

ABSTRACT
During HIV-1 assembly, the Gag viral proteins are targeted and assemble at the inner leaflet of the cell plasma membrane. This process could modulate the cortical actin cytoskeleton, located underneath the plasma membrane, since actin dynamics are able to promote localized membrane reorganization. In addition, activated small Rho GTPases are known for regulating actin dynamics and membrane remodeling. Therefore, the modulation of such Rho GTPase activity and of F-actin by the Gag protein during virus particle formation was considered. Here, we studied the implication of the main Rac1, Cdc42, and RhoA small GTPases, and some of their effectors, in this process. The effect of small interfering RNA (siRNA)-mediated Rho GTPases and silencing of their effectors on Gag localization, Gag membrane attachment, and virus-like particle production was analyzed by immunofluorescence coupled to confocal microscopy, membrane flotation assays, and immunoblot assays, respectively. In parallel, the effect of Gag expression on the Rac1 activation level was monitored by G-LISA, and the intracellular F-actin content in T cells was monitored by flow cytometry and fluorescence microscopy. Our results revealed the involvement of activated Rac1 and of the IRSp53-Wave2-Arp2/3 signaling pathway in HIV-1 Gag membrane localization and particle release in T cells as well as a role for actin branching and polymerization, and this was solely dependent on the Gag viral protein. In conclusion, our results highlight a new role for the Rac1-IRSp53-Wave2-Arp2/3 signaling pathway in the late steps of HIV-1 replication in CD4 T lymphocytes.
IMPORTANCE During HIV-1 assembly, the Gag proteins are targeted and assembled at the inner leaflet of the host cell plasma membrane. Gag interacts with specific membrane phospholipids that can also modulate the regulation of cortical actin cytoskeleton dynamics. Actin dynamics can promote localized membrane reorganization and thus can be involved in facilitating Gag assembly and particle formation. Activated small Rho GTPases and effectors are regulators of actin dynamics and membrane remodeling. We thus studied the effects of the Rac1, Cdc42, and RhoA GTPases and their specific effectors on HIV-1 Gag membrane localization and viral particle release in T cells. Our results show that activated Rac1 and the IRSp53-Wave2-Arp2/3 signaling pathway are involved in Gag plasma membrane localization and viral particle production. This work uncovers a role for cortical actin through the activation of Rac1 and the IRSp53/Wave2 signaling pathway in HIV-1 particle formation in CD4 T lymphocytes.
INTRODUCTION
The HIV-1 replication cycle leads to the formation of new viral particles, which assemble in specific microdomains located at the plasma membrane or in some intracellular compartments, according to cell type (1–4). These particles are then released from the host cell membrane by budding or from intracellular compartments by exocytosis (5). When expressed in cells, the pr55Gag precursor is both necessary and sufficient for the assembly and production of virus-like particles (VLPs). This protein has an NH2-terminal myristate and four major domains: matrix (MA), capsid (CA), nucleocapsid (NC), and p6. After translation, Gag recruits the dimeric positive-strand RNA viral genome to the cytoplasm and then assembles at the inner leaflet of the plasma membrane (6). Gag multimerizes on the viral RNA via its NC and CA domains (7) and assembles on a specific plasma membrane phospholipid, phosphatidylinositol bisphosphate [PI(4,5)P2], via its highly basic MA domain (8). The C-terminal p6 domain and a part of the NC domain then allow the recruitment of the ESCRT complex to induce particle budding (9, 10).
The MA domain of retroviral Gag proteins binds specifically to acidic lipids located at the inner leaflet of the plasma membrane and more specifically to PI(4,5)P2 (11–20). In cells, Gag can be found in several types of membrane microdomains, named lipid raft domains, which are enriched in cholesterol and sphingomyelin (21–24), or tetraspanin-enriched microdomains (TEMs) when they contain some membrane-organizing proteins such as CD81, CD63, and CD9, etc. (25–27). However, Gag can also be found in liquid-disordered-phase membranes (17, 28). All these plasma membrane microdomains are associated with the cortical actin network (29), and it has been shown that plasma membrane deformations require the remodeling of the cytoskeleton and the cooperation of signaling proteins such as Rho GTPases and effectors. Therefore, it is possible that Gag assembly and particle release require the modulation of actin cytoskeleton dynamics and membrane curvature effectors.
In fact, several studies indicate a role for the actin network in the HIV-1 assembly process. First, actin is found inside HIV-1 virions, which can contain up to a 15% equimolar level of actin in comparison to Gag (30). Other related actin-binding proteins (like cofilin, Moesin, or Ezrin) are also actively or passively incorporated into the particles (31). Moreover, it was shown previously by immunoprecipitation and fractionation that actin interacts with the NC domain of Gag (32, 33), and another study suggested that actin-enriched structures could be localized underneath viral assembly sites (34). However, these results are discussed in the scientific community, because in another cell type, it was recently observed by live-cell imaging microscopy that F-actin seems not to be recruited at the HIV Gag assembly site (35). Nonetheless, the actin cytoskeleton might play an important role in virus assembly and production, since its drug-mediated inhibition changes the intracellular localization of Gag and diminishes virus production in T cells (36–38). In addition, small interfering RNA (siRNA) depletion of Filamin A, a host cell protein that regulates actin network dynamics, impairs viral particle assembly and release (39). Other cell effectors that regulate actin turnover, such as LIMK1 and ROCK1, were recently described to play a role in retrovirus release (40). Altogether, these results tend to show, directly or indirectly, a role for the regulation of the cortical actin cytoskeleton during HIV assembly and/or release, especially in CD4 T cells. This effect has also been described for the related retrovirus equine infectious anemia virus (EIAV), for which a role for actin in virus release has been reported (41, 42). It has been proposed that polymerization of actin-enriched structures at HIV-1 assembly sites could also promote cell polarization, virological synapse formation, and HIV-1 cell-to-cell transmission (36, 43). The influence of the Env glycoprotein on this mechanism is important (36, 43), but other viral proteins could also be involved. For example, the Nef regulatory viral protein acts on actin cytoskeleton remodeling and the Pak2-cofilin signaling pathway (44, 45) to inhibit infected-T-cell migration and promote cortical actin polymerization (46).
The cortical actin cytoskeleton is composed of a network of connected actin filaments, which are polymerized and depolymerized very quickly depending on the cellular context. In T cells, actin dynamics can be initiated by the signaling phospholipid PI(4,5)P2 through the activation of Rho GTPase-mediated pathways (47–49). During HIV-1 assembly, Gag also interacts specifically with PI(4,5)P2 at the cell plasma membrane and thus could modulate Rho GTPase-mediated actin reorganization signaling pathways. In mammals, the Rho GTPases comprise a family of at least 20 members, including the main Rac1, Cdc42, and RhoA GTPases. Most of these proteins switch between an inactive GDP-bound form and an active GTP-bound form. The cycling between these two states is regulated by three sets of proteins, guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs), and guanine nucleotide dissociation inhibitors (GDIs). Activated Rho GTPases interact with downstream effector proteins, thereby stimulating a variety of cell signaling pathways (50). The RhoA-Dia1, Rac1-Pak1 (or Pak2), and Rac1-IRSp53-Wave2 pathways have been reported to be involved in the regulation of actin cytoskeleton and membrane dynamics in T lymphocytes (51–54). Interestingly, it has also been reported that inhibition of Rac1, Cdc42, and RhoA GTPases is able to diminish virus production, while the ability of Gag to oligomerize remains unaffected (55, 56). Thus, at least one of these Rho GTPases and its signaling pathway seem to be required for HIV-1 release. However, the specific signaling pathway involved in the late phases of HIV replication remains to be identified.
In the present study, we first investigated which one of the three main Rho GTPases, Rac1, Cdc42, or RhoA, was involved in the regulation of viral particle production (50). For this purpose, we used siRNA knockdown (KD) to deplete the endogenous Rho GTPases of interest in CD4 T cells. The phenotype of virus-like particle production was analyzed by immunoblotting, and Gag intracellular localization was analyzed by immunofluorescence coupled to confocal microscopy. Our results show the importance of the Rac1-derived signaling pathway in Gag membrane attachment and VLP release. Second, we focused on the Rac1 effectors regulating actin cytoskeleton dynamics that could have an impact on Gag VLP release. With this aim, a siRNA screen against selected Rac1 effectors was performed, and the impact on VLP production was evaluated in the context of the expression of Gag/Gag-Pol, HIV-1ΔEnv (Env-deleted HIV-1), and Gag alone. We targeted the Pak2, IRSp53, Wave2, and Arp3 proteins because they are Rac1 downstream effectors that are able to modulate actin cytoskeleton and membrane remodeling. Pak2 is also known to be activated by HIV-1 Nef and consequently to interfere with actin reorganization and T-cell migration (44, 45, 57). Wave2 can activate the Arp2/3 complex through IRSp53 (53, 58), an adaptor protein that also binds Rac1 and Wave2, inducing actin branching and lamellipodium formation (58, 59). IRSp53 is also known to contain an I-BAR domain, which is able to induce membrane curvature (58). Interestingly, it has been reported that the Rac1-IRSp53-Wave2 signaling complex has an effect on HIV entry: it is activated by HIV-1 Env to promote membrane fusion and HIV-1 entry into host cells (60). Here, we studied the roles of these effectors in HIV-1 Gag assembly and VLP release, in the absence of any viral Env proteins, to focus on the late steps of HIV replication. We found that Rac1 is activated upon Gag expression and that the IRSp53-Wave2-Arp2/3 signaling pathway is involved in HIV-1 Gag VLP production in a Jurkat T-cell line and primary blood lymphocytes (PBLs), along with an increase in the intracellular F-actin content. To our knowledge, this is the first time that the Wave2 complex, which promotes F-actin polymerization, is reported to be involved in the late steps of the HIV-1 replication cycle in T cells.
MATERIALS AND METHODS
Cell culture.
Jurkat T cells (human T-cell leukemia cell line) were grown in RPMI 1640 plus Glutamax (Gibco) supplemented with 10% fetal calf serum (FCS) and antibiotics (penicillin-streptomycin). PBLs were purified from the blood of healthy donors (Etablissement Français du Sang, Montpellier, with Institutional Review Board [IRB] approval for the use of anonymized samples) on Ficoll gradients. PBLs were stimulated with phytohemagglutinin (PHA) (1 μg/ml; Sigma) and interleukin 2 (IL-2) (150 U/ml; AIDS Reagents and Reference Program, NIH) for 48 h prior to transfection.
DNA plasmids.
The plasmid expressing HIV-1 Gag alone (pCMVGag [named pGag]), the plasmid expressing HIV-1 Gag, Gag-Pol, and accessory proteins except Vpu (pCMVΔ8.2 [named p8.2]), and the plasmid expressing Env-deleted HIV-1 (named pNL4.3ΔEnv) were described previously (see references 61 and 62, respectively). Plasmids pCMV-LacZ and pCMV-GFP were used as control plasmids and were derived from a pCMV tag plasmid (Stratagene, Agilent Technologies, Madrid, Spain) that expresses green fluorescent protein (GFP) or LacZ under the control of the cytomegalovirus (CMV) promoter.
DNA transfection, small interfering RNA knockdown, and cell viability.
Jurkat T cells (2 × 106) were microporated with 5 μg of p8.2 or pNL4.3ΔEnv or 4 μg of pGag, pCMV-LacZ, or pCMV-GFP (as control plasmids), together with 150 pmol Stealth RNA interference (RNAi) siRNA against Rho GTPases (Invitrogen); 360 pmol Stealth RNAi siRNA against the Rho GTPase effector Wave2, Vav1, Dia1, or Pak2 (Invitrogen); or 250 pmol siRNA against IRSp53 or Arp3 (Santa Cruz) or the corresponding siRNA controls. The Neon transfection system, the Microporator MP-100 (Invitrogen), and the Amaxa system (Lonza) were used. After microporation, T cells were plated in RMPI complete medium, washed 24 h later with 1× phosphate-buffered saline (PBS), and harvested at 48 h posttransfection. Under each condition, the percent cell viability was measured with a BD FACSCalibur flow cytometer and by trypan blue cell counting. The efficiency of cell transfection was calculated by fluorescence microscopy: the number of “green” fluorescent Gag-labeled cells divided by the total number of cells was determined by using the ImageJ Analyzed Particles plug-in. The siRNA transfection efficiency was evaluated as the percentage of protein depletion relative to the siRNA control (adjusted to the tubulin loading control) by immunoblot quantification analysis using ImageJ software.
