experiment (n = 4). *, P < 0.001 compared to the control. (G) HEK293T cells were seeded and treated as described for panel E, but they were transfected with 25 μM Cullin-5 or control siRNA for 24 h and then immunoblotted for Cullin-5 expression. (H) Cullin-5 siRNA reduces MC132-stimulated p65 ubiquitination. Cells were treated as described for panel C, except that Cullin-5 or control siRNA was transfected prior to MG132 treatment and harvest. Representative blots are shown (n = 4).
FIG 8.
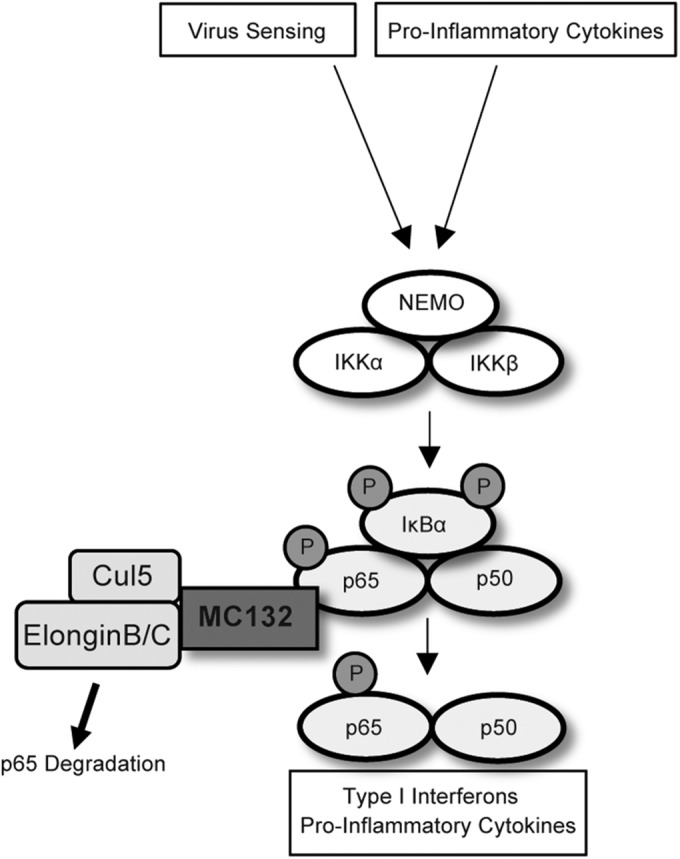
Model for MC132-mediated inhibition of human NF-κB signaling. Innate virus-sensing and cytokine signaling pathways converge on the IKK complex, leading to IKK-dependent phosphorylation of p65 and IκBα. MC132 recruits a Cullin-5/Elongin B/Elongin C complex to p65, causing ubiquitin-mediated degradation of p65 and subsequent suppression of NF-κB activity.
DISCUSSION
MCV is a common, dermatotropic poxvirus that causes benign skin neoplasms in humans, with a more serious presentation in immunocompromised patients (6). Compared to VACV, which causes local inflammation in human skin lesions (20), MCV displays an apparent ability to silence the human antiviral response, suggesting that it is better equipped than VACV to suppress human innate immunity as a result of long-term coevolution and adaptation to human infection. NF-κB has a critical role in initiating both virus-induced inflammation and antiviral immunity, and notwithstanding the ability of VACV to trigger local inflammation in skin lesions, VACV possesses numerous immunomodulatory proteins, including at least 10 inhibitors of NF-κB, with evidence of others yet to be identified (21). In contrast, prior to this study, fewer than five MCV immunomodulators had been reported, and only two of these affect NF-κB activation, through partially characterized mechanisms (22–24). Although MCV research has been hampered by the lack of an animal or cell line infection model, analysis of the functions of isolated ORFs expressed in cell lines previously revealed important insights into how MCV proteins suppress host immunity (6).
