

ABSTRACT
Ovine pulmonary adenocarcinoma is a naturally occurring lung cancer in sheep induced by the Jaagsiekte sheep retrovirus (JSRV). Its envelope glycoprotein (Env) carries oncogenic properties, and its expression is sufficient to induce in vitro cell transformation and in vivo lung adenocarcinoma. The identification of cellular partners of the JSRV envelope remains crucial for deciphering mechanisms leading to cell transformation. We initially identified RALBP1 (RalA binding protein 1; also known as RLIP76 or RIP), a cellular protein implicated in the ras pathway, as a partner of JSRV Env by yeast two-hybrid screening and confirmed formation of RALBP1/Env complexes in mammalian cells. Expression of the RALBP1 protein was repressed in tumoral lungs and in tumor-derived alveolar type II cells. Through its inhibition using specific small interfering RNA (siRNA), we showed that RALBP1 was involved in envelope-induced cell transformation and in modulation of the mTOR (mammalian target of rapamycin)/p70S6K pathway by the retroviral envelope.
IMPORTANCE JSRV-induced lung adenocarcinoma is of importance for the sheep industry. While the envelope has been reported as the oncogenic determinant of the virus, the cellular proteins directly interacting with Env are still not known. Our report on the formation of RALBP/Env complexes and the role of this interaction in cell transformation opens up a new hypothesis for the dysregulation observed upon virus infection in sheep.
INTRODUCTION
Ovine pulmonary adenocarcinoma is a contagious tumor that originates from the distal lung upon infection by the Jaagsiekte sheep retrovirus (JSRV). It is now clearly established that JSRV induces tumors via the oncogenic properties of its envelope (1), which is necessary and sufficient to induce transformation (1–3). The oncogenic property of the JSRV envelope has been evidenced in various cell lines (reviewed in reference 4) and in vivo in mice (5, 6) and in sheep (7). Beside the transmembrane (TM) region, deletions of surface (SU) glycoproteins from the signal peptide to the junction between the SU and TM subunits can abolish the envelope glycoprotein (Env)-induced cell transformation (1). The cytoplasmic tail of TM is essential for cell transformation (8, 9). This region contains an YXXM motif (3) corresponding to a potential consensus site linked to the SH2 domain of the p85 subunit of phosphatidylinositol 3-kinase (PI3K), a kinase that activates the serine/threonine kinase Akt. The PI3K/Akt signaling pathway is essential in cell proliferation, survival, and metabolism (10, 11). Mechanisms potentially involved in tumor formation include extensive cell division as a result of oncogenic mutations, inactivation of cellular senescence, tumor suppressor pathways, or apoptosis mechanisms that may otherwise arrest proliferation or induce death of potential cancer cells (12). Telomerase activation is considered mandatory for tumor cells to escape cell senescence and to gain increased proliferative capacities (13). Complex regulation of telomerase activity may include the PI3K pathway through phosphorylation of telomerase reverse transcriptase (TERT) by Akt (14). Telomerase activity is significantly higher in ovine pulmonary adenocarcinomas compared to control lungs; this suggests that inhibition of cell senescence may be involved in the tumoral process in sheep and in the accumulation of tumoral cells within the lung (15). The regulatory Akt kinase is constitutively activated in ovine tumors and deregulated in primary cultures derived from JSRV-induced cancers; therefore, Akt may be involved in telomerase activation in a proportion of tumors (15). Akt is constitutively activated in various human tumors, including lung cancer (16). In vitro experiments that mimic cellular transformation by JSRV Env expression have implicated Akt as well as Ras/MEK/MAPK (mitogen-activated protein kinase) pathways but in a cell-dependent manner (4, 17, 18).
While the role of the envelope in JSRV-mediated transformation is now well established, the early mechanisms that lead to initiation of cell transformation are still unknown. The importance of HYAL-2, the cellular receptor for JSRV (19), remains unclear and might be cell dependent; it plays no role in transformation of murine cells, but human HYAL-2 suppresses envelope-mediated transformation by increasing its degradation (20, 21). The identification of cellular partners of the JSRV envelope remains crucial for deciphering mechanisms that lead to cell transformation. We identified RALBP1 (RalA binding protein 1; also known as RLIP76 or RIP), a cellular protein implicated in the ras pathway and an effector of RalA (Ras-like protein A) (22), as a partner of the JSRV envelope by yeast two-hybrid screening and confirmed formation of RALBP1/Env complexes in mammalian cells. Through inhibition of RALBP1 expression using specific small interfering RNA (siRNA), we showed that the cellular protein is involved in envelope-induced cell transformation.
MATERIALS AND METHODS
Biological material.
The tumor tissues used in this study were collected immediately postmortem from 10 sheep from milk farms with clinical signs suggestive of lung adenocarcinoma such as dyspnea, altered general status, and evacuation of mucoid fluid through the nostrils. The control lungs were collected from 12 lambs (∼3 months of age) with no clinical signs of respiratory disease at the Corbas slaughterhouse (France). Formal authorization for access to the facility was obtained, and access was granted under the supervision of a veterinarian. None of the animals used in this study were engaged in an experimental protocol. Clinical status was confirmed by pathological examination (F. Thivolet-Béjui, Service d'Anatomopathologie Clinique, Louis Pradel Hospital, Lyon, France). The presence of JSRV in the tumors and its absence in healthy lungs was confirmed by detection of viral genome using seminested PCR. Briefly, DNA was extracted from lung tissues (Fast DNA kit; MP Biomedicals, France) as recommended. The PCRs were performed using 100 ng of DNA with 1× KAPA2G Robust HotStart Ready Mix 2× (Clinisciences, France), 0.2 μM each primer, and PCR-grade H2O up to 25 μl. Primers specific to the exogenous form of JSRV were used, namely, JSRV-53 (5′-GGATTCTTACACAATCACC-3′) in U3-LTR (long terminal repeat) and JSRV-98 (5′-GAGTTGAAATGCTGCATATG-3′) in env for the first round (290 bp for the expected size of the amplicon) of amplification and JSRV-52 (5′-CACCGGATTCTTATATAATC-3′) in U3-LTR and JSRV-98 for the second round (274 bp for the expected size of the amplicon). The initial denaturation step at 95°C for 2 min was followed by 35 cycles, with 1 cycle consisting of 10 s at 95°C, 10 s at 55°C, and 10 s at 72°C.
Ovine primary alveolar type II cells (AECIIs) were isolated from 10 tumors and 8 healthy lungs, phenotypically characterized and cultured as previously described (23). Briefly, cells were plated onto cell culture plates coated with type I and type IV collagens (Sigma) and fibronectin (Millipore) with Quantum 286 synthetic medium (PAA Laboratories, France) selective for epithelial cells complemented with 10 ng/ml human keratinocyte growth factor (KGF) (Abcys, France), 5 ng/ml human hepatocyte growth factor (HGF) (Abcys), 10 U/ml penicillin, and 10 μg/ml streptomycin (PAA). The cells were grown at 37°C in a humidified atmosphere with 5% CO2. MDCK (Madin-Darby canine kidney) and HEK 293T (human embryonic kidney 293 with simian virus 40 [SV40] large T-antigen) cells were grown in complete Dulbecco's modified Eagle's medium (DMEM) (PAA) supplemented with 10% inactivated fetal calf serum (FCS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 1% nonessential amino acids (PAA) at 37°C in a humidified atmosphere with 5% CO2.
Yeast two-hybrid screening for identification of cellular interactants of the cytoplasmic tail.
The cytoplasmic tail (CT) domain sequence of JSRV Env, nucleotides (nt) 7063 to 7197 from the full-length sequence of the South African strain of JSRV (GenBank accession no. NC_001494.1; kindly provided by Gilles Quérat, Marseille, France) (24), was PCR amplified with KOD polymerase (Novagen) using attB1.1 and attB2.1 recombination sites, respectively, fused to forward and reverse primers. The amplified product was then cloned into pDONR207 (25) using Gateway BP clonase II enzyme mix (Invitrogen) as recommended. After sequencing, the sequence of the CT domain was transferred from pDONR207 into the bait vector (pPC97) using Gateway LR clonase II enzyme mix (Invitrogen) as recommended so that it would be expressed as Gal4-DB fusion in yeast. Bait vectors were introduced in Saccharomyces cerevisiae AH109 strain, and a human spleen activation domain (AD) cDNA library (Invitrogen) was screened by transformation as previously described (26). AD cDNAs were PCR amplified, and inserts were sequenced.
Generation of RALBP1 and Env expression plasmids.
A RALBP1 expression plasmid carrying an N-terminal (N-ter) Myc-tagged RALBP1 (referred to as “N-ter Myc RALBP1” in this study) was generated using Gateway cloning technology (Invitrogen, USA) as recommended. Briefly, the complete open reading frame of human RALBP1 was amplified from the IRATp970B0836D clone (ImaGenes; GenBank accession no. NM_006788.3) using attB1.1 and attB2.1 recombination sites that were fused to forward and reverse primers, respectively. The ∼2-kb amplified full-length RALBP1 was introduced into the pDON207 donor vector using Gateway BP clonase II enzyme mix (Invitrogen) and transferred by homologous recombination into the pCMV-myc destination vector (Invitrogen) using Gateway LR clonase II enzyme mix (Invitrogen). The resulting construct allowed expression of a 90-kDa myc-RALBP1 fusion protein with the complete amino acid sequence of the human RALBP1 fused in the N terminus with the myc epitope under the control of the cytomegalovirus (CMV) promoter. Similarly, a human TRIM23 (tripartite motif-containing protein 23) expression plasmid that carries an N-ter Myc-tagged TRIM23 (referred to “N-ter Myc TRIM23”) was constructed from the complete cDNA (ImaGenes; GenBank accession no. NM_033227.2). The expression plasmid for the JSRV envelope fused to Flag in the C terminus (named “C-ter Flag Env” in this study; kindly provided by M. Palmarini, University of Glasgow Veterinary School, United Kingdom) produced an ∼72-kDa fusion protein with a C-ter Flag tag under the control of the CMV promoter. The N-ter Flag TM expression plasmid, used solely for immunoprecipitation, was generated using the Gateway Cloning technology (Invitrogen) as recommended. Briefly, the complete open reading frame of JSRV TM (GenBank accession no. NC_001494.1; from nt 6484 to 7197) was introduced into the donor vector pDON207 using attB1.1 and attB2.1 recombination sites that were fused to forward and reverse primers, respectively, and transferred by homologous recombination into the pCI-Neo3XFlag destination vector (Invitrogen) for the expression of a 32-kDa N-ter Flag TM fusion protein. A plasmid named “v-mos” in this study (kindly provided by Hung Fan, Cancer Research Institute, University of California Irvine, USA) allowed expression of the v-mos oncogene. All plasmid DNAs were extracted with the Nucleobond Xtra Midi EF kit (Macherey-Nagel) as recommended and checked by digestion with the appropriate restriction enzymes.
Cell transfection.
HEK 293T cells were transfected with N-ter myc RALBP1 or N-ter Flag TM expression plasmid alone or concomitantly. After 24 h of incubation of six-well plates with 8 × 105 HEK 293T cells per well, cells were transfected with a mix of DNA and jetPEI (Polyplus Transfection SA). Briefly, jetPEI and DNA (between 1 and 5 μg per condition according to the plasmid used) were diluted in 150 mM NaCl and then were mixed and incubated at room temperature for 30 min. The DNA-jetPEI mix was gently added to the cells, and transfected cells were grown at 37°C in a humidified atmosphere with 5% CO2 in DMEM supplemented with FCS. MDCK cells were transfected with the C-ter Flag Env, N-ter Myc TRIM23, or v-mos expression vector using the Neon transfection system kit (Invitrogen) and the MP100 Labtech Microporator (Life Technologies, USA) as recommended. Briefly, 5 × 105 cells were resuspended in R buffer (Invitrogen) at 37°C before the addition of 3 μg of the C-ter Flag Env, 3 μg of the N-ter Myc TRIM23, or 5 μg of the v-mos constructs. Transfected cells were grown at 37°C in a humidified atmosphere with 5% CO2 in complete DMEM. RALBP1 knockdown was realized using 25 pmol of a mix of three RALBP1-specific siRNAs targeting different parts of the human RALBP1 mRNA (Stealth siRNA duplexes; Invitrogen). Irrelevant siRNA (Stealth siRNA negative-control duplexes; Invitrogen), referred to as “scramble siRNA” in this study, were designed to minimize sequence homology with the known vertebrate transcripts and were used as negative controls. The siRNAs were transfected into MDCK cells by electroporation as described above.
Immunoprecipitation of RALBP1/TM complexes in HEK 293T cells.
Total proteins were extracted from HEK 293T cells cotransfected with N-ter Myc RALBP1 and N-ter Flag TM expression vectors. Fifty microliters of magnetic microbeads coated with G proteins (Miltenyi Biotech) was added to a mix of 2 μg of mouse anti-Myc antibodies (M4439; Sigma-Aldrich) or mouse anti-Flag antibodies (F3165; Sigma-Aldrich) for the reverse immunoprecipitation (IP), and 1 mg of total proteins diluted in 500 μl of lysis buffer (20 mM Tris HCl [pH 8], 10% glycerol, 150 mM NaCl, 1% Triton X-100, 5 mM EDTA, 1 mM Na3VO4, 1 mM phenylmethanesulfonyl fluoride [PMSF], and 10 μg/ml of aprotinin and leupeptin). After 30 min at 4°C, mixtures were loaded onto μMACS columns (Miltenyi Biotech) previously rinsed with 200 μl of lysis buffer and placed in a magnetic field using the μMACS separator (Miltenyi Biotech). The flowthrough was collected. After four washes with lysis buffer, columns were loaded with 20 μl of preheated loading buffer (50 mM Tris HCl [pH 6.8], 50 mM dithiothreitol [DTT], 1% SDS, 0.005% bromophenol blue, 10% glycerol). After 5-min incubation at room temperature, protein complexes were eluted with 50 μl of preheated loading buffer. The flowthrough and eluates were analyzed for detection of TM or RALBP1 using Western blot analysis.
Measurement of cell transformation by spheroid formation in soft agar.
Twenty-four hours after transfection with C-ter Flag Env or v-mos expression plasmid and scramble siRNA or RALBP1-specific siRNA, the MDCK cells were detached and cultured in 10× CytoSelect agar matrix (Cell Biolabs) diluted in complete DMEM at 25 × 103 cells per well in 24-well plates. The cells were grown at 37°C, and the number and size of the spheroids formed were monitored daily for 12 days. Twenty-four hours after the cells were cultured in soft agar, we ensured that there were no cell clumps that could be mistaken for spheroids. All spheroids were measured with the Image J software (W. S. Rasband, National Institutes of Health, Bethesda, MD, USA). Cells transfected with C-ter Flag Env alone, v-mos alone, or nontransfected cells were used as controls. Each transformation experiment was performed in triplicate and repeated twice.
Analysis of mRNA expression of RALBP1, Env, and v-mos.
RNA from MDCK cells transfected with C-ter Flag Env or v-mos expression plasmid and scramble siRNA or RALBP1-specific siRNA were extracted 48 h posttransfection and analyzed by reverse transcriptase PCR (RT-PCR) to confirm RALPB1, env, and v-mos mRNA expression during the transformation tests. Briefly, RNAs were extracted with the Pure Link RNA minikit (Life Technologies) as recommended; 500 ng of total RNAs was reverse transcribed using Moloney murine leukemia virus reverse transcriptase (M-MLV RT) RNase H minus and random hexamers (Promega, France). A control minus RT was subjected to the procedure in the absence of M-MLV RT. Fifty nanograms of cDNA was amplified using the Kapa Taq DNA polymerase kit (Clinisciences) as recommended using JSRV env-specific primers (forward [FOR] primer, 5′-TCTTATCAATCGCAGCATCC-3′; reverse [REV] primer, 5′-GAGTGTCTATGCCTATGCCG-3′), canine RALBP1-specific primers (FOR primer, 5′-GGAGGAGATCAGAAGACAGGAGT-3′; REV primer, 5′-ACTGGCGATCTCTTGTGCAA-3′), v-mos-specific primers (FOR primer, 5′-ATCAGTGACTTCGGCTGCTC-3′; REV primer, 5′-TGCCACAGGGTGATTCCAAA-3′), and β-actin-specific primers (FOR primer, 5′-CCAACCGTGAGAAGATGACC-3′; REV primer, 5′-CCAGAGGCGTACAGGGACAG-3′). The initial denaturation step at 95°C for 3 min was followed by 25 cycles for RALBP1 or 35 cycles for v-mos, Env, and β-actin, with one cycle consisting of 30 s at 95°C, 30 s at 60°C, and 30 s at 72°C.
Immunodetection of RALBP1, CDC42, or Env on fixed cells.
MDCK cells were transfected with the C-ter Flag Env construct and cultured in eight-well chamber slides for 48 h. The cells were then rinsed twice with 1× phosphate-buffered saline (PBS) for 5 min, fixed, and permeabilized with ice-cold acetone for 10 min at −20°C. After rehydration for 20 min in 1× PBS, the cells were incubated overnight at 4°C with mouse anti-Flag (F3165; Sigma-Aldrich) and rabbit anti-RALBP1 (Ab133549; Abcam) or anti-CDC42 (antibody against cell division control protein 42 [CDC42]) (C5747; Sigma-Aldrich) antibodies, respectively, diluted at 1/5,000, 1/1,000, and 1/2,000 in PBS-BSA (1× PBS, 1% bovine serum albumin [BSA]). After washes with 1× PBS, the cells were incubated for 1 h at room temperature with anti-mouse IgG antibody labeled with Dylight 488 (anti-mouse IgG Dylight 488) and anti-rabbit IgG Dylight 594 antibodies (Eurobio) diluted at 1/500 in PBS-BSA. Fixed cells were rinsed with 1× PBS, and nuclei were stained with 1 μg/ml 4′,6′-diamidino-2-phenylindole (DAPI) for 10 min and mounted with Fluoromount G (Electron Microscopy Sciences). Healthy MDCK cells and cells incubated solely with the secondary antibody were used as negative controls. Microscopic examinations were performed using an Axioimager Z1 epifluorescence microscope and an LSM710 confocal microscope, and images were analyzed using the Zen software (Zeiss).
Proximity ligation assay (PLA).
Transfected MDCK cells, once fixed and incubated with primary RALBP1, CDC42, Myc, or Flag antibodies, as described above, were washed with 1× PBS and incubated for 1 h at 37°C with Duolink PLA rabbit plus and PLA mouse minus proximity probes (Olink Bioscience, Sweden). Proximity ligation was performed using the Duolink detection kit (Olink Bioscience) according to the manufacturer's instructions. Controls were subjected to the same conditions on MDCK cells in the absence of Env expression. Microscopic examinations were performed as described above.
Western blot analysis.
MDCK cell pellets were resuspended in 200 μl of lysis buffer per 1 × 106 cells and incubated for 15 min on ice. Lysates were homogenized with four sonication cycles for 20 s at 300 W each (Bioblock; Fisher Scientific). After centrifugation at 11,000 × g for 15 min at 4°C, supernatants were harvested. Lung tissue samples were immersed in 400 μl of lysis buffer per 50 mg of tissue in a lysing matrix D tube (MP Biomedicals) as recommended. After tissue dissociation with the FastPrep device (MP Biomedicals) and incubation for 30 min on ice, four sonication cycles of 20 s at 300 W each were applied to the lysates. Supernatants were harvested after centrifugation for 30 min at 15,000 × g at 4°C. The amount of total proteins was measured with the Quick Start Bradford 1× dye reagent kit (Bio-Rad) and read on a Victor II spectrophotometer (PerkinElmer). After heat denaturation, total proteins (15 μg) were separated by migration on an SDS-polyacrylamide gel and transferred onto a nitrocellulose membrane (Bio-Rad). The membranes were preincubated with TBST-milk (TBST stands for Tris-buffered saline with Tween 20) (25 mM Tris [pH 7.6], 0.15 M NaCl, 0.05% Tween 20, 5% nonfat dry milk) for 1 h at room temperature. After three washes in TBST, the membranes were incubated overnight at 4°C with specific antibodies. RALBP1 protein was detected with a 1/20,000 dilution in TBST-milk of rabbit anti-RALBP1 antibodies (Ab133549; Abcam) in canine MDCK cells or with a 1/250 dilution in TBST-milk of rabbit polyclonal anti-RALBP1 antibodies (sc28575; Santa Cruz) in tissues and cells from sheep. When the N-ter Myc RALBP1 vector was used, expression of the fusion protein was detected with a 1/1,000 dilution in PBS with 0.05% Tween 20 of mouse anti-Myc antibodies (M4439; Sigma-Aldrich). Analysis of the Akt/mTOR (mammalian target of rapamycin) pathway was performed using antibodies directed against Akt (9272; Cell Signaling), phospho-Akt (9271; Cell Signaling), mTOR (2976; Cell Signaling), phospho-mTOR (2971; Cell Signaling), and phospho-p70S6K (9205; Cell Signaling) at a 1/500 dilution in TBST with 5% BSA (TBST—5% BSA) and against CDC42 (Sigma) at a 1/300 dilution in TBST with 5% milk (TBST–5% milk). After overnight incubation with primary antibodies, the membranes were washed in TBST and incubated for 1 h at room temperature with a 1/20,000 dilution of anti-rabbit IgG (whole molecule) antibodies (A0545; Sigma) labeled with peroxidase. Detection of the C-ter Flag Env and β-actin were performed with appropriate antibodies using the SNAP id system (Millipore). Briefly, the membranes were blocked with Superblock T20 buffer (Thermo Scientific), incubated for 10 min with a 1/8,000 dilution of mouse anti-Flag antibodies labeled with peroxidase (anti-Flag peroxidase antibodies) (A8592; Sigma) or a 1/75,000 dilution of anti-β-actin peroxidase antibodies (Ab A3854; Sigma), and washed in TBST. Immunoreactive bands were detected using the Super Signal reagent (Pierce). Western blots were performed three times for one transfection. The protein expression was semiquantified by densitometry using the image processing software UN-SCAN-IT (Silk Scientific Corporation). The ratio of a protein of interest to β-actin (protein of interest/β-actin ratio) was calculated.
Statistical analysis.
Statistical analyses were performed using the Wilcoxon, Student, or Welsh test using R software (27). All tests were done with a significance threshold of α = 0.05.
RESULTS
RALBP1 and Env proteins interact in cellulo.
As part of the identification of cellular partners of the JSRV oncogenic envelope, RALBP1 was identified as a cellular interactant of the CT domain by yeast two-hybrid screening. The yeast two-hybrid screen revealed seven cellular proteins interacting with the CT domain. Of these proteins, RALBP1 was the most frequently detected interactant with 15 hits. Interaction in eukaryotic cells was confirmed by formation of viral-cellular protein complexes in HEK 293T cells cotransfected with N-ter Flag TM and N-ter Myc RALBP1 (Fig. 1A). By immunoprecipitation, TM was detected in the fraction retained by the protein G-coated beads coupled to anti-Myc antibodies when N-ter Myc RALBP1 proteins were present in cell lysates (Fig. 1A). The same interaction was evidenced in the reversed IP with detection of RALBP1 retained by the beads coupled with anti-Flag antibodies when N-ter Flag TM was present in lysates (Fig. 1A).
FIG 1.

Env and RALBP1 formed protein-protein complexes. (A) Coimmunoprecipitation of RALBP1 and TM in HEK 293T cells. Lysates of HEK 293T cells overexpressing N-ter Flag TM (+) in the presence (+) or absence (−) of N-ter Myc RALBP1 were incubated with protein G-coated beads coupled with anti-Myc antibody (RALBP1 pulldown) or anti-Flag (TM pulldown) antibodies. TM (32 kDa) or RALBP1 (90 kDa) was detected by Western blotting using anti-Flag antibodies or anti-Myc antibodies, respectively. (B) Colocalization of RALBP1 and Env proteins in MDCK cells. C-ter Flag Env was overexpressed by transfection in MDCK cells, spontaneously expressing high levels of RALBP1. RALBP1 and Env were detected using anti-RALBP1 or anti-Flag antibodies, respectively. The overlay shows the colocalization of Env and RALBP1 (indicated by white arrows) in cells. The nuclei were stained with DAPI (blue). (C) Proximity ligation assay (PLA). An in situ proximity ligation assay was performed on MDCK cells overexpressing C-ter Flag Env. Primary anti-RALBP1, anti-Myc for TRIM23, and anti-Flag antibodies for Env were combined with secondary PLA probes (Olink Bioscience). The interaction events between Env and RALBP1 are visible as green dots in the cytoplasm. Negative controls were performed in the same conditions on MDCK cells in the absence of Env expression (w/o Env) or using TRIM23, which we know does not interact with Env. Nuclei were stained with DAPI. Magnification, ×200.
The cellular localization of RALBP1 and Env was analyzed by immunostaining on fixed MDCK cells transfected with the C-ter Flag Env expression construct. During the preliminary steps of our study, we observed that MDCK cells constitutively expressed high levels of RALBP1 (Fig. 1B); this made these cells suitable for our study without the need of RALBP1 overexpression by transfection. JSRV Env was expressed at the cell membrane, in the cytoplasm, and at the periphery of the nucleus (Fig. 1B). RALBP1 was expressed both in the cytoplasm and at the cell membrane (Fig. 1B). RALBP1 and Env colocalized at the cell membrane and in the cytoplasm (Fig. 1B).
In MDCK cells transfected with the C-ter Flag Env, a proximity ligation assay (PLA) demonstrated the interaction between RALBP1 and Env in the cytoplasm, as visualized by green dots (Fig. 1C). As a negative control, the same assay gave no signal on MDCK cells transfected with the C-ter Flag Env and N-ter Myc TRIM23, a cellular protein that we knew does not interact with Env. Taken together, these data confirmed that RALBP1 and Env were able to form protein-protein complexes in mammalian cells; therefore, our initial observation in yeast was validated.
The RALBP1/Env complexes are involved in Env-mediated transformation.
To study the role of RALBP1/Env complexes during Env-induced cell transformation, we experimentally controlled RALBP1 expression in MDCK cells using siRNA targeting this cell protein. We monitored the expression of RALBP1 mRNAs and proteins in MDCK cells transfected with RALBP1-specific siRNAs or scramble siRNAs as negative controls. Two days after transfection, there was no detectable expression of RALBP1 mRNA or protein in MDCK cells transfected with the RALBP1 siRNAs, unlike cells transfected with scramble siRNAs (Fig. 2). This inhibition was maintained for up to 5 days after transfection with a low but detectable level of protein and mRNA expression compared to controls (Fig. 2). By day 9, the level of expression was restored (Fig. 2). The siRNA targeting RALBP1 proved to efficiently extinguish expression of RALBP1 mRNAs and proteins in MDCK cells.
FIG 2.
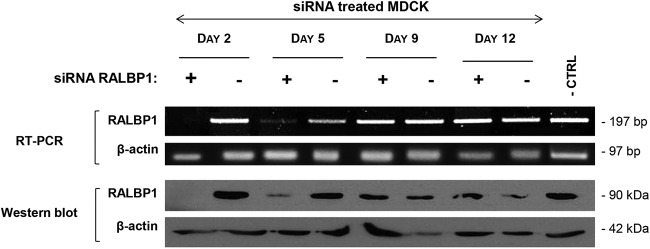
RALBP1 expression was efficiently reduced by specific siRNAs. MDCK cells spontaneously expressing high levels of RALBP1 were cotransfected with a mixture of three siRNAs targeting RALBP1 (+) or irrelevant scramble siRNA (−) as controls. The expression of mRNA and RALBP1 protein was monitored for 12 days posttransfection. Untreated MDCK cells were used as a negative control (− CTRL). β-Actin was used as a positive control for RNA expression and protein load.
To evaluate the contribution of RALBP1/Env complexes during Env-mediated transformation, we measured the induction of MDCK spheroids in soft agar upon Env expression in the absence (RALBP1-specific siRNAs) or presence (scramble siRNA or none) of RALBP1 (Fig. 3). The number and size of spheroids indicative of cell transformation were recorded over 12 days. When cultured in soft agar, control MDCK cells did not form spheroids after 12 days (data not shown). When the cells were transfected with C-ter Flag Env, spheroids were readily visible by day 9 (Fig. 3A). Transfection of Env-expressing MDCK cells with control scramble siRNA had no effect on the size and number of spheroids (Fig. 3A and B) compared to Env-transfected cells. In clear contrast, silencing of RALBP1 significantly increased the number of spheroids (Fig. 3B) and reduced their size (Fig. 3A and C) compared to nontransfected or scramble siRNA-transfected cells. We concluded that RALBP1 had a dual effect during the early steps of transformation: on one hand, RALBP1 inhibition resulted in an increased number of spheroids compared to the controls, suggesting that RALBP1 prevents cell transformation; on the other hand, RALBP1 inhibition slowed down the spheroid expansion in Env-transfected MDCK cells, suggesting that RALBP1 participates in the growth of spheroids.
FIG 3.

RALBP1/Env complexes are involved in Env-mediated cell transformation. (A) Changes over time in the number and size of spheroids were monitored in MDCK cells transformed by C-ter Flag Env or v-mos plasmid upon inhibition of RALBP1 expression (by RALBP1-specific siRNA) or in control conditions (in the presence or absence of irrelevant scramble siRNA) over 12 days. Spheroids are indicated by black arrowheads. (B) Number of spheroids after 12 days in MDCK cells transfected with JSRV Env or v-mos and treated with RALBP1 siRNA or scramble siRNA or not treated (none). Statistical analysis was performed using the Wilcoxon test, and values that are significantly different are indicated by bars and asterisks as follows: *, P ≤ 0.05; **, P ≤ 0.01. (C) Size of spheroids (in micrometers) after 12 days in MDCK cells transfected with C-ter Flag Env or v-mos and treated with RALBP1 siRNA (◆) or scramble siRNA (□) or not treated (○). Statistical analysis was performed with the Welsh test; *, P ≤ 0.05; **, P ≤ 0.01.
In order to control that the observed effects were specifically attributable to the formation of RALBP1/Env complexes, the same tests were performed using the v-mos oncogene as an inductor of MDCK cell transformation (Fig. 3A). In the presence of v-mos, RALBP1 inhibition using specific siRNA did not alter the number or size of spheroids (Fig. 3B and C). We concluded that RALBP1 inhibition had no effect on the transformation mediated by v-mos; this suggests that RALBP1 is not a ubiquitous partner of the viral oncogenes but rather that it interacts specifically with JSRV Env during cell transformation.
RALBP1 is downregulated in Env-expressing cells, tumoral AECIIs, and tumoral lung tissues.
The level of RALBP1 expression was analyzed in healthy or Env-transfected MDCK cells. Each experiment was performed in triplicate. In the presence of JSRV envelope, there was a twofold decrease in RALBP1 expression (P = 0.0095) from the level in healthy MDCK cells (Fig. 4A and B); this suggests that downregulation of RALBP1 occurs upon envelope expression. Similarly, downregulation of RALBP1 expression was highlighted in HEK 293T cells in the presence of Env (data not shown). We then investigated the level of RALBP1 expression in JSRV-induced tumors and in tumor-derived AECIIs. We compared the level of RALBP1 expression measured as the RALBP1/β-actin ratio in lung cancers (n = 10) and healthy lungs (n = 12) and AECIIs derived from lung tumors (n = 10) or healthy lungs (n = 8) (Fig. 6). Interestingly, as shown in Env-expressing MDCK cells (Fig. 4), there was a twofold reduction of RALBP1 protein expression (P = 0.003) in lung tumors and in tumor-derived AECIIs compared to normal conditions (see Fig. 6). In conclusion, lung tumors and AECIIs derived from JSRV-induced lung tumors expressed less RALBP1 proteins than their healthy counterparts.
FIG 4.

Modulation of RALBP1 and Akt/mTOR/p70S6K pathway by JSRV Env. (A) In MDCK cells not transfected (control [CTRL]) or transfected by C-ter Flag Env, total and phosphorylated proteins were analyzed by Western blotting 48 h after transfection using antibodies directed against total Akt, phosphorylated Akt (P-Akt), mTOR, phosphorylated mTOR (P-mTOR), phosphorylated p70S6K (P-p70S6K), RALBP1, and β-actin. Upon transfection with the C-ter Flag Env expression vector, both the full-length Env and TM proteins were detected; only the 72-kDa full-length protein is shown. (B) Total protein expression was semiquantified by the protein of interest/β-actin ratio and showed as the percentage of the control cells in the absence of Env used as the 100% threshold (indicated by the dashed line). Statistical analysis was performed with the t test; **, P ≤ 0.01.
FIG 6.
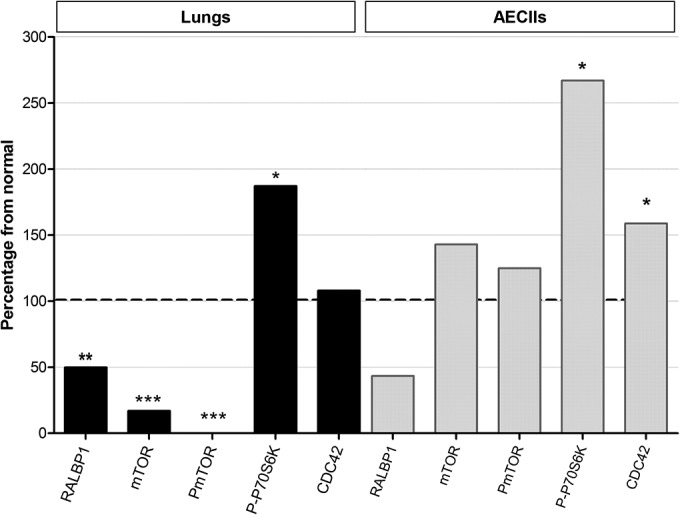
Modulation of RALBP1 and Akt/mTOR pathway in lungs and primary AECIIs. Expression of RALBP1, mTOR, P-mTOR, P-p70S6K, and CDC42 was analyzed by Western blotting in lungs (10 lungs with tumors and 12 healthy lungs) and AECIIs (10 derived from JSRV-induced tumors and 8 derived from healthy lungs). Protein expression was semiquantified by the protein of interest/β-actin ratio and is shown as a percentage from the nontumoral lungs (black bars) and AECIIs (gray bars) used as the 100% threshold (indicated by the horizontal dashed line). Statistical analysis was performed with the Wilcoxon, Welsh, and t tests. Values that are significantly different are indicated by asterisks as follows: *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
The Akt/mTOR/p70S6K pathway is activated by the JSRV envelope in MDCK cells.
We then studied the activation of the Akt/mTOR/p70S6K pathway in Env-transfected MDCK cells by measuring expression of total and phosphorylated Akt (P-Akt), mTOR (P-mTOR), and p70S6K (P-p70S6K) proteins. Each experiment was performed in triplicate, and the signals were semiquantified. The quantity of total proteins was constant in all tested conditions as determined by expression of β-actin (Fig. 4A). We observed a twofold decrease in expression of total Akt and mTOR upon Env expression (Fig. 4A and B). In the presence of JSRV envelope, we observed a twofold increase in the expression of P-mTOR and a moderate increase in the expression of P-Akt and P-p70S6K (Fig. 4A and B). These results highlight activation of the Akt/mTOR/p70S6K pathway mediated by JSRV envelope.
RALBP1 is involved in Env-driven modulation of the Akt/mTOR/p70S6K pathway.
To determine the role of RALBP1 in activation of the Akt/mTOR/p70S6K pathway, expression of key proteins was evaluated after siRNA silencing of RALBP1 in Env-transfected MDCK cells (Fig. 5A). Each experiment was performed in triplicate. The quantity of total proteins was constant in all tested conditions as determined by expression of β-actin. While the levels of total Akt and mTOR were not markedly changed upon inhibition of RALPB1 in Env-expressing MDCK cells (Fig. 5B), their phosphorylated forms were negatively modulated with a marked twofold reduction of P-mTOR (P = 0.015) (Fig. 5B). Interestingly, inhibition of RALBP1 in Env-expressing MDCK cells completely abolished the phosphorylation of P-p70S6K (Fig. 5B). These results highlighted that in Env-expressing MDCK cells, RALBP1 regulates the expression of proteins of the Akt/mTOR/p70S6K pathway and stimulates activation of the Akt, mTOR, and p70S6K key proteins.
FIG 5.

Involvement of RALBP1 in modulation of the Akt/mTOR/p70S6K pathway by JSRV Env. (A) MDCK cells were transfected with C-ter Flag Env and siRNA targeting RALBP1 or scramble siRNA. Total proteins were extracted and analyzed by Western blotting 48 h after transfection using antibodies directed against Akt, P-Akt, mTOR, P-mTOR, P-p70S6K, and RALBP1. (B) Protein expression was semiquantified using the protein of interest/β-actin ratio and expressed as a percentage of the control cells transfected with the scramble siRNA used as the 100% threshold (indicated by the dashed line). Statistical analysis was performed with the t test; *, P ≤ 0.05; ***, P ≤ 0.001.
The Akt/mTOR/p70S6K pathway is modulated in JSRV-induced tumors and tumor-derived AECIIs.
We investigated the activation of Akt, mTOR, and p70S6K in JSRV-induced lung tumors and tumor-derived AECIIs. The expression of protein was semiquantified using the protein of interest/β-actin ratio. In lung tissues, the dysregulation of the Akt/mTOR/p70S6K pathway was noticeable with a marked and significant decrease of mTOR expression (P = 0.0002) down to 20% of the level observed in the healthy lungs and a complete abolishment of its activation (P = 0.0008) (Fig. 6). The mTOR or P-mTOR protein expression was not significantly disturbed in tumor-derived AECIIs compared to healthy cells (Fig. 6). Remarkably, P-p70S6K was significantly overexpressed in tumors and in tumor-derived AECIIs: there was almost a 2-fold increase in lungs (P = 0.0015) and a 2.5-fold increase in AECIIs (P = 0.02) compared to normal conditions (Fig. 6). To summarize, we demonstrated a dysregulation of the Akt/mTOR/p70S6K pathway with a marked activation of p70S6K in lung tumors.
CDC42 is overexpressed in epithelial cells derived from ovine tumors and formed complexes with Env.
We analyzed the level of total CDC42 protein in ovine tumors and in AECIIs isolated from tumors. If no major difference of expression was noticeable between tumoral and healthy lungs, a significant overexpression (P < 0.05) of CDC42 was evidenced in AECIIs derived from tumors (Fig. 6). We then analyzed the localization of CDC42 and envelope proteins in MDCK overexpressing the C-ter Flag Env and demonstrated that the two proteins were colocalized in the cytoplasm of the cells (Fig. 7A). Finally, CDC42 and the JSRV envelope formed protein-protein complexes as shown by PLA (Fig. 7B).
FIG 7.

Formation of Env/CDC42 complexes. (A) Colocalization of endogenous CDC42 and Env proteins in MDCK cells. C-ter Flag Env was overexpressed by transfection in MDCK cells. CDC42 and Env were detected using anti-CDC42 or anti-Flag antibodies, respectively. The overlay shows the colocalization of Env and CDC42 in cells (indicated by white arrows). (B) PLA was performed on MDCK cells transfected with C-ter Flag Env. Primary anti-CDC42 and anti-Flag antibodies were combined with secondary PLA probes (Olink Bioscience). The interaction events between Env and CDC42 are visible as green dots in the cytoplasm. Nuclei were stained with DAPI. Magnification, ×200. (C) Putative regulation of the Akt/mTOR/p70S6K pathway upon Env expression based on our results. The pathway in healthy cells (light blue boxes) is shown in the left panel and the observed dysregulation in Env-expressing cells is shown in the right panel. Upregulated proteins (red boxes) and downregulated proteins (green boxes) are indicated.
DISCUSSION
To decipher the early steps of JSRV-induced cell transformation, we identified cellular partners of the cytoplasmic tail of its envelope by yeast two-hybrid screening. Among the identified cell partners, we focused on the interaction between the envelope of JSRV and RALBP1, a cellular protein implicated in the ras pathway and effector of RalA (Ras-like protein A) (22). We demonstrated the formation of RALBP1/Env complexes in mammalian cells by visualizing their colocalization as well as protein-protein interaction using a proximity ligation assay. We studied how the disruption of the RALBP1/Env complexes might modify the transformation process and subsequent activation of signaling pathways.
RALBP1 is ubiquitously expressed and acts as a multifunctional cellular protein. It contains a region of homology with the GTPase-activating protein (GAP) domain involved in regulation of GTPases of the Rho family. By recruitment of CDC42 (cell division control protein 42) through its GAP domain, RALBP1 participates in the remodeling of the actin cytoskeleton (28). RALBP1 regulates various endocytic pathways, including those mediated by receptors for epidermal growth factor, transferrin, or insulin (29, 30). RALBP1 is involved in cell cycle progression; it promotes mitochondrial fission during mitosis via its interaction with CDK1 (31). It also acts as a non-ABC transporter, and thus, it participates in resistance to small chemotherapeutic drugs (32). Importantly, RALBP1 has been identified as an important neoplastic player by its crucial role in some of the key cellular functions. Depletion of RALBP1 in xenografts has efficiently cured skin (33), lung (34), colon (35), prostate (36), and kidney (37) cancers. Inhibition of RAL-A, an activator of RALBP1, leads to reduction of cell proliferation in cells derived from non-small cell lung cancers (38). RALBP1 has an important antiapoptotic role with the transport of glutathione conjugates of electrophilic proapoptotic compounds produced during oxidative stress (32, 39) and is involved in the oxidative stress response through its interaction with HSF1 (heat shock factor 1) (40).
Interestingly, in our study, RALBP1 was underexpressed in MDCK cells expressing the JSRV envelope and in ovine lung tumors and tumor-derived AECIIs. These results are in contrast with reports on RALBP1 overexpression in lung, prostate, or ovary cancers (41), among others, as well as in cell lines derived from non-small cell lung cancers (38). The RALBP1 down-expression that we observed in ovine pulmonary adenocarcinomas might be specifically associated with JSRV and associated with envelope expression as shown by the negative regulation of endogenous RALPB1 in MDCK cells expressing Env.
We analyzed activation of the Akt/mTOR/p70S6K pathway in cells overexpressing the envelope and in tissues and AECIIs derived from JSRV-induced tumors. We report the activation of p70S6K in MDCK cells overexpressing Env and in tumor-derived AECIIs and its downregulation upon siRNA-mediated inhibition of RALBP1. The Akt/mTOR/p70S6K signaling pathway is crucial for cell growth and survival in physiological and pathological situations and is activated by multiple factors, e.g., hormones, growth factors, or mutations of components of the tyrosine kinase pathways (11, 42, 43). When phosphorylated, Akt activates the mTOR kinase (44), which controls cell growth by promoting translation and ribosome biogenesis by phosphorylation of p70S6K and 4E-BP1 (45). The p70S6K kinase controls cellular growth by promoting mRNA translation (45). Activation of the PI3K/Akt pathway has been reported in various JSRV-transformed cell lines (8, 46, 47) and confers a growth advantage. Overall, the role of the PI3K/Akt pathway appears to be cell line dependent with murine cells depending on Akt activation, while chicken cell lines can be transformed by mutant cells with mutations in the YXXM motifs that fail to activate Akt (47).
In this report, we studied mechanisms of Env-mediated transformation in MDCK cells when maintained in a three-dimensional (3D) semisolid matrix (soft agar). When MDCK cells were transfected with Env, we observed the formation of MDCK spheroids due to the ability of these cells to grow in the absence of anchorage, a hallmark of cellular transformation and uncontrolled cellular growth (48). As previously reported (9, 49), we showed activation of the Akt/mTOR pathway with moderate activation of Akt and strong activation of mTOR and p70S6K in Env-expressing cells. The mTOR and p70S6K proteins were activated in AECIIs derived from JSRV-induced tumors; this suggested the role of JSRV in activation of these key proteins. The activation of the Akt/mTOR/p70S6K pathway may participate in telomerase activation that leads to inhibition of apoptosis in AECIIs derived from tumors (15). Interestingly, phosphorylated Akt was not detected (47) or was detected only in one-third (15) of the tested ovine lung adenocarcinomas. We previously demonstrated that primary AECIIs were insensitive to stimulation by epidermal growth factor, an activator of the PI3K/Akt/mTOR pathway, suggesting impairment of this pathway in cells transformed by JSRV (15). Taken together, these results reinforce the role of the PI3K/Akt/mTOR pathway in the JSRV Env-induced transformation and suggest that mTOR and p70S6K might be more important than Akt during the transformation process. Note that many viruses, including Epstein-Barr virus, papillomaviruses, polyomaviruses, human herpesvirus 8, hepatitis B virus, hepatitis C virus, human immunodeficiency viruses, cytomegalovirus, respiratory syncytial virus, or rubella virus require upregulation of the PI3K/Akt pathway to sustain long-standing infections and to create a favorable environment for cell transformation or virus replication (42).
We observed that envelope overexpression induced decreases in RALBP1 proteins in MDCK cells in parallel with activation of the Akt/mTOR pathway. Visualization of Env-RALBP1 interactions 2 days posttransfection by the proximity ligation assay, which enables visualization of individual protein-protein interactions in cells, evidenced a punctiform and cytoplasmic localization of the protein-protein complexes in MDCK cells expressing JSRV Env. We demonstrated that extinction of RALBP1 expression using RALBP1-specific siRNAs turned down the activation of mTOR and p70S6K in Env-transfected cells, while activation of Akt was not significantly altered compared to controls. Moreover, the reduction of RALBP1 expression was associated with a dual effect on the appearance and development of spheroids in Env-transformed MDCK cells. On one hand, the absence of RALBP1 increased the number of transformation events induced by Env, while on the other hand, it prevented expansion of the foci in terms of size. The capacity of Env to transform cells and to generate spheroids was amplified in the absence of RALBP1 expression. This suggests that Env may downregulate or interfere with the multifunctional RALBP1 protein in order to be more efficient at initiating transforming events. It would be interesting to look at the efficiency of RALBP1 overexpression to prevent the transformation process. On the other hand, when inhibiting expression of RALBP1 with siRNA, the size of growing spheroids was reduced at 12 days posttransfection, which implies the involvement of RALBP1 in the proliferation and/or maintenance of Env-transformed cells. After a few days posttransfection, RALBP1 tends to recover its normal expression level (siRNA being efficient up to 5 days posttransfection). This suggests that development in the size of the spheroid may not be under the control of Env expression, but rather it may be modulated by RALBP1 reexpression. This is in agreement with the reported capacity of RALBP1 to suppress apoptosis and to promote cell proliferation (50).
RALBP1 interacts with a number of cellular proteins. By its GAP domain, it inactivates Rho GTPases such as CDC42 (a small GTPase of the Rho family) by hydrolyzing its GTP into GDP (51, 52). In its active form (bound to GTP), CDC42 is able to mediate signaling cascades leading to activation of more than 20 downstream effectors involved in proliferation, migration, adhesion, and cytoskeleton remodeling (51); mTOR and p70S6K are among the effectors activated by CDC42 (53, 54). From our results, we propose that RALBP1 plays a role in JSRV Env-mediated transformation through CDC42, which in turn activates p70S6K (Fig. 7C). Moreover, CDC42 is upregulated in various tumors, such as colon, breast, and lung cancers, and its overexpression has been associated with carcinogenesis and progression (55, 56). As in other cancers, we reported the upregulation of CDC42 in ovine tumor-derived epithelial cells.
In conclusion, our study shows that the PI3K/Akt/mTOR pathway plays a critical role in JSRV-induced transformation and that activation of mTOR and p70S6K is a determinant event for envelope-mediated transformation. RALBP1 is an important player in this process, as its inhibition reduces mTOR activation and abolishes p70S6K activation, limiting the expansion of spheroids. We showed that RALBP1/Env complexes have a dual effect during the early steps of cell transformation. In Env-transfected cells, RALBP1 reduced the number of transformation events but promotes expansion of the foci in terms of size. Therefore, RALBP1 has a positive effect on the progression of cell transformation triggered by the JSRV envelope. Interactions of RALBP1 with viruses are poorly described, and for the first time, this work emphasizes its involvement in tumors associated with a retroviral oncogene. In accordance with the multifunctional properties attributed to the RALBP1 pathway, we hypothesize that CDC42 might be a major key player in this process through activation of p70S6K during envelope-induced transformation associated with alteration of cytoskeleton or modification of endocytosis processes.
ACKNOWLEDGMENTS
This work has been funded by the Ligue Nationale de Recherche sur le Cancer (Ardèche committee), the Plan Cancer, and the Rhône Alpes Region. The imaging platform Platim is part of the SFR BioSciences Gerland-Lyon Sud (UMS 3444/US8). M.M. was the recipient of a Ph.D. fellowship from the French Ministry of Research.
We thank François Guiguen, Jean-Christophe Natorp, and Rémy Falguière for the collection of postmortem lungs and Guillaume Poupy for preliminary experiments.
REFERENCES
- 1.Hofacre A, Fan H. 2004. Multiple domains of the Jaagsiekte sheep retrovirus envelope protein are required for transformation of rodent fibroblasts. J Virol 78:10479–10489. doi: 10.1128/JVI.78.19.10479-10489.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chow YH, Alberti A, Mura M, Pretto C, Murcia P, Albritton LM, Palmarini M. 2003. Transformation of rodent fibroblasts by the Jaagsiekte sheep retrovirus envelope is receptor independent and does not require the surface domain. J Virol 77:6341–6350. doi: 10.1128/JVI.77.11.6341-6350.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hull S, Fan H. 2006. Mutational analysis of the cytoplasmic tail of Jaagsiekte sheep retrovirus envelope protein. J Virol 80:8069–8080. doi: 10.1128/JVI.00013-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leroux C, Girard N, Cottin V, Greenland T, Mornex JF, Archer F. 2007. Jaagsiekte sheep retrovirus (JSRV): from virus to lung cancer in sheep. Vet Res 38:211–228. doi: 10.1051/vetres:2006060. [DOI] [PubMed] [Google Scholar]
- 5.Wootton SK, Halbert CL, Miller AD. 2005. Sheep retrovirus structural protein induces lung tumours. Nature 434:904–907. doi: 10.1038/nature03492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Linnerth-Petrik NM, Santry LA, Yu DL, Wootton SK. 2012. Adeno-associated virus vector mediated expression of an oncogenic retroviral envelope protein induces lung adenocarcinomas in immunocompetent mice. PLoS One 7:e51400. doi: 10.1371/journal.pone.0051400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caporale M, Cousens C, Centorame P, Pinoni C, De las Heras M, Palmarini M. 2006. Expression of the Jaagsiekte sheep retrovirus envelope glycoprotein is sufficient to induce lung tumors in sheep. J Virol 80:8030–8037. doi: 10.1128/JVI.00474-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palmarini M, Maeda N, Murgia C, De-Fraja C, Hofacre A, Fan H. 2001. A phosphatidylinositol 3-kinase docking site in the cytoplasmic tail of the Jaagsiekte sheep retrovirus transmembrane protein is essential for envelope-induced transformation of NIH 3T3 cells. J Virol 75:11002–11009. doi: 10.1128/JVI.75.22.11002-11009.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu SL, Miller AD. 2005. Transformation of Madin-Darby canine kidney epithelial cells by sheep retrovirus envelope proteins. J Virol 79:927–933. doi: 10.1128/JVI.79.2.927-933.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cully M, You H, Levine AJ, Mak TW. 2006. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer 6:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 11.Porta C, Paglino C, Mosca A. 2014. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol 4:64. doi: 10.3389/fonc.2014.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Mathon NF, Lloyd AC. 2001. Cell senescence and cancer. Nat Rev Cancer 1:203–213. doi: 10.1038/35106045. [DOI] [PubMed] [Google Scholar]
- 14.Kang SS, Kwon T, Kwon DY, Do SI. 1999. Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J Biol Chem 274:13085–13090. doi: 10.1074/jbc.274.19.13085. [DOI] [PubMed] [Google Scholar]
- 15.Suau F, Cottin V, Archer F, Croze S, Chastang J, Cordier G, Thivolet-Bejui F, Mornex JF, Leroux C. 2006. Telomerase activation in a model of lung adenocarcinoma. Eur Respir J 27:1175–1182. doi: 10.1183/09031936.06.00125105. [DOI] [PubMed] [Google Scholar]
- 16.Shah A, Swain WA, Richardson D, Edwards J, Stewart DJ, Richardson CM, Swinson DE, Patel D, Jones JL, O'Byrne KJ. 2005. Phospho-akt expression is associated with a favorable outcome in non-small cell lung cancer. Clin Cancer Res 11:2930–2936. doi: 10.1158/1078-0432.CCR-04-1385. [DOI] [PubMed] [Google Scholar]
- 17.Linnerth-Petrik NM, Santry LA, Petrik JJ, Wootton SK. 2014. Opposing functions of Akt isoforms in lung tumor initiation and progression. PLoS One 9:e94595. doi: 10.1371/journal.pone.0094595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu SL, Miller AD. 2007. Oncogenic transformation by the Jaagsiekte sheep retrovirus envelope protein. Oncogene 26:789–801. doi: 10.1038/sj.onc.1209850. [DOI] [PubMed] [Google Scholar]
- 19.Rai SK, Duh FM, Vigdorovich V, Danilkovitch-Miagkova A, Lerman MI, Miller AD. 2001. Candidate tumor suppressor HYAL2 is a glycosylphosphatidylinositol (GPI)-anchored cell-surface receptor for Jaagsiekte sheep retrovirus, the envelope protein of which mediates oncogenic transformation. Proc Natl Acad Sci U S A 98:4443–4448. doi: 10.1073/pnas.071572898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu SL, Duh FM, Lerman MI, Miller AD. 2003. Role of virus receptor Hyal2 in oncogenic transformation of rodent fibroblasts by sheep betaretrovirus Env proteins. J Virol 77:2850–2858. doi: 10.1128/JVI.77.5.2850-2858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller AD. 2008. Hyaluronidase 2 and its intriguing role as a cell-entry receptor for oncogenic sheep retroviruses. Semin Cancer Biol 18:296–301. doi: 10.1016/j.semcancer.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kashatus DF. 2013. Ral GTPases in tumorigenesis: emerging from the shadows. Exp Cell Res 319:2337–2342. doi: 10.1016/j.yexcr.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Archer F, Jacquier E, Lyon M, Chastang J, Cottin V, Mornex JF, Leroux C. 2007. Alveolar type II cells isolated from pulmonary adenocarcinoma: a model for JSRV expression in vitro. Am J Respir Cell Mol Biol 36:534–540. doi: 10.1165/rcmb.2006-0285OC. [DOI] [PubMed] [Google Scholar]
- 24.York DF, Vigne R, Verwoerd DW, Querat G. 1992. Nucleotide sequence of the Jaagsiekte retrovirus, an exogenous and endogenous type D and B retrovirus of sheep and goats. J Virol 66:4930–4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rual JF, Hirozane-Kishikawa T, Hao T, Bertin N, Li S, Dricot A, Li N, Rosenberg J, Lamesch P, Vidalain PO, Clingingsmith TR, Hartley JL, Esposito D, Cheo D, Moore T, Simmons B, Sequerra R, Bosak S, Doucette-Stamm L, Le Peuch C, Vandenhaute J, Cusick ME, Albala JS, Hill DE, Vidal M. 2004. Human ORFeome version 1.1: a platform for reverse proteomics. Genome Res 14:2128–2135. doi: 10.1101/gr.2973604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li S, Armstrong CM, Bertin N, Ge H, Milstein S, Boxem M, Vidalain PO, Han JD, Chesneau A, Hao T, Goldberg DS, Li N, Martinez M, Rual JF, Lamesch P, Xu L, Tewari M, Wong SL, Zhang LV, Berriz GF, Jacotot L, Vaglio P, Reboul J, Hirozane-Kishikawa T, Li Q, Gabel HW, Elewa A, Baumgartner B, Rose DJ, Yu H, Bosak S, Sequerra R, Fraser A, Mango SE, Saxton WM, Strome S, Van Den Heuvel S, Piano F, Vandenhaute J, Sardet C, Gerstein M, Doucette-Stamm L, Gunsalus KC, Harper JW, Cusick ME, Roth FP, Hill DE, Vidal M. 2004. A map of the interactome network of the metazoan C. elegans. Science 303:540–543. doi: 10.1126/science.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.R Development Core Team. 2010. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org. [Google Scholar]
- 28.Boissel L, Houssin N, Chikh A, Rynditch A, Van Hove L, Moreau J. 2007. Recruitment of Cdc42 through the GAP domain of RLIP participates in remodeling of the actin cytoskeleton and is involved in Xenopus gastrulation. Dev Biol 312:331–343. doi: 10.1016/j.ydbio.2007.09.027. [DOI] [PubMed] [Google Scholar]
- 29.Jullien-Flores V, Mahe Y, Mirey G, Leprince C, Meunier-Bisceuil B, Sorkin A, Camonis JH. 2000. RLIP76, an effector of the GTPase Ral, interacts with the AP2 complex: involvement of the Ral pathway in receptor endocytosis. J Cell Sci 113:2837–2844. [DOI] [PubMed] [Google Scholar]
- 30.Nakashima S, Morinaka K, Koyama S, Ikeda M, Kishida M, Okawa K, Iwamatsu A, Kishida S, Kikuchi A. 1999. Small G protein Ral and its downstream molecules regulate endocytosis of EGF and insulin receptors. EMBO J 18:3629–3642. doi: 10.1093/emboj/18.13.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kashatus DF, Lim KH, Brady DC, Pershing NL, Cox AD, Counter CM. 2011. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol 13:1108–1115. doi: 10.1038/ncb2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Awasthi S, Cheng J, Singhal SS, Saini MK, Pandya U, Pikula S, Bandorowicz-Pikula J, Singh SV, Zimniak P, Awasthi YC. 2000. Novel function of human RLIP76: ATP-dependent transport of glutathione conjugates and doxorubicin. Biochemistry 39:9327–9334. doi: 10.1021/bi992964c. [DOI] [PubMed] [Google Scholar]
- 33.Singhal SS, Awasthi YC, Awasthi S. 2006. Regression of melanoma in a murine model by RLIP76 depletion. Cancer Res 66:2354–2360. doi: 10.1158/0008-5472.CAN-05-3534. [DOI] [PubMed] [Google Scholar]
- 34.Singhal SS, Singhal J, Nair MP, Lacko AG, Awasthi YC, Awasthi S. 2007. Doxorubicin transport by RALBP1 and ABCG2 in lung and breast cancer. Int J Oncol 30:717–725. [PubMed] [Google Scholar]
- 35.Singhal SS, Singhal J, Yadav S, Dwivedi S, Boor PJ, Awasthi YC, Awasthi S. 2007. Regression of lung and colon cancer xenografts by depleting or inhibiting RLIP76 (Ral-binding protein 1). Cancer Res 67:4382–4389. doi: 10.1158/0008-5472.CAN-06-4124. [DOI] [PubMed] [Google Scholar]
- 36.Singhal SS, Roth C, Leake K, Singhal J, Yadav S, Awasthi S. 2009. Regression of prostate cancer xenografts by RLIP76 depletion. Biochem Pharmacol 77:1074–1083. doi: 10.1016/j.bcp.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singhal SS, Singhal J, Yadav S, Sahu M, Awasthi YC, Awasthi S. 2009. RLIP76: a target for kidney cancer therapy. Cancer Res 69:4244–4251. doi: 10.1158/0008-5472.CAN-08-3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Male H, Patel V, Jacob MA, Borrego-Diaz E, Wang K, Young DA, Wise AL, Huang C, Van Veldhuizen P, O'Brien-Ladner A, Williamson SK, Taylor SA, Tawfik O, Esfandyari T, Farassati F. 2012. Inhibition of RalA signaling pathway in treatment of non-small cell lung cancer. Lung Cancer 77:252–259. doi: 10.1016/j.lungcan.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 39.Awasthi S, Singhal SS, Awasthi YC, Martin B, Woo JH, Cunningham CC, Frankel AE. 2008. RLIP76 and cancer. Clin Cancer Res 14:4372–4377. doi: 10.1158/1078-0432.CCR-08-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu Y, Mivechi NF. 2003. HSF-1 interacts with Ral-binding protein 1 in a stress-responsive, multiprotein complex with HSP90 in vivo. J Biol Chem 278:17299–17306. doi: 10.1074/jbc.M300788200. [DOI] [PubMed] [Google Scholar]
- 41.Vatsyayan R, Lelsani PC, Awasthi S, Singhal SS. 2010. RLIP76: a versatile transporter and an emerging target for cancer therapy. Biochem Pharmacol 79:1699–1705. doi: 10.1016/j.bcp.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cooray S. 2004. The pivotal role of phosphatidylinositol 3-kinase-Akt signal transduction in virus survival. J Gen Virol 85:1065–1076. doi: 10.1099/vir.0.19771-0. [DOI] [PubMed] [Google Scholar]
- 43.Datta SR, Brunet A, Greenberg ME. 1999. Cellular survival: a play in three Akts. Genes Dev 13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 44.Liao Y, Hung MC. 2010. Physiological regulation of Akt activity and stability. Am J Transl Res 2:19–42. [PMC free article] [PubMed] [Google Scholar]
- 45.Hay N, Sonenberg N. 2004. Upstream and downstream of mTOR. Genes Dev 18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 46.Liu SL, Lerman MI, Miller AD. 2003. Putative phosphatidylinositol 3-kinase (PI3K) binding motifs in ovine betaretrovirus Env proteins are not essential for rodent fibroblast transformation and PI3K/Akt activation. J Virol 77:7924–7935. doi: 10.1128/JVI.77.14.7924-7935.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zavala G, Pretto C, Chow YH, Jones L, Alberti A, Grego E, De las Heras M, Palmarini M. 2003. Relevance of Akt phosphorylation in cell transformation induced by Jaagsiekte sheep retrovirus. Virology 312:95–105. doi: 10.1016/S0042-6822(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 48.Chitra E, Lin YW, Davamani F, Hsiao KN, Sia C, Hsieh SY, Wei OL, Chen JH, Chow YH. 2010. Functional interaction between Env oncogene from Jaagsiekte sheep retrovirus and tumor suppressor Sprouty2. Retrovirology 7:62. doi: 10.1186/1742-4690-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnson C, Sanders K, Fan H. 2010. Jaagsiekte sheep retrovirus transformation in Madin-Darby canine kidney epithelial cell three-dimensional culture. J Virol 84:5379–5390. doi: 10.1128/JVI.02323-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Q, Wang JY, Zhang XP, Lv ZW, Fu D, Lu YC, Hu GH, Luo C, Chen JX. 2013. RLIP76 is overexpressed in human glioblastomas and is required for proliferation, tumorigenesis and suppression of apoptosis. Carcinogenesis 34:916–926. doi: 10.1093/carcin/bgs401. [DOI] [PubMed] [Google Scholar]
- 51.Arias-Romero LE, Chernoff J. 2013. Targeting Cdc42 in cancer. Expert Opin Ther Targets 17:1263–1273. doi: 10.1517/14728222.2013.828037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mott HR, Owen D. 2014. Structure and function of RLIP76 (RalBP1): an intersection point between Ras and Rho signalling. Biochem Soc Trans 42:52–58. doi: 10.1042/BST20130231. [DOI] [PubMed] [Google Scholar]
- 53.Chou MM, Masuda-Robens JM, Gupta ML. 2003. Cdc42 promotes G1 progression through p70 S6 kinase-mediated induction of cyclin E expression. J Biol Chem 278:35241–35247. doi: 10.1074/jbc.M305246200. [DOI] [PubMed] [Google Scholar]
- 54.Fang Y, Park IH, Wu AL, Du G, Huang P, Frohman MA, Walker SJ, Brown HA, Chen J. 2003. PLD1 regulates mTOR signaling and mediates Cdc42 activation of S6K1. Curr Biol 13:2037–2044. doi: 10.1016/j.cub.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 55.Fritz G, Just I, Kaina B. 1999. Rho GTPases are overexpressed in human tumors. Int J Cancer 81:682–687. doi:. [DOI] [PubMed] [Google Scholar]
- 56.Tucci MG, Lucarini G, Brancorsini D, Zizzi A, Pugnaloni A, Giacchetti A, Ricotti G, Biagini G. 2007. Involvement of E-cadherin, beta-catenin, Cdc42 and CXCR4 in the progression and prognosis of cutaneous melanoma. Br J Dermatol 157:1212–1216. doi: 10.1111/j.1365-2133.2007.08246.x. [DOI] [PubMed] [Google Scholar]