

ABSTRACT
There are two subgroups of respiratory syncytial virus (RSV), A and B, and within each subgroup, isolates are further divided into clades. Several years ago, multiple subgroup B isolates which contained a duplication of 60 nucleotides in the glycoprotein (G) gene were described. These isolates were given a new clade designation of BA based on the site of isolation, Buenos Aires, Argentina. BA RSV strains have since become the predominant circulating clade of RSV B viruses. We hypothesized that the duplicated region in G serves to enhance the function of G in the virus life cycle. We generated recombinant viruses that express a consensus BA G gene or a consensus BA G gene lacking the duplication (GΔdup). We determined that the duplicated region functions during virus attachment to cells. Additionally, we showed that in vitro, the virus containing the duplication has a fitness advantage compared to the virus without the duplication. Our data demonstrate that the duplicated region in the BA strain G protein augments virus attachment and fitness.
IMPORTANCE Respiratory syncytial virus (RSV) is an important pathogen for infants for which there is no vaccine. Different strains of RSV circulate from year to year, and the predominating strains change over time. Subgroup B RSV strains with a duplication in the attachment glycoprotein (G) emerged and then became the dominant B genotype. We found that a recombinant virus harboring the duplication bound more efficiently to cells and was more fit than a recombinant strain lacking the duplication. Our work advances a mechanism for an important natural RSV mutation.
INTRODUCTION
Respiratory syncytial virus (RSV) is the most common cause of lower respiratory tract infections in infants worldwide. The attachment glycoprotein (G) is the most variable RSV protein (1). The G protein is a type II transmembrane protein, consisting of an N-terminal cytoplasmic tail, a transmembrane domain, and a C-terminal ectodomain. The ectodomain of RSV G is characterized by two hypervariable, mucin-like domains which flank a central conserved region. As the name implies, most of the variability between G proteins of different strains is located in the hypervariable regions. The final 270 nucleotides (nt) of the G gene, encompassing part of the second hypervariable region, are commonly used to determine classification of RSV strains as belonging to subgroup A or B. Phylogenetic analysis of this region of G also classifies clades within each subgroup (2).
In the late 1990s, several isolates of RSV subgroup B strains from Buenos Aires, Argentina, were identified to contain an as yet undescribed duplication of 60 nt in the second hypervariable region of the G gene (3). The duplication was exact, such that an additional sequence of 20 amino acids followed the first run of those residues. The specific genotype of strains containing the duplication was termed “BA” for Buenos Aires, the site of the first described isolation events (4). Since the identification of those original isolates, B strains of RSV harboring the same duplication or derivatives thereof have been isolated globally (5–7). In multiple studies, the only B strains isolated contained the duplicated region in G (8, 9). Due to the global spread of these strains, and the decrease in the isolation events of B strains lacking the duplication, RSV BA strains are recognized as the predominant circulating RSV subgroup B strains (5).
Perhaps as interesting as the emergence of BA strains as the predominant circulating RSV B strains was the appearance of an RSV A genotype harboring a similar duplication in G (10). These strains, termed “ON1” for their original isolation in Ontario, Canada, were first described in 2012 (10). As with BA strains, since their original description, ON1 RSV strains have been isolated globally and are trending toward predominating among circulating A strains (11–13).
While multiple investigators have hypothesized what the potential advantage of the duplicated region is for these strains, the functional role of the duplication has not been reported. The most conventional role of G is in virus attachment to host cells (14), but G also serves an immunomodulatory role during RSV infection (15–17) and has been shown to contribute to the pathogenesis of recombinant strain A2-line19F (18). The original description of the second hypervariable region of G identified the possibility of positive selection occurring in this region of the protein (19), and a more recent study described positively selected sites in this region of G (20). These reports imply that RSV G mutates to contain residue changes that are advantageous for the virus. We chose to study whether the duplication found in BA RSV strains had an advantage for RSV, in vitro and in vivo.
We hypothesized that the duplicated region serves to enhance one or more of the functions of the G protein. Due to the small number of full-length BA G sequences, and an even smaller number of full-length BA strain genomes available in the GenBank database, we designed a consensus BA G nucleotide sequence and cloned it into the A2-line19F genetic background. Additionally, we generated a BA G with the duplicated sequence deleted, termed BA GΔdup, to directly address the role of the duplication in RSV infection. Using these recombinant viruses, we were able to show that the duplication plays a role in virus attachment to cells, which is associated with a slight advantage of the virus in vitro. Our results suggest a role for the duplication in enhancing the function of the G protein to aid in the spread and infectivity of RSV.
MATERIALS AND METHODS
Cloning, rescue, and purification of recombinant viruses.
For generating the consensus sequence of BA G, the full G gene nucleotide sequences deposited under GenBank accession numbers AB117522, DQ270227, HQ699287-HQ699310, JN032115-JN032117, and JN032119-JN032120 were obtained. The sequences were aligned using MegAlign from the DNASTAR Lasergene Suite. The alignment figure (see Fig. 1) was generated using Geneious software version 5.5 (21). A construct harboring the SacI to SacII fragment of pSynkRSVline19F (described in reference 22) with the consensus sequence generated by the BA G alignment in place of A2 G was obtained from GeneArt. For BA GΔdup, the same consensus sequence was used, except that the duplicated 60 nt were deleted from the consensus sequence. A similar construct was obtained from GeneArt. The SacI-SacII fragment carrying the A2 G gene was excised from the pSynkRSVline19F construct and replaced with either the BA G SacI-SacII fragment or the BA GΔdup SacI-SacII fragment. Both constructs were confirmed to contain the correct G constructs by restriction fragment length polymorphism (RFLP) analysis.
FIG 1.

Recombinant virus design. (A) Amino acid alignment used to generate the consensus sequences. Numbering is based on the consensus. Amino acid disagreements from the consensus are indicated. The final QKL residues were not included in the consensus used for cloning. The duplication spans residues 258 to 277. (B) Subclones harboring the designed consensus sequences of BA G or BA GΔdup were obtained from GeneArt. SacI and SacII restriction sites flanking the G genes were utilized to clone the G constructs into pSynkRSVline19F in place of A2 G. Important domains of G are labeled, and the amino acid sequence of the duplication is shown, with the initial run of amino acids in regular type and the inexact duplication in italics.