PBLs (3 × 106) were microporated with 15 μg of pCMVΔ8.2 or 15 μg of pNL4.3ΔEnv, together with 450 pmol Stealth RNAi siRNA against Rac1 (Invitrogen), 1,080 pmol Stealth RNAi siRNA against Wave2 (Invitrogen),750 pmol siRNA against IRSp53 or Arp3 (Santa Cruz Biotechnologies), or the corresponding amount of siRNA controls from Invitrogen or Santa Cruz Biotechnologies. The Amaxa system (Lonza) was used for cell microporation. PBLs were then plated in RPMI complete medium. Eight hours later, they were washed once in 1× PBS and replated in RPMI complete medium. Finally, PBLs were harvested 48 h after transfection. Under each condition, the percent cell viability was measured with a BD FACSCalibur flow cytometer and/or by trypan blue cell counting.
Antibodies.
Immunoblottings were performed by using the following antibodies: anti-MAp17 and anti-CAp24 (NIH AIDS Reagent Program); HIV-1 p17 (VU47) rabbit and HIV-1 p24Gag monoclonal (24-4) mouse antisera, mouse anti-RhoA (ARH03), mouse anti-Rac1 (ARC03), and mouse anti-Cdc42 (ACD03) antisera (Cytoskeleton); rabbit anti-Wave2 (D2C8), goat anti-IRSp53 (W-20), goat anti-Dia1 (C-20), and mouse anti-Arp3 (51) (Santa-Cruz); rabbit anti-Pak2 (2608) (Cell Signaling); mouse anti-Vav1 (39-2050) (Invitrogen); mouse anti-Lamp2 (human lysosome-associated membrane protein 2) (H4B4) (Santa Cruz Biotechnologies); anti-mouse and anti-rabbit antibodies coupled to horseradish peroxidase (HRP) (Dako); and anti-β-tubulin antibody coupled to HRP (ab21058) (Abcam).
For immunofluorescence staining, mouse anti-CAp24 (24-4) and rabbit anti-MAp17 (VU47) (NIH), mouse anti-PI(4,5)P2 (2C11) (Abcam) (63), and fluorescent Alexa 488- or 546-conjugated secondary antibodies (Molecular Probes, Invitrogen) were used.
Viral particle purification and immunoblotting.
To monitor viral particle production, culture medium containing virus-like particles was harvested at 48 h posttransfection. After filtration (0.45-μm pore size), the viral supernatant was purified by ultracentrifugation through a sucrose cushion, i.e., 25% (wt/vol) in TNE buffer (10 mM Tris-HCl [pH 7.4], 100 mM NaCl, 1 mM EDTA), at 35,000 rpm for 1 h 30 min in a Beckman SW60Ti rotor. The pellet was resuspended overnight at 4°C in TNE buffer and stored at −80°C. To analyze the intracellular viral protein content, cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 20 mM Tris-HCl [pH 8], 1% NP-40, 0.1% SDS, 0.2 mM EDTA) and sonicated. The cell lysate was then clarified for 10 min at 13,000 rpm, and the protein concentration in the cell lysate was determined by the Bradford assay (64). For Western blot analysis, proteins from the viral pellet or the cell lysate (50 μg of total proteins) or from membrane flotation assay fractions (20 μl) were loaded and separated on a 10% SDS-PAGE gel and transferred onto a polyvinylidene difluoride transfer membrane (Thermo Fischer), and immunoblotting was performed by using the corresponding antibodies. Finally, HRP signals were revealed by using the SuperSignal West Pico substrate (Thermo Scientific).
Transfection efficiency and VLP release calculation.
The efficiency of plasmid transfection in T cells was evaluated by measuring the percentage of fluorescent cells by immunofluorescence or flow cytometry analysis. For the G-LISA, the pCMV-GFP plasmid was cotransfected with the plasmid encoding Gag, and prior to the G-LISA, the percentage of GFP-positive (GFP+) cells under each condition was monitored by fluorescence-activated cell sorter (FACS) analysis in order to determine the efficacy of microporation. This value was used to normalize the results of the G-LISA as a function of the transfection efficiency.
For VLP release, the HRP signals from immunoblot membranes were imaged by using the G:Box system (Syngene), and the viral Gag or CAp24 protein signals were quantified by using ImageJ software. The percentage of VLP release (48 h after T-cell microporation with plasmids and siRNA) relative to the tubulin loading sample was then estimated as (Vsv − Vblanc)/[(Vsv − Vblanc)|(Vec − Vblanc)] × 100, where Vsv is the viral supernatant, taking into account the quantification of the blot signals for pr55Gag and CAp24; Vec is the quantification of blot signals for pr55Gag, p49, p41, and CAp24 found in the cell extract; and Vblanc is the background of the immunoblot membrane. Tubulin served as a loading control.
Membrane flotation assay.
Jurkat T cells were transfected (microporated) with the pGag or p8.2 plasmid together with the corresponding siRNA by using Lonza Nucleoporator kit V. After 24 h, cells were washed with 1× PBS, transferred to fresh RPMI medium, and then collected at 48 h posttransfection. For membrane flotation assays, cells were washed with 1× PBS and resuspended in cold 1× TE (10 mM Tris-HCl [pH 7.4], 1 mM EDTA) with a Complete protease inhibitor cocktail (Roche). Cells were disrupted by sonication with 2 pulses for 25 s in a Bioruptor instrument at 4°C. The cell lysates were then centrifuged at 370 × g for 3 min at 4°C to pellet the nuclei and unlysed cells. The resulting postnuclear supernatant (PNS) was adjusted to 150 mM NaCl, loaded under a discontinuous 10 to 75% sucrose gradient made in TNE buffer, and centrifuged to equilibrium in a Beckmann SW60Ti rotor overnight at 125,000 × g at 4°C (as described in reference 15). After ultracentrifugation, fractions were collected from the top to the bottom of the density gradient. Equal volumes of samples from each fraction were loaded onto an SDS-PAGE gel and immunoblotted as described above.
Immunofluorescence staining and confocal microscopy imaging and analysis.
Immunofluorescence experiments were performed on Jurkat T cells. Briefly, cells were transfected with pGag or p8.2, together with the corresponding siRNA (see above). Twenty-four hours later, cells were then seeded onto polylysine-coated coverslips and fixed in 3% paraformaldehyde (PFA)–PBS for 15 min. The fixative was then removed, and free aldehydes were quenched with 50 mM NH4Cl. Cells were then permeabilized with 0.2% Triton X-100 for 5 min, blocked in 1% bovine serum albumin (BSA)–PBS, and incubated with primary antibodies for 1 h at room temperature. The cells were then washed 3 times with 1% BSA–PBS and further incubated for 1 h with the corresponding secondary fluorescent antibodies. The slides were mounted with Fluoromount (Sigma). Images were acquired on an Axioplan 2 Zeiss LSM510 (at PLATIM ENS Lyon) or LSM780 (at MRI CNRS Montpellier) confocal microscope with 488/458-nm argon and 543-nm HeNe lasers and a Plan Apochromat 63× 1.4-numerical-aperture oil objective lens, supplied with LSM510 or LSM780 software.
In order to assess the difference between the “punctate” and the “diffuse” patterns of the Gag signal, confocal laser scanning microscopy (CLSM) images (Fig. 1F to I) were analyzed by using ImageJ software (NIH, Bethesda, MD, USA). In order to segment the images, simple normalized threshold processing was used. These images were then converted to binary images, followed by watershed processing, allowing a better separation of poorly distinguishable peak and valley intensities. All the obtained segmented dots were classified by their measured areas and quantified (Fig. 1F to I).
FIG 1.

Effect of Rho GTPase depletion on VLP production, Gag intracellular localization, and Gag membrane attachment in T cells. (A to E) Effect of Rac1, RhoA, and Cdc42 depletion on VLP production. Jurkat T cells were transfected with p8.2 (expression of Gag, Gag-Pol, and accessory viral proteins) and with the siRNA control or siRNA against Rac1, RhoA, or Cdc42. (A) Immunoblot analysis for detection of the HIV-1 proteins pr55Gag and CAp24 in cell lysates and in VLPs. Tubulin was used as a loading control. (B) Extracellular virus production measured by quantification of immunoblot images, i.e., the ratio between extracellular CAp24 and intracellular Pr55 Gag + CAp24. Bars show mean values and standard deviations resulting from three independent experiments. The statistical significances of differences were calculated by an unpaired t test. **, P value of <0.01; *, P value of <0.05. (C) Cell viability measured by flow cytometry analysis. (D) Quantification of Rho GTPase depletion after siRNA treatment. (E) Percent transfection measured by flow cytometry analysis. Bars show mean values and standard deviations resulting from three independent experiments. (F to J) Effect of Rac1, RhoA, and Cdc42 depletion on Gag intracellular localization. Jurkat T cells were transfected with p8.2 and with the siRNA control (F), siRNA against Rac1 (G), siRNA against RhoA (H), or siRNA against Cdc42 (I). Cells were fixed at 48 h posttransfection, permeabilized, stained for HIV-1 Gag, and analyzed by confocal microscopy. (J) Percentage of cells with each phenotype, calculated for 50 cells. Bars show mean values and standard deviations resulting from three independent experiments. (K and L) Effect of Rac1 or RhoA depletion on Gag cell membrane attachment. Jurkat T cells were microporated with p8.2 and with the siRNA control or siRNA against Rac1 or against RhoA. Cells were then lysed, and the PNS was used for membrane flotation assays. Intracellular proteins of each gradient fraction were loaded onto an SDS-PAGE gel. (K) The viral Gag and Lamp2 proteins were then revealed by immunoblotting, as indicated. (L) Rac1 and RhoA depletion by siRNA knockdown, in the PNS, shown by anti-Rac1 and anti-RhoA immunoblots, respectively.
Measurement of intracellular PI(4,5)P2 levels.
Measurement of intracellular PI(4,5)P2 levels was performed by immunofluorescence coupled to confocal microcopy analysis. Briefly, Jurkat T cells were transfected with p8.2 or the pCMV-LacZ, or the pCMV-Pleckstrin homology (PH) domain of phospholipase C delta fused to GFP (pPH-PLCd-GFP), washed once 48 h later with 1× PBS, and immobilized on polylysine-coated coverslips. All subsequent stages were performed in a cold room. Cells were washed with PBS-CaMg buffer (1 mM CaCl2, 1 mM MgCl2) and fixed in 4% PFA–Dulbecco's modified Eagle's medium (DMEM) for 3 h at 4°C. The fixative was then removed, and free aldehydes were quenched with 50 mM NH4Cl for 15 min at 4°C. Jurkat T cells were blocked in REV buffer (1 mM MgCl2, 0.2% saponin, 50 mM glycin, 0.1% BSA, 1% fetal bovine serum, 1 × PBS) for 20 min and incubated with primary antibodies for 16 h at 4°C. The cells were then washed once with REV buffer for 45 min and further incubated with the corresponding secondary fluorescent antibodies for 2 h. After another wash in REV buffer for 45 min, the slides were mounted with Fluoromount (Sigma). Images were acquired on an Axioplan 2 Zeiss LSM510 confocal microscope with 488/458-nm argon and 543-nm HeNe lasers and a Plan Apochromat 63× 1.4-numerical-aperture oil objective lens, supplied with LSM510 software. The fluorescence intensity of the total intracellular PI(4,5)P2 signal was quantified for each cell (n = 20 cells) by image analysis (ImageJ) and divided by the cell area.
Rac1 and Rho inhibitor assay.
Jurkat T cells (1 × 106) were microporated with 5 μg of pCMVΔ8.2 and treated 24 h later with different concentrations of the Rac1-specific inhibitor NSC23766 (65) or the Rho inhibitor CT04, which targets ROCK. After 10 h of Rac1 inhibitor treatment or CT04 treatment, cell supernatants containing virus-like particles were harvested, and virus production under each condition was analyzed by a reverse transcription assay. Cell lysates in RIPA buffer were analyzed for Gag content by immunoblot analysis. The percent cell viability under each condition was also measured by trypan blue cell counting.
Reverse transcription assay.
