

Abstract
Kinase-catalyzed protein phosphorylation is involved in a wide variety of cellular events. Development of methods to monitor phosphorylation is critical to understand cell biology. Our lab recently discovered kinase-catalyzed biotinylation, where ATP-biotin is utilized by kinases to label phosphopeptides or phosphoproteins with a biotin tag. To exploit kinase-catalyzed biotinylation for phosphoprotein purification and identification in a cellular context, the susceptibility of the biotin tag to phosphatases was characterized. We found that the phosphorylbiotin group on peptide and protein substrates was relatively insensitive to protein phosphatases. To understand how phosphatase stability would impact phosphoproteomics research applications, kinase-catalyzed biotinylation of cell lysates was performed in the presence of kinase or phosphatase inhibitors. We found that biotinylation with ATP-biotin was sensitive to inhibitors, although with variable effects compared to ATP phosphorylation. The results suggest that kinase-catalyzed biotinylation is well suited for phosphoproteomics studies, with particular utility towards monitoring low abundance phosphoproteins or characterizing the influence of inhibitor drugs on protein phosphorylation.
Keywords: kinases, biotinylation, phosphatases, HeLa cells, protein labeling
Introduction
Protein phosphorylation is a fundamental and well-studied posttranslational modification occurring in cells.[1] Phosphorylation is catalyzed by kinases with adenosine 5′- triphosphate (ATP (1), Scheme 1A) as cosubstrate.[2] Phosphorylation attaches a phosphate group onto amino acid residues such as serine, threonine, and tyrosine (Scheme 1B)[3] and can affect protein activity and cell biology.[4] Many biological processes involve kinases and protein phosphorylation, including cell signaling, immunosuppression and cancer formation, for examples.[5] Protein phosphorylation is a reversible process whereby phosphatases catalyze the dephosphorylation reaction (Scheme 1B).[6]
Scheme 1.

Kinase-catalyzed phosphorylation and biotinylation. (A) Chemical structure of ATP (1), ATP-biotin (2) and ATP-γS (3). (B) Peptides or proteins (4) undergo phosphorylation with kinases and ATP to give phosphopeptides or phosphoproteins products (5), which are dephosphorylated with phosphatases. (C) Biotinylation with kinases and ATP-biotin gives biotinylated phosphopeptide or phosphoprotein products (6). The sensitivity of biotinylated phosphopeptides and phosphoproteins to phosphatases is studied in this work.
Phosphopeptide and phosphoprotein detection has become important in the proteomics field. Techniques to monitor phosphorylation involve 32P-radiolabeling,[7] immobilizing metal affinity chromatography,[8] 2-D gel analysis,[9] mass spectrometric (MS) analysis,[7b, 10] covalent modification of phosphates,[11] phosphate staining (such as with Pro-Q diamond),[11a] and specific antibodies.[12] Even though a number of different methods are available to detect phosphoproteins in complex mixtures, each has its own advantages and disadvantages. For example, 32P radiolabeling is sensitive and widely used, but involves hazardous materials. Metal affinity chromatography is successfully coupled with MS analysis to identify phosphopeptides, but is bias towards purification of acidic peptides.[13] Additional phosphoprotein detection methods will provide needed alternatives to strengthen the study of phosphopeptides and phosphoproteins.
Recently we reported the use of γ-phosphate modified ATP analogs for studying phosphorylation.[14] In particular, we demonstrated that an ATP analog with biotin attached to the γ-phosphate (ATP-biotin (2), Scheme 1A) acts as a cosubstrate for kinases and transfers a phosphorylbiotin group to peptides and proteins (Scheme 1C).[14d] The biotinylation reaction was successful with synthetic peptide and full-length protein substrates. Importantly, proteins in cell lysates were labeled using kinase-catalyzed biotinylation. With these successes, kinase-catalyzed biotinylation has application towards characterizing the complete phosphoproteome. However, cellular experiments are complicated by the presence of protein phosphatases that could alter the biotinylation state of the labeled phosphoproteins (Scheme 1C). To assess the suitability of kinase-catalyzed biotinylation for phosphoproteomics applications, the sensitivity of the phosphorylbiotin tag to phosphatase activity must be characterized.