The two antigenomic plasmids harboring BA G or BA GΔdup were used to rescue the corresponding viruses as previously described (22). Briefly, the antigenome-containing bacterial artificial chromosomes were transfected into BSR-T7/5 cells in 6-well plates along with codon-optimized expression plasmids encoding RSV N, P, M2-1, and L. After 48 h, the cells were passaged into T-25 flasks and subsequently passaged every other day until 70% cytopathic effect (CPE) and mKate2 fluorescence were visible throughout the flask. At that time, the cells were scraped into their media and frozen at −80°C. Master and working stocks were generated in HEp-2 cells as previously described (23). After the working stocks were generated, viral RNA was isolated using the QIAamp viral RNA minikit (Qiagen, Valencia, CA) in order to sequence the genomes of the working stocks. Reverse transcription (RT) was performed using G- or F-specific primers, and PCR primers specific to G or F were used to amplify the G and F genes. For the rest of the genome, reverse transcription was performed using random hexamer primers. PCR primers for seven amplicons were designed based on the expected sequences and were used for amplification of regions of the virus other than G and F. Primer sequences are available upon request. The sequences of the G and F genes from the working stocks of A2-K-BAG-line19F and A2-K-BAGΔdup-line19F were determined to be the expected sequences. Additionally, for A2-K-BAG-line19F and A2-K-BAGΔdup-line19F, there was no evidence of recombination of viral genome with codon-optimized helper plasmid-derived RNA, as all genes for the four helper proteins were devoid of codon-optimized sequence.
Purified virus stocks were generated by infecting subconfluent HEp-2 cells in T-182 flasks with virus master stocks. When CPE and mKate2 fluorescence were evident throughout at least 70% of the flasks, the flasks were placed at −80°C. Material in the flasks was thawed in a 37°C water bath, with vigorous shaking, before the contents were transferred to conical tubes. Cell debris was removed by centrifugation at 931 × g for 10 min at 4°C. The supernatant was layered onto a cushion of minimal essential medium (MEM) containing 20% sucrose and centrifuged at 23,000 rpm in an SW32 rotor (Beckman Coulter Optima L-90K ultracentrifuge) at 4°C for 3 h, prior to overnight storage at 4°C. The supernatants were removed, and virus pellets were resuspended in 500 μl MEM, aliquoted, and snap-frozen for storage at −80°C until further use.
Cells and mice.
HEp-2 and 293T cells obtained from the ATCC were maintained in Eagle's modified essential medium (EMEM) containing 10% fetal bovine serum (FBS) and a 1-μg/ml solution of penicillin, streptomycin, and amphotericin B (PSA). BEAS-2B cells were maintained in RPMI medium containing 10% FBS and 1 μg/ml PSA. CHO-K1 and CHO pgsD-677 cells were maintained in F12-K with 10% FBS and 1 μg/ml PSA. CHO pgsD-677 cells were also supplemented with 1.5 g/liter sodium bicarbonate.
Seven- to 8-week old-female BALB/c mice were obtained from the Jackson Laboratories (Bar Harbor, ME). Mice were housed in specific-pathogen-free facilities, and all experiments were performed according to the rules and regulations set by the Emory University Institutional Animal Care and Use Committee (IACUC).
In vitro growth analyses.
Subconfluent BEAS-2B cells in 6-well plates were infected, in duplicate, at a multiplicity of infection (MOI) of 0.01 of A2-K-BAG-line19F or A2-K-BAGΔdup-line19F. After 1 h of rocking at room temperature, the inoculum was washed from the cells, and 2 ml of complete growth medium was added to each well. At 12, 24, 48, 72, and 96 h postinfection, cells were scraped into the medium, aliquoted, and frozen at −80°C. Titration of virus in the samples was performed by use of a focus-forming unit assay on HEp-2 cells in 96-well plates, as described previously (24). Samples were titrated in triplicate.
The infectivity of A2-K-BAG-line19F and A2-K-BAGΔdup-line19F was determined in CHO-K1 cells and in CHO pgs-D677 cells by a fluorescent focus-forming assay, as described previously (24). Infectivity of three vials of each virus was determined in duplicate, and the experiment was repeated three times.
Binding assays.
Subconfluent BEAS-2B cells in 6-well plates were inoculated, in duplicate, at an MOI of 1.0 of A2-K-BAG-line19F or A2-K-BAGΔdup-line19F. Virus was adsorbed to the cells for 2 h at 4°C. Extra inoculum was frozen at −20°C for further use. After the 2-h incubation, the inoculum was removed, and the cells were washed three times in cold phosphate-buffered saline (PBS). Two hundred microliters of cold radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich, St. Louis, MO; catalog number R0278) containing HALT protease inhibitor cocktail (Thermo Scientific, Waltham, MA) was added to each well. Cells were removed from the wells by pipetting in lysis buffer, and lysates were transferred to microcentrifuge tubes and frozen at −20°C until used for Western blot analysis. For binding assays in CHO-K1 and CHO pgsD-677 cells in 12-well plates, purified virus stocks were used. Inocula for the two viruses were normalized based on the N levels present in each stock, as determined by Western blotting (see “Western blots” below). All other steps were carried out as for BEAS-2B cells, except that 100 μl lysis buffer was used to lyse the cells in each well.
Western blots.
Prior to Western blot analysis, lysates were cleared by centrifugation at 12,000 × g for 5 min. Protein in binding assay lysates, binding assay inoculum samples, or purified virus stocks were separated via sodium dodecyl sulfide-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred to polyvinylidene fluoride (PVDF) membranes. Western blot analyses of purified virus stocks were performed by sequentially probing with monoclonal antibody 131-2G (anti-RSV G; Millipore catalog number MAB858-2), motavizumab (anti-RSV F; generously provided by Nancy Ulbrandt, MedImmune), or D-14 (anti-RSV N; a gift from Edward Walsh, University of Rochester), followed by the appropriate peroxidase-conjugated anti-mouse or anti-human secondary antibodies (Jackson ImmunoResearch, West Grove, PA). Chemiluminescent signal was developed using Western Bright Quantum substrate (Advansta, Menlo Park, CA) and detected on a ChemiDoc XRS analyzer (Bio-Rad, Hercules, CA). Antibodies were stripped from the membrane using Restore Western protein stripping buffer (Thermo Scientific) and reprobed for the next protein in the sequence.
