

ABSTRACT
The E2 protein of classical swine fever virus (CSFV) is an envelope glycoprotein that is involved in virus attachment and entry. To date, the E2-interacting cellular proteins and their involvement in viral replication have been poorly documented. In this study, thioredoxin 2 (Trx2) was identified to be a novel E2-interacting partner using yeast two-hybrid screening from a porcine macrophage cDNA library. Trx2 is a mitochondrion-associated protein that participates in diverse cellular events. The Trx2-E2 interaction was further confirmed by glutathione S-transferase (GST) pulldown, in situ proximity ligation, and laser confocal assays. The thioredoxin domain of Trx2 and the asparagine at position 37 (N37) in the E2 protein were shown to be critical for the interaction. Silencing of the Trx2 expression in PK-15 cells by small interfering RNAs significantly promotes CSFV replication, and conversely, overexpression of Trx2 markedly inhibits viral replication of the wild-type (wt) CSFV and to a greater extent that of the CSFV N37D mutant, which is defective in binding Trx2. The wt CSFV but not the CSFV N37D mutant was shown to reduce the Trx2 protein expression in PK-15 cells. Furthermore, we demonstrated that Trx2 increases nuclear factor kappa B (NF-κB) promoter activity by promoting the nuclear translocation of the p65 subunit of NF-κB. Notably, activation of the NF-κB signaling pathway induced by tumor necrosis factor alpha (TNF-α) significantly inhibits CSFV replication in PK-15 cells, whereas blocking the NF-κB activation in Trx2-overexpressing cells no longer suppresses CSFV replication. Taken together, our findings reveal that Trx2 inhibits CSFV replication via the NF-κB signaling pathway.
IMPORTANCE Thioredoxin 2 (Trx2) is a mitochondrion-associated protein that participates in diverse cellular events, such as antioxidative and antiapoptotic processes and the modulation of transcription factors. However, little is known about the involvement of Trx2 in viral replication. Here, we investigated, for the first time, the role of Trx2 in the replication of classical swine fever virus (CSFV), a devastating pestivirus of pigs. By knockdown and overexpression, we showed that Trx2 negatively regulates CSFV replication. Notably, we demonstrated that Trx2 inhibits CSFV replication by promoting the nuclear translocation of the p65 subunit of NF-κB, a key regulator of the host's innate immunity and inflammatory response. Our findings reveal a novel role of Trx2 in the host's antiviral response and provide new insights into the complex mechanisms by which CSFV interacts with the host cell.
INTRODUCTION
Classical swine fever (CSF), caused by classical swine fever virus (CSFV), is a highly contagious, often fatal porcine disease with significant economic losses. CSFV belongs to the Pestivirus genus within the Flaviviridae family (1). It is an enveloped virus with a single-stranded, positive-sense RNA genome of approximately 12.3 kb in length. The RNA genome contains a single large open reading frame (ORF). This ORF is translated into a polyprotein, which is further processed into 12 mature proteins (Npro-C-Erns-E1-E2-p7-NS2-3-NS4A-NS4B-NS5A-NS5B) by viral and cellular proteases (2). The C, Erns, E1, and E2 proteins represent the structural components of the virion. The E1 and E2 glycoproteins are anchored to the envelope by their carboxyl termini, while Erns is loosely associated with the envelope. Erns presents as homodimers linked by disulfide bridges in virus-infected cells and in virions (3, 4). The C terminus of E2 functions as a membrane-spanning domain anchoring the E2-E1 or E2-E2 dimer to the viral envelope (5). E2 is also involved in virus attachment to and entry into the target cell (6). Furthermore, as a virulence determinant in pigs (7), the E2 protein can efficiently induce protective immune responses (8–13). Additionally, we recently showed that host β-actin interacts with E2 and modulates the early life cycle of CSFV (14).
Thioredoxins (Trxs) are a class of ubiquitously expressed redox proteins that contain a conserved consensus amino acid sequence (Cys-Gly-Pro-Cys) in the catalytic center. There are two distinct forms of Trxs (Trx1 and Trx2), which act as antioxidants facilitating the reduction of other proteins by cysteine thiol-disulfide exchange. Trx2 consists of 157 amino acid (aa) residues, and its thioredoxin domain is located in the C terminus of the protein. Trxs are involved in diverse biological processes, such as cell growth, proliferation, apoptosis, and gene regulation (15–18). Additionally, they interact with several transcription factors and modulate their activities, including nuclear factor kappa B (NF-κB) and glucocorticoid receptor (GR) (19).
To date, most studies have been focused on the roles of the glycoprotein E2 in virus attachment, entry, and virulence, but knowledge of the CSFV E2-interacting host proteins and their impacts on the outcome of CSFV replication is limited. In this study, we identified the cellular protein Trx2 to be a novel E2-interacting partner that negatively modulates CSFV replication.
MATERIALS AND METHODS
Cells, viruses, and virus titer assay.
PK-15 (porcine kidney cells) and HEK293T cells (human embryonic kidney cells) were cultured in Dulbecco's minimal essential medium (DMEM) (Gibco) with 10% fetal bovine serum (FBS) (HyClone). The CSFV Shimen strain was propagated in PK-15 cells and titrated according to the Reed-Muench formula (20). The CSFV C-strain is a lapinized live attenuated vaccine that was developed by hundreds of passages of a highly virulent CSFV strain in rabbits (21).
Construction of expression vectors.
The pEGFP-p65 plasmid expressing the enhanced green fluorescent protein (EGFP)-tagged p65 of NF-κB was kindly provided by Shaobo Xiao (22). The E2 gene of the CSFV Shimen strain or C-strain with a Flag tag in the C terminus was amplified by PCR and ligated into the pCAGGS vector (Addgene) to generate pCAGGS-SME2-Flag or pCAGGS-CE2-Flag, respectively. The porcine Trx2 gene was amplified by PCR and cloned into pCMV-Myc (Clontech), pDsRed2-N1 (Clontech), pGEX-6p-1 (GE Healthcare), or pFUGW (Addgene) to generate pMyc-Trx2 (expressing Myc-tagged Trx2), pDsRed-Trx2 (expressing red fluorescent protein [RFP]-tagged Trx2), pGEX-Trx2 (expressing glutathione S-transferase [GST]-tagged Trx2), or pFUGW-Trx2 (for packaging Trx2-expressing lentivirus), respectively. To construct the plasmids expressing chimeric E2 mutants, various regions of E2 from the Shimen strain and C-strain were amplified by overlapping PCR and cloned into pCAGGS. The primers for amplifying the E2 and Trx2 genes are listed in Table 1.
TABLE 1.