Since the study of poxvirus immunoregulators that inhibit and fine-tune the host response has been highly informative in defining host-pathogen interactions and revealing new aspects of innate immunity (3), MCV is unparalleled as a model poxvirus worthy of further analysis, since it coevolved to specifically antagonize the human immune system. As such, elucidating how MCV inhibitors of immune responses function will be especially relevant to understanding viral pathogenesis in humans and may also reveal new insights into the human immune signaling mechanisms which the inhibitors have evolved to inhibit. Thus, we screened isolated MCV ORFs for effects on known antiviral innate immune signaling pathways in human cells, and we report the discovery of MC132 as an inhibitor of NF-κB. The gene for MC132 is located on the right terminus of the MCV genome, and MC132 exhibits no similarity to other proteins, aside from a portion of the N terminus (residues 6 to 40) that is similar to mammalian IRF5 (51% identity within residues 237 to 271 of Sus scrofa IRF5 isoform X1; 60% identity with conservative substitutions). Given the specificity of MC132 inhibition of the NF-κB signaling pathway, the significance of this homology is currently unclear.
Research on poxviruses, such as VACV and MVA, has shown that the TLR and cytosolic nucleic acid detection PRRs are involved in initial viral sensing and type I IFN induction, while IL-1 and TNF production and subsequent signaling regulate virus-induced inflammation (3). Compellingly, MC132 limited both PRR and inflammatory cytokine signaling to NF-κB activation, as well as suppressing NF-κB activation following poxviral or RNA virus infection.
We characterized in detail how MC132 exerts its effect on NF-κB. This showed that MC132 bound p65 via the RHD and recruited it to the host Elongin B/Elongin C/Cullin-5 E3 ligase complex to cause p65 ubiquitination and subsequent degradation, since in the absence of MC132, p65 and the Elongin B/Elongin C/Cullin-5 complex did not interact (Fig. 7B). This is reminiscent of the mechanism employed by the KSHV protein LANA-1 and the related MuHV-4 ORF73 protein to suppress NF-κB activation (18, 19), although how LANA-1 binds to p65 is not yet clear. Since small deletions in the sequence of MC132 abolish its activity, we were unable to determine precisely where p65 and the Elongin B/Elongin C/Cullin-5 complex bind within its sequence or whether this occurs through distinct domains. Interestingly, although we found that MC132 possesses a partial consensus sequence for the Cullin-5 binding box and one complete consensus for Elongin B/C binding (25), extensive targeted deletion of these regions had no effect on the ability of MC132 to drive ubiquitin-mediated degradation of p65 (data not shown), suggesting that recruitment of this complex by MC132 occurs in a manner different from that previously described for other bridging proteins. Although depletion of Elongin B had a profound effect on p65 ubiquitination, depletion of Cullin-5 had only a mild effect, suggesting that this protein may be less essential for MC132 inhibition. Since the homologous Cullin-2 protein has also been shown to similarly utilize the Elongin B/C adapter complex to degrade target proteins (26) and is highly expressed in HEK293 cells (27), we suspect that this family member may compensate for the loss of Cullin-5 in this process (28).
Interestingly, we observed that depletion of Elongin B was also associated with a slight increase in TRAF6-driven NF-κB activation (Fig. 7F), suggesting that MC132 may coopt an endogenous Elongin B-dependent process that negatively regulates NF-κB activity, although further research would be necessary to confirm this hypothesis. Given the key role of the RHD in the activity of p65, binding of MC132 to this region may also acutely inhibit p65 function prior to degradation, which would explain the potent inhibition of p65 phosphorylation even in the absence of a proportional level of degradation (Fig. 5A).
Host cell ubiquitin-proteasome systems are commonly exploited by viruses, particularly members of the Poxviridae family (29). However, this mechanism of NF-κB inhibition through p65 depletion is a unique finding in poxvirus biology, and as it does not appear to require previously identified consensus sequences for binding of the complex, further analysis of MC132 may reveal novel insights into the biology of Elongin B/Elongin C/Cullin-5 complexes and their regulation of immune processes. Furthermore, the presence of a convergently evolved viral mechanism to target human p65 for degradation, involving the same E3 ligase complex by both poxviruses and herpesviruses, suggests that this mechanism is an effective means of immunosuppression that deserves further characterization to assess possible avenues for its use in therapeutic intervention.