Kinases utilize γ-thio-ATP (ATP-γS (3), Scheme 1A) to generate thiophosphorylated proteins. Prior work revealed that the thiophosphoryl group is insensitive to phosphatases[15] and provided the foundation for use of ATP-γS and thiophosphorylation in phosphoproteomics applications.[16] With this precedent, we examined whether the phosphorylbiotin modification is similarly insensitive to phosphatases. If the biotin tag is stable under cellular conditions, then kinase-catalyzed biotinylation would be an excellent tool to visualize and purify phosphorylated proteins in lysates, with the ability to monitor low abundance phosphoproteins.
Herein we test the stability of the phosphorylbiotin product of kinase-catalyzed biotinylation towards protein phosphatases. The data indicated that the phosphorylbiotin modification is stable to phosphatases. To expand this work, inhibitors of kinases and phosphatases were studied in HeLa cell lysates and the results showed that both phosphorylation and biotinylation are sensitive to inhibitors and can be used to monitor changes in phosphorylation.
Results and Discussion
ATP-biotin synthesis
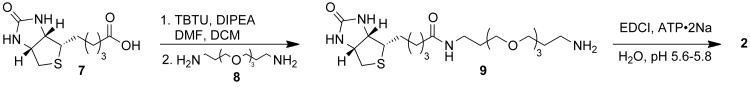
To study the phosphatase sensitivity of a kinase-catalyzed biotinylation, we synthesized ATP-biotin 2 (Scheme 2) using a similar strategy as previously published.[17] Commercially available biotin 7 was activated by TBTU, and then treated with 4, 7, 10-trioxa-1-13-tridecanediamine 8 to yield biotin amine 9. Biotin amine 9 was then coupled with the disodium salt of ATP using EDCI in water while maintaining the pH between 5.6-5.8 to give ATP-biotin 2. Intermediate biotin amine 9 and the final ATP-biotin 2 were characterized by HRMS, 1H, 13C and 31P NMR (Figures S1-9).
Scheme 2. ATP-biotin (2) Synthesis.
The cosubstrate compatibility of synthesized ATP-biotin 2 was tested with three kinases/substrate pairs: PKA (Protein Kinase A) and serine-containing substrate (LRRASLG), CK2 (Casein Kinase 2) and a threonine-containing substrate (RRREEETEEE), and Abl (Abl Protein Tyrosine Kinase) and a tyrosine-containing substrate (EAIYAAPFAKKK). The percentage conversion was calculated relative to ATP using quantitative MS, as previously reported.[18] The quantitative MS data indicated that peptides were biotinylated with 53% (CK2), 81% (PKA), and 78% (Abl) conversion (Figures S10-12). The efficiencies of the kinase reaction with ATP-biotin 2 were comparable to those previously reported with a similar ATP-biotin analog that has two fewer methylene groups in the linker (56-80%).[18]
Phosphatase activity with biotinylated phosphopeptides
Phosphatases are divided into two major classes-[19] serine/threonine phosphatases[20] and tyrosine phosphatases [21], which hydrolyzes phosphoserine/threonine or phosphotyrosine, respectively. Unlike kinases, phosphatases have differing active sites and mechanisms depending on their substrate.[21-22] Serine/threonine phosphatases have an active site that utilizes catalytic metals to bind the phosphate[23] and activates water for phosphate ester hydrolysis.[24] In contrast, tyrosine phosphatases hydrolyze the phosphate ester by employing a nucleophillic cysteine.[21, 25] To characterize the compatibility of the two active sites with phosphoryl biotin degradation, we tested two serine/threonine phosphatases (protein phosphatase 1 (PP1) and calf intestinal phosphatase (CIP)) and one tyrosine phosphatase (T-cell protein tyrosine phosphatase (TCPTP)).