Primers used in this study
| Primer | Sequence (5′→3′) | Purpose |
|---|---|---|
| Flag-SME2-F | CGGAATTCGCCACCATGGTATTAAGAGGACAGATCGTGC | Amplification of SME2 |
| Flag-CE2-F | CGGAATTCGCCACCATGGTATTAAGGGGACAGATCGTGC | Amplification of CE2 |
| Flag-(SM/C)E2-R | CATCTCGAGCTAAACAAATTCTGCGAAGTAATC | Amplification of SM/CE2 |
| E2(N37D mutant)-1F | GCCCCTCTAGAGTTTTTCAATGAG | Amplification of SME2(N37D) |
| E2(N37D mutant)-1R | CGGCCCCGAGTAGCCCTATCTCATCGGTTGATGATATTGCGTACCTG | Amplification of SME2(N37D) |
| E2(N37D mutant)-2F | CAGGTACGCAATATCATCAACCGATGAGATAGGGCTACTCGGGGCCG | Amplification of SME2(N37D) |
| E2(N37D mutant)-2R | CTTTGTTAACTCGTAAGTGG | Amplification of SME2(N37D) |
| Myc-Trx2-F | CTGGAATTCATATGGCTCAGCGTCTTCTCCTG | Amplification of Trx2 |
| Myc-Trx2-R | GTCCTCGAGTCAGCCAATCAGCTTCTTCAG | |
| Flag-Trx2-F | GAATTCAATGGCTCAGCGTCTTCTCCTG | Amplification of Trx2 |
| Flag-Trx2-R | GGTACCTCAGCCAATCAGCTTCTTC | |
| mCherry-Trx2-F | GAATTCAATGGCTCAGCGTCTTCTCCTG | Amplification of Trx2 |
| mCherry-Trx2-R | GGTACCTCAGCCAATCAGCTTCTTC | |
| GST-Trx2-F | CTGGAATTCATGGCTCAGCGTCTTCTCCTG | Amplification of Trx2 |
| GST-Trx2-R | GTCCTCGAGTCAGCCAATCAGCTTCTTCAG | |
| GST-Trx2-F (aa 1) | CTGGAATTCATGGCTCAGCGTCTTCTCCTG | Amplification of Trx2 (aa 1–65) |
| GST-Trx2-R (aa 65) | GTCCTCGAGTCACTGGATGTTAAAGGTTGTTG | |
| GST-Trx2-F (aa 46) | CTGGAATTCCCCAGCCAAGCCCGGTCATTATAC | Amplification of Trx2 (aa 46–100) |
| GST-Trx2-R (aa 100) | GTCCTCGAGTCATAACCTTGGCCCCAGGATCTTGC | |
| GST-Trx2-F (aa 66) | CTGGAATTCGATGGACCTGACTTTCAAG | Amplification of Trx2 (aa 66–157) |
| GST-Trx2-R (aa 157) | GTCCTCGAGTCAGCCAATCAGCTTCTTCAG | |
| FUGW-Trx2-F | ACAGGCCATTACGGCCATGGCTCAGCGTCTTCTCCT | Amplification of Trx2 |
| FUGW-Trx2-R | TACGGCCGAGGCGGCCTCAGCCAATCAGCTTCTTC | |
| mCherry-calnexin-F | GAATTCTATGGAAGGGAAGTGGTTGC | Amplification of calnexin |
| mCherry-calnexin-R | GGTACCTCACTCTCTTCGTGGCTTTC | |
| IL-8-F | AGCCCGTGTCAACATGACTTCC | Quantitative real-time PCR for detection of IL-8 |
| IL-8-R | GAAGTTGTGTTGGCATCTTTACTGA | |
| GAPDH-F | ACATGGCCTCCAAGGAGTAAGA | Quantitative real-time PCR for detection of GAPDH |
| GAPDH-R | GATCGAGTTGGGGCTGTGACT |
Yeast two-hybrid (Y2H) screening.
A Matchmaker two-hybrid system (Clontech) was used to screen the host proteins that interact with E2. Briefly, the plasmid encoding E2 fused with the DNA-binding domain (BD) (BD-E2) was transformed into the yeast strain Y2HGold and used as a bait to screen from a porcine primary macrophage cDNA library cloned into the pGADT7-Rec vector (AD) according to the manufacturer's instructions. Transformants were screened for growth on synthetically defined medium lacking His, Leu, Trp, and Ade (SD/−4). The colonies were then transferred to SD/−4 plates containing X-alpha-galactosidase (X-α-Gal) and aureobasidin A (SD/−4/X-α-Gal-Aba). Blue colonies were selected and cultured in the SD/−4 medium for plasmid extraction. The prey plasmids were extracted, and the target inserts were verified by sequencing. To confirm the interaction between E2 and the cellular proteins, Y2HGold was cotransformed with the bait and prey plasmids. The transformants were grown on synthetically defined medium lacking Leu and Trp (SD/−2), SD/−4, and SD/−4/X-α-Gal-Aba plates. Subsequently, a β-galactosidase assay was performed according to the manufacturer's instructions. Cotransformations with BD-lamin (human lamin C protein)/AD-T (simian virus 40 [SV40] large T antigen), BD-p53/AD-T, and BD/AD served as negative, positive, and blank controls, respectively.
GST pulldown assay.
GST-tagged Trx2 and Trx2 mutants were expressed in Escherichia coli BL21(DE3) cells and incubated with a glutathione-Sepharose 4B resin (catalog no. 10049253; GE Healthcare). The resin was washed twice with phosphate-buffered saline (PBS) and incubated with the lysate of HEK293T cells transfected with pCAGGS-based plasmids expressing Flag-tagged Shimen E2 (SME2), C-strain E2 (CE2), or chimeric E2 mutants for 2 h at 4°C. The resin was washed six times with PBS, and the bound proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting analysis. GST-E2 expressed in E. coli and Trx2 expressed in HEK293T cells were also used in a GST pulldown assay to further verify the interaction as described above.
Confocal microscopy.
PK-15 cells were infected with the CSFV Shimen strain at a multiplicity of infection (MOI) of 0.1 at 37°C for 48 h. The CSFV-infected or mock-treated cells were fixed with 4% paraformaldehyde in PBS for 30 min and permeabilized with 0.1% Triton X-100 for 15 min. The cells were first incubated with a mouse anti-E2 monoclonal antibody (MAb) (1:100) (23) and rabbit anti-Trx2 polyclonal antibody (PAb) (1:100) (sc50036; Santa Cruz) for 2 h, followed by a 2-h incubation with an anti-mouse IgG (whole-molecule)-fluorescein isothiocyanate (FITC) antibody (F2012; Sigma-Aldrich) and an anti-rabbit IgG (whole-molecule)-tetramethyl rhodamine isothiocyanate (TRITC) antibody (T6778; Sigma-Aldrich). The cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) for 15 min and examined using a Leica SP2 confocal system (Leica Microsystems). The colocalization signal was analyzed using Colocalizer Pro software (Colocalization Research Software, Boise, ID) as described previously (24).
To investigate whether E2 somehow contacts or comes close to Trx2 located in the mitochondria, voltage-dependent anion-selective channel protein 1 (VDAC1), a mitochondrial marker, and calnexin, an endoplasmic reticulum (ER) marker, were included in laser confocal analysis. Briefly, HEK293T cells were cotransfected with pCAGGS-SME2-EGFP and pmCherry-Trx2, pCAGGS-SME2-EGFP and pmCherry-VDAC1, pCAGGS-SME2-EGFP and pmCherry-calnexin, or pCAGGS-VDAC1-EGFP and pmCherry-Trx2. After a 48-h incubation, the transfected cells were fixed with 4% paraformaldehyde in PBS for 30 min and permeabilized with 0.1% Triton X-100 for 15 min. The cells were stained with DAPI and examined with a Leica SP2 confocal system as described above.
In situ PLA.
In situ proximity ligation assay (PLA) was performed with a Duolink in situ PLA kit (Olink Bioscience) as recommended by the manufacturer. Briefly, PK-15 cells grown in 12-well plates (Nunc) were cotransfected with 1 μg of pCAGGS-SME2-Flag and 1 μg of pMyc-Trx2 or were infected with the CSFV Shimen strain at an MOI of 0.1. After 48 h of incubation, the transfected or infected cells were fixed with 4% paraformaldehyde in PBS for 30 min and permeabilized with 0.1% Triton X-100 for 15 min. Subsequently, the cotransfected cells were incubated with a mouse anti-Flag MAb (1:100) (F1804; Sigma-Aldrich) or a rabbit anti-Myc PAb (1:100) (C3956; Sigma-Aldrich), and the cells infected with CSFV were incubated with a mouse anti-E2 MAb (1:100) or a rabbit anti-Trx2 PAb (1:100). After removing unbound antibodies, the cells were incubated with anti-mouse Plus and anti-rabbit Minus proximity probes for 2 h at 37°C. Thereafter, all of the steps were followed as recommended by the manufacturer. The cells were stained with DAPI for 15 min and visualized under an immunofluorescence microscope (Nikon) using suitable filters for Texas Red (PLA) and DAPI (nuclear) staining.
Construction of a CSFV mutant defective in binding Trx2.
The CSFV infectious cDNA clone pBRCISM (25) was used as a backbone to construct the CSFV N37D mutant, in which the asparagine at position 37 (N37, which is critical for Trx2 binding [see below]) in the E2 protein was replaced with aspartic acid by site-directed mutagenesis using overlapping PCR. The primers for amplifying the mutated E2 genes are listed in Table 1. The CSFV mutant defective in Trx2 binding was rescued and identified as described previously (25).