Overall, our analysis of the MCV genome for determination of ORFs that affect human signaling networks culminating in the activation of NF-κB revealed MC132, an inhibitor of PRR- and cytokine-activated NF-κB that is unique to MCV among poxviruses, and delineated the detailed mechanism of its action. Further screening of the MCV genome for novel inhibitors of human innate immunity will likely reveal additional inhibitors of innate immune signaling and novel details of the signaling pathways they antagonize, and it may present new strategies for selective inhibition of sensing and inflammatory events in disease.
ACKNOWLEDGMENTS
This work was supported by Science Foundation Ireland grant 11/PI/1056 (to A.G.B.), the Max-Planck Free Floater program (to A.P.), the ERC (StG 311339-iViP, to A.P.), and Marie Curie Intra-European Fellowship no. 332057 (to G.B. and A.G.B). P.J.F. was supported by the Imperial College Trust.
REFERENCES
- 1.Gurtler C, Bowie AG. 2013. Innate immune detection of microbial nucleic acids. Trends Microbiol 21:413–420. doi: 10.1016/j.tim.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bowie AG, Unterholzner L. 2008. Viral evasion and subversion of pattern-recognition receptor signalling. Nat Rev Immunol 8:911–922. doi: 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brady G, Bowie AG. 2014. Innate immune activation of NFkappaB and its antagonism by poxviruses. Cytokine Growth Factor Rev 25:611–620. doi: 10.1016/j.cytogfr.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 4.Senkevich TG, Koonin EV, Bugert JJ, Darai G, Moss B. 1997. The genome of molluscum contagiosum virus: analysis and comparison with other poxviruses. Virology 233:19–42. doi: 10.1006/viro.1997.8607. [DOI] [PubMed] [Google Scholar]
- 5.Struzik J, Szulc-Dabrowska L, Niemialtowski M. 2014. Modulation of NF-κB transcription factor activation by Molluscum contagiosum virus proteins. Postepy Hig Med Dosw 68:129–136. doi: 10.5604/17322693.1088053. [DOI] [PubMed] [Google Scholar]
- 6.Chen X, Anstey AV, Bugert JJ. 2013. Molluscum contagiosum virus infection. Lancet Infect Dis 13:877–888. doi: 10.1016/S1473-3099(13)70109-9. [DOI] [PubMed] [Google Scholar]
- 7.Hubner NC, Bird AW, Cox J, Splettstoesser B, Bandilla P, Poser I, Hyman A, Mann M. 2010. Quantitative proteomics combined with BAC TransgeneOmics reveals in vivo protein interactions. J Cell Biol 189:739–754. doi: 10.1083/jcb.200911091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McDermott EP, O'Neill LA. 2002. Ras participates in the activation of p38 MAPK by interleukin-1 by associating with IRAK, IRAK2, TRAF6, and TAK-1. J Biol Chem 277:7808–7815. doi: 10.1074/jbc.M108133200. [DOI] [PubMed] [Google Scholar]
- 9.Hutchens M, Luker KE, Sottile P, Sonstein J, Lukacs NW, Nunez G, Curtis JL, Luker GD. 2008. TLR3 increases disease morbidity and mortality from vaccinia infection. J Immunol 180:483–491. doi: 10.4049/jimmunol.180.1.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Samuelsson C, Hausmann J, Lauterbach H, Schmidt M, Akira S, Wagner H, Chaplin P, Suter M, O'Keeffe M, Hochrein H. 2008. Survival of lethal poxvirus infection in mice depends on TLR9, and therapeutic vaccination provides protection. J Clin Invest 118:1776–1784. doi: 10.1172/JCI33940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ku JK, Kwon HJ, Kim MY, Kang H, Song PI, Armstrong CA, Ansel JC, Kim HO, Park YM. 2008. Expression of Toll-like receptors in verruca and molluscum contagiosum. J Korean Med Sci 23:307–314. doi: 10.3346/jkms.2008.23.