Two steps were employed to assess the phosphatase sensitivity of biotinylated phosphopeptides. First, biotinylphosphopeptides were prepared by incubating ATP-biotin with the same peptide/kinase pairs used in quantitative MS experiments. After generating the biotinylphosphopeptides, the second step involved heat denaturation of the kinase, followed by incubation with PP1, CIP, or TCPTP. As a control, identical experiments were carried out with ATP as a cosubstrate. All reactions were analyzed using high performance liquid chromatography (HPLC). In the presence of acidic HPLC Buffer A (0.1% TFA in water, pH ∼2), the phosphoramidate bond of the biotinylphosphopeptide was cleaved to remove the biotin group and yield a phosphopeptide product.[26] Therefore, ATP and ATP-biotin reactions produced HPLC traces containing identical unmodified (Figure 1A) or phosphorylated peptide products (Figure 1B and C).
Figure 1.
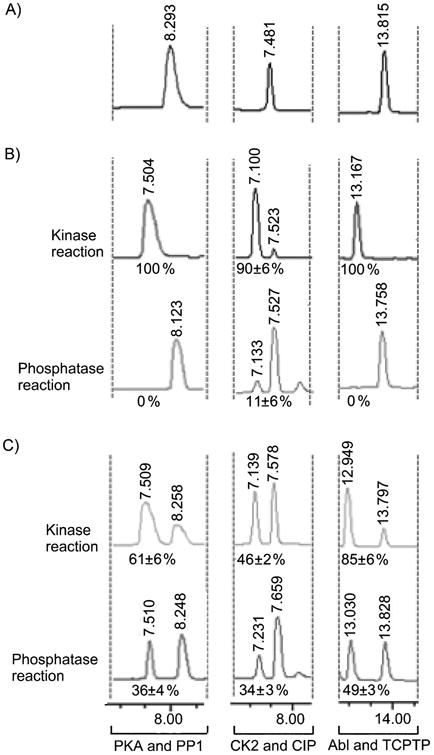
A) HPLC analysis of unmodified peptides (PKA peptide at ∼8.3 min; CK2 peptide at ∼7.5 min; Abl peptide at ∼13.8 min). B) HPLC analysis of ATP-phosphorylated peptides (top; PKA phosphopeptide at ∼7.5 min; CK2 phosphopeptide at ∼7.1 min; Abl peptide at ∼13.2 min) after phosphatase treatment (bottom). C) HPLC analysis of ATP-biotin phosphopeptides (top; PKA phosphopeptide at ∼7.5 min; CK2 phosphopeptide at ∼7.1 min; Ablphosphopeptide at ∼12.9 min) after phosphatase treatment (bottom). The percentages in B) and C) represent the percentage phosphopeptide observed. The complete HPLC traces are shown in Figure S13 of supporting information.
Under conditions where ATP control reactions demonstrated 90% to 100% kinase conversion to phosphopeptide products (Figure 1B, top), the ATP-biotin reactions demonstrated 46-85% conversion (Figure 1C, top). The efficiencies of the kinase reactions with ATP-biotin were comparable to the quantitative mass spectrometric analysis reported here (53-81%) and previously (56-80%),[14a] demonstrating the reliability of the HPLC analysis. After phosphatase treatment, ATP phosphorylated peptides were almost completely hydrolyzed; only 0-11% phosphopeptides remained by HPLC (Figure 1B, bottom), indicating that the phosphatase maintains 89-100% activity. In contrast, a significant amount of biotinylated phosphopeptide products were observed after phosphatase treatment; 34-49% phosphopeptides were still observed after phosphatase reaction (Figure 1C, bottom). Comparing the percentage of biotinylated product before and after phosphatase incubation (Figure 1C, bottom versus top) revealed that 59% (PP1), 74% (CIP), and 58% (TCPTP) of the modification remained intact. Therefore, while phosphatases maintained 89-100% activity with phosphopeptide substrates, only 26-42% activity was seen against biotinylated phosphopeptides. Of the three phosphatases tested, CIP was the least active (26%) with PP1 and TCPTP maintaining similar activities (41% and 42%). These studies indicate that the phosphorylbiotin modification is less susceptible than a phosphate modification to phosphatase hydrolysis.
Phosphatase activity with biotinylated phosphoproteins
Our next goal was to study the sensitivity of full-length biotinylated phosphoproteins to phosphatase activity. While the kinase/phosphatase pairing was the same as with the peptides experiments, the full-length substrates used were myelin basic protein (MBP) with PKA and Abl and β-casein with CK2. Similar to the peptide experiments, a two-step reaction series was employed with kinase reaction first, followed by phosphatase incubation. In this case, the crude products were analyzed by gel methods.