RNA interference assay.
Small interfering RNAs (siRNAs) against the porcine Trx2 and p65 genes were synthesized by GenePharma. The siRNA sequences targeting Trx2 were GCC CGG UCA UUA UAC ACU ATT (siTrx2-1), CAG UGA GAC GCC AGU GGU UTT (siTrx2-2), and GGA UGG ACC UGA CUU UCA ATT (siTrx2-3). The siRNA sequences targeting p65 were GCA UCC AGA CCA ACA ACA ATT (sip65-1), GCA CCG GAU UGA GGA GAA ATT (sip65-2), and CCC UAU CCC UUU ACG CCA UTT (sip65-3). The nontargeting control siRNA (siNC) sequence was UUC UCC GAA CGU GUC ACG UTT. Approximately 2 × 105 PK-15 cells were seeded into 12-well plates. On the following day, the cells were transfected with 200 nM pooled specific siRNAs (siTrx2 or sip65) or siNC using the X-tremeGENE siRNA transfection reagent (catalog no. 4476093001; Roche) according to the manufacturer's instructions. At 36 h posttransfection (hpt), the transfected cells were incubated with the CSFV Shimen strain at an MOI of 0.1 at 37°C for 2 h, followed by washing three times with DMEM and incubation at 37°C for 48 h or 72 h. At various time points postinfection, cell-free culture supernatant and cell lysate were harvested for analysis.
Construction of a stable cell line overexpressing Trx2.
Recombinant lentiviruses were produced and titrated as described previously (26). Briefly, HEK293T cells were cotransfected with 21 μg of pFUGW-Trx2 or pFUGW, 14 μg of psPAX2, and 7 μg of pMD2.G (Addgene). At 6 hpt, the medium was replaced with fresh DMEM containing 5% PBS. After a 48-h incubation, the supernatant of the transfected cells was harvested and filtered through a 0.22-μm-pore-size membrane. The filtrates were ultracentrifuged to concentrate the recombinant lentivirus expressing EGFP (Lenti-EGFP) or EGFP-Trx2 (Lenti-EGFP-Trx2). Subsequently, PK-15 cells were transduced with the lentiviruses at 10 transduction units (TU) per cell. The expression of EGFP-Trx2 or EGFP in the transduced cells (PK-EGFP-Trx2 or PK-EGFP, respectively) was examined by flow cytometry on a FACSAria instrument (BD Biosciences) and immunoblotting analysis.
NF-κB luciferase reporter assay.
HEK293T cells (105) in 24-well plates were transfected with 500 ng of pNF-κB-Luc, 15 ng of pRL-TK (Promega) (as an internal control), and different amounts (0, 100, 200, and 500 ng) of pMyc-Trx2. At 24 hpt, the cells were stimulated with 20 ng/ml tumor necrosis factor alpha (TNF-α) (catalog no. H8916; Sigma-Aldrich) or left untreated for 24 h, and the whole-cell extracts were prepared for the analysis of dual-luciferase activities. The activities of the reporter genes, including firefly luciferase and renilla luciferase, were determined using a dual-luciferase reporter 1000 assay system (Promega) according to the manufacturer's instructions. The data represented the firefly luciferase activity normalized to the renilla luciferase activity. Three independent experiments were carried out in duplicate.
Real-time RT-PCR.
The viral genome copies in CSFV-infected PK-15 cells were quantified by real-time reverse transcription-PCR (RT-PCR) (27). Briefly, the total RNA was extracted from CSFV-infected cells with TRIzol (catalog no. 15596026; Invitrogen) and treated with DNase I to remove potential genomic DNA contaminants. The isolated RNA was then reverse transcribed to cDNA with Moloney murine leukemia virus reverse transcriptase (TaKaRa) according to the manufacturer's instructions. Quantification of genome copies of CSFV was performed as described previously (27).
The expression of interleukin 8 (IL-8) was examined by a relative quantitative real-time RT-PCR. PK-15 cells were stimulated with 20 or 50 ng/ml TNF-α (catalog no. H8916; Sigma-Aldrich) for 6 h at 37°C. Total RNA of the TNF-α-treated cells was extracted using TRIzol as described above. Synthesis of cDNA was performed with 1,000 ng of total RNA. The transcriptional level of IL-8 mRNA was quantified by real-time RT-PCR with SYBR Permix Ex Taq II (catalog no. DRR081A; TaKaRa) in a LightCycler 480 II real-time PCR system (Roche). The target gene expression was normalized to the expression of a reference gene, the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene. The primers for amplifying GAPDH and IL-8 genes are listed in Table 1. Relative fold changes in gene expression were determined by the threshold cycle (2−ΔΔCT) method (28).
Western blotting.
Total cellular proteins were extracted with the cell lysis buffer (catalog no. P0013; Beyotime), and the concentrations were determined with a Tiangen Bradford protein assay kit (catalog no. PA102; Tiangen). The total proteins (100 μg) were subjected to SDS-PAGE, and separated protein bands were electrotransferred onto a nitrocellulose membrane (catalog no. 66485; Pall) using a semidry blotter (Bio-Rad). The membrane was soaked in blocking buffer (PBS containing 5% nonfat milk) for 1 h and probed with specific antibodies. The proteins were revealed by IRDye 800CW secondary antibodies, and thereafter the blots were visualized using an Odyssey infrared imaging system (Li-Cor Biosciences).
Statistical analysis.
Statistical analysis was performed using the SPSS 17.0 software. Student's t test or one-way analysis of variance was used to compare viral titers and genome copies. A P value of <0.05 was considered significant.
RESULTS
The CSFV E2 interacts with the cellular Trx2 protein.
In the yeast two-hybrid screening, 8 proteins were identified as potential binding partners of the CSFV E2 protein (Table 2). One of these preys, Trx2 (Fig. 1A), was chosen for further study considering its involvement in diverse cellular events such as apoptosis, antioxidation, and the modulation of NF-κB and GR transcription factors (16, 19).
TABLE 2.
Cellular proteins interacting with the CSFV E2 protein in the Y2H screen
| Protein(s) | Maximum identity (%) | No. of clones |
|---|---|---|
| Sus scrofa lectin, galactoside binding, soluble, 3 (galectin 3) | 100 | 10 |
| Pig Ig gamma 3 chain constant region | 96 | 7 |
| Guanine nucleotide-binding protein subunit beta-2-like (RACK1) | 99 | 8 |
| Immunoglobulin lambda-like polypeptide 5-like (IGLL5) | 97 | 6 |
| Sus scrofa cytochrome P450 4B1-like | 96 | 4 |
| Homo sapiens quaking homolog, KH domain RNA (QKI) | 100 | 3 |
| Sus scrofa eukaryotic translation elongation factor 2 (EEF2) | 99 | 2 |
| Thioredoxin 2 (Trx2) | 99 | 5 |
FIG 1.