For the binding assays, membranes were probed for N as described above, followed by probing for GAPDH (glyceraldehyde-3-phosphate dehydrogenase) using antibody clone 6C5 (Life Technologies) and a peroxidase-conjugated anti-mouse secondary antibody. Signal was detected using Western Bright Quantum and a ChemiDoc XRS. Densitometry was performed using Image Lab software (Bio-Rad). Densitometry of N in lysates (bound virus) was normalized to N in inoculum as well as to GAPDH in lysates prior to comparison between groups.
Quantification of lung viral load.
Seven- to 8-week-old female BALB/c mice were inoculated intranasally with 1 × 105 PFU of A2-K-BAG-line19F or A2-K-BAGΔdup-line19F. On day 4, 6, 8, or 10 postinfection, the mice were euthanized and the left lung from each mouse was collected and homogenized for viral load determination as previously described (23, 25). Titration was performed on HEp-2 cells in 24-well plates using an immunodetection plaque assay.
Competitive infection assay.
Subconfluent BEAS-2B cells in 6-well plates were infected singly with A2-K-BAG-line19F or A2-K-BAGΔdup-line19F at an MOI of 1.0 or were infected with both viruses at a ratio of 1:1 (MOI, 0.5 of each), 10:1 (MOIs, 1 and 0.1, respectively), or 1:10 (MOIs, 0.1 and 1.0, respectively). Twenty-four or 48 h postinfection, supernatant was removed, and RNA was extracted from cells using TRIzol (Life Technologies) reagent according to the manufacturer's instructions. After RNA extraction, RNA was quantified and 1 μg total RNA was used as the template in a reverse transcription reaction for G messenger or antigenomic RNA using a G-specific primer, Gr (23), and SuperScript III reverse transcriptase. Equal volumes of each cDNA generated from reverse transcription were used as templates in PCR amplification of a portion of the G gene encoding the duplicated region. PCR was performed using Platinum PCR supermix (Life Technologies) and primers Gr and BA-5801F (5′-CCACCAATCCACACAAACTCAGCC-3′). Expected PCR product sizes were 709 nt for A2-K-BAG-line19F and 649 nt for A2-K-BAGΔdup-line19F. Equal volumes of PCR products were separated by agarose gel electrophoresis in a 1.5% agarose gel.
Microneutralization assay.
A microneutralization assay based on counting focus-forming units (FFU) has been described previously (24). Briefly, the F-specific monoclonal antibody motavizumab was serially diluted in PBS. Serial dilutions of antibody were mixed with an equivalent volume of either A2-K-BAG-line19F or A2-K-BAGΔdup-line19F at a concentration of 2 FFU/μl. The antibody-virus mixtures were incubated for 1 h at 37°C. After incubation, 50-μl volumes of the mixtures were used to infect subconfluent HEp-2 cells in 96-well plates in duplicate by spinoculation at 2,900 × g for 30 min at 4°C. Infected cells were overlaid with complete growth medium supplemented with 0.75% methylcellulose. FFU were counted approximately 36 h postspinoculation, and the level of neutralization was determined by comparing each antibody-virus condition to a PBS-virus negative control.
RESULTS
Generation of recombinant viruses.
To design a consensus nucleotide sequence of the BA G gene, we aligned the full-length G nucleotide sequences from 30 BA isolates present in the GenBank nucleotide database which displayed the full-length G gene, and we excluded the sequences of the original BA isolates. The protein sequence of our BA consensus strain and those strains used in its design are shown as an amino acid alignment in Fig. 1A. Our rationale for excluding the original isolates from the design was that the duplication has mutated since those original strains were isolated. Initially, the duplicated region was an exact copy of 60 nt (3). Over time, the duplicated region in most BA isolates harbored sequences coding for one to four mutated amino acids that differed from the original sequences. We chose to generate our consensus based on sequences that were most closely related to strains that were currently circulating at the time we initiated our study.
We obtained BA G and BA GΔdup gene subclones from GeneArt and cloned the genes into our bacterial artificial chromosome construct containing the antigenome of strain A2-line19F (clone construction shown in Fig. 1B). We generated two viruses with these constructs, A2-K-BAG-line19F and A2-K-BAGΔdup-line19F.
After rescuing the recombinant viruses, we purified stocks of each through a 20% sucrose cushion. We used the purified stocks to determine whether the two viruses incorporated equivalent amounts of glycoproteins into the virions. Western blots of the purified viruses indicated that both viruses incorporated similar amounts of the fusion glycoprotein (F) but that A2-K-BAGΔdup-line19F incorporated greater amounts of G than A2-K-BAG-line19F (Fig. 2). Additionally, the G protein from A2-K-BAG-line19F was of a greater molecular mass than that from A2-K-BAGΔdup-line19F, likely due to an increase in glycosylation of the mature form of G (Fig. 2). There are 10 additional serines and threonines in the duplicated region, which could serve as a new O-glycosylation anchor residue.
FIG 2.
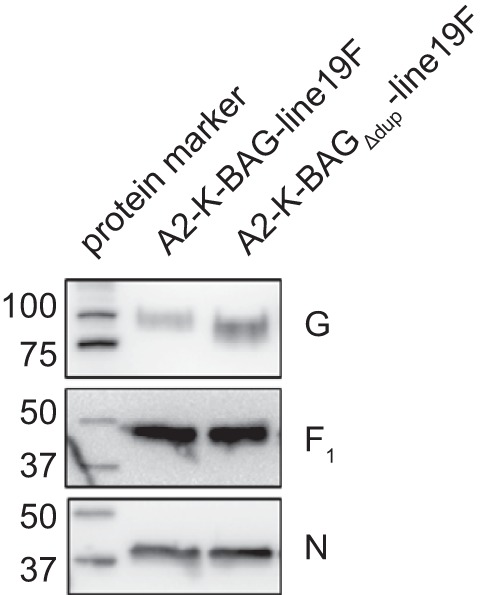
Glycoprotein incorporation into recombinant viruses. Virus stocks of each recombinant virus were purified through a 20% sucrose cushion and subjected to SDS-PAGE and Western blotting for the G or F glycoproteins or the RSV nucleoprotein as a loading control. Images shown are from Western blot analyses performed on a single membrane probed for each protein sequentially and represent results from the purification of two independent sets of virus stocks. Molecular masses of marker bands are shown in kilodaltons.