Ten microliters of the supernatant containing virus-like particles was added to 50 μl of a reverse transcription mix {60 mM Tris (pH 8.0), 180 mM KCl, 6 mM MgCl2, 0.6 mM EGTA (pH 8.0), 0.12% Triton X-100, 6 mM dithiothreitol, 6 μg/ml oligo(dT), 12 μg/ml poly(rA), and 20 μCi/ml [α-32P]dTTP (specific activity, 3,000 Ci/mmol)}. After 1 h of incubation at 37°C, 5 μl was loaded onto DEAE paper (DE-81; Whatman) and then rinsed with 2× SSC (0.3 M NaCl, 0.03 M sodium citrate [pH 5]). The radioactivity (X rays) of the sample was recorded by using a storage phosphorscreen (Molecular Dynamics), measured with a phosphorimager (Fuji), and quantified by using MultiGauge software (Fuji).
Rac1 and RhoA GTPase activation assay.
Jurkat cells (107) were microporated with pCMV-LacZ (control), pGag, or pCMVΔ8.2, together with the pCMV-GFP vector, by using the Neon transfection system and the MP-100 microporator (Invitrogen). Cells were resuspended in RPMI complete medium, and 10 h later, the microporation efficiency was estimated by measuring the percentage of cells expressing GFP by flow cytometry (Becton Dickinson).
Rac1 and RhoA GTPase activation was measured by using a G-LISA Biochem Rac1 activation assay kit and a G-LISA Biochem RhoA activation assay kit (Cytoskeleton), according to the manufacturer's instructions. Briefly, cells were serum starved for 2 h, washed with cold 1× PBS, and lysed in 250 μl of lysis buffer containing protease inhibitors. The same amount of protein was used under each condition. Extracts were incubated in the Rac1 or RhoA affinity plate for 30 min at 4°C with agitation at 400 rpm. After washing and incubation with the antigen-presenting buffer at room temperature, the primary and secondary antibodies were incubated for 45 min at room temperature with agitation at 400 rpm. The luminescence signal representing Rac1 or RhoA GTPase activation was measured in a luminescence reader after 3 min of incubation with horseradish peroxidase detection reagent. Relative light units (RLUs) from RhoA and Rac1 GTPase activation, obtained from Jurkat cells microporated with the pGag or pCMVΔ8.2 vector, were normalized by considering the efficiency factor for each microporation.
F-actin cell content measurement.
F-actin measurement was performed by flow cytometry and immunofluorescence coupled to confocal microscopy image analysis, as previously described (66). The levels of polymerized actin were measured in 106 microporated cells (see above), in the absence of any stimuli. Briefly, T cells were microporated with pCMV-LacZ (control), pGag, or p8.2, together with the pCMV-GFP plasmid. The cells were then simultaneously fixed, permeabilized, and stained by incubation for 15 min at 37°C with a mixture containing 2% PFA, 0.1% Triton X-100, and phalloidin-Alexa 546 (2 U/ml) (Invitrogen). The percentage of cells expressing F-actin was measured by flow cytometry and was corrected with the transfection efficiency factor, regarding GFP expression for each microporation (T cells were microporated with pGag or p8.2 and the pCMV-GFP control vector expressing GFP).
Analysis by fluorescence microscopy coupled to confocal microscopy was performed on Jurkat T cells that were transfected with pGag, p8.2, or pCMV-LacZ; washed once 24 or 48 h later with 1× PBS; fixed with 3% PFA; and immobilized on polylysine-coated coverslips. The immunofluorescence protocol is similar to the one described above. HIV-1 Gag was revealed by anti-MAp17 or anti-CAp24 antibodies (NIH) and Alexa 488-labeled secondary antibodies. F-actin was revealed by staining with phalloidin-Alexa 546 (2 U/ml) (Invitrogen). Images of samples were acquired on a Zeiss LSM780 confocal microscope with a 63×/1.4-numerical-aperture oil differential interference contrast (DIC) Plan-Apo objective lens. The total cell F-actin/phalloidin-Alexa 546 fluorescence intensity was measured in whole cells by performing a z-projection of the cell z-stack, normalized to the cell area, by using ImageJ Z project plug-in software. The results are averages of data from 15 to 20 cells under each condition (see Fig. 9G).
FIG 9.

Effect of Gag on Rac1 and RhoA activation and on F-actin content in T cells. Jurkat T cells were microporated with a control plasmid or pGag (HIV-1 Gag expression) or p8.2 (expression of Gag, Gag-Pol, and viral accessory proteins), together with the pCMV-GFP plasmid, which was used to normalize the microporation efficiency. (A and B) Effect of Gag on Rac1 and RhoA activation. Data shown are representative of data from three independent experiments. Rac1 (A) and RhoA (B) activation was measured by G-LISA. Data shown indicate the fold increases of GTPase activation regarding activation in Jurkat T cells expressing the control plasmid. * and ** indicate P values of <0.05 and 0.01, respectively. (C to E) Effect of Gag on F-actin content in Jurkat T cells as determined by flow cytometry. F-actin expression was measured by flow cytometry analysis in cells simultaneously fixed and permeabilized and then stained with phalloidin-Alexa 546. (C) Median percentages of cells expressing F-actin and standard deviations, which were calculated for Gag- or p8.2-microporated T cells compared to the GFP expression levels in control cells (microporated with pCMV-GFP), from seven independent experiments. (D) Data from a representative flow cytometry experiment. The continuous line represents F-actin fluorescence in T cells microporated with pGag or p8.2, and the discontinuous line is the control (T cells microporated with pCMV-GFP). * and ** indicate P values of <0.05 and 0.01, respectively. (E) Transfection efficiency was monitored by the percentage of GFP expression in each experiment. Values are averages of data from seven independent experiments. (F and G) Effect of Gag on F-actin content in Jurkat T cells, as determined by immunofluorescence imaging. Cells were transfected with pCMV-LacZ (control), pGag (encoding HIV-1 Gag), or p8.2 (encoding Gag, Gag-Pol, and accessory proteins). At 24 h posttransfection, Jurkat T cells were harvested, fixed, permeabilized, and stained with anti-MAp17 antibodies and phalloidin-Alexa 546. The left panels show images from a representative experiment. (G)The phalloidin intensity in Jurkat T cells under each condition was measured by image analysis (ImageJ) (n = 20 cells). The histogram shows the results obtained. * and ** indicate P values of <0.05 and 0.01, respectively.
Gag and F-actin localization determined by confocal microscopy and cell size measurement.
The cells were prepared and imaged as described above, and for evaluating F-actin and Gag localization at the cell periphery, we performed sectioning through the cell center; the corresponding plot profile of fluorescence intensities is shown (see Fig. 8A). From the cell sections, performed on 9 to 21 cells under each condition, the maximum Gag intensity signal and its distance from the cell edge (as determined by the actin signal) were determined (see Fig. 8B). For cell size measurements, the confocal section at the z-cell center was used to measure the cell diameter by using the ImageJ “measure and label” plug-in on the F-actin images, and the average cell diameter was determined (n = 60 to 100 cells) (the P value was measured with the Student test, with a bilateral distribution of heteroscedastic samples) (see Fig. 8C).
FIG 8.

Effect of a partial double knockdown of IRSp53/Wave2 by siRNA on HIV-1 Gag localization and VLP release in T cells. (A) Jurkat T cells were transfected with the vector pGag (HIV-1 Gag expression) (a to d) or pCMV-LacZ (control) (e and f), together with a mix of siRNA (Wave2 and IRSp53) (a and f) or a mix of control siRNAs (d). The cells were fixed and stained for F-actin (phalloidin-Alexa 546) (red) and Gag (anti-MAp17-Alexa 488) (green). Merge and transmission images are presented. The cell sections show Gag and F-actin fluorescent signals at the cell periphery. (B) Distance of the maximum Gag fluorescent signal from the cell edge (determined by the actin signal) under each condition (n = 9 to 21 cells). (C) Cell mean diameters (in micrometers) of the different cell transfection conditions, as indicated. The effect of the depletion of IRSp53 and/or Wave2 on Jurkat T-cell sizes compared to those of siRNA control-treated cells, in the presence of Gag, is shown. (D) Immunoblot analysis of pr55Gag in T-cell lysates and in VLPs. On the right are the average percentages of depletion of each protein and of inhibition of VLP release from three independent experiments. Tubulin was used as a loading control.
RESULTS
Rac1 knockdown modulates HIV-1 Gag VLP release and Gag intracellular localization and membrane attachment in Jurkat T cells.
A previous study has shown that drug inhibition of Rho GTPases is able to reduce HIV-1 production (55). Thus, our initial goal was to determine which one of the three main Rho GTPases, Rac1, Cdc42, or RhoA, was required for virus production and release. It is known that some Rac1 and RhoA signaling pathways are activated by the viral envelope glycoprotein during the early phases of infection (56, 67, 68). Therefore, to restrict our study and focus on the late steps of the viral cycle, only HIV-1 Gag, Gag-Pol, and the accessory proteins (Tat, Rev, Nef, Vpr, and Vif) were expressed by plasmid DNA transfection in Jurkat T lymphocytes. Using these cells, we studied the effect of siRNA depletion of Rac1, Cdc42, and RhoA proteins on VLP production and Gag intracellular localization.
VLP production was monitored by ultracentrifugation of the clarified cell supernatant followed by antiviral immunoblotting and was normalized to the amount of intracellular Gag produced (Fig. 1A and B). We first checked the siRNA treatment efficiency and its effect on cell viability (Fig. 1C and D). We show that individual Rho GTPase siRNA treatment of Jurkat T cells was able to decrease endogenous Rac1 by 70%, RhoA by 80 to 90%, and Cdc42 by 90% (Fig. 1A and D). The cell transfection efficiency was evaluated to be 60 to 65% regarding the number of Gag(+) cells over total cells by immunofluorescence (Fig. 1E to I). In addition, Rho GTPase silencing did not induce strong cell mortality after microporation (average of 80 to 85% living-cell recovery) (Fig. 1C). Second, we studied the effect of Rho GTPase depletion on VLP assembly and release (as explained in Materials and Methods) (Fig. 1B), and for each condition, we checked Gag localization by immunofluorescence microscopy (Fig. 1F to I). For each image, the distributions of the object areas for each Gag signal pattern (i.e., “punctate” versus “diffuse”) under each condition were compared by using ImageJ (Fig. 1F to I): it can clearly be seen that the punctate pattern of Gag exhibits a larger amount of small objects than does the diffuse phenotype, under each of the conditions, justifying the existence of two different phenotypes (Fig. 1F to I). These two different phenotypes were then quantified for several cells under each condition (Fig. 1J).
The depletion of endogenous Rac1 by siRNA decreased virus production by 50%, as shown by immunoblotting (Fig. 1A and B), for a 70% Rac1 knockdown (KD) efficiency (Fig. 1A and D). In this case, we observed that the intracellular Gag localization was modified (Fig. 1G). Indeed, while the wild-type Gag phenotype was punctate in 96% of the cells in the presence of the siRNA control (Fig. 1F), we observed that the Gag phenotype became diffuse in 75% of the cells when Rac1 was depleted (Fig. 1G) and in 42% and 27% of the cells in the cases of RhoA and Cdc42 KDs, respectively. The depletion of endogenous RhoA by siRNA, with an 85% gene KD (Fig. 1D) and 80% cell viability (Fig. 1C), diminished virus production by only ∼20% (Fig. 1A and B). In this case, Gag localization was slightly modified in comparison to that of the control (Fig. 1B) but remained punctate in 58% of the cells (Fig. 1H and J). Similarly, the depletion of Cdc42 by siRNA, with a 90% gene KD (Fig. 1D) and 70% cell viability (Fig. 1C), did not significantly change VLP release (Fig. 1A) and intracellular Gag localization (Fig. 1I and J), suggesting that Cdc42 was not involved in VLP release in these cells.
This first result suggested that the absence of Rac1 could impair Gag trafficking, membrane localization, or membrane attachment. In order to check this hypothesis, we performed a membrane flotation assay on Jurkat T cells microporated with p8.2 and the siRNA control or siRNA against Rac1 or RhoA (Fig. 1K). In this case, we first checked, by anti-Rac1 or anti-RhoA immunoblotting, that Rac1 or RhoA was depleted (Fig. 1L). Second, with the siRNA control, we were able to detect Gag in the membrane fractions (Fig. 1K, p8.2 + siRNA Control, lanes 2 and 3). However, when Rac1 was depleted, all Gag proteins were detected in the cytoplasmic fractions (Fig. 1K, p8.2 + siRNA Rac1, lanes 7 and 8), while the Lamp2 membrane protein was located in the upper gradient fraction (as shown by Lamp2 immunoblotting) (Fig. 1K, p8.2 + siRNA Rac1, lane 2). As a control, we performed the same experiment with the siRNA against RhoA, and we observed that under these conditions, RhoA was depleted (Fig. 1L), and Gag was found in the cytosol (Fig. 1K, lanes 7 and 8) and in the gradient membrane fraction (fractions 2 and 3), in which the membrane protein Lamp2 was detected (Fig. 1K, p8.2 + siRNA RhoA). These results indicated a role for Rac1 in Gag membrane attachment, compared to RhoA. Note that in the membrane flotation assay performed on Jurkat T cells expressing Gag, it was difficult to detect a strong signal for Gag in the membrane fraction of the PNS gradient, certainly due to the fact that it was difficult to collect enough membrane in T cells, compared to other cell lines, or it was due to the limit of detection of this assay.