Interaction of E2 with Trx2. (A) Reactivity of CSFV E2 with Trx2 in a yeast two-hybrid system. The yeast strain Y2HGold was cotransformed with the bait plasmid, BD-E2, and the prey plasmid, pGADT7-Trx2 (AD-Trx2), which encodes Trx2 fused to the Gal4 activation domain. Cotransformations with BD-Lamin/AD-T, BD-p53/AD-T, and BD/AD were used as negative, positive, and blank controls, respectively. (B) GST-Trx2 pulldown assay. The GST and GST-Trx2 proteins expressed in E. coli BL21(DE3) were purified with a glutathione-Sepharose 4B resin (catalog no. 10049253; GE Healthcare). The resin conjugated with GST or GST-Trx2 was incubated with the recombinant E2-Flag protein. After washing with cold PBS, the bound proteins were separated by SDS-PAGE (12%) and detected using a mouse anti-GST PAb (1:1,000) (catalog no. AB101; Tiangen) and a mouse anti-Flag MAb (1:1,000) (catalog no. F1804; Sigma-Aldrich) in Western blotting. (C) GST-E2 pulldown assay. The GST or GST-E2 expressed in E. coli BL21(DE3) was purified with a glutathione-Sepharose 4B resin (catalog no. 10049253; GEHealthcare). The resin conjugated with GST or GST-E2 was incubated with the recombinant Flag-Trx2 protein. (D) Colocalization of the E2 protein with Trx2 in CSFV-infected PK-15 cells or transfected HEK293T cells. PK-15 cells were mock infected or infected with the CSFV Shimen strain. Cells were fixed at 48 h postinfection (hpi) and subjected to an indirect immunofluorescence assay to detect E2 protein (green) and Trx2 (red) with mouse anti-E2 and rabbit anti-Trx2 antibodies. HEK293T cells were cotransfected with the indicated plasmids for colocalization assay. The position of the nucleus is indicated by DAPI (blue) staining in the merged image. (E) In situ proximity ligation assay (PLA). PK-15 cells were cotransfected with pCAGGS-SME2-Flag and pMyc-Trx2 constructs or were infected with the CSFV Shimen strain. After incubation with the primary antibodies, the species-specific oligonucleotide-conjugated secondary antibodies (PLA probes) were used to bind to the primary antibodies. Following a DNA ligation step, synthetic DNA was amplified by rolling circle polymerization and detected by the hybridization of “red” oligonucleotide probes complementary to the synthetic DNA. The hybridized probe is visible as “red foci” under a fluorescence microscope.
To confirm the interaction between E2 and Trx2, GST pulldown experiments were performed with the GST-tagged Trx2 protein expressed in E. coli and the Flag-tagged E2 protein of the Shimen strain (SME2) expressed in HEK293T cells. The results showed that GST-Trx2, but not GST, interacts with Flag-SME2 (Fig. 1B). To further verify the interaction between E2 and Trx2, GST-SME2 expressed in E. coli BL21(DE3) cells and the Flag-Trx2 expressed in HEK293T cells were also used in the pulldown assay. The results indicated that GST-SME2, but not GST, interacts with Flag-Trx2 (Fig. 1C).
To examine whether the endogenous Trx2 protein colocalizes with E2, PK-15 cells were infected with the CSFV Shimen strain for 48 h. Confocal images of the cells immunostained with anti-Trx2 and anti-E2 antibodies showed the colocalization of Trx2 and E2 in the CSFV-infected cells (Fig. 1D). Based on the digital analysis of multiple cell images, the colocalization coefficient of E2 and Trx2 was 0.93. Confocal images of the HEK293T cells cotransfected with the indicated plasmids showed that the E2 protein was localized not only in the ER but also in the mitochondria (Fig. 1D).
The in situ PLA also exhibited positive signals (red foci) of the E2-Trx2 interaction in the cotransfected or CSFV-infected PK-15 cells (Fig. 1E). Collectively, these findings demonstrate that Trx2 is an interacting partner of E2.
The thioredoxin domain of Trx2 is critical for its interaction with E2.
To determine which domain(s) of Trx2 is necessary for its interaction with the E2 protein, a series of GST-tagged Trx2 mutants were generated (Fig. 2A) and tested for the interaction with CSFV E2 by a GST pulldown assay. Notably, the thioredoxin domain (amino acids [aa] 66 to 157) of Trx2 was found to be critical for its interaction with the CSFV E2 protein (Fig. 2B).
FIG 2.
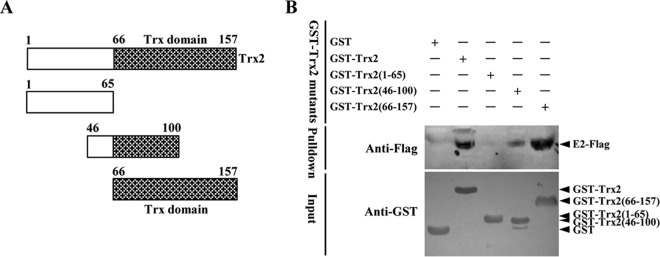
The thioredoxin domain of Trx2 is required for its interaction with E2. (A) Schematic representation of the swine Trx2 protein domains and the individual Trx2 deletion mutants tested in this study. (B) GST pulldown analysis of the association of GST-Trx2 or its mutants with the Flag-tagged E2 expressed in HEK293T cells. The recombinant GST-Trx2 and GST-tagged Trx2 mutants expressed in Escherichia coli BL21(DE3) were incubated with a glutathione-Sepharose 4B resin (catalog no. 10049253; GE Healthcare). After washing twice with PBS, the resin was incubated with the lysate of HEK293T cells transfected with pCAGGS-SME2-Flag. The bound proteins were subjected to SDS-PAGE and immunoblotting analysis.
The asparagine at position 37 of the E2 protein is indispensable to its interaction with Trx2.
The CSFV C-strain is a lapinized modified live vaccine that was attenuated by hundreds of passages of a highly virulent CSFV strain in the rabbits (21). The sequence identity of the E2 protein between the C-strain and Shimen strain is approximately 94.5% with 20 amino acid differences (Fig. 3A). We showed that the E2 protein of the Shimen strain, but not that of the C-strain, interacts with Trx2 (Fig. 3C). To define which part of E2 mediates its interaction with Trx2, we generated an array of chimeric E2 mutants between the Shimen strain and C-strain (Fig. 3B) and determined their ability to interact with Trx2 by a GST pulldown assay. We found that the chimeric E2 with the N-terminal 45 aa replaced by the counterpart of the C-strain E2 in the Shimen E2 backbone abrogated the ability of E2 to interact with Trx2. However, the chimeric E2 with the N-terminal 36 aa replaced by the corresponding region of the C-strain E2 in the Shimen E2 backbone restored the interaction, and the replacement of asparagine at position 37 with aspartic acid (N37D) in the Shimen E2 backbone abolished the ability of E2 to interact with Trx2 (Fig. 3C), indicating that the N37 is indispensable to the interaction. The N37 in the E2 protein is highly conserved among different virulent strains, whereas the N37D mutation is consistently found in the attenuated vaccine strains (Fig. 3D).
FIG 3.

The N37 of E2 is critical for its interaction with Trx2. (A) Sequence alignment of the E2 protein between the CSFV Shimen strain and C-strain. (B) Schematic representation of the chimeric E2 mutants between Shimen strain and C-strain expressed in HEK293T cells. (C) GST pulldown analysis of the interaction of GST-Trx2 with the Flag-tagged chimeric E2 mutants expressed in HEK293T cells. The glutathione-Sepharose 4B resin (catalog no. 10049253; GE Healthcare) conjugated with GST or GST-Trx2 was incubated with the recombinant Flag-tagged SME2, CE2, or chimeric E2 expressed in HEK293T cells. After washing with PBS, the bound proteins were subjected to SDS-PAGE and immunoblotting analysis. (D) Sequence alignment of the Trx2-binding sites in the E2 proteins of different CSFV strains.
Knockdown of Trx2 by siRNAs enhances CSFV replication.
To test the functional relevance of Trx2 in CSFV replication, specific siRNAs were used to inhibit Trx2 expression in PK-15 cells, resulting in efficient knockdown of the Trx2 expression (Fig. 4A). Thirty-six hours after siRNA transfection, PK-15 cells were infected with the CSFV Shimen strain and viral replication was analyzed by real-time RT-PCR, virus titration, and Western blotting. Compared with siNC-treated cells, the viral genome copies in the Trx2-downregulated cells were increased by 6.0- and 12.1-fold at 48 and 72 h postinfection (hpi), respectively (Fig. 4B), and the infectious virus titers in the culture supernatant of siTrx2-treated cells were increased by 10.2- and 23.9-fold at 48 and 72 hpi, respectively (Fig. 4C). Similarly, the expression of Npro protein in siTrx2-treated cells was increased compared with the siNC control (Fig. 4D). The results indicate that knockdown of the cellular Trx2 enhances the CSFV viral genome replication and progeny virus production.
FIG 4.