Control reactions with ATP demonstrated the sensitivity of phosphoproteins to degradation by phosphatases; only 6-7% phosphoproteins were observed by ProQ staining (Figure 2, lanes 2, 6, and 10, ProQ), indicating that phosphatases maintain 93-94% activity at removing phosphates. In contrast, a significant amount of biotinylated phosphoproteins was still detected after phosphatase treatment; 72-83% biotinylated phosphoproteins were observed (Figure 2, lanes 4, 8, and 12, SA-Cy5), indicating that the phosphatases demonstrated only 17-28% activity at removing the biotin tag. Consistent with the stability of the biotin tag to phosphatases, 37-66% biotinylated phosphoproteins were detected after phosphatase incubation using Pro-Q staining (Figure 2, lanes 4, 8, and 12, Pro-Q). Comparing the percentage of Pro-Q-visualized biotinylphosphoproteins after and before phosphatase incubation (Figure 2, lanes 4 vs 3, 8 vs 7, and 12 vs 11, ProQ) revealed that 85% (PP1), 66% (CIP), and 90% (TCPTP) of the modification remained intact. The Pro-Q analysis indicated that phosphatases maintained 10-34% activity against biotinylated phosphopeptides. The differences in activity comparing biotin (17-28%) and phosphate (10-34%) detection (Figure 3, SA-Cy5 versus ProQ) may be due to the presence of phosphorylation sites on the substrates prior to kinase reaction, which would skew the Pro-Q staining data.
Figure 2.

Gel analysis and quantification of phosphorylated and biotinylated proteins before and after phosphatase treatment. The contents of each reaction are indicated below each lane. Pro-Q staining visualizes phosphoproteins, SA-Cy5 staining detects biotin-labeled proteins, and Syproruby stains for total protein content. The percentages of phosphorylated (ProQ) or biotinylated (SA-Cy5) relative to lanes 1, 5, or 9 (ProQ, 100%) or lanes 3, 7, or 11 (SA-Cy5, 100%). The full gel images, along with additional control reactions, are shown in Figure S14 of supporting information.
Figure 3.

Gel analysis of phosphoproteins (ProQ, top) or biotinylated phosphoproteins (SA-Cy5, bottom) from HeLa cell lysates incubated with ATP or ATP-biotin in the absence or presence of kinase or phosphatase inhibitors.
A comparison of the activity data suggests that full-length biotinylated proteins are less sensitive to phosphatases than biotinylated peptides (17-28% versus 26-42%). The protein data represents the aggregate reactivities of all phosphorylation sites in the protein to phosphatases. As a result, the data suggest that the majority of phosphorylation siteson the full length protein are less susceptible to phosphatase hydrolysis than the isolated peptides. The steric bulk of full-length proteins may prevent phosphatase binding and catalysis. The significant conclusion from the experiments is that the phosphorylbiotin group, particularly on full length proteins, shows significant stability to phosphatase degradation.
Inhibitor studies in cell lysates
With an eye towards future phosphoproteomics applications, we were interested in probing how the phosphatase stability of the phosphorylbiotin tag would impact lysate labelling. Because cell lysates naturally contain phosphatases, typical phosphoproteomics experiments include a phosphatase inhibitor in the cell lysates to stabilize phosphorylation.[27] With the stability of the phosphorylbiotin tag, we hypothesized that robust kinase-catalyzed labelling will occur in lysates without the need for phosphatase inhibitor treatment. In addition, we wondered how phosphatase inhibitors would impact kinase-catalyzed labeling. Given the interest in kinases and phosphatase inhibitors as drug targets,[28] inhibitor experiments would also establish the utility of kinase-catalyzed labelling as a drug characterization tool.