Replication of the recombinant BA strains in vitro.
We tested the ability of the two viruses to replicate in a human bronchial epithelial cell line, BEAS-2B, by performing multistep growth curve analyses with an initial multiplicity of infection (MOI) of 0.01 for each virus. There was no distinguishable difference in replication when the virus expressing BA G and the one expressing BA GΔdup were compared (Fig. 3A).
FIG 3.
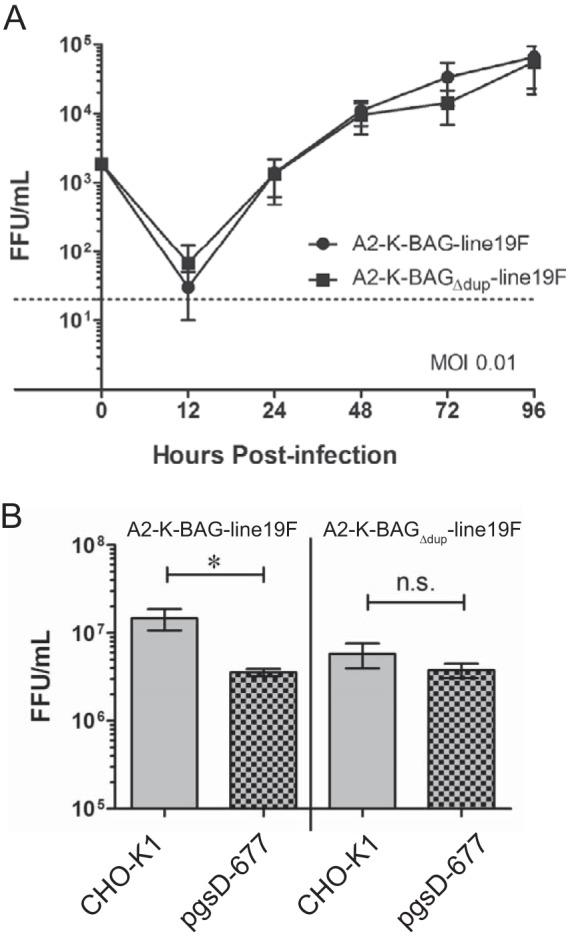
Recombinant BA strain replication in vitro. (A) BEAS-2B cells were infected with the two viruses indicated at an MOI of 0.01. Cells were scraped into medium at the indicated time points, and virus was titrated by a fluorescent focus-forming assay in HEp-2 cells. The data are a combination of three experiments. There were no significant differences based on a two-way analysis of variance (ANOVA). (B) Single vials of virus stocks were titrated in both CHO-K1 and pgsD-677 cell lines. Each experiment was done in triplicate, and the graph shows a combination of results of three experiments. *, P < 0.05 (by one-way ANOVA and Tukey's multiple-comparison test). n.s., not significant.
RSV G has been reported to bind many cell factors, including the fractalkine receptor (CX3CR1) (26), surfactant protein A (27), annexin II (28), DC-SIGN, and L-SIGN (16), but the majority of G binding to cells in vitro is thought to occur through attachment to heparan sulfate (HS)-containing glycosaminoglycans (GAGs) (29, 30). We measured the GAG dependency of A2-K-BAG-line19F and A2-K-BAGΔdup-line19F by testing the infectivities of the viruses in Chinese hamster ovary (CHO-K1) cells and CHO cells deficient in generating heparan sulfate (pgsD-677). A2-K-BAG-line19F exhibited statistically significantly greater infectivity in CHO-K1 cells than in pgsD-677 cells (Fig. 3B). The virus with G lacking the duplication, A2-K-BAGΔdup-line19F, showed no difference in infectivity between the two cell lines (Fig. 3B). These data show that the virus with the duplication is more dependent on HS for infectivity.
Role of the G duplicated region in virus-cell binding.
We next sought to determine if the duplicated region played a role in the function of G as the RSV attachment protein. We inoculated BEAS-2B cells at an MOI of 1.0 at 4°C to allow virus binding to cells but prevent virus fusion with cells. After sufficient time for virus binding, the cells were lysed and the amounts of bound virus were compared by Western blotting for RSV N levels in the cell lysates. Lysate N levels were normalized to N levels in the inoculum, as well as to GAPDH levels in the cell lysates, to control for equal protein loading. We found that A2-K-BAG-line19F bound more efficiently to BEAS-2B cells than A2-K-BAGΔdup-line19F (Fig. 4A and B). These data demonstrate a role for the duplicated region of BA G in enhancing the in vitro attachment function of the RSV BA G protein.
FIG 4.

Recombinant BA strain binding to cells in vitro. (A and B) BEAS-2B cells were inoculated with the indicated viruses at an MOI of 1.0, and virus was allowed to adsorb to cells at 4°C. Inoculum was removed, and cells were washed three times in cold PBS to remove unbound virus. Cells were lysed, and lysates, along with an aliquot of original inoculum, was subjected to SDS-PAGE and Western blotting with an anti-N monoclonal antibody. GAPDH was also probed as a loading control. (A) Representative Western blots of binding assay. (B) Combination of densitometry results of the three experiments illustrated in panel A. The amount of bound N was normalized to N in the inoculum as well as GAPDH prior to comparison between groups. *, P < 0.05 (by paired t test). (C) Representative Western blots from binding assay performed with CHO-K1 and pgsD-677 cell lines. For experiments in CHO-K1 and pgsD-677 cells, sucrose-purified virus stocks were used, and the infection inoculum was normalized based on N protein levels in purified stocks. (D) Combination of densitometry results of the three experiments illustrated in panel C. *, P < 0.05 (by paired t test). ns, not significant.
Because our infectivity data for CHO-K1 and pgsD-677 cells suggested a role for the duplication in binding to HS, we also determined the binding efficiencies of the viruses on these two cell lines. We found that A2-K-BAG-line19F bound more efficiently than A2-K-BAGΔdup-line19F to CHO-K1 (HS-expressing) cells but that there was no difference in binding efficiency between the two viruses to pgsD-677 (HS-deficient) cells (Fig. 4C and D). These data implicate the duplicated region in BA G in binding HS on the cell surface.