Knockdown of Trx2 increases CSFV growth. (A) Knockdown of Trx2 protein expression by siRNAs. PK-15 cells transfected with different concentrations (100, 200, or 300 nM) of siTrx2 or control siRNA (siNC) were harvested at 36 h posttransfection (hpt). Endogenous Trx2 was detected by immunoblotting with antibodies against the indicated proteins. (B) CSFV genome copies in Trx2-knockdown cells. PK-15 cells treated with 200 nM siTrx2 or siNC for 36 h were infected with the CSFV Shimen strain at an MOI of 0.1 for 48 h or 72 h. The CSFV genome copy numbers were assessed using a quantitative RT-PCR assay as described previously (27). (C) CSFV titers in Trx2-knockdown cells. Virus titers in supernatant collected at 48 and 72 hpi were determined and expressed as 50% tissue culture infective doses (TCID50)/ml. Error bars represent the standard deviations of the means from three independent experiments. P values are indicated above the bars. (D) Increased expression of the CSFV Npro protein in Trx2-knockdown cells. The endogenous Trx2 and the CSFV Npro protein were detected by Western blotting using anti-Trx2 and anti-Npro PAb, respectively.
Overexpression of Trx2 inhibits the replication of the wild-type (wt) CSFV or the Trx2-binding-defective CSFV N37D mutant.
The observation that the depletion of Trx2 resulted in increased CSFV replication prompted us to examine the effects of the Trx2 overexpression on CSFV replication. To this end, stable PK-15 cell lines expressing EGFP (PK-EGFP) or EGFP-tagged Trx2 (PK-EGFP-Trx2) were generated and infected with the CSFV Shimen strain or the Trx2-binding-defective CSFV N37D mutant. Flow cytometry analysis demonstrated that nearly all cells showed fluorescence (Fig. 5A).
FIG 5.

Overexpression of Trx2 inhibits the replication of CSFV N37D mutant more efficiently than that of wt CSFV. PK-15 cells were transduced with Lenti-EGFP-Trx2 or Lenti-EGFP for 48 h followed by infection with the CSFV Shimen strain or N37D mutant. (A) Ectopic expression of EGFP-Trx2 in PK-15 cells. Flow cytometry assay was used to detect the cellular expression of EGFP in PK-EGFP-Trx2, PK-EGFP, or PK-15 cells. (B) Expression of the CSFV Npro protein in the stable cell line overexpressing Trx2. Western blotting of the Npro expression in the cell lysate collected at the indicated time points after infection. β-Tubulin was used as a loading control. The experiment was carried out in three replicates. The quantification analysis of Npro expression in the cell lysate was carried out using the Odyssey application software version 3.0. (C) Viral genome copies of Trx2-overexpressing cells infected with CSFV Shimen or N37D mutant. CSFV genome copy numbers in Trx2-overexpressing cells were assessed using a quantitative RT-PCR assay. (D) Infectious progeny virus titers in the supernatant of Trx2-overexpressing cells infected with CSFV Shimen or N37D mutant. Virus titers in the supernatant collected at 48 and 72 hpi were determined and expressed as 50% tissue culture infective doses (TCID50)/ml. Error bars represent the standard deviations of the means from three independent experiments. P values are indicated above the bars.
The expression of the Npro protein in the PK-EGFP-Trx2 cells infected with the wild-type (wt) CSFV or the N37D mutant (MOI of 0.1) was decreased compared with the PK-EGFP cells (Fig. 5B). The expression of Npro was markedly lower in the PK-EGFP-Trx2 cells infected with N37D mutant than in the cells infected with the wt CSFV (Fig. 5B). The viral genome copies in PK-EGFP-Trx2 cells infected with the wt CSFV or the N37D mutant were reduced by 58.9- and 141.3-fold at 48 hpi and reduced by 112.2- or 354.8-fold at 72 hpi, respectively, compared with PK-EGFP cells (Fig. 5C). The yields of infectious virus in culture supernatant of the wt CSFV- or the N37D mutant-infected PK-EGFP-Trx2 cells were decreased by 20.4- or 57.5-fold at 48 hpi and reduced by 14.8- or 72.4-fold at 72 hpi, respectively (Fig. 5D). These data suggest that Trx2 is a cellular antiviral factor against CSFV infection and shows greater inhibitory effects on the viral replication of the CSFV N37D mutant than the wt CSFV.
CSFV inhibits the expression of the cellular Trx2 protein.
To examine the potential effects of E2 on Trx2 expression, HEK293T cells were cotransfected with pCAGGS-SME2-Flag and pMyc-Trx2. Immunoblotting analysis demonstrated that the exogenous Trx2 level was reduced with increased E2 expression in a dose-dependent manner (Fig. 6A). To test the effects of CSFV infection on Trx2 expression, PK-15 cells were infected with the CSFV Shimen strain or the N37D mutant and examined by immunoblotting analysis. The results showed that the wt CSFV (Fig. 6B) but not the CSFV mutant (Fig. 6C) induced the downregulation of Trx2 in PK-15 cells.
FIG 6.

Wild-type but not N37D mutant CSFV induces downregulation of Trx2. (A) Western blotting of Trx2 expression in pCAGGS-SME2-Flag and pMyc-Trx2-cotransfected HEK293T cells. HEK293T cells were cotransfected with increased amounts of pCAGGS-SME2-Flag (0, 0.5, 1.0, and 1.5 μg) and decreased amounts of pCAGGS (1.5, 1, 0.5, and 0 μg) and indicated quality of pMyc-Trx2 (0.5 μg). At 48 hpt, the cell lysate was analyzed by Western blotting using mouse anti-Flag MAb (1:1,000) (catalog no. F1804; Sigma-Aldrich), mouse anti-Myc MAb (1:500) (catalog no. M4439; Sigma-Aldrich), and mouse anti-GAPDH MAb (1:500) (catalog no. sc-166574; Santa Cruz). GAPDH was used as a loading control. (B) Immunoblotting analysis of Trx2 in CSFV-infected PK-15 cells. PK-15 cells were infected with CSFV at an MOI of 0.1 or were mock infected, and the cell lysates at 48 and 72 hpi were analyzed by Western blotting using rabbit anti-Trx2 PAb (1:200) (catalog no. sc-50036; Santa Cruz), mouse antisera against Npro (1:500) (produced in-house), and mouse anti-GAPDH MAb (1:500) (catalog no. sc-166574; Santa Cruz). (C) Western blotting of Trx2 expression in PK-15 cells infected with the CSFV N37D mutant. PK-15 cells were infected with the CSFV mutant N37D at an MOI of 0.1 for 48 and 72 h. The cell lysate was subjected to SDS-PAGE and Western blotting as described above. The experiment was carried out in three replicates. The quantification analysis of Trx2 expression in the cell lysate was conducted using the Odyssey application software version 3.0. Error bars represent the standard deviations of the means from three independent experiments. P values are indicated above the bars.
Trx2 activates the NF-κB signaling pathway.
To clarify the role of Trx2 in regulating the cellular antiviral signaling pathway, the effects of Trx2 on the NF-κB signaling pathway were examined. The results indicated that Trx2 increased the NF-κB promoter transcription level in a dose-dependent manner in HEK293T cells with or without TNF-α treatment (Fig. 7A and B). Activation of NF-κB is usually characterized by the nuclear translocation of NF-κB (29). To investigate the potential mechanism of NF-κB activation by Trx2, we examined the NF-κB expression level in cytoplasmic and nuclear extracts using Western blotting. The results showed that the nuclear p65 protein was increased, while the amount of total p65 was not altered (Fig. 7C). To further validate the effects of Trx2 overexpression on the NF-κB nuclear translocation, HEK293T cells cotransfected with pEGFP-p65 and pDsRed-Trx2 were used to monitor the translocation of p65 under a confocal fluorescence microscope. The results showed that the p65 protein accumulated in the nucleus when coexpressed with Trx2, while it was detained in the cytoplasm when pEGFP-p65 and the empty vector pDsRed2-N1 were coexpressed (Fig. 7D). Immunoblotting analysis showed that the fusion protein EGFP-p65 was intact in the HEK293T cells cotransfected with pEGFP-p65 and pDsRed-Trx2 (Fig. 7E). Taken together, these results clearly demonstrate that Trx2 induces NF-κB activation through promoting p65 nuclear translocation.