Our results show that Rac1 plays a role in regulating VLP production and Gag membrane localization in Jurkat T lymphocytes, while RhoA and Cdc42 seem less involved or dispensable.
A Rac1-specific inhibitor is able to decrease VLP release.
To confirm the role of Rac1, we assayed the effects of two drugs targeting Rho GTPases on VLP production in Jurkat T cells (Fig. 2). The specific Rac1 inhibitor NSC23766 was able to decrease VLP production in a dose-dependent manner (Fig. 2A), while CT04, a Rho inhibitor targeting ROCK, was not (Fig. 2C), under conditions which preserved cell viability (Fig. 2B and D). In only one experiment was the cell viability decreased to 65% at the highest NSC23766 concentration (i.e., 200 μM); otherwise, the cell viability was 80%, as measured by trypan blue exclusion. In addition, at 50 and 100 μM NSC23766, the decrease in VLP release correlated to a 2.5- ± 0.6-fold increase in the intracellular Gag content (data not shown). Thus, under these conditions, the decrease in VLP release is dependent on a specific drug effect (Fig. 2A). In contrast, with CT04 treatment, we did not observe any decrease in VLP release (Fig. 2C), no increase in intracellular Gag content was observed, and, if anything, there was a slight decrease (0.7- ± 0.2-fold compared to the control without drug) (data not shown). These results confirm that Rac1 plays an important role in VLP production in Jurkat T lymphocytes, contrary to Rho GTPases regulated by ROCK.
FIG 2.
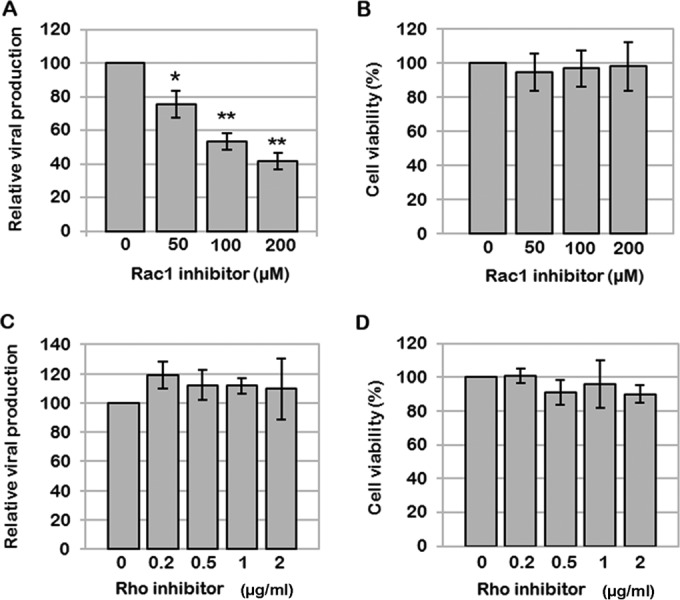
Effects of the Rac1 inhibitor NSC23766 and the Rho inhibitor CT04 on VLP production. Jurkat T cells were transfected with p8.2 and treated 24 h later with different concentrations of a Rac1-specific inhibitor or the Rho inhibitor CT04. (A and B) Effect of the Rac1-specific inhibitor NSC23766 on VLP production. (A) After 10 h of treatment, virus production was analyzed by a reverse transcription assay. Bars show mean values and standard deviations resulting from three independent experiments. The statistical significances of differences were calculated by an unpaired t test. **, P value of <0.01; *, P value of <0.05. (B) Cell viability measured by using trypan blue. (C and D) Effect of the Rho inhibitor CT04 on VLP production. (C) After drug treatment, virus production was analyzed by a reverse transcription assay. Bars show mean values and standard deviations resulting from three independent experiments. (D) Cell viability measured by using trypan blue.
An intact Rac1-Wave2-IRSp53-Arp2/3 pathway is required for optimal virus production.
Based on the data presented above, it appears that a Rac1-derived signaling pathway is required for virus assembly and release in CD4 T lymphocytes. However, Rac1 regulates many signaling pathways in T cells. Indeed, on one hand, Rac1 can activate the phosphatidylinositol 4-phosphate-5-kinase, which induces PI(4,5)P2 synthesis (69, 70). On the other hand, Rac1 can also regulate actin cytoskeleton dynamics (49, 50) through several signaling pathways, such as Rac1-Pak2-cofilin or Rac1-IRSp53-Wave2-Arp3 (Fig. 3A). Therefore, using a siRNA approach, we analyzed which of these effectors might be required for VLP assembly and release in T cells.
FIG 3.

Effect of siRNA-mediated depletion of Rac1 on intracellular PI(4,5)P2 levels in Jurkat T cells. (A) Rac1-dependent cell signaling pathways having an effect on actin filament dynamics. In T cells, Rac1 GTPase plays an important role in the regulation of actin cytoskeleton rearrangement. Indeed, on one hand, in response to extracellular signals, Rac1 is activated and could stimulate actin filament turnover by the intermediate of the Pak1 (or Pak2)-LIMK-cofilin pathway. On the other hand, activated Rac1 could also stimulate actin filament branching in lamellipodia. In the latter case, IRSp53 is recruited by Rac1 and binds the proline-rich region of Wave2. As a result, Wave2 is activated and induces actin branching via Arp2/3 complex recruitment. In a third case, Rac1-GTP could also activate the phosphatidylinositol-4-phosphate 5-kinase (PIP5K), which catalyzes the synthesis of PI(4,5)P2 at the cell plasma membrane. (B to D) Effect of siRNA-mediated depletion of Rac1 on intracellular PI(4,5)P2 levels. (B) Cells were transfected with pCMV-LacZ, p8.2 (Gag, Gag-Pol, and viral accessory proteins), or PH-phospholipase C delta (PLCδ)-GFP, together with the siRNA control or siRNA against Rac1, as indicated. At 48 h posttransfection, cells were fixed, permeabilized, and stained with anti-MAp17 and anti-PI(4,5)P2 antibodies. The colocalization of the GFP-tagged PH domain of phospholipase C delta and the PI(4,5)P2 antibody is indicated as a control for PI(4,5)P2-enriched membrane domains (white arrows). (C) The ratio of the PI(4,5)P2 signal intensity to the cell area was measured by image analysis (ImageJ) (n = 20 cells). (D) Immunoblot analysis of Rac1 GTPase and HIV-1 Gag in cell lysates and/or in virus particles. Tubulin was used as a loading control.
First, we analyzed whether the impact of Rac1 on virus production was related to its regulating role in PI(4,5)P2 synthesis. Indeed, during HIV-1 assembly, the MA domain of Gag binds specifically to the acidic phospholipids, especially PI(4,5)P2, located at the inner leaflet of the cell plasma membrane, thus targeting Gag to the plasma membrane for assembly into VLPs. It is therefore possible that Rac1 silencing might induce a decrease in the intracellular PI(4,5)P2 level and consequently a decrease in Gag membrane binding and virus production. To check this hypothesis, we performed an analysis of the intracellular localization and level of PI(4,5)P2 in Rac1-depleted cells by immunofluorescence and confocal microscopy (Fig. 3B and C). We checked that the PI(4,5)P2 antibody was able to recognize the Jurkat T-cell plasma membrane and to colocalize with a GFP-tagged PH domain of phospholipase C, which is known to specifically recognize PI(4,5)P2-enriched membrane domains (Fig. 3B). We then analyzed the ratio of the PI(4,5)P2 fluorescence intensity signal to the cell area in T cells expressing Gag, or not, in the presence of Rac1 siRNA or the siRNA control (Fig. 3C). Despite efficient Rac1 silencing (Fig. 3D), no deleterious effect on the localization of PI(4,5)P2 at the plasma membrane (Fig. 3B) or on its intracellular level (Fig. 3C) was observed, while VLP production was decreased (Fig. 3D). This result suggested that the effect of Rac1 on Gag membrane localization and VLP release seemed not to be primarily related to its role in the regulation of PI(4,5)P2 synthesis. This may be explained by the fact that PI(4,5)P2 was present in large amounts at the cell plasma membrane.
As it has been reported in several studies that the actin cytoskeleton might have a role in HIV-1 assembly and release (26, 38, 39), particularly in T cells, we next analyzed whether Rac1 was modulating VLP release via the downstream effectors Pak2, IRSp53, Wave2, and Arp2/3, which have a role in cortical actin dynamics (Fig. 3A). Indeed, Pak2 regulates actin filament turnover, while the IRSp53-Wave2 complex activates the Arp2/3 proteins and consequently actin filament branching in lamellipodia (49, 59). Moreover, these proteins are known to be involved in the regulation of HIV-1 replication. Pak2 is activated by HIV-1 Nef to interfere with T-cell migration (44, 45, 57). With this aim, silencing of these Rac1 effectors was performed, and the impact on cell viability and virus production was evaluated in Jurkat T cells and primary T cells (PBLs) (see Fig. 5 and 6). As it was reported previously that the Rac1-Wave2-IRSp53 signaling complex is activated by HIV-1 Env to promote membrane fusion and HIV-1 entry into cells (60), the role of Rac1 effectors in VLP release in the absence of HIV-1 Env was studied to focus on the late steps of the viral replication cycle.
FIG 5.

Effect of Rac1-dependent cell signaling and the Wave2 multicomplex on virus assembly and mature VLP production. (A to D) Effect of Dia1, Pak2, Wave2, IRSp53, and Arp3 depletion on virus assembly and mature particle production. Jurkat T cells were transfected with p8.2 (Gag, Gag-Pol, and viral accessory proteins) and with the siRNA control or siRNA against Dia1, Pak2, Wave2, IRSp53, or Arp3. (A) Immunoblot analysis of HIV-1 pr55Gag and CAp24 in cell lysates and in virus particles. Tubulin was used as a loading control. (B) Extracellular virus production measured by quantification of immunoblot images, corresponding to the ratio of extracellular CAp24 to intracellular pr55Gag and CAp24. Bars show mean values and standard deviations resulting from three independent experiments. The statistical significances of differences were calculated by an unpaired t test. **, P value of <0.01; *, P value of <0.05. (C) Cell viability measured by using trypan blue. (D) Quantification of protein depletion after siRNA treatment. Bars show mean values and standard deviations resulting from three independent experiments. (E to H) Effect of the Rac1-Wave2-IRSp53-Arp3 cell signaling pathway on virus assembly and mature particle production in primary T lymphocytes (PBLs). PBLs were transfected with p8.2 and with the siRNA control or siRNA against Rac1, IRSp53, Wave2, or Arp3. (E) Immunoblot analysis of HIV-1 pr55Gag and CAp24 in cell lysates and in virus particles. Tubulin was used as a loading control. (F) Extracellular virus production measured as described above for panel C. Bars show mean values and standard deviations resulting from two independent experiments. (G) Cell viability measured by using trypan blue. (H) Quantification of protein depletion after siRNA treatment.
FIG 6.

Effect of Rac1-dependent cell signaling on production of Env-deleted HIV-1 particles. (A to D) Effect of Rac1, Dia1, Wave2, IRSp53, or Arp3 depletion on production of mature particles. Jurkat T cells were transfected with pNL4.3ΔEnv and with the siRNA control or siRNA against Rac1, Dia1, Wave2, IRSp53, or Arp3. (A) Immunoblot analysis of pr55Gag and CAp24 in cell lysates and in virus particles. Tubulin was used as a loading control. (B) Extracellular virus production measured by quantification of immunoblot images, corresponding to the ratio of extracellular CAp24 to intracellular pr55Gag and CAp24. Bars show mean values and standard deviations resulting from two independent experiments. The statistical significances of differences were calculated by an unpaired t test. **, P value of <0.01; *, P value of <0.05. (C) Cell viability measured by using trypan blue. (D) Quantification of protein depletion after siRNA treatment by using ImageJ software, relative to the tubulin loading control. Bars show mean values and standard deviations resulting from three independent experiments. (E to H) Effect of Rac1, Dia1, Wave2, IRSp53, or Arp3 depletion on production of mature particles in PBLs. PBLs were transfected with pNL4.3ΔEnv and with the siRNA control or siRNA against Rac1, Dia1, Wave2, IRSp53, or Arp3. (E) Immunoblot analysis of HIV-1 pr55Gag and CAp24 in cell lysates and in virus particles. Tubulin was using as a loading control. (F) Extracellular virus production calculated as described above for panel B. Bars show mean values and standard deviations resulting from two independent experiments. (G) Cell viability measured by dead cell counting using trypan blue. (H) Quantification of protein depletion after siRNA treatment.