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nourbakhsh M, Hoffmann K, Hauser H. 1993. Interferon-beta promoters contain a DNA element that acts as a position-independent silencer on the NF-kappa B site. EMBO J 12:451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dai P, Wang W, Cao H, Avogadri F, Dai L, Drexler I, Joyce JA, Li XD, Chen Z, Merghoub T, Shuman S, Deng L. 2014. Modified vaccinia virus Ankara triggers type I IFN production in murine conventional dendritic cells via a cGAS/STING-mediated cytosolic DNA-sensing pathway. PLoS Pathog 10:e1003989. doi: 10.1371/journal.ppat.1003989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, Hornung V. 2013. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 503:530–534. doi: 10.1038/nature12640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valentine R, Smith GL. 2010. Inhibition of the RNA polymerase III-mediated dsDNA-sensing pathway of innate immunity by vaccinia virus protein E3. J Gen Virol 91:2221–2229. doi: 10.1099/vir.0.021998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 18.Li X, Liang D, Lin X, Robertson ES, Lan K. 2011. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen reduces interleukin-8 expression in endothelial cells and impairs neutrophil chemotaxis by degrading nuclear p65. J Virol 85:8606–8615. doi: 10.1128/JVI.00733-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodrigues L, Filipe J, Seldon MP, Fonseca L, Anrather J, Soares MP, Simas JP. 2009. Termination of NF-kappaB activity through a gammaherpesvirus protein that assembles an EC5S ubiquitin-ligase. EMBO J 28:1283–1295. doi: 10.1038/emboj.2009.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wlodaver CG, Palumbo GJ, Waner JL. 2004. Laboratory-acquired vaccinia infection. J Clin Virol 29:167–170. doi: 10.1016/S1386-6532(03)00118-5. [DOI] [PubMed] [Google Scholar]
- 21.Sumner RP, Maluquer de Motes C, Veyer DL, Smith GL. 2014. Vaccinia virus inhibits NF-kappaB-dependent gene expression downstream of p65 translocation. J Virol 88:3092–3102. doi: 10.1128/JVI.02627-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Randall CM, Jokela JA, Shisler JL. 2012. The MC159 protein from the molluscum contagiosum poxvirus inhibits NF-kappaB activation by interacting with the IkappaB kinase complex. J Immunol 188:2371–2379. doi: 10.4049/jimmunol.1100136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nichols DB, Shisler JL. 2006. The MC160 protein expressed by the dermatotropic poxvirus molluscum contagiosum virus prevents tumor necrosis factor alpha-induced NF-kappaB activation via inhibition of I kappa kinase complex formation. J Virol 80:578–586. doi: 10.1128/JVI.80.2.578-586.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murao LE, Shisler JL. 2005. The MCV MC159 protein inhibits late, but not early, events of TNF-alpha-induced NF-kappaB activation. Virology 340:255–264. doi: 10.1016/j.virol.2005.06.036. [DOI] [PubMed] [Google Scholar]
- 25.Okumura F, Matsuzaki M, Nakatsukasa K, Kamura T. 2012. The role of Elongin BC-containing ubiquitin ligases. Front Oncol 2:10. doi: 10.3389/fonc.2012.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sarikas A, Hartmann T, Pan ZQ. 2011. The cullin protein family. Genome Biol 12:220. doi: 10.1186/gb-2011-12-4-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liakopoulos D, Busgen T, Brychzy A, Jentsch S, Pause A. 1999. Conjugation of the ubiquitin-like protein NEDD8 to cullin-2 is linked to von Hippel-Lindau tumor suppressor function. Proc Natl Acad Sci U S A 96:5510–5515. doi: 10.1073/pnas.96.10.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mahrour N, Redwine WB, Florens L, Swanson SK, Martin-Brown S, Bradford WD, Staehling-Hampton K, Washburn MP, Conaway RC, Conaway JW. 2008. Characterization of Cullin-box sequences that direct recruitment of Cul2-Rbx1 and Cul5-Rbx2 modules to Elongin BC-based ubiquitin ligases. J Biol Chem 283:8005–8013. doi: 10.1074/jbc.M706987200. [DOI] [PubMed] [Google Scholar]
- 29.Barry M, van Buuren N, Burles K, Mottet K, Wang Q, Teale A. 2010. Poxvirus exploitation of the ubiquitin-proteasome system. Viruses 2:2356–2380. doi: 10.3390/v2102356. [DOI] [PMC free article] [PubMed] [Google Scholar]