FIG 7.

Trx2 activates the NF-κB signaling pathway by promoting p65 subunit nuclear translocation. (A) Trx2 activates NF-κB signaling pathway in a dose-dependent manner. HEK293T cells were transfected with the NF-κB reporter and increased amounts of the Trx2 plasmid (0, 0.1, 0.2, and 0.5 μg) for 24 h and were untreated or treated with TNF-α for 24 h before the luciferase assay. (B) Immunoblotting analysis of the expression of Myc-Trx2 in HEK293T cells. HEK293T cells were transfected with increased amounts of pMyc-Trx2 and the indicated reporter plasmid. β-Tubulin was used as a loading control. (C and D) Trx2 promotes the nuclear translocation of the NF-κB p65 subunit. HEK293T cells on 6-well plates were cotransfected with pEGFP-p65 (1.5 μg) and pMyC-Trx2 (1.5 μg). At 48 hpt, the cytoplasm and nucleus proteins were extracted using a CelLytic nuclear extraction kit (catalog no. Nxtract; Sigma-Aldrich). Immunoblotting analysis of the p65 and Trx2 expression in the cytoplasm and nucleus was performed using rabbit anti-p65 PAb (1:500) and rabbit anti-Myc PAb (1:1,000) (catalog. no. C3956; Sigma-Aldrich). HEK293T cells were cotransfected with RFP-tagged Trx2 expression plasmid pDsRed-Trx2 (1.0 μg) and the EGFP-tagged p65 expression plasmid pEGFP-p65 (0.5 μg). Cells cotransfected with pDsRed2-N1 and EGFP-tagged p65 served as a negative control. Cells were fixed and permeabilized at 48 hpt. The cell nuclei were counterstained with DAPI. Nuclear translocation of NF-κB was observed under a Leica SP2 confocal system (Leica Microsystems). (E) Western blotting of EGFP-tagged p65 and RFP-tagged Trx2 in HEK293T cells cotransfected with pEGFP-p65 and pDsRed-Trx2. HEK293T cells on 6-well plates were cotransfected with pEGFP-p65 and pDsRed-Trx2 as described above. At 48 hpt, the cell lysate was subjected to immunoblotting analysis using the anti-EGFP MAb (1:500) (catalog no. A00185; GenScript) and anti-RFP MAb (1:500) (catalog no. sc-101526; Santa Cruz).
Activation of NF-κB signaling pathway inhibits CSFV replication in PK-15 cells.
To investigate the relevance of the activation of the NF-κB signaling pathway in CSFV replication, the infectious progeny virus of the CSFV Shimen strain in TNF-α-treated PK-15 cells was examined at 48 and 72 hpi. The transcriptional level of IL-8, a proinflammatory cytokine that is modulated by NF-κB, was increased in the TNF-α-treated PK-15 cells (Fig. 8A), indicating that the activation of the NF-κB signaling pathway was induced by TNF-α. Compared with the mock control, the viral genome copies (Fig. 8B), the progeny virus titers (Fig. 8C), and the expression of Npro protein (Fig. 8D) were decreased in the TNF-α-treated PK-15 cells, suggesting that the activation of the NF-κB signaling pathway inhibits CSFV replication in PK-15 cells.
FIG 8.

TNF-α inhibits CSFV replication in PK-15 cells via the NF-κB signaling pathway. (A) TNF-α treatment increased the transcriptional level of the endogenous IL-8 mRNA in PK-15 cells. After treatment with TNF-α for 6 h, IL-8 mRNA abundance in PK-15 cells was assessed using a relative quantitative RT-PCR assay. The data represent three independent experiments. (B) Reduced CSFV genome copies in TNF-α-stimulated PK-15 cells. Viral genome copies in TNF-α-stimulated cells at 48 and 72 hpi were assessed using a real-time RT-PCR assay. (C) Reduced viral growth in TNF-α-stimulated PK-15 cells. Virus titers in supernatant collected at 48 and 72 hpi were determined and expressed as 50% tissue culture infective doses (TCID50)/ml. (D) Decreased viral protein expression in TNF-α-stimulated cells. The CSFV Npro protein expression at 48 and 72 hpi was examined by immunoblotting analysis. GAPDH was used as a loading control. PK-15 cells were transfected with sip65, subsequently treated with TNF-α, and infected with the CSFV Shimen strain. (E and F) Analysis of CSFV genome copies (E) and virus titers (F) in the p65-knockdown and TNF-α-treated cells. (G) Viral protein expression in the p65-knockdown and TNF-α-treated cells.
To further investigate whether TNF-α inhibits CSFV replication via the NF-κB signaling pathway, we examined CSFV replication in TNF-α-treated PK-15 cells with p65 being silenced by siRNAs. The results showed that the viral genome copies (Fig. 8E), progeny virus titers (Fig. 8F), or the expression of Npro protein (Fig. 8G) was not reduced in the p65-silenced PK-15 cells, suggesting that TNF-α inhibits CSFV replication via the NF-κB signaling pathway.
Trx2 did not inhibit CSFV replication in the absence of p65 in PK-15 cells.
We investigated whether Trx2 is able to inhibit CSFV replication after blockage of the NF-κB signaling pathway. PK-EGFP-Trx2 or PK-EGFP cells were transfected with p65-specific siRNAs (sip65) and infected with the CSFV Shimen strain. Compared with the CSFV-infected PK-EGFP cells, the viral genome copies and progeny virus titers in the PK-EGFP-Trx2 cells were not altered significantly at 48 or 72 hpi, respectively (Fig. 9A and B), and the expression of the Npro protein was not markedly changed at 48 or 72 hpi (Fig. 9C and D). The results indicated that Trx2 was unable to suppress CSFV replication after the blockade of the NF-κB signaling pathway. These data further demonstrate that the host Trx2 inhibits CSFV replication via the NF-κB signaling pathway.
FIG 9.

Overexpression of Trx2 cannot inhibit CSFV replication after blockade of the NF-κB signaling pathway by silencing p65 expression in PK-15 cells. (A) CSFV genome copies in p65-knockdown cells. The CSFV genome copies were assessed using a quantitative RT-PCR assay in p65-knockdown cells. (B) CSFV titers in p65-knockdown cells. Virus titers in the supernatant collected at 48 and 72 h postinfection were determined and expressed as 50% tissue culture infective doses (TCID50)/ml. (C) Knockdown of p65 protein levels by siRNA treatment. PK-EGFP-Trx2 or PK-EGFP cells transfected with 200 nM sip65 or siNC were harvested at 36 hpt. Endogenous p65 and GAPDH were detected by immunoblotting analysis with antibodies directed against the indicated proteins. (D) Npro expression in p65-knockdown cells infected with CSFV. Western blotting was used to analyze the p65 or Npro expression in the cell lysate collected at the indicated time points postinfection. GAPDH was used as a loading control.
DISCUSSION
Trx2 is involved in diverse cellular processes, such as apoptosis, antioxidation, and regulation of transcription factors. It has been reported that the absence of Trx2 causes massive apoptosis and early embryonic lethality in homozygous mice (30). Overexpression of Trx2 decreases the accumulation of nitric oxide-evoked hypoxia-inducible factor 1 (HIF-1) alpha and the transactivation of HIF-1 in HEK293 cells (31). Furthermore, Trx2 interacts with several transcription factors, including the p65 subunit of NF-κB and GR (19). A recent study demonstrates that exogenous administration of recombinant human Trx-1 significantly improves the survival rate and attenuates lung histological changes in a murine influenza pneumonia model (32). We show here that the cellular protein Trx2 interacts with the E2 protein of CSFV and inhibits viral replication. The thioredoxin domain of Trx2 and the N37 in the E2 protein were shown to be critical for the interaction. Interestingly, the N37 in the E2 protein that is critical for Trx2 interaction is highly conserved among different virulent strains but not in the attenuated vaccine strains, implying that it may be a potential target of anti-CSFV strategies. To the best of our knowledge, this is the first study that has indicated that Trx2 is involved in the life cycle of a virus.