HeLa cell lysates were incubated with ATP-biotin in the presence of a kinase inhibitor (staurosporine) or phosphatase inhibitors (sodium vanadate and sodium fluoride). As a control, inhibitor experiments were also performed with ATP incubation. As expected, reactions performed in the presence of kinase inhibitors showed only trace levels of phosphorylated (Figure 3, lane 3, Pro-Q,) and biotinylated (Figure 3, lane 6, SA-Cy5) proteins. Quantification and comparison between kinase inhibitor treated and untreated lanes revealed that only 18±2% of phosphorylated (Figure 3, lanes 3 versus 2, Pro-Q) and 27±5% of biotinylated (Figure 3, lanes 6 versus 5, SA-Cy5) proteins remained. The slightly elevated level of biotinylation remaining with kinase inhibition may be due to the natural presence of biotinylated proteins in cell lysates. These experiments indicate that kinase inhibitors similarly reduce phosphorylation or biotinylation. Because monitoring changes in phosphorylation after inhibitor treatment has been used as a drug characterization tool,[29] a future direction is use of ATP-biotin labeling to characterize kinase inhibitor activities.
In contrast to the kinase inhibitor experiments, phosphatase inhibitors had differing effects on phosphorylation and biotinylation. As expected with ATP phosphorylation, reactions containing phosphatase inhibitors showed robust phosphoprotein signal (Figure 3, lane 4, Pro-Q). Comparison of the phosphatase treated and untreated reaction lanes showed that the phosphoprotein signal was elevated to 118±9% (Figure 3, lane 4 versus 2, Pro-Q). These results are consistent with the use of phosphatase inhibitors to augment phosphorylation levels in phosphoproteomics applications.[27] In the case of the ATP-biotin reactions, a biotin signal was also observed in phosphatase inhibitor treated reactions (Figure 3, lane 7, SA-Cy5). However, comparison of the treated and untreated reaction lanes revealed that the biotin signal was reduced to 81±6% (Figure 3, lanes 7 versus 5, SA-Cy5). The decrease in biotinylation can be rationalized considering the kinase/phosphatase equilibrium in lysates. Because phosphorylated amino acids will be unavailable for ATP-biotin labeling, active phosphatases are needed to remove phosphoryl groups and promote kinase-catalyzed biotinylation. If phosphatase activity is reduced by inhibitors, ATP-biotin labeling will not occur on already phosphorylated sites, reducing the level of biotinylation. Therefore, these experiments are consistent with the hypothesis that active phosphatases are critical for robust kinase-catalyzed labeling in lysates.
To further test the sensitivity of biotinylation to phosphatase inhibitors, two dimensional (2-D) gel analysis was performed (Figure 4). 2-D gel analysis is a classical method that offers visualization of many more proteins in lysates than 1D methods.[30] In addition, 2-D gel methods have been used widely in phosphoproteomics analysis.[31] HeLa cell lysates were incubated with either ATP or ATP-biotin in the absence or presence of phosphatase inhibitors (sodium vanadate and sodium fluoride). After reaction, proteins were separated by their isoelectric points (pI) in the first dimension, followed by molecular weight in the second dimension. As expected, ATP-labeled lysates treated with phosphatase inhibitors contained more phosphoproteins compared to untreated reactions (Figure 4A, see boxed area). In contrast, ATP-biotin-labeled lysates showed a greater number of biotinylated proteins in the untreated reaction than the phosphatase inhibitor-treated reaction (Figure 4B, see boxed area). The reduction in ATP-biotin labeling upon phosphatase inhibitor treatment is consistent with the 1-D gel analysis (Figure 3), which provide further evidence for the hypothesis that active phosphatases are needed for robust kinase-catalyzed biotinylation.
Figure 4.

(A) Reactions of HeLa cell lysates and ATP without (untreated) or with phosphatase inhibitors (phosphatase inhibitor treated). Proteins were visualized with Pro-Q diamond phosphoprotein stain. (B) Reactions of HeLa cell lysates and ATP-biotin without (untreated) or with phosphatase inhibitors (phosphatase inhibitor treated). Biotinylated proteins were visualized with SA-Cy5. The boxed region of the images highlights changes due to inhibitor treatment.