As a control, we checked that siRNA microporation in Jurkat T cells and PBLs did not affect VLP production (Fig. 4). Indeed, when Jurkat T cells (Fig. 4A and B) or PBLs (Fig. 4C and D) were microporated with a plasmid expressing either p8.2 or Gag without any siRNA, after 48 h, the cells were able to produce mature and immature VLPs, respectively (Fig. 4A and C). The same experiment was performed by microporation of Jurkat T cells, or PBLs, with p8.2, or pGag, in the presence of the control siRNA: it was observed that the presence of the siRNA slightly increased the intracellular Gag content but also VLP release (Fig. 4A and C). However, the calculations of VLP release (Fig. 4B and D) showed no difference in VLP release from Jurkat T cells, or PBLs, microporated with or without siRNA. Second, to check that Jurkat T cells, upon microporation with pGag and siRNA, were indeed producing VLPs and not cellular debris containing Gag resulting from the experimental microporation procedure, we monitored Gag VLP release versus MACA release (Fig. 4F). MACA is a Gag protein that is deleted of its C-terminal NC-p1-p6 region and deleterious in VLP release. Under our conditions, i.e., microporated Jurkat T cells with plasmids and siRNA, we could not detect any MACA VLP released in the cell supernatant in the presence of either Rac1 siRNA or control siRNA, as shown by anti-CA immunoblotting (Fig. 4F), compared to Gag VLPs. This was for 60% of cells expressing MACA-mEOS2 (a monomeric variant of green-to-red photoswitchable fluorescent protein) and 80% cell viability, as measured by FACS analysis (data not shown), while the protein was well expressed in cells, as seen by green fluorescence microscopy (Fig. 4E).
FIG 4.

Effect of siRNA microporation on production of mature and immature VLPs. Jurkat T cells or PBLs were microporated with p8.2 or pGag without any siRNA (−) or with control siRNA (+). (A and C) Immunoblot analysis of pr55Gag and CAp24 in cell lysates and in VLPs of Jurkat T cells (A) or primary T lymphocytes (C). Tubulin was using as a loading control. (B and D) Extracellular VLP production was measured by quantification of immunoblot images, corresponding to the ratio of extracellular pr55Gag or CAp24 to intracellular pr55Gag. Bars show mean values and standard deviations resulting from three independent experiments. p.d.u., protein detectable unit. (E and F) Control analysis of Gag and MACA VLP production. (E) Visualization of Jurkat T cells expressing mEOS2-tagged Gag or MACA with the corresponding siRNA by fluorescence and transmission microscopy (bar = 100 μm). (F) Immunoblots (anti-CAp24) of VLPs of Jurkat T cells expressing mEOS2-tagged Gag (82 kDa) or mEOS2-tagged MACA (68 kDa) together with the siRNA control or siRNA against Rac1, at 48 h postmicroporation.
In addition to the above-mentioned Rac1 effectors, the role of a RhoA effector, Dia1, in VLP production in Jurkat T cells (Fig. 5A and D) and in PBLs (Fig. 5E and H) was analyzed. Dia1 is an effector of RhoA that activates the nucleation and elongation of actin filaments (71). As RhoA had only a minor impact on virus production (Fig. 1A and 2C), Dia1 silencing by siRNA should not alter Gag VLP production in T cells, as shown in Fig. 5 and 6. Indeed, in Jurkat T cells expressing Gag, in the presence of Gag-Pol and accessory proteins (i.e., p8.2), siRNA depletion of Dia1 did not significantly modify VLP production (Fig. 5A and B), whereas cell viability was preserved (Fig. 5C). Similarly, Pak2 silencing had no effect (Fig. 5D). On the contrary, siRNA depletion of IRSp53, Wave2, and Arp3 decreased virus production by 55% ± 2%, 50% ± 10%, and 25% ± 6%, respectively (Fig. 5A and B), in Jurkat T cells, under conditions where cell viability was preserved (Fig. 5C), with a KD efficiency of 60 to 70% (Fig. 5D). The same experiment was performed on PBLs (Fig. 5E to H) purified from uninfected patient blood cells and activated with PHA/IL-2. Similarly to what was observed for Jurkat T cells, siRNA depletion of Wave2, IRSp53, and Arp3 in PBLs microporated with p8.2 was able to decrease VLP production by 50% ± 8%, 60% ± 12%, and 50% ± 10%, respectively (Fig. 5E and F), while cell viability was maintained for 60 to 75% of the cells (Fig. 5G), for a KD efficiency of 65 to 80% (Fig. 5H). These results demonstrated for the first time the involvement of Rac1, IRSp53, Wave2, and Arp3 in HIV-1 particle production in primary T lymphocytes.
We then studied the role of Rac1 and its effectors in the context of an HIV-1 provirus depleted of the Env gene to focus on the late steps of virus replication and to prevent the activation of the Rac1 signaling pathway during virus entry. These experiments were conducted with HIV-1ΔEnv-expressing Jurkat T cells (Fig. 6A to D) and PBLs (Fig. 6E to H). While the Dia1 siRNA control had no significant effect on HIV-1ΔEnv production, as expected, the KDs of Rac1, Wave2, IRSp53, and Arp3 decreased HIV-1ΔEnv particle production by 38% ± 20%, 48% ± 13%, 85% ± 7%, and 62% ± 30%, respectively, in Jurkat T cells (Fig. 6A and B) and by 45% ± 7%, 46% ± 14%, 62% ± 8%, and 70% ± 5%, respectively, in PBLs (Fig. 6E and F). Under these conditions, cell viability was preserved for at least 85% of the cells (Fig. 6C and G), and the KD gene efficiency varied from 50 to 80% (Fig. 6D and H). It is worth noting that IRSp53 gene silencing by siRNA had more of an effect on VLP release (up to a 5-fold reduction in virus particle production, for 70% protein depletion) than did silencing of other genes, underlining the involvement of this cellular protein (Fig. 6A and B). Importantly, the same results were observed with PBLs (Fig. 6E and F). Overall, these data show the involvement of the cellular proteins Rac1, Wave2, IRSp53, and Arp3 in optimal VLP release, independently of the Env protein, in both a T-cell line and primary blood lymphocytes.
The involvement of the Rac1-Wave2-IRSp53-Arp3 pathway in VLP production is dependent on Gag.
The literature has reported an effect of the HIV-1 Tat and Nef accessory proteins on several Rac1-derived signaling pathways (44, 45, 66). We thus decided to study if the effect of the Rac1-Wave2-IRSp53-Arp3 signaling pathway on virus production observed here was mediated by HIV-1 accessory proteins or solely by the Gag viral protein. For this purpose, we performed siRNA screening targeting Rac1 effectors on Jurkat T cells expressing immature HIV-1 Gag and producing VLPs (Fig. 7). As shown in Fig. 7A, the depletion of Rac1, Wave2, IRSp53, and Arp3 by siRNA decreased Gag VLP production by 50% ± 7%, 45% ± 12%, 55% ± 4%, and 34% ± 2%, respectively (Fig. 7B), while Pak2 depletion had no effect. Under these conditions, cell viability was preserved (Fig. 7C), and the KD efficiency was up to 80% (Fig. 7D). Altogether, these results suggested that the effect of the Rac1-Wave2-IRSp53-Arp2/3 signaling pathway on viral particle production was mediated mainly by the Gag viral protein. It was interesting to notice that, as observed with the HIV-1 provirus, the most pronounced effects on Gag VLP release were observed with IRSp53 and Wave2 siRNA treatments, even if these cellular proteins were not completely depleted, strongly suggesting that these two proteins might be important cofactors for HIV-1 Gag particle release. For the first time, here, we describe a role for two cellular proteins, IRSp53 and Wave2, that are cofactors of cell membrane curvature and cortical actin dynamics in HIV-1 Gag particle release, due solely to the presence of Gag in T cells.
FIG 7.

Effect of siRNA targeting the Wave2 multicomplex on Gag VLP production. Jurkat T cells were transfected with pGag and with the siRNA control or siRNA against Rac1, Pak2, Wave2, IRSp53, or Arp3. (A) Immunoblot analysis of pr55Gag in cell lysates and in viral particles. Tubulin was using as a loading control. (B) Extracellular virus production measured by quantification of immunoblot images, corresponding to the ratio of extracellular pr55Gag to intracellular pr55Gag. Bars show mean values and standard deviations resulting from four independent experiments. The statistical significances of differences were calculated by an unpaired t test. **, P value of <0.01. (C) Cell viability evaluated by dead cell counting using trypan blue. (D) Quantification of protein depletion after siRNA treatment. Bars show mean values and standard deviations resulting from four independent experiments.
Double Wave2 and IRSp53 knockdowns, although partial, modulate intracellular Gag localization and strongly decrease VLP production.
In order to check if IRSp53 and Wave2 acted in synergy in the same signaling pathway, the effect of the double knockdown of these two proteins was tested following treatment of Gag-expressing Jurkat T cells with a double-siRNA mix against Wave2 and IRSp53 proteins. Gag localization, compared to F-actin, as well as cell sizes were analyzed by confocal microscopy imaging (Fig. 8A to C). Gag VLP release and cellular protein depletion were monitored by immunoblotting (Fig. 8D). We found that the transfection efficiency of the HIV-1 pGag plasmid in Jurkat T cells was 72% ± 7% (for a total number of cells of 140 ≤ n ≤187), as analyzed by Gag immunofluorescence (Gag-positive “green” fluorescent cells over nonfluorescent cells). Strikingly, we observed that the double-knockdown T cells could be identified by cell size measurements (Fig. 8C). We then found that 55% ± 9% of total cells were transfected with siRNA and that 48% ± 3% of total cells were transfected with both pGag and siRNA. Indeed, the Wave2 or IRSp53 siRNA-treated cells presented a defect in their cellular F-actin phenotype: instead of being regularly located underneath the cell plasma membrane, cortical F-actin staining became irregular, in patches in the cytosol (Fig. 8A), and a slight increase in cell size was observed, i.e., ∼9 μm in diameter (IRSp53 or Wave2 siRNA), instead of ∼7 μm on average for the siRNA control cells (Fig. 8C). For the double siRNA (IRSp53 and Wave2), the cells exhibited a cellular volume that increased significantly (Fig. 8A), i.e., ∼12 μm in diameter, instead of ∼7 μm on average for the siRNA control cells (Fig. 8C). The double-knockdown cells were easily identified by their loss of F-actin shape (more diffuse in the cytosol) (Fig. 8A), in comparison to wild-type Jurkat T cells, in which the F-actin formed a uniform, well-defined ring underneath the cell plasma membrane (Fig. 8A). In addition, it is worth noting that the partial depletion of the IRSp53 and Wave2 siRNA (Fig. 8A, panel a) seemed to prevent Gag localization to the cell plasma membrane, as Gag appeared highly localized in the cytosol compared to its localization in siRNA control cells (Fig. 8A, panel d). Several images of double-knockdown (IRSp53 and Wave2 siRNA) cells expressing Gag showed a loss of Gag membrane labeling and mainly a cytosolic Gag localization (Fig. 8Aa to c), in comparison to the control (Fig. 8Ad). As shown in Fig. 8B, we were able to quantify the distance between the maximum intensity of the intracellular Gag fluorescent signal and the cell border (determined by the F-actin fluorescent signal): one can observe that this distance (for an average of 9 to 11 cells) was ∼833 nm when Gag was expressed in the double-KD (IRSp53 and Wave2) cells and was ∼145 to 172 nm in Wave2 or IRSp53 single-KD cells, compared to the control wild-type cells (Fig. 8B). These results suggested that in the double-KD cells, Gag was unable to reach or attach to the cell plasma membrane. Unfortunately, we were unable to verify this observation by membrane flotation assays with Jurkat T cells expressing Gag only, as the latter was not detectable in the membrane fractions under these conditions (in contrast to p8.2 in Fig. 1K) (data not shown). However, upon the partial depletion of both IRSp53 and Wave2 (38% ± 6% and 44% ± 15% protein depletion, respectively), we observed that Gag VLP release was strongly reduced (by 75% ± 5%), in comparison to the control double siRNA (Fig. 8D). Overall, these results showed that a partial double depletion of Wave2 and IRSp53 impaired Gag membrane localization, resulting in a greater decrease in Gag VLP release than with individual knockdowns. These data support a role for the Wave2 complex in intracellular Gag localization and VLP production.