A number of host factors have been identified as being involved in CSFV replication. The cellular membrane-associated heparan sulfate (HS), CD46, and laminin receptor have been shown to mediate virus attachment (33–35). Recently, annexin 2 has been identified as an E2-binding protein that promotes CSFV growth in PK-15 cells (36). Moreover, it has been reported that several CSFV-interacting partners, such as interferon (IFN) regulatory factor 3 (IRF3), IRF7, and hemoglobin subunit beta (HB), could activate the type I IFN signaling pathway (37–39). Gladue and coworkers screened 59 cellular proteins that interact with the CSFV E2 protein using yeast two-hybrid (Y2H) analysis (40). However, the involvement of the E2-interacting proteins in CSFV replication has not been elucidated. Some proteins (including Trx2) were consistently identified in that work and in our study, indicating that Y2H analysis is reliable. In the present study, we demonstrate that Trx2 interacts with the CSFV E2 and inhibits CSFV replication via the NF-κB signaling pathway. Moreover, we show that TNF-α activates the NF-κB signaling pathway in PK-15 cells and subsequently inhibits CSFV replication, indicating that CSFV is possibly sensitive to the inflammatory cytokines modulated by NF-κB. Notably, Trx2 reduces the replication of the CSFV N37D mutant more efficiently than that of the wt CSFV. A possible explanation is that the mutant virus is inefficient to suppress the NF-κB signaling pathway due to the inability of the mutated E2 to interact with and thus antagonize Trx2, and the precise mechanism needs to be further investigated.
During evolution, viruses have acquired numerous strategies to evade or subvert key elements of the host's antiviral response. Previous studies have shown that CSFV counteracts the alpha/beta interferon (IFN-α/β) induction by mediating the proteasomal degradation of IRF3 or mRNA turnover of IRF7, allowing the virus to establish a productive infection (37, 38). The NF-κB activation induced by CSFV infection varies among different cell types. CSFV infection does not activate NF-κB in dendritic cells (41). In contrast, CSFV triggers NF-κB activation in porcine alveolar macrophages (42). The CSFV Npro protein has been found to interact with IκBα, an inhibitor of NF-κB, but the interaction does not regulate the NF-κB signaling pathway (43). To date, how CSFV antagonizes the NF-κB signaling pathway remains unclear. In line with previous work (19), our study shows that overexpression of Trx2 activates the NF-κB signaling pathway. In addition, we demonstrate that the wt CSFV or E2 protein induces downregulation of Trx2 in PK-15, whereas the CSFV N37D mutant defective in binding Trx2 does not affect the expression of Trx2, indicating that the downregulation of Trx2 induced by E2 plays an important role in the inhibition of the NF-κB signaling pathway during CSFV infection. Whether CSFV infection induces the proteasomal degradation of Trx2 or inhibits the translation of Trx2 requires further investigation.
In conclusion, we show that Trx2 inhibits CSFV replication via the NF-κB signaling pathway. Our results provide insights into the mechanisms of CSFV replication. Although the precise mechanism needs to be further elucidated, the present study identifies Trx2 as a potential target for the inhibition of CSFV replication.
ACKNOWLEDGMENTS
We are grateful to Lihong Liu at the National Veterinary Institute, Sweden, for his helpful suggestions.
This study was supported by the Natural Science Foundation of China (grant 31201921) and the Natural Science Foundation of Heilongjiang Province of China (grants ZD201410 and C2015066).
REFERENCES
- 1.Pletnev A, Gould E, Heinz FX, Meyers G, Thiel HJ, Bukh J, Stiasny K, Collett MS, Becher P, Simmonds P, Rice CM, Monath TP. 2011. Flaviviridae, p 1003–1020. In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (ed), Virus taxonomy: classification and nomenclature of viruses. Ninth report of the International Committee on Taxonomy of Viruses Academic Press, London, United Kingdom. [Google Scholar]
- 2.Lindenbach BD, Murray CL, Thiel HJ. 2013. Flaviviridae, p 712–746. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Thiel HJ, Stark R, Weiland E, Rümenapf T, Meyers G. 1991. Hog cholera virus: molecular composition of virions from a pestivirus. J Virol 65:4705–4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weiland E, Stark R, Haas B, Rümenapf T, Meyers G, Thiel HJ. 1990. Pestivirus glycoprotein which induces neutralizing antibodies forms part of a disulfide-linked heterodimer. J Virol 64:3563–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weiland F, Weiland E, Unger G, Saalmuller A, Thiel HJ. 1999. Localization of pestiviral envelope proteins E(rns) and E2 at the cell surface and on isolated particles. J Gen Virol 80:1157–1165. [DOI] [PubMed] [Google Scholar]
- 6.Wang Z, Nie Y, Wang P, Ding M, Deng H. 2004. Characterization of classical swine fever virus entry by using pseudotyped viruses: E1 and E2 are sufficient to mediate viral entry. Virology 330:332–341. doi: 10.1016/j.virol.2004.09.023. [DOI] [PubMed] [Google Scholar]
- 7.Risatti GR, Borca MV, Kutish GF, Lu Z, Holinka LG, French RA, Tulman ER, Rock DL. 2005. The E2 glycoprotein of classical swine fever virus is a virulence determinant in swine. J Virol 79:3787–3796. doi: 10.1128/JVI.79.6.3787-3796.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.König M, Lengsfeld T, Pauly T, Stark R, Thiel HJ. 1995. Classical swine fever virus: independent induction of protective immunity by two structural glycoproteins. J Virol 69:6479–6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu X, Tu C, Li H, Hu R, Chen C, Li Z, Zhang M, Yin Z. 2001. DNA-mediated protection against classical swine fever virus. Vaccine 19:1520–1525. doi: 10.1016/S0264-410X(00)00334-0. [DOI] [PubMed] [Google Scholar]
- 10.Li N, Qiu HJ, Zhao JJ, Li Y, Wang MJ, Lu BW, Han CG, Hou Q, Wang ZH, Gao H, Peng WP, Li GX, Zhu QH, Tong GZ. 2007. A Semliki Forest virus replicon vectored DNA vaccine expressing the E2 glycoprotein of classical swine fever virus protects pigs from lethal challenge. Vaccine 25:2907–2912. doi: 10.1016/j.vaccine.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 11.Sun Y, Li HY, Tian DY, Han QY, Zhang X, Li N, Qiu HJ. 2011. A novel alphavirus replicon-vectored vaccine delivered by adenovirus induces sterile immunity against classical swine fever. Vaccine 29:8364–8372. doi: 10.1016/j.vaccine.2011.08.085. [DOI] [PubMed] [Google Scholar]
- 12.Reimann I, Depner K, Trapp S, Beer M. 2004. An avirulent chimeric pestivirus with altered cell tropism protects pigs against lethal infection with classical swine fever virus. Virology 322:143–157. doi: 10.1016/j.virol.2004.01.028. [DOI] [PubMed] [Google Scholar]
- 13.Eblé PL, Geurts Y, Quak S, Moonen-Leusen HW, Blome S, Hofmann MA, Koenen F, Beer M, Loeffen WL. 2013. Efficacy of chimeric pestivirus vaccine candidates against classical swine fever: protection and DIVA characteristics. Vet Microbiol 162:437–446. doi: 10.1016/j.vetmic.2012.10.030. [DOI] [PubMed] [Google Scholar]