A comparison of the boxed areas within the 2-D gel images shows that the phosphatase inhibitor-treated ATP labeling (Figure 4A, right) and untreated ATP-biotin labeling (Figure 4B, left) showed similarities. We note that the differences in charge state of the phosphorylated and biotinylated proteins (see Scheme 1B and C) likely influences migration in the gels, making direct comparison of ATP and ATP-biotin labeled reactions difficult by 2-D methods. The comparison is further complicated by the use of different visualization methods (Pro-Q versus SA-Cy5). However, the combined data from 1-D and 2-D methods is consistent with the hypothesis that robust kinase-catalyzed biotinylation occurs without the need for phosphatase inhibitor treatment. Therefore, kinase-catalyzed biotinylation is appropriate for phosphoproteomics application using untreated lysates.
Conclusion
We have shown that the phosphorylbiotin product of kinase-catalyzed biotinylation is stable to phosphatases. In vitro experiments suggest that both Ser/Thr and Tyr phosphatase are similarly ineffective at phosphorylbiotin hydrolysis. These results are consistent with prior work documenting that the product of ATP-γS labeling, a thiophosphorylated protein, is also insensitive to phosphatase degradation.[15, 32] The combined results suggest that phosphoproteins containing a phosphate modification will generally display phosphatase insensitivity. ATP-γS has been used for phosphoprotein labeling and detection directly in lysates, without the need for phosphatase inhibitors.[16] The use of untreated lysates combined with the added functionality of the biotin group makes ATP-biotin labeling attractive for phosphoproteomics applications. In particular, the likelihood of detecting low abundance phosphoproteins is enhanced due to the stability of the biotin tag to phosphatases and the ability to purify labeled proteins directly. Use of kinase-catalyzed biotinylation for detection of low abundance phosphoproteins is currently under investigation. In addition, the sensitivity of ATP-biotin labeling to both kinase and phosphatase inhibitors indicates that kinase-catalyzed labeling has future application to phosphoproteomic-based drug characterization efforts. In conclusion, the stability of the phosphorylbiotin product of ATP-biotin labeling to phosphatases suggests that kinase-catalyzed biotinylation will be an excellent tool for phosphoproteomics research.
Experimental Section
Synthesis of biotin-PEG amine (9)
Biotin (977.24 mg, 4 mmol) was dissolved in DMF (5 mL). TBTU (1.5412 g, 4.8 mmol) and DIPEA (0.836 mL, 4.8 mmol) were added and the mixture was stirred at room temperature for 30 min. The mixture was added dropwise to a solution of 4,7,10-trioxa-1-13-tridecanediamine (2.2 mL, 10 mmol) in DCM (250 mL) at 4 °C. After addition was complete, the reaction mixture was stirred overnight at room temperature. The reaction progress was monitored by TLC (3:1:0.5 EtOH: DCM: NH4OH, Rf =0.60). The solvent was removed in vacuo. The product was purified by flash chromatography on silica gel with EtOH/DCM (1:1→3:1 v/v) to yield 82% of biotin-PEG-amine 9 as dark brown solid (1.463 g, 3.28 mmol). Figures S1-4; IR (CHCl3, cm-1): 3457, 3206, 3052, 2939, 1658, 1434, 1184, 1129, 847, 798, 724. 1H NMR (400 MHz, D2O): δ 4.43 (dd, J=8.0 and 4.8 Hz, 1H), 4.25 (dd, J=8 and 4.4 Hz, 1H), 3.51 (s, 8H), 3.43 (t, J=6.4 Hz, 2H), 3.40 (t, J=6.0 Hz, 2H), 3.18-3.12 (m, 1H), 3.09 (t, J=6.4 Hz, 2H), 2.82 (dd, J=12.8 and 4.8 Hz, 2H), 2.56 (t, J=6.8 Hz, 2H), 2.08 (t, J=7.2 Hz, 2H), 1.65-1.36 (m, 8H), 1.27-1.19 (m, 2H). 13C NMR (100 MHz, D2O): δ 25.1, 27.6, 27.9 28.2, 30.7, 35.4, 36.2, 37.6, 39.6, 55.3, 60.1, 61.9, 68.3, 68.8, 69.2, 69.3 69.5, 165.1, 176.5. HRMS (ESI): m/z calculated for C20H39N4O5S [M+H]+ 447.2641, observed 447.2633.