Advantage of A2-K-BAG-line19F in vitro.
While growth curve analyses are one means to determine virus replicative fitness, we sought to determine whether the duplication in G provides a competitive advantage to RSV. To address this by a more direct approach, we performed a competitive infection experiment by infecting HEp-2 cells simultaneously with A2-K-BAG-line19F and A2-K-BAGΔdup-line19F and assessing G RNA levels in the cells by reverse transcription followed by PCR. When cells were infected with both viruses at an MOI of 0.5 (1:1 ratio), even as early as 24 h postinfection, most of the G-specific RNA in the cells was from the virus harboring BA G, not BA GΔdup (Fig. 5). During infection with a 10:1 ratio of A2-K-BAG-line19F to A2-K-BAGΔdup-line19F, virtually no BA GΔdup G RNA was detectable, whereas with a 1:10 infection ratio of the two viruses, BA G-specific G RNA was readily visible (Fig. 5). In addition to the results shown in Fig. 5, we performed control experiments to confirm that the reaction conditions in our PCR did not favor one template over the other (data not shown). These results show that A2-K-BAG-line19F has an advantage when in direct competition with A2-K-BAGΔdup-line19F.
FIG 5.
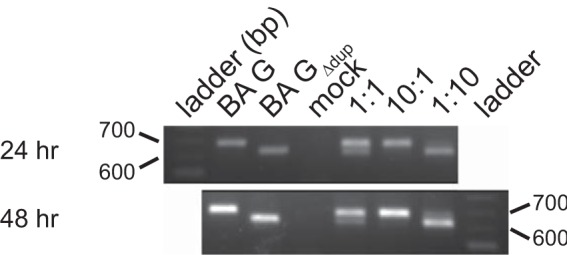
Competitive-infection assay. HEp-2 cells were infected with A2-K-BAG-line19F, A2-K-BAGΔdup-line19F, or both viruses at a ratio of 1:1, 10:1, or 1:10. Twenty-four and 48 h postinfection, RNA was harvested from cells and subjected to reverse transcription (RT)-PCR to amplify a portion of the G gene. Equal amounts of RNA were used for the reverse transcription, and equal volumes of cDNA were used as templates for the PCR. Gels are representative of two experiments. Expected PCR product sizes are 709 bp for A2-K-BAG-line19F and 649 bp for A2-K-BAGΔdup-line19F.
No effect of the duplicated region in G on mouse lung viral load.
We investigated whether the attachment enhancement provided by the duplication in BA G contributes to advantages in vivo. We measured the lung viral load in BALB/c mice infected with either A2-K-BAG-line19F or A2-K-BAGΔdup-line19F. In three independent experiments, A2-K-BAG-line19F exhibited lung viral loads equivalent to those of A2-K-BAGΔdup-line19F at every time point tested (Fig. 6). We found no statistically significant difference in the rate of clearance of the two viruses in BALB/c mice (Fig. 6, middle panel). Taken together, these results show that the duplication in G present in RSV BA strains, while enhancing virus binding to cells and fitness in vitro, does not augment viral load in the BALB/c mouse model.
FIG 6.
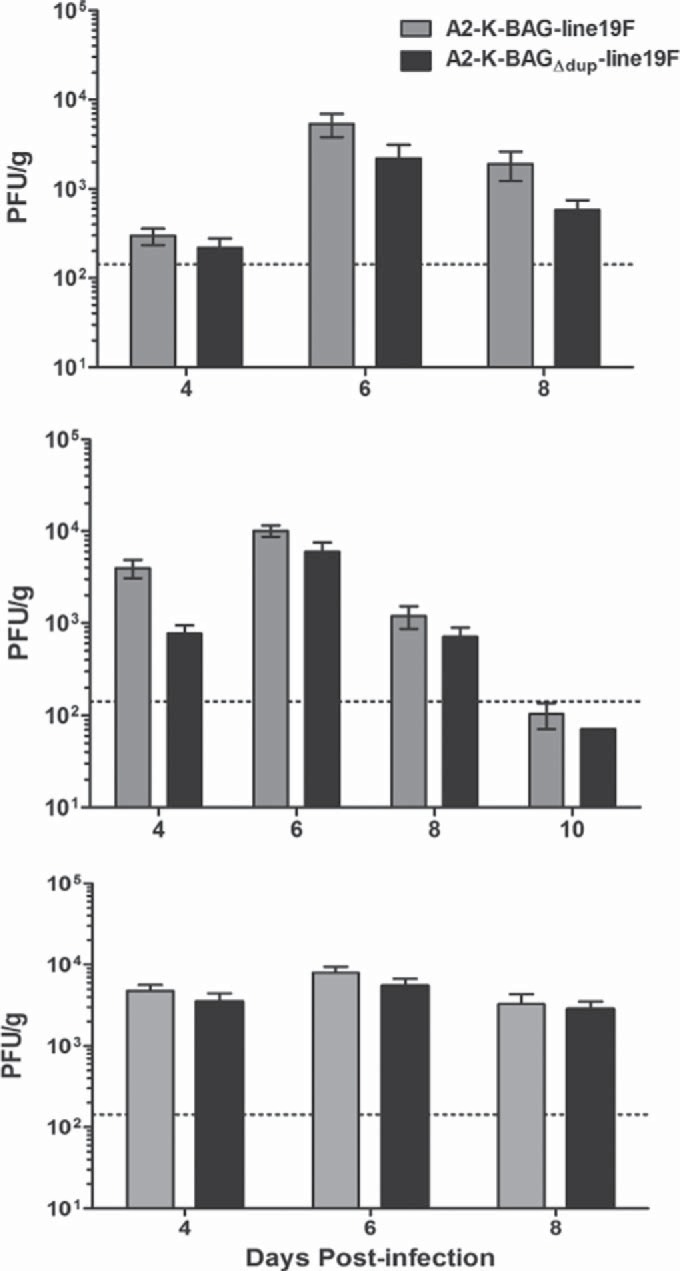
Recombinant BA strain lung viral load in BALB/c mice. Groups of 5 BALB/c mice were infected with 1 × 105 PFU of either A2-K-BAG-line19F or A2-K-BAGΔdup-line19F, and lungs were harvested on day 4, 6, 8, or 10 postinfection. Viral load was quantified by an immunodetection plaque assay. Each graph is an independent experiment. There were no significant differences. A horizontal dotted line indicates the limit of detection.
No effect of the duplicated region in G on in vitro neutralization.
Additional glycosylation events were shown to contribute to escape from neutralizing antibodies by both human immunodeficiency virus type 1 (31) and influenza A virus (32). Because the duplication in BA virus G proteins results in additional glycosylation sites, we hypothesized that a more heavily glycosylated G protein would shield F from neutralizing antibodies. To test this, we performed microneutralization assays using the F-specific monoclonal antibody motavizumab. Our data showed no difference in neutralization of A2-K-BAG-line19F and A2-K-BAGΔdup-line19F by motavizumab (Fig. 7). We also tested neutralization by a polyclonal antibody and found no difference between A2-K-BAG-line19F and A2-K-BAGΔdup-line19F (data not shown), suggesting that the duplication in BA G relative to BA GΔdup has no effect on antibody neutralization.
FIG 7.
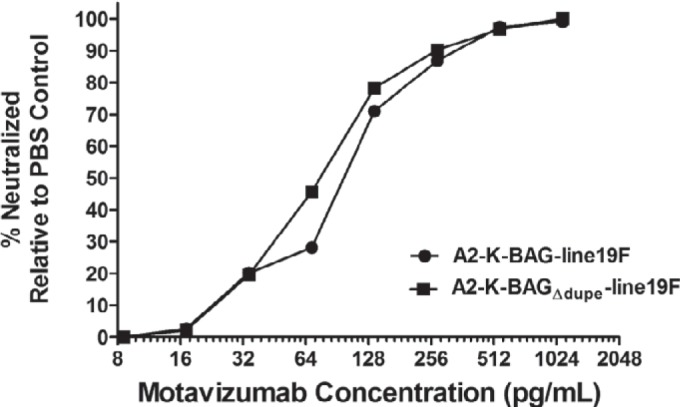
Microneutralization assay of recombinant BA viruses. A2-K-BAG-line19F and A2-K-BAGΔdup-line19F were incubated with PBS or with serial dilutions of motavizumab, an F-specific monoclonal antibody, and then used to infect HEp-2 cells. The level of neutralization of each virus was calculated by comparing the number of FFU present when the virus was mixed with antibody dilutions to the number of FFU present when the virus was mixed with PBS. The results shown are representative of three replicate experiments.
DISCUSSION
The RSV G glycoprotein is capable of tolerating many mutations, with perhaps the most striking change occurring in the last 2 decades exemplified by the emergence of a 60-nt duplication in B strains and a 72-nt duplication in A strains and the subsequent increased prevalence of these genotypes in nature. The role of these mutations in RSV infection has not previously been reported. We generated recombinant virus strains expressing consensus G proteins from the BA subgroup with or without the 60-nt duplication to determine a role for the duplication in the virus life cycle.
In the context of viruses expressing line 19F, we were able to show a role for the duplication in G in virus attachment to cells (Fig. 4). We also showed that this enhanced attachment by BA G-expressing viruses relative to BA GΔdup-expressing viruses was dependent on heparan sulfate-containing GAGs (Fig. 3 and 4). Although the duplicated region does not encompass the linear heparin binding domain characterized on RSV G (33), there are 10 potential O-glycosylation sites in the duplicated region (serine and threonine residues). In the study first describing strains with the duplication, Western blot analyses were performed in the presence and absence of tunicamycin to identify the size difference in G conferred by the additional 20 amino acids (3). It was unclear in these blots whether additional glycosylation events occurred in the presence of the duplication, but our Western blot of purified virions indicates that the two viruses harbor mature G proteins of different apparent molecular masses (Fig. 2). One study suggested that heparin-like structures on G were critical for the role of G in attachment progressing to viral infection (34). These two characteristics of G lead to our working hypothesis that the extra glycans likely present on BA G are responsible for the enhanced binding of BA G relative to BA GΔdup. Studies are needed to determine whether mutagenesis of the additional 10 glycosylation sites abrogates the BA G enhanced cell-binding phenotype. Additionally, while heparan sulfate-containing GAGs are important for RSV infectivity in vitro (29, 30, 35), heparan sulfate-containing GAGs are not the most common GAGs found on the luminal surface of human airways or in mucus (36). Heparan sulfate GAGs may instead be one of multiple GAGs bound by RSV, and further work may identify the role of the duplicated region in BA G in binding to other GAGs, especially those found at higher frequency on airway cells.
In addition to a general enhancement of binding efficiency to BEAS-2B cells, we showed that the duplication provides an advantage in a competitive infection assay (Fig. 5). While we did not find a significant difference in viral load in BALB/c mice (Fig. 6), it is possible that a more striking difference would be present in humans, where RSV replicates to higher titers. The advantages in attachment and competition identified in vitro suggest a mechanism for how BA strains became predominant after their emergence. It is conceivable that a slight growth advantage in vivo could lead to greater transmission of a virus strain in the human population. Due to limitations of RSV animal models, direct-contact transmission studies with RSV are not reported but might provide further clues to the selective advantage of the duplication in BA strain G genes if they could be achieved.
ON1 subgroup A RSV strains also harboring a duplication in the G protein are beginning to predominate among circulating RSV strains (11–13). The duplicated region in G of ON1 strains encodes 11 additional potential O-glycosylation sites (10). We speculate that a binding enhancement and mechanism similar to those of BA strains may underlie the emergence of the ON1 strains. Further studies specifically on the duplication in subgroup A strains are needed to confirm this but could easily be performed based on the results and reagents established here. Further, a recombinant B strain of RSV was recently described (37) and would provide an additional tool to investigate the role of the duplication in RSV spread and pathogenesis.
ACKNOWLEDGMENTS
This work was supported by NIH grants 1R01AI087798 and 1U19AI095227 (to M.L.M.) and the Children's Center for Immunology and Vaccines.
We thank Nancy Ulbrandt (MedImmune LLC) for the motavizumab MAb. We thank Ursula Buchholz and Karl-Klaus Conzelmann for the BSR-T7/5 cells. We thank Edward Walsh for the D14 monoclonal antibody.
REFERENCES
- 1.Collins PL, Crowe JE Jr. 2007. Respiratory syncytial virus and metapneumovirus, p 1601–1646. In Knipe DM, Howley PM (ed), Fields virology, vol 2 Lippincott, Williams, and Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Peret TC, Hall CB, Schnabel KC, Golub JA, Anderson LJ. 1998. Circulation patterns of genetically distinct group A and B strains of human respiratory syncytial virus in a community. J Gen Virol 79(Pt 9):2221–2229. [DOI] [PubMed] [Google Scholar]
- 3.Trento A, Galiano M, Videla C, Carballal G, Garcia-Barreno B, Melero JA, Palomo C. 2003. Major changes in the G protein of human respiratory syncytial virus isolates introduced by a duplication of 60 nucleotides. J Gen Virol 84:3115–3120. doi: 10.1099/vir.0.19357-0. [DOI] [PubMed] [Google Scholar]
- 4.Trento A, Viegas M, Galiano M, Videla C, Carballal G, Mistchenko AS, Melero JA. 2006. Natural history of human respiratory syncytial virus inferred from phylogenetic analysis of the attachment (G) glycoprotein with a 60-nucleotide duplication. J Virol 80:975–984. doi: 10.1128/JVI.80.2.975-984.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trento A, Casas I, Calderon A, Garcia-Garcia ML, Calvo C, Perez-Brena P, Melero JA. 2010. Ten years of global evolution of the human respiratory syncytial virus BA genotype with a 60-nucleotide duplication in the G protein gene. J Virol 84:7500–7512. doi: 10.1128/JVI.00345-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y, Xu W, Shen K, Xie Z, Sun L, Lu Q, Liu C, Liang G, Beeler JA, Anderson LJ. 2007. Genetic variability of group A and B human respiratory syncytial viruses isolated from 3 provinces in China. Arch Virol 152:1425–1434. doi: 10.1007/s00705-007-0984-3. [DOI] [PubMed] [Google Scholar]
- 7.Zlateva KT, Vijgen L, Dekeersmaeker N, Naranjo C, Van Ranst M. 2007. Subgroup prevalence and genotype circulation patterns of human respiratory syncytial virus in Belgium during ten successive epidemic seasons. J Clin Microbiol 45:3022–3030. doi: 10.1128/JCM.00339-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Almajhdi FN, Farrag MA, Amer HM. 2014. Group B strains of human respiratory syncytial virus in Saudi Arabia: molecular and phylogenetic analysis. Virus Genes 48:252–259. doi: 10.1007/s11262-013-1030-z. [DOI] [PubMed] [Google Scholar]
- 9.Agoti CN, Mayieka LM, Otieno JR, Ahmed JA, Fields BS, Waiboci LW, Nyoka R, Eidex RB, Marano N, Burton W, Montgomery JM, Breiman RF, Nokes DJ. 2014. Examining strain diversity and phylogeography in relation to an unusual epidemic pattern of respiratory syncytial virus (RSV) in a long-term refugee camp in Kenya. BMC Infect Dis 14:178. doi: 10.1186/1471-2334-14-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eshaghi A, Duvvuri VR, Lai R, Nadarajah JT, Li A, Patel SN, Low DE, Gubbay JB. 2012. Genetic variability of human respiratory syncytial virus A strains circulating in Ontario: a novel genotype with a 72 nucleotide G gene duplication. PLoS One 7:e32807. doi: 10.1371/journal.pone.0032807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agoti CN, Otieno JR, Gitahi CW, Cane PA, Nokes DJ. 2014. Rapid spread and diversification of respiratory syncytial virus genotype ON1, Kenya. Emerg Infect Dis 20:950–959. doi: 10.3201/eid2006.131438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Avadhanula V, Chemaly RF, Shah DP, Ghantoji SS, Azzi JM, Aideyan LO, Mei M, Piedra PA. 2015. Infection with novel respiratory syncytial virus genotype Ontario (ON1) in adult hematopoietic cell transplant recipients, Texas, 2011-2013. J Infect Dis 211:582–589. doi: 10.1093/infdis/jiu473. [DOI] [PubMed] [Google Scholar]
- 13.Pierangeli A, Trotta D, Scagnolari C, Ferreri ML, Nicolai A, Midulla F, Marinelli K, Antonelli G, Bagnarelli P. 2014. Rapid spread of the novel respiratory syncytial virus A ON1 genotype, central Italy, 2011 to 2013. Euro Surveill 19:pii=20843. [DOI] [PubMed] [Google Scholar]
- 14.Levine S, Klaiber-Franco R, Paradiso PR. 1987. Demonstration that glycoprotein G is the attachment protein of respiratory syncytial virus. J Gen Virol 68(Pt 9):2521–2524. doi: 10.1099/0022-1317-68-9-2521. [DOI] [PubMed] [Google Scholar]
- 15.Bukreyev A, Yang L, Fricke J, Cheng L, Ward JM, Murphy BR, Collins PL. 2008. The secreted form of respiratory syncytial virus G glycoprotein helps the virus evade antibody-mediated restriction of replication by acting as an antigen decoy and through effects on Fc receptor-bearing leukocytes. J Virol 82:12191–12204. doi: 10.1128/JVI.01604-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson TR, McLellan JS, Graham BS. 2012. Respiratory syncytial virus glycoprotein G interacts with DC-SIGN and L-SIGN to activate ERK1 and ERK2. J Virol 86:1339–1347. doi: 10.1128/JVI.06096-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Melendi GA, Bridget D, Monsalvo AC, Laham FF, Acosta P, Delgado MF, Polack FP, Irusta PM. 2011. Conserved cysteine residues within the attachment G glycoprotein of respiratory syncytial virus play a critical role in the enhancement of cytotoxic T-lymphocyte responses. Virus Genes 42:46–54. doi: 10.1007/s11262-010-0545-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boyoglu-Barnum S, Gaston KA, Todd SO, Boyoglu C, Chirkova T, Barnum TR, Jorquera P, Haynes LM, Tripp RA, Moore ML, Anderson LJ. 2013. A respiratory syncytial virus (RSV) anti-G protein F(ab′)2 monoclonal antibody suppresses mucous production and breathing effort in RSV rA2-line19F-infected BALB/c mice. J Virol 87:10955–10967. doi: 10.1128/JVI.01164-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cane PA, Matthews DA, Pringle CR. 1991. Identification of variable domains of the attachment (G) protein of subgroup A respiratory syncytial viruses. J Gen Virol 72(Pt 9):2091–2096. doi: 10.1099/0022-1317-72-9-2091. [DOI] [PubMed] [Google Scholar]
- 20.Zlateva KT, Lemey P, Vandamme AM, Van Ranst M. 2004. Molecular evolution and circulation patterns of human respiratory syncytial virus subgroup a: positively selected sites in the attachment g glycoprotein. J Virol 78:4675–4683. doi: 10.1128/JVI.78.9.4675-4683.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drummond A, Ashton B, Buxton S, Cheung M, Cooper A, Duran C, Field M, Heled J, Kearse M, Markowitz S, Moir R, Stones-Havas S, Sturrock S, Thierer T, Wilson A. 2011. Geneious, v.5.5. Biomatters, Auckland, New Zealand. [Google Scholar]
- 22.Hotard AL, Shaikh FY, Lee S, Yan D, Teng MN, Plemper RK, Crowe JE Jr, Moore ML. 2012. A stabilized respiratory syncytial virus reverse genetics system amenable to recombination-mediated mutagenesis. Virology 434:129–136. doi: 10.1016/j.virol.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stokes KL, Chi MH, Sakamoto K, Newcomb DC, Currier MG, Huckabee MM, Lee S, Goleniewska K, Pretto C, Williams JV, Hotard A, Sherrill TP, Peebles RS Jr, Moore ML. 2011. Differential pathogenesis of respiratory syncytial virus (RSV) clinical isolates in BALB/c mice. J Virol 85:5782–5793. doi: 10.1128/JVI.01693-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meng J, Lee S, Hotard AL, Moore ML. 2014. Refining the balance of attenuation and immunogenicity of respiratory syncytial virus by targeted codon deoptimization of virulence genes. mBio 5(5):e01704-14. doi: 10.1128/mBio.01704-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hotard AL, Lee S, Currier MG, Crowe JE Jr, Sakamoto K, Newcomb DC, Peebles RS Jr, Plemper RK, Moore ML. 2015. Identification of residues in the human respiratory syncytial virus fusion protein that modulate fusion activity and pathogenesis. J Virol 89:512–522. doi: 10.1128/JVI.02472-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tripp RA, Jones LP, Haynes LM, Zheng H, Murphy PM, Anderson LJ. 2001. CX3C chemokine mimicry by respiratory syncytial virus G glycoprotein. Nat Immunol 2:732–738. doi: 10.1038/90675. [DOI] [PubMed] [Google Scholar]
- 27.Hickling TP, Malhotra R, Bright H, McDowell W, Blair ED, Sim RB. 2000. Lung surfactant protein A provides a route of entry for respiratory syncytial virus into host cells. Viral Immunol 13:125–135. doi: 10.1089/vim.2000.13.125. [DOI] [PubMed] [Google Scholar]
- 28.Malhotra R, Ward M, Bright H, Priest R, Foster MR, Hurle M, Blair E, Bird M. 2003. Isolation and characterisation of potential respiratory syncytial virus receptor(s) on epithelial cells. Microbes Infect 5:123–133. doi: 10.1016/S1286-4579(02)00079-5. [DOI] [PubMed] [Google Scholar]
- 29.Hallak LK, Spillmann D, Collins PL, Peeples ME. 2000. Glycosaminoglycan sulfation requirements for respiratory syncytial virus infection. J Virol 74:10508–10513. doi: 10.1128/JVI.74.22.10508-10513.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Techaarpornkul S, Collins PL, Peeples ME. 2002. Respiratory syncytial virus with the fusion protein as its only viral glycoprotein is less dependent on cellular glycosaminoglycans for attachment than complete virus. Virology 294:296–304. doi: 10.1006/viro.2001.1340. [DOI] [PubMed] [Google Scholar]
- 31.Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, Salazar-Gonzalez JF, Salazar MG, Kilby JM, Saag MS, Komarova NL, Nowak MA, Hahn BH, Kwong PD, Shaw GM. 2003. Antibody neutralization and escape by HIV-1. Nature 422:307–312. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- 32.Job ER, Deng YM, Barfod KK, Tate MD, Caldwell N, Reddiex S, Maurer-Stroh S, Brooks AG, Reading PC. 2013. Addition of glycosylation to influenza A virus hemagglutinin modulates antibody-mediated recognition of H1N1 2009 pandemic viruses. J Immunol 190:2169–2177. doi: 10.4049/jimmunol.1202433. [DOI] [PubMed] [Google Scholar]
- 33.Feldman SA, Hendry RM, Beeler JA. 1999. Identification of a linear heparin binding domain for human respiratory syncytial virus attachment glycoprotein G. J Virol 73:6610–6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bourgeois C, Bour JB, Lidholt K, Gauthray C, Pothier P. 1998. Heparin-like structures on respiratory syncytial virus are involved in its infectivity in vitro. J Virol 72:7221–7227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donalisio M, Rusnati M, Cagno V, Civra A, Bugatti A, Giuliani A, Pirri G, Volante M, Papotti M, Landolfo S, Lembo D. 2012. Inhibition of human respiratory syncytial virus infectivity by a dendrimeric heparan sulfate-binding peptide. Antimicrob Agents Chemother 56:5278–5288. doi: 10.1128/AAC.00771-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Monzon ME, Casalino-Matsuda SM, Forteza RM. 2006. Identification of glycosaminoglycans in human airway secretions. Am J Respir Cell Mol Biol 34:135–141. doi: 10.1165/rcmb.2005-0256OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lemon K, Nguyen DT, Ludlow M, Rennick LJ, Yuksel S, van Amerongen G, McQuaid S, Rima BK, de Swart RL, Duprex WP. 2015. Recombinant subgroup B human respiratory syncytial virus expressing enhanced green fluorescent protein efficiently replicates in primary human cells and is virulent in cotton rats. J Virol 89:2849–2856. doi: 10.1128/JVI.03587-14. [DOI] [PMC free article] [PubMed] [Google Scholar]