Rac1 is activated upon expression of HIV-1 Gag in Jurkat T cells, and intracellular F-actin content is increased.
In the literature, it has been reported that Wave2 is an essential regulator of the actin cytoskeleton through its ability to stimulate Arp2/3-dependent actin polymerization downstream of activated Rac1 (72). The regulation of Wave2 by Rac1 is complicated because it can require other signaling molecules, such as IRSp53 (58). As we observed that the depletion of these proteins mediating the Rac1-IRSp53-Wave2-Arp2/3 signaling pathway was able to modulate HIV-1 Gag production in T cells, we thus studied whether HIV-1 Gag was able to activate Rac1 and F-actin polymerization. We focused on these parameters because Rac1 activation is the signal that initiates the signaling pathway, and F-actin polymerization is the cellular response to this signal.
First, we measured the level of endogenous Rac1 activation (Fig. 9A), or RhoA as a control (Fig. 9B), in Jurkat T cells by G-LISA (see Materials and Methods). Rac1 and RhoA GTPases are able to switch between an active GTP-bound state and an inactive GDP-bound state. The G-LISAs allowed the detection and quantification of the amount of the intracellular GTP-bound forms of Rac1 and RhoA by specific antibody detection and luminescence and thus allowed us to monitor the intracellular levels of activated Rac1 and RhoA. In Jurkat T cells expressing Gag, in the presence of Gag-Pol and accessory proteins, the activation level of Rac1 was increased by 9- ± 1.7-fold (Fig. 9A), in comparison to the negative control (Fig. 9A). This effect might be due to the presence of the viral accessory proteins Tat and/or Nef, as it was reported previously that they could modulate Rac1 activity during the late steps of HIV-1 replication (66, 73, 74). Therefore, to determine the role of HIV-1 Gag alone in Rac1 activation, the same experiment was performed following pGag microporation in Jurkat T cells. In this case, Rac1 activation was increased by 4.3- ± 1.9-fold relative to the control (Fig. 9A) and normalized to the amount of transfected cells (GFP+ cells), as measured by flow cytometry. These results indicated that Rac1-GTP activation could be induced by the expression of only Gag in Jurkat T cells. In contrast, the effect of Gag alone on the activation level of RhoA (1.2- ± 0.4-fold) was equivalent to that of the control (Fig. 9B) and thus was negligible compared to that of Rac1 activation. The level of activation of RhoA was slightly increased (by 2- ± 1.4-fold) when Gag and accessory proteins were expressed but not when Gag alone was expressed (Fig. 9B). These data showed that the role of RhoA in the regulation of the late steps of virus replication in Jurkat T cells seemed minor compared to that of Rac1 and was dependent on HIV-1 accessory proteins. In these cells, an activated form of Rac1 seemed to be required for efficient particle formation and release, and this activation could be mediated by Gag alone, although it was enhanced by the presence of viral accessory proteins.
Second, we studied the effect of HIV-1 Gag expression in Jurkat T cells on the intracellular F-actin content. Here, we quantified the intracellular F-actin content by flow cytometry analysis and by fluorescence microscopy. The F-actin level was measured by flow cytometry analysis of phalloidin-Alexa 546-stained Jurkat T cells expressing Gag or not (Fig. 9C and D). The expression of Gag alone and in the presence of Gag-Pol and accessory proteins resulted in significant increases of the intracellular F-actin content, by 6.3% ± 2% and 8.4 ± 3.3%, respectively (Fig. 9C). To confirm these results, we also measured the F-actin level by fluorescent phalloidin-Alexa 546 staining and semiquantitative fluorescence microscopy analysis (Fig. 9F and G). In this case, we observed an increase of the intracellular F-actin level when Gag was expressed in Jurkat T cells (Fig. 9F). We measured the F-actin fluorescence intensity in the total cell volume (as described in Materials and Methods) for cells expressing Gag labeled by phalloidin-Alexa 546, and we observed a 2- to 3-fold increase in the F-actin content in Jurkat T cells expressing Gag compared to the control (pCMV-LacZ) (Fig. 9G). These data suggested that both Gag alone and Gag with accessory proteins could activate F-actin polymerization in T cells, as suggested in previous studies (44, 45, 66). Altogether, our data showed that Gag expression in Jurkat T cells activated Rac1 and increased the intracellular F-actin content.
DISCUSSION
The aim of our study was to characterize which one of the main small Rho GTPases Rho, Rac1, and Cdc42 and which associated specific downstream effectors were involved in HIV-1 particle release in host CD4+ T cells. Indeed, HIV assembly could occur either at the plasma membrane of infected cells or in virus-containing compartments, depending on the cell type (2, 3, 5, 75). It has been reported that in CD4+ T lymphocytes, viral particles are released mainly by direct budding from the plasma membrane (76). HIV-1 particle assembly is driven by the viral Gag protein, which targets specifically the PI(4,5)P2 phosphoinositide, located at the inner leaflet of the cell plasma membrane (18). In addition, this phosphoinositide is a critical second messenger that regulates a myriad of cellular activities, including actin remodeling and branching. In the latter case, the PI(4,5)P2 phosphoinositides are the sites where activated Rho GTPases are recruited to initiate cell signaling pathways (47, 49). As small Rho GTPases and their effectors are key regulators of actin remodeling and membrane dynamics (77), their involvement at the HIV-1 assembly site was considered.
Several studies have shown that activated Rac1 as well as downstream signaling pathways, including the Wave2 multicomplex (78), are involved in the early steps of HIV-1 replication (56, 67, 68), such as virus entry and virus-cell fusion. However, the requirement for activated Rac1 in the late steps of HIV replication had not been assessed thoroughly. It was recently reported that the hematopoietic cell-specific Rho GTPase inhibitor Arhgdib/d4gdi limited HIV-1 replication, but the GTPase involved was not identified (56). In our study, we first checked the implications of the main small Rho GTPases Rac1, RhoA, and Cdc42 in VLP release and intracellular Gag localization and membrane attachment in CD4 T cells. We then identified the downstream effectors involved in the context of Gag, Gag/Gag-Pol, or HIV-1ΔEnv VLP production. Our work was performed by using a noninfectious system devoid of the viral envelope glycoproteins in order to prevent interference with the early steps of HIV-1 infection, as a role for the Rac1-Wave2-IRSp53 pathway in HIV-1 entry was reported previously (60). Using a siRNA strategy, we clearly showed the involvement of the Rac1-Wave2-IRSp53-Arp2/3 signaling pathway in the late phases of HIV-1 replication, both in a T-cell line and in PBLs. Our results revealed an essential role of activated Rac1, and of RhoA to a lesser extent, in HIV-1 Gag VLP production in Jurkat T cells. The minor effect of RhoA could be explained by a potential role of Citron-K, an effector of RhoA, in enhancing HIV-1 production by stimulating the endosomal compartments and exocytosis, as observed previously for adherent cells (79). Here, using nonadherent Jurkat T cells and PBLs, for which it has been shown that HIV-1 buds mostly from the cell plasma membrane (5, 75), we reported a major involvement of Rac1 (Fig. 1, 5 to 7, and 9).
As activated Rac1 is known to be located at the cell plasma membrane and able to modulate several signaling pathways (Fig. 3A), our results strongly suggested the involvement of a Rac1-dependent signaling pathway in HIV-1 assembly and particle release in T cells. These pathways are involved in the regulation of actin cytoskeleton and membrane dynamics in T lymphocytes (52, 53, 58, 80), including the Vav1-Rac1-Pak1 (or Pak2)-cofilin and the Rac1-IRSp53-Wave2-Arp3 pathways, which were therefore explored. Interestingly, depletion of Pak2 did not decrease Gag VLP production, i.e., in the absence of Nef (Fig. 5), while depletion of Pak1 decreased Gag VLP production by ∼25% (data not shown). Vav1 is an important Rac1 activator involved in the formation of the immunological synapse by promoting F-actin nucleation and accumulation at the cell edge (80). Furthermore, during the last steps of viral replication, Vav1 can be activated by the HIV-1 accessory protein Nef (74, 81) as well as by the cellular Zap70 protein (82), which regulates cell-to-cell spread of HIV-1 (83). Thus, we tested the effect of siRNA depletion of Vav1 on virus production, and we observed a 2-fold decrease of virus production in Jurkat T cells expressing Gag, Gag-Pol, and accessory proteins (including Nef) but no decrease of Gag VLP production with Gag alone (data not shown). These results were in agreement with data from previous studies showing that Pak1 (or Pak2), Vav1, and HIV-1 Nef form a complex during the last steps of viral replication, which modulates actin dynamics and induces cytoskeleton rearrangements (57, 74, 81) and which might regulate particle production.
Here, our data showed, for the first time, that another Rac1-mediated signaling pathway, the Wave2-IRSp53-Arp2/3 multiprotein complex, is involved in HIV-1 particle release (Fig. 5 and 6). This effect was mediated mainly by HIV-1 Gag alone (Fig. 7). In addition, IRSp53/Wave2 double-siRNA treatment of T cells expressing Gag significantly increased the inhibitory effect on VLP release in comparison to single knockdowns (Fig. 8D) and induced the mislocalization of Gag in Jurkat T cells (Fig. 8A). This finding strongly supports the involvement of this pathway in virus production through the viral Gag protein. The fact that the observed siRNA effect on Gag VLP release never exceeded 4-fold might be due to the fact that the targeted proteins, i.e., IRSp53 and Wave2, are required for cell survival and cell membrane and cytoskeleton integrities and therefore cannot be further silenced without affecting cell viability. However, by adapting experimental conditions to maintain cell survival with a partial knockdown of targeted genes, it was possible to monitor significant effects on Gag membrane localization and on virus release, as shown by several control experiments (Fig. 4). Therefore, our results suggest that the Wave2 complex could be essential for HIV-1 Gag membrane localization and virus production (Fig. 8).
Furthermore, the activation of Rac1 was induced by the sole presence of Gag in Jurkat T cells (Fig. 9A). The hypothesis of a recruitment of a Rac1-activated Wave2 multicomplex at the Gag assembly sites was therefore possible. Indeed, we were able to observe a significant modulation of the intracellular F-actin content in Jurkat T cells expressing Gag (Fig. 9C and G). These measurements were used to quantify intracellular F-actin contents in another study, and the results shown were of the same order of magnitude (66). Activation of the Wave2 complex at the cell plasma membrane requires the simultaneous presence of activated Rac1 (GTP-bound Rac1) together with PI(3,4,5)P3 or PI(4,5)P2 phospholipids (72), the IRSp53 protein as a linker between Rac1 and Wave2 (58), and the Arp2/3 branching factor. Thus, it is conceivable that the Wave2/IRSp53 complex and Gag may recognize these acidic phospholipids in the same membrane domains during virus assembly to promote local F-actin branching. Actually, the depletion of the Arp2/3 actin nucleator by siRNA impaired HIV-1ΔEnv and Gag VLP production (Fig. 5 to 7), and a significant increase in the level of F-actin in T cells expressing Gag was observed (Fig. 9C to G). In the literature, roles for the regulatory HIV-1 Tat protein (66) and Nef (44, 45) in F-actin cytoskeleton remodeling, during the early and the late steps of the viral cycle, respectively, have already been described. Here, we showed that, in addition to Tat and Nef, the structural Gag protein itself could have an effect on F-actin cell content (Fig. 9C, F, and G).
To our knowledge, this work reports, for the first time, the involvement of activated Rac1 and the IRSp53-Wave2-Arp2/3 signaling pathway during the late steps of the HIV replication cycle concomitant with an increase in the F-actin T-cell content dependent solely on the Gag viral protein. Further investigations are needed to determine whether Gag and Wave2 interact directly or indirectly, whether they are located within the same membrane domains or not, and if Gag could trigger its recruitment to the cell plasma membrane.
In conclusion, a potential role of actin polymerization during HIV-1 particle formation in T cells can be considered, involving the activation of Rac1 and the IRSp53-Wave2-Arp2/3 signaling pathway. We thus hypothesize that a temporary increase in the F-actin level at the Gag assembly site could occur to maintain Gag multimers in cell membrane domains by preventing their return to the cytosol, therefore stabilizing the multi-Gag assembling bud. An alternative hypothetical scenario could be that actin branching may favor viral budding by creating a template in the vicinity of the viral budding site, acting as a mechanical force for particle release, as suggested by previous cryo-electron microscopy (cryo-EM) studies (84). Actually, cryo-EM quantification of F-actin filaments within or in the vicinity of viral buds (for adherent cells expressing HIV-1 Gag/Gag-Pol) was recently reported, indicating that almost half of the budding sites analyzed were associated with F-acting branching in the viral bud in formation (85). This observation is in agreement with our results. A potential implication of the NC domain of Gag was proposed (32, 33), but this has recently been challenged, as that study reported no difference in virus release when the NC domain of Gag was replaced by a leucine zipper domain (85). Those same authors concluded that the NC domain of HIV-1 Gag was dispensable for actin incorporation into HIV-1 particles (85). Therefore, the domain(s) of Gag responsible for the F-actin association with viral buds remains to be identified.
Finally, several studies indicated a role for actin or related actin-binding proteins in HIV-1 assembly or release, which could be actively or passively incorporated into the particles (31), and an atomic force microscopy study suggested that large actin-enriched structures could localize underneath virus assembly sites (34). However, a recent study failed to detect F-actin at the HIV-1 Gag assembly sites by live-cell imaging microscopy (35). Of note, F-actin was labeled through Life-actin-mCherry, with the uncertainty of the incorporation of such a molecule in filamentous actin, and this was done in adherent HeLa cells (35). Nonetheless, by using Jurkat T cells or PBLs, this study, as well as others (36–38), clearly showed that drug-mediated or siRNA-mediated inhibition of actin cytoskeleton dynamics changed the intracellular Gag membrane localization and impaired virus production. This suggests that short and dynamic F-actin oligomers can act temporally at the HIV-1 assembly sites to favor Gag membrane localization and particle release, at least in nonadherent T cells.
ACKNOWLEDGMENTS
We thank D. Décimo and J. L. Darlix for scientific advice and Caroline Goujon for review. We thank the PLATIM microscopy platform (ENS Lyon) and MRI (Montpellier). We are also grateful to the CNRS, INSERM, and ENS Lyon.
A.T. received fellowship support from the ANRS and FRM. We are very thankful for grant funding from SIDACTION and ANRS, France.
REFERENCES
- 1.Corbin A, Grigorov B, Roingeard P, Darlix J, Muriaux D. 2008. Revisiting HIV-1 assembly. Med Sci (Paris) 24:49–55. (In French.) doi: 10.1051/medsci/200824149. [DOI] [PubMed] [Google Scholar]
- 2.Grigorov B, Arcanger F, Roingeard P, Darlix J, Muriaux D. 2006. Assembly of infectious HIV-1 in human epithelial and T-lymphoblastic cell lines. J Mol Biol 359:848–862. doi: 10.1016/j.jmb.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 3.Joshi A, Ablan SD, Soheilian F, Nagashima K, Freed EO. 2009. Evidence that productive human immunodeficiency virus type 1 assembly can occur in an intracellular compartment. J Virol 83:5375–5387. doi: 10.1128/JVI.00109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jouve M, Sol-Foulon N, Watson S, Schwartz O, Benaroch P. 2007. HIV-1 buds and accumulates in “nonacidic” endosomes of macrophages. Cell Host Microbe 2:85–95. doi: 10.1016/j.chom.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 5.Ono A, Freed EO. 2004. Cell-type-dependent targeting of human immunodeficiency virus type 1 assembly to the plasma membrane and the multivesicular body. J Virol 78:1552–1563. doi: 10.1128/JVI.78.3.1552-1563.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kutluay SB, Bieniasz PD. 2010. Analysis of the initiating events in HIV-1 particle assembly and genome packaging. PLoS Pathog 6:e1001200. doi: 10.1371/journal.ppat.1001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muriaux D, Darlix J. 2010. Properties and functions of the nucleocapsid protein in virus assembly. RNA Biol 7:744–753. doi: 10.4161/rna.7.6.14065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ono A. 2010. Relationships between plasma membrane microdomains and HIV-1 assembly. Biol Cell 102:335–350. doi: 10.1042/BC20090165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dussupt V, Javid MP, Abou-Jaoudé G, Jadwin JA, de La Cruz J, Nagashima K, Bouamr F. 2009. The nucleocapsid region of HIV-1 Gag cooperates with the PTAP and LYPXnL late domains to recruit the cellular machinery necessary for viral budding. PLoS Pathog 5:e1000339. doi: 10.1371/journal.ppat.1000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.von Schwedler UK, Stuchell M, Müller B, Ward DM, Chung H, Morita E, Wang HE, Davis T, He G, Cimbora DM, Scott A, Kräusslich H, Kaplan J, Morham SG, Sundquist WI. 2003. The protein network of HIV budding. Cell 114:701–713. doi: 10.1016/S0092-8674(03)00714-1. [DOI] [PubMed] [Google Scholar]
- 11.Chen K, Bachtiar I, Piszczek G, Bouamr F, Carter C, Tjandra N. 2008. Solution NMR characterizations of oligomerization and dynamics of equine infectious anemia virus matrix protein and its interaction with PIP2. Biochemistry 47:1928–1937. doi: 10.1021/bi701984h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chukkapalli V, Hogue IB, Boyko V, Hu W, Ono A. 2008. Interaction between the human immunodeficiency virus type 1 gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient gag membrane binding. J Virol 82:2405–2417. doi: 10.1128/JVI.01614-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chukkapalli V, Oh SJ, Ono A. 2010. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc Natl Acad Sci U S A 107:1600–1605. doi: 10.1073/pnas.0908661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamard-Peron E, Muriaux D. 2011. Retroviral matrix and lipids, the intimate interaction. Retrovirology 8:15. doi: 10.1186/1742-4690-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamard-Peron E, Juillard F, Saad JS, Roy C, Roingeard P, Summers MF, Darlix JL, Picart C, Muriaux D. 2010. Targeting of murine leukemia virus gag to the plasma membrane is mediated by PI(4,5)P2/PS and a polybasic region in the matrix. J Virol 84:503–515. doi: 10.1128/JVI.01134-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inlora J, Chukkapalli V, Derse D, Ono A. 2011. Gag localization and virus-like particle release mediated by the matrix domain of human T-lymphotropic virus type 1 Gag are less dependent on phosphatidylinositol-(4,5)-bisphosphate than those mediated by the matrix domain of HIV-1 Gag. J Virol 85:3802–3810. doi: 10.1128/JVI.02383-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kerviel A, Thomas A, Chaloin L, Favard C, Muriaux D. 2013. Virus assembly and plasma membrane domains: which came first? Virus Res 171:332–340. doi: 10.1016/j.virusres.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 18.Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. 2004. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci U S A 101:14889–14894. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prchal J, Srb P, Hunter E, Ruml T, Hrabal R. 2012. The structure of myristoylated Mason-Pfizer monkey virus matrix protein and the role of phosphatidylinositol-(4,5)-bisphosphate in its membrane binding. J Mol Biol 423:427–438. doi: 10.1016/j.jmb.2012.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saad JS, Ablan SD, Ghanam RH, Kim A, Andrews K, Nagashima K, Soheilian F, Freed EO, Summers MF. 2008. Structure of the myristylated human immunodeficiency virus type 2 matrix protein and the role of phosphatidylinositol-(4,5)-bisphosphate in membrane targeting. J Mol Biol 382:434–447. doi: 10.1016/j.jmb.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brügger B, Glass B, Haberkant P, Leibrecht I, Wieland FT, Kräusslich H. 2006. The HIV lipidome: a raft with an unusual composition. Proc Natl Acad Sci U S A 103:2641–2646. doi: 10.1073/pnas.0511136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dick RA, Goh SL, Feigenson GW, Vogt VM. 2012. HIV-1 Gag protein can sense the cholesterol and acyl chain environment in model membranes. Proc Natl Acad Sci U S A 109:18761–18766. doi: 10.1073/pnas.1209408109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguyen DH, Hildreth JE. 2000. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J Virol 74:3264–3272. doi: 10.1128/JVI.74.7.3264-3272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ono A, Freed EO. 2001. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc Natl Acad Sci U S A 98:13925–13930. doi: 10.1073/pnas.241320298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grigorov B, Attuil-Audenis V, Perugi F, Nedelec M, Watson S, Pique C, Darlix J, Conjeaud H, Muriaux D. 2009. A role for CD81 on the late steps of HIV-1 replication in a chronically infected T cell line. Retrovirology 6:28. doi: 10.1186/1742-4690-6-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jolly C, Sattentau QJ. 2007. Human immunodeficiency virus type 1 assembly, budding, and cell-cell spread in T cells take place in tetraspanin-enriched plasma membrane domains. J Virol 81:7873–7884. doi: 10.1128/JVI.01845-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thali M. 2011. Tetraspanin functions during HIV-1 and influenza virus replication. Biochem Soc Trans 39:529–531. doi: 10.1042/BST0390529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keller H, Kräusslich H, Schwille P. 2013. Multimerizable HIV Gag derivative binds to the liquid-disordered phase in model membranes. Cell Microbiol 15:237–247. doi: 10.1111/cmi.12064. [DOI] [PubMed] [Google Scholar]
- 29.Ridley A. 2011. Life at the leading edge. Cell 145:1012–1022. doi: 10.1016/j.cell.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 30.Ott DE, Coren LV, Kane BP, Busch LK, Johnson DG, Sowder RC II, Chertova EN, Arthur LO, Henderson LE. 1996. Cytoskeletal proteins inside human immunodeficiency virus type 1 virions. J Virol 70:7734–7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ott DE, Coren LV, Johnson DG, Kane BP, Sowder RC II, Kim YD, Fisher RJ, Zhou XZ, Lu KP, Henderson LE. 2000. Actin-binding cellular proteins inside human immunodeficiency virus type 1. Virology 266:42–51. doi: 10.1006/viro.1999.0075. [DOI] [PubMed] [Google Scholar]
- 32.Liu B, Dai R, Tian CJ, Dawson L, Gorelick R, Yu XF. 1999. Interaction of the human immunodeficiency virus type 1 nucleocapsid with actin. J Virol 73:2901–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilk T, Gowen B, Fuller SD. 1999. Actin associates with the nucleocapsid domain of the human immunodeficiency virus Gag polyprotein. J Virol 73:1931–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gladnikoff M, Shimoni E, Gov NS, Rousso I. 2009. Retroviral assembly and budding occur through an actin-driven mechanism. Biophys J 97:2419–2428. doi: 10.1016/j.bpj.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rahman SA, Koch P, Weichsel J, Godinez WJ, Schwarz U, Rohr K, Lamb DC, Kräusslich H, Müller B. 2014. Investigating the role of F-actin in human immunodeficiency virus assembly by live-cell microscopy. J Virol 88:7904–7914. doi: 10.1128/JVI.00431-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jolly C, Mitar I, Sattentau QJ. 2007. Requirement for an intact T-cell actin and tubulin cytoskeleton for efficient assembly and spread of human immunodeficiency virus type 1. J Virol 81:5547–5560. doi: 10.1128/JVI.01469-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muriaux D, Darlix JL, Cimarelli A. 2004. Targeting the assembly of the human immunodeficiency virus type I. Curr Pharm Des 10:3725–3739. doi: 10.2174/1381612043382701. [DOI] [PubMed] [Google Scholar]
- 38.Sasaki H, Nakamura M, Ohno T, Matsuda Y, Yuda Y, Nonomura Y. 1995. Myosin-actin interaction plays an important role in human immunodeficiency virus type 1 release from host cells. Proc Natl Acad Sci U S A 92:2026–2030. doi: 10.1073/pnas.92.6.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cooper J, Liu L, Woodruff EA, Taylor HE, Goodwin JS, D'Aquila RT, Spearman P, Hildreth JEK, Dong X. 2011. Filamin A protein interacts with human immunodeficiency virus type 1 Gag protein and contributes to productive particle assembly. J Biol Chem 286:28498–28510. doi: 10.1074/jbc.M111.239053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wen X, Ding L, Wang J, Qi M, Hammonds J, Chu H, Chen X, Hunter E, Spearman P. 2014. ROCK1 and LIM kinase modulate retrovirus particle release and cell-cell transmission events. J Virol 88:6906–6921. doi: 10.1128/JVI.00023-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen C, Jin J, Rubin M, Huang L, Sturgeon T, Weixel KM, Stolz DB, Watkins SC, Bamburg JR, Weisz OA, Montelaro RC. 2007. Association of gag multimers with filamentous actin during equine infectious anemia virus assembly. Curr HIV Res 5:315–323. doi: 10.2174/157016207780636542. [DOI] [PubMed] [Google Scholar]
- 42.Chen C, Weisz OA, Stolz DB, Watkins SC, Montelaro RC. 2004. Differential effects of actin cytoskeleton dynamics on equine infectious anemia virus particle production. J Virol 78:882–891. doi: 10.1128/JVI.78.2.882-891.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lehmann M, Nikolic DS, Piguet V. 2011. How HIV-1 takes advantage of the cytoskeleton during replication and cell-to-cell transmission. Viruses 3:1757–1776. doi: 10.3390/v3091757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stolp B, Abraham L, Rudolph JM, Fackler OT. 2010. Lentiviral Nef proteins utilize PAK2-mediated deregulation of cofilin as a general strategy to interfere with actin remodeling. J Virol 84:3935–3948. doi: 10.1128/JVI.02467-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stolp B, Reichman-Fried M, Abraham L, Pan X, Giese SI, Hannemann S, Goulimari P, Raz E, Grosse R, Fackler OT. 2009. HIV-1 Nef interferes with host cell motility by deregulation of cofilin. Cell Host Microbe 6:174–186. doi: 10.1016/j.chom.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 46.Nobile C, Rudnicka D, Hasan M, Aulner N, Porrot F, Machu C, Renaud O, Prévost M, Hivroz C, Schwartz O, Sol-Foulon N. 2010. HIV-1 Nef inhibits ruffles, induces filopodia, and modulates migration of infected lymphocytes. J Virol 84:2282–2293. doi: 10.1128/JVI.02230-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chichili GR, Rodgers W. 2009. Cytoskeleton-membrane interactions in membrane raft structure. Cell Mol Life Sci 66:2319–2328. doi: 10.1007/s00018-009-0022-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sechi AS, Wehland J. 2000. The actin cytoskeleton and plasma membrane connection: PtdIns(4,5)P(2) influences cytoskeletal protein activity at the plasma membrane. J Cell Sci 113(Part 21):3685–3695. [DOI] [PubMed] [Google Scholar]
- 49.Tybulewicz VLJ, Henderson RB. 2009. Rho family GTPases and their regulators in lymphocytes. Nat Rev Immunol 9:630–644. doi: 10.1038/nri2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heasman SJ, Ridley AJ. 2008. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- 51.Burbach BJ, Medeiros RB, Mueller KL, Shimizu Y. 2007. T-cell receptor signaling to integrins. Immunol Rev 218:65–81. doi: 10.1111/j.1600-065X.2007.00527.x. [DOI] [PubMed] [Google Scholar]
- 52.Knaus UG, Bokoch GM. 1998. The p21Rac/Cdc42-activated kinases (PAKs). Int J Biochem Cell Biol 30:857–862. doi: 10.1016/S1357-2725(98)00059-4. [DOI] [PubMed] [Google Scholar]
- 53.Takenawa T, Miki H. 2001. Wasp and wave family proteins: key molecules for rapid rearrangement of cortical actin filaments and cell movement. J Cell Sci 114:1801–1809. [DOI] [PubMed] [Google Scholar]
- 54.Vicente-Manzanares M, Rey M, Pérez-Martínez M, Yáñez-Mó M, Sancho D, Cabrero JR, Barreiro O, de la Fuente H, Itoh K, Sánchez-Madrid F. 2003. The RhoA effector mDia is induced during T cell activation and regulates actin polymerization and cell migration in T lymphocytes. J Immunol 171:1023–1034. doi: 10.4049/jimmunol.171.2.1023. [DOI] [PubMed] [Google Scholar]
- 55.Audoly G, Popoff MR, Gluschankof P. 2005. Involvement of a small GTP binding protein in HIV-1 release. Retrovirology 2:48. doi: 10.1186/1742-4690-2-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Watanabe T, Urano E, Miyauchi K, Ichikawa R, Hamatake M, Misawa N, Sato K, Ebina H, Koyanagi Y, Komano J. 2012. The hematopoietic cell-specific Rho GTPase inhibitor ARHGDIB/D4GDI limits HIV type 1 replication. AIDS Res Hum Retroviruses 28:913–922. doi: 10.1089/aid.2011.0180. [DOI] [PubMed] [Google Scholar]
- 57.Fackler OT, Lu X, Frost JA, Geyer M, Jiang B, Luo W, Abo A, Alberts AS, Peterlin BM. 2000. p21-activated kinase 1 plays a critical role in cellular activation by Nef. Mol Cell Biol 20:2619–2627. doi: 10.1128/MCB.20.7.2619-2627.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abou-Kheir W, Isaac B, Yamaguchi H, Cox D. 2008. Membrane targeting of wave2 is not sufficient for wave2-dependent actin polymerization: a role for irsp53 in mediating the interaction between rac and wave2. J Cell Sci 121:379–390. doi: 10.1242/jcs.010272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee SH, Dominguez R. 2010. Regulation of actin cytoskeleton dynamics in cells. Mol Cells 29:311–325. doi: 10.1007/s10059-010-0053-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harmon B, Campbell N, Ratner L. 2010. Role of Abl kinase and the Wave2 signaling complex in HIV-1 entry at a post-hemifusion step. PLoS Pathog 6:e1000956. doi: 10.1371/journal.ppat.1000956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. 1997. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol 15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]
- 62.Burniston MT, Cimarelli A, Colgan J, Curtis SP, Luban J. 1999. Human immunodeficiency virus type 1 Gag polyprotein multimerization requires the nucleocapsid domain and RNA and is promoted by the capsid-dimer interface and the basic region of matrix protein. J Virol 73:8527–8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thomas CL, Steel J, Prestwich GD, Schiavo G. 1999. Generation of phosphatidylinositol-specific antibodies and their characterization. Biochem Soc Trans 27:648–652. [DOI] [PubMed] [Google Scholar]
- 64.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 65.Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. 2004. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A 101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.López-Huertas MR, Callejas S, Abia D, Mateos E, Dopazo A, Alcamí J, Coiras M. 2010. Modifications in host cell cytoskeleton structure and function mediated by intracellular HIV-1 Tat protein are greatly dependent on the second coding exon. Nucleic Acids Res 38:3287–3307. doi: 10.1093/nar/gkq037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harmon B, Ratner L. 2008. Induction of the Galpha(q) signaling cascade by the human immunodeficiency virus envelope is required for virus entry. J Virol 82:9191–9205. doi: 10.1128/JVI.00424-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pontow SE, Heyden NV, Wei S, Ratner L. 2004. Actin cytoskeletal reorganizations and coreceptor-mediated activation of rac during human immunodeficiency virus-induced cell fusion. J Virol 78:7138–7147. doi: 10.1128/JVI.78.13.7138-7147.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Halstead JR, Savaskan NE, van den Bout I, Van Horck F, Hajdo-Milasinovic A, Snell M, Keune W, Ten Klooster J, Hordijk PL, Divecha N. 2010. Rac controls PIP5K localisation and PtdIns(4,5)P2 synthesis, which modulates vinculin localisation and neurite dynamics. J Cell Sci 123:3535–3546. doi: 10.1242/jcs.062679. [DOI] [PubMed] [Google Scholar]
- 70.Weernink PAO, Meletiadis K, Hommeltenberg S, Hinz M, Ishihara H, Schmidt M, Jakobs KH. 2004. Activation of type I phosphatidylinositol 4-phosphate 5-kinase isoforms by the Rho GTPases RhoA, Rac1, and Cdc42. J Biol Chem 279:7840–7849. doi: 10.1074/jbc.M312737200. [DOI] [PubMed] [Google Scholar]
- 71.Goode BL, Eck MJ. 2007. Mechanism and function of formins in the control of actin assembly. Annu Rev Biochem 76:593–627. doi: 10.1146/annurev.biochem.75.103004.142647. [DOI] [PubMed] [Google Scholar]
- 72.Lebensohn AM, Kirschner MW. 2009. Activation of the wave complex by coincident signals controls actin assembly. Mol Cell 36:512–524. doi: 10.1016/j.molcel.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lu X, Wu X, Plemenitas A, Yu H, Sawai ET, Abo A, Peterlin BM. 1996. CDC42 and Rac1 are implicated in the activation of the Nef-associated kinase and replication of HIV-1. Curr Biol 6:1677–1684. doi: 10.1016/S0960-9822(02)70792-6. [DOI] [PubMed] [Google Scholar]
- 74.Rauch S, Pulkkinen K, Saksela K, Fackler OT. 2008. Human immunodeficiency virus type 1 nef recruits the guanine exchange factor vav1 via an unexpected interface into plasma membrane microdomains for association with p21-activated kinase 2 activity. J Virol 82:2918–2929. doi: 10.1128/JVI.02185-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jouvenet N, Neil SJD, Bess C, Johnson MC, Virgen CA, Simon SM, Bieniasz PD. 2006. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol 4:e435. doi: 10.1371/journal.pbio.0040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mariani C, Desdouits M, Favard C, Benaroch P, Muriaux DM. 2014. Role of Gag and lipids during HIV-1 assembly in CD4(+) T cells and macrophages. Front Microbiol 5:312. doi: 10.3389/fmicb.2014.00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gowrishankar K, Ghosh S, Saha S, C R, Mayor S, Rao M. 2012. Active remodeling of cortical actin regulates spatiotemporal organization of cell surface molecules. Cell 149:1353–1367. doi: 10.1016/j.cell.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 78.Spear M, Guo J, Turner A, Yu D, Wang W, Meltzer B, He S, Hu X, Shang H, Kuhn J, Wu Y. 2014. HIV-1 triggers WAVE2 phosphorylation in primary CD4 T cells and macrophages, mediating Arp2/3-dependent nuclear migration. J Biol Chem 289:6949–6959. doi: 10.1074/jbc.M113.492132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Loomis RJ, Holmes DA, Elms A, Solski PA, Der CJ, Su L. 2006. Citron kinase, a RhoA effector, enhances HIV-1 virion production by modulating exocytosis. Traffic 7:1643–1653. doi: 10.1111/j.1600-0854.2006.00503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tybulewicz VLJ, Ardouin L, Prisco A, Reynolds LF. 2003. Vav1: a key signal transducer downstream of the TCR. Immunol Rev 192:42–52. doi: 10.1034/j.1600-065X.2003.00032.x. [DOI] [PubMed] [Google Scholar]
- 81.Fackler OT, Luo W, Geyer M, Alberts AS, Peterlin BM. 1999. Activation of vav by nef induces cytoskeletal rearrangements and downstream effector functions. Mol Cell 3:729–739. doi: 10.1016/S1097-2765(01)80005-8. [DOI] [PubMed] [Google Scholar]
- 82.García-Bernal D, Parmo-Cabañas M, Dios-Esponera A, Samaniego R, Hernán de la Ossa DH-P, Teixidó J. 2009. Chemokine-induced Zap70 kinase-mediated dissociation of the Vav1-talin complex activates alpha4beta1 integrin for T cell adhesion. Immunity 31:953–964. doi: 10.1016/j.immuni.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 83.Sol-Foulon N, Sourisseau M, Porrot F, Thoulouze M, Trouillet C, Nobile C, Blanchet F, di Bartolo V, Noraz N, Taylor N, Alcover A, Hivroz C, Schwartz O. 2007. Zap-70 kinase regulates HIV cell-to-cell spread and virological synapse formation. EMBO J 26:516–526. doi: 10.1038/sj.emboj.7601509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Carlson L, de Marco A, Oberwinkler H, Habermann A, Briggs JAG, Kräusslich H, Grünewald K. 2010. Cryo electron tomography of native HIV-1 budding sites. PLoS Pathog 6:e1001173. doi: 10.1371/journal.ppat.1001173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stauffer S, Rahman SA, de Marco A, Carlson L, Glass B, Oberwinkler H, Herold N, Briggs JAG, Müller B, Grünewald K, Kräusslich H. 2014. The nucleocapsid domain of Gag is dispensable for actin incorporation into HIV-1 and for association of viral budding sites with cortical F-actin. J Virol 88:7893–7903. doi: 10.1128/JVI.00428-14. [DOI] [PMC free article] [PubMed] [Google Scholar]