Synthesis of ATP-biotin (2)
Adenosine 5′- triphosphate disodium salt (27.5 mg, 0.05 mmol) was dissolved in water (5 mL) and the pH of the solution was raised to 7.0 by adding 1M sodium hydroxide. 1-(3-Dimethylaminpropyl)-3-ethylcarbodiimide hydrochloride (EDCI, 383.42 mg, 2 mmol) dissolved in water (1 mL) was added to the reaction mixture. The pH of the solution was adjusted to 5.6 with 1M hydrochloric acid and a pH range between 5.6-5.8 was maintained throughout the reaction. Biotin-PEG-amine 9 (894.44 mg, 2 mmol) was added to the ATP solution and progress of the reaction was monitored by TLC (6:3:1 iPrOH: NH4OH: H2O Rf =0.69). The reaction was stirred for 4 hours and then treated with triethylamine (TEA) to reach pH 8.0. The product was purified on DEAE Sephadex-A25 anion exchange column. The column was run with flow rate of 4 mL/min. A stepwise elution using 5%, 10%, 25%, 37.5%, 50%, 75% and 100% triethylammonium bicarbonate buffer (TEAB) was applied. The product eluted between 37.5-50% TEAB. Purified product was lyophilized to yield 20% of ATP-biotin 2 as white TEA salt (10.2 mg, 0.01 mmol). Figures S5-9; UV (MeOH): λ 259 nm. 1H NMR (400 MHz, D2O): δ8.92 (s, 1H), 8.63 (s, 1H), 6.48 (t, J=6.0 Hz, 1H), 4.92-4.89 (m, 2H), 4.74-4.72 (m, 2H), 4.59-4.57 (m, 2H), 4.01-3.84 (m, 12H), 3.64-3.58 (m, 4H), 3.46-3.24 (m, 6H), 2.57 (t, J=7.2 Hz, 2H), 2.16-1.68 (m, 8H). 13C NMR (100 MHz, D2O): δ25.1, 26.4, 27.8, 28.0, 36.2, 37.5, 38.5, 39.6, 42.1, 55.2, 60.1, 61.9, 65.1, 66.6, 68.2, 68.7, 69.2, 69.3, 69.4, 70.3, 74.2, 84.0, 86.6, 110.8, 118.5, 140.0, 149.1, 152.3, 164.1, 176.6. 31P NMR (400 MHz, D2O): δ -0.06 (d, γ-P), -10.35 (d, α-P), -21.78 (t, β-P). HRMS (ESI): m/z calculated for C30H51N9O17P3S [M-H]-934.2337, observed 934.2329.
Quantitative Mass spectrometric analysis
Phosphorylated peptides or biotinylated phosphopeptides were created by incubating ATP 1 or ATP-biotin 2 (2 mM) with PKA (20 units/μL) and Kemptide peptide substrate (40 μM, LRRASLG), CK2 (20 units/μL) and CK2 peptide substrate (40 μM, RRREEETEEE), or Abl (20 units/μL) with Abl peptide substrate (40 μM, EAIYAAPFAKKK). The final volume of the reactions was 10 μL and the manufacturer-supplied buffers were used at a 17times; concentration. The reaction mixtures were incubated at 30 °C for 2 hours. Quantitative mass spectrometric analysis was performed, as described previously.[18, 33]
Kinase-catalyzed phosphorylation and biotinylation
Phosphorylated peptides or biotinylated phosphopeptides were generated as described above except in a reaction volume of 20 μL. After reaction, the mixtures were heated at 95°C for 1 minute to denature the kinase, and split into two 10 μL portions. One 10 μL sample was used directly for HPLC analysis, as described below, while the second 10 μL sample was treated with phosphatase, as described below.
Phosphorylated or biotinylated proteins were created by incubating ATP (1) or ATP-biotin (2) (2 mM) with PKA (25 units/μL) and myelin basic protein (MBP, 0.2 mM), CK2 (5 units/μL) and β-casein (0.2 mM), or Abl (5 units/μL) and MBP (0.2 mM) in the manufacturer-provided buffer (1×, NEB). As a control, reactions were performed without kinases (see Figure S14 of supporting information). The final volume of the reactions was 10 μL. The reaction mixtures were incubated at 30°C for 2 hours. After the reaction, the mixture was heated at 95 °C for 1 minute to denature the kinase. Only with CK2 reactions, ATP or ATP-biotin was removed using a 10 kDa Centriprep spin column (Millipore); we found that ATP or ATP-biotin interfered with subsequent treatment with CIP. The reactions were split into two 5 μL portions. One 5 μL sample was used directly for SDS-PAGE analysis, as described below, while the second 5 μL sample was treated with phosphatase, as described below.
Phosphatase-catalyzed dephosphorylation
Protein or peptide phosphorylation and biotinylation were performed as described above. Phosphatase was added to the appropriate reaction mixtures to create a final volume of 20 μL. For PKA reactions, PP1 (1.0 units/μL) in the manufacturer-provided buffer (1×, NEB) was added and the reaction mixture was incubated at 30°C for 2 hours. For CK2 reactions, CIP (2.5 units/μL) in the manufacturer-provided buffer (1×, NEB) was added and the reaction was incubated at 37 °C for 2 hours. For Abl reactions, TCPTP (2.5 units/μL) in the manufacturer-provided buffer (1×, NEB) was added and the reaction was incubated at 30°C for 2 hours. The crude protein products were analyzed by SDS-PAGE with subsequent gel imaging, as described below, while the peptide reactions were analyzed by HPLC analysis, as described below.
HPLC analysis
For HPLC analysis, peptides were pre-equilibrated in Buffer A (50 μL; 99.9% water with 0.1% trifluoroacetic acid). Reverse phase chromatographic separation of peptides was performed using a C18 column (YMC America INC; 250×4.6 mm, 4μm, 8 nm). The elution gradient used started at 95% Buffer Ain Buffer B(99.9% acetonitrile with 0.1% trifluoroacetic acid), and decreased to 70% Buffer A over 15 min. Due to overlap of several peaks, the PKA/PP1 reaction used a different gradient starting at 90% Buffer A in Buffer B, and increasing to 70% Buffer A over 15 min. The flow rate was 1 mL/min and the peptide absorbance was detected at 214 nm. The percentage of observed phosphopeptide was calculated by dividing the area under the phosphopeptide peak by the total area under both the phosphopeptide and unmodified peptide peaks. Figure 2B and 2C show the average percentage and standard error of observed phosphopeptide from three independent trials.
Gel imaging and Quantification
After SDS-PAGE, total proteins were visualized with SYPRO Ruby stain, while phosphoproteins were detected using Pro-Q diamond stain. For biotinylated protein detection, proteins separated by SDS-PAGE gel were transferred to a polyvinyldene fluoride (PVDF, Immobilon P) membrane before staining withSA-Cy5. The stained gels and membranes were visualized using a Typhoon scanner. Gel bands were quantified using ImageQuant 5.2 by drawing the same size rectangular shape around comparable bands. The percentage of phosphoproteins or biotinylated phosphoproteins observed after phosphatase treatment was calculated by dividing the protein signal after treatment to the protein signal before treatment. The percentages displayed in Figure 3 are average percentages and standard error of observed phosphoprotein or biotinylated phosphoprotein from three independent trials. To calculate phosphatase activity, the average percentage of phosphoproteins or biotinylated phosphoproteins observed after phosphatase treatment was subtracted from 100%.
HeLa cell lysis procedure
Frozen HeLa cells (National Cell Culture Center) were thawed and kept on ice. 20 × 106 HeLa cells were lysed in lysis buffer (1 mL; 50 mM Tris, pH 8.0, 150 mM NaCl, 10% glycerol, 0.5% Triton X-100 and 1× protease inhibitor cocktail V). Cells were incubated with rotation at 4°C for 10 minutes. The soluble fraction was separated from the cell debris by spinning at 12,000 rpm at 4°C for 15 minutes. The supernatant was collected and the concentration of total protein in the lysate was determined via a Bradford assay (generally 5-6 mg/mL total protein). The lysate supernatant was stored at -80°C.
Kinase and phosphatase inhibitor studies
For the reactions consisting of lysates, HeLa cell lysates (100 μg) were incubated with either ATP or ATP-biotin (2 mM) in the presence kinase inhibitor (Staurosporine, 0.10 mM) or phosphatase inhibitors (sodium fluoride and sodium