- 14.He F, Ling L, Liao Y, Li S, Han W, Zhao B, Sun Y, Qiu HJ. 2014. Beta-actin interacts with the E2 protein and is involved in the early replication of classical swine fever virus. Virus Res 179:161–168. doi: 10.1016/j.virusres.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 15.Tanaka T, Hosoi F, Yamaguchi-Iwai Y, Nakamura H, Masutani H, Ueda S, Nishiyama A, Takeda S, Wada H, Spyrou G, Yodoi J. 2002. Thioredoxin-2 (Trx-2) is an essential gene regulating mitochondria-dependent apoptosis. EMBO J 21:1695-1703. doi: 10.1093/emboj/21.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshioka J, Schreiter ER, Lee RT. 2006. Role of thioredoxin in cell growth through interactions with signaling molecules. Antioxid Redox Signal 8:2143–2151. doi: 10.1089/ars.2006.8.2143. [DOI] [PubMed] [Google Scholar]
- 17.Lillig CH, Holmgren A. 2007. Thioredoxin and related molecules from biology to health and disease. Antioxid Redox Signal 9:25–47. doi: 10.1089/ars.2007.9.25. [DOI] [PubMed] [Google Scholar]
- 18.Sengupta R, Holmgren A. 2012. The role of thioredoxin in the regulation of cellular processes by S-nitrosylation. Biochim Biophys Acta 1820:689–700. doi: 10.1016/j.bbagen.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 19.Psarra AM, Hermann S, Panayotou G, Spyrou G. 2009. Interaction of mitochondrial thioredoxin with glucocorticoid receptor and NF-kappaB modulates glucocorticoid receptor and NF-kappaB signalling in HEK-293 cells. Biochem J 422:521–531. doi: 10.1042/BJ20090107. [DOI] [PubMed] [Google Scholar]
- 20.Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoint. Am J Hyg (Lond) 27:493–497. [Google Scholar]
- 21.Qiu HJ, Tong GZ, Shen RX. 2005. The lapinized Chinese strain of classical swine fever virus: a retrospective review spanning half a century. J Integr Agric 38:1675–1685. [Google Scholar]
- 22.Fang Y, Fang LR, Wang Y, Lei YY, Luo R, Wang D, Chen HC, Xiao SB. 2012. Porcine reproductive and respiratory syndrome virus nonstructural protein 2 contributes to NF-κB activation. Virol J 9:83. doi: 10.1186/1743-422X-9-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng WP, Hou Q, Xia ZH, Chen D, Li N, Sun Y, Qiu HJ. 2008. Identification of a conserved linear B-cell epitope at the N-terminus of the E2 glycoprotein of classical swine fever virus by phage-displayed random peptide library. Virus Res 135:267–272. doi: 10.1016/j.virusres.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 24.Li D, Li S, Sun Y, Dong H, Li Y, Zhao B, Guo D, Weng C, Qiu HJ. 2013. Poly(C)-binding protein 1, a novel Npro-interacting protein involved in classical swine fever virus growth. J Virol 87:2072–2080. doi: 10.1128/JVI.02807-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li C, Huang JH, Li YF, He F, Li D, Sun Y, Han W, Li S, Qiu HJ. 2013. Efficient and stable rescue of classical swine fever virus from cloned cDNA using an RNA polymerase II system. Arch Virol 158:901–907. doi: 10.1007/s00705-012-1548-8. [DOI] [PubMed] [Google Scholar]
- 26.Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. 1997. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol 15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]
- 27.Zhao JJ, Cheng D, Li N, Sun Y, Shi Z, Zhu QH, Tu C, Tong GZ, Qiu HJ. 2008. Evaluation of a multiplex real-time RT-PCR for quantitative and differential detection of wild-type viruses and C-strain vaccine of classical swine fever virus. Vet Microbiol 126:1–10. doi: 10.1016/j.vetmic.2007.04.046. [DOI] [PubMed] [Google Scholar]
- 28.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 4:402–408. [DOI] [PubMed] [Google Scholar]
- 29.Hayden MS, Ghosh S. 2004. Signaling to NF-κB. Genes Dev 18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 30.Nonn L, Williams RR, Erickson RP, Powis G. 2003. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol Cell Biol 23:916–922. doi: 10.1128/MCB.23.3.916-922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou J, Eleni C, Spyrou G, Brüne B. 2008. The mitochondrial thioredoxin system regulates nitric oxide-induced HIF-1alpha protein. Free Radic Biol Med 44:91–98. doi: 10.1016/j.freeradbiomed.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 32.Yashiro M, Tsukahara H, Matsukawa A, Yamada M, Fujii Y, Nagaoka Y, Tsuge M, Yamashita N, Ito T, Yamada M, Masutani H, Yodoi J, Morishima T. 2013. Redox-active protein thioredoxin-1 administration ameliorates influenza A virus (H1N1)-induced acute lung injury in mice. Crit Care Med 41:171-181. doi: 10.1097/CCM.0b013e3182676352. [DOI] [PubMed] [Google Scholar]
- 33.Hulst MM, van Gennip HG, Moormann RJ. 2000. Passage of classical swine fever virus in cultured swine kidney cells selects virus variants that bind to heparan sulfate due to a single amino acid change in envelope protein Erns. J Virol 4:9553–9561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dräger C, Beer M, Blome S. 2015. Porcine complement regulatory protein CD46 and heparan sulfates are the major factors for classical swine fever virus attachment in vitro. Arch Virol 160:739–746. doi: 10.1007/s00705-014-2313-y. [DOI] [PubMed] [Google Scholar]
- 35.Chen J, He WR, Shen L, Dong H, Yu J, Wang X, Yu S, Li Y, Li S, Luo Y, Sun Y, Qiu HJ. 2015. The laminin receptor is a cellular attachment receptor for classical swine fever virus. J Virol 89:4894–4906. doi: 10.1128/JVI.00019-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Z, Shi Z, Guo H, Qu H, Zhang Y, Tu C. 2015. Annexin 2 is a host protein binding to classical swine fever virus E2 glycoprotein and promoting viral growth in PK-15 cells. Virus Res 201:16–23. doi: 10.1016/j.virusres.2015.02.013. [DOI] [PubMed] [Google Scholar]
- 37.Bauhofer O, Summerfield A, Sakoda Y, Tratschin JD, Hofmann MA, Ruggli N. 2007. Classical swine fever virus Npro interacts with interferon regulatory factor 3 and induces its proteasomal degradation. J Virol 81:3087–3096. doi: 10.1128/JVI.02032-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fiebach AR, Guzylack-Piriou L, Python S, Summerfield A, Ruggli N. 2011. Classical swine fever virus Npro limits type I interferon induction in plasmacytoid dendritic cells by interacting with interferon regulatory factor 7. J Virol 85:8002–8011. doi: 10.1128/JVI.00330-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li D, Dong H, Li S, Munir M, Chen J, Luo Y, Sun Y, Liu L, Qiu HJ. 2013. Hemoglobin subunit beta interacts with the capsid protein and antagonizes the growth of classical swine fever virus. J Virol 87:5707–5717. doi: 10.1128/JVI.03130-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gladue DP, Baker-Bransetter R, Holinka LG, Fernandez-Sainz IJ, O'Donnell V, Fletcher P, Lu Z, Borca MV. 2014. Interaction of CSFV E2 protein with swine host factors as detected by yeast two-hybrid system. PLoS One 9:e85324. doi: 10.1371/journal.pone.0085324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen LJ, Dong XY, Shen HY, Zhao MQ, Ju CM, Yi L, Zhang XT, Kang YM, Chen JD. 2012. Classical swine fever virus suppresses maturation and modulates functions of monocyte-derived dendritic cells without activating nuclear factor kappa B. Res Vet Sci 93:529–537. doi: 10.1016/j.rvsc.2011.06.026. [DOI] [PubMed] [Google Scholar]
- 42.Dong XY, Liu WJ, Zhao MQ, Wang JY, Pei JJ, Luo YW, Ju CM, Chen JD. 2013. Classical swine fever virus triggers RIG-I and MDA5-dependent signaling pathway to IRF-3 and NF-κB activation to promote secretion of interferon and inflammatory cytokines in porcine alveolar macrophages. Virol J 10:286. doi: 10.1186/1743-422X-10-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doceul V, Charleston B, Crooke H, Reid E, Powell PP, Seago J. 2008. The Npro product of classical swine fever virus interacts with Ikappa B alpha, the NF-kappaB inhibitor. J Gen Virol 89:1881–1889. doi: 10.1099/vir.0.83643-0. [DOI] [PubMed] [Google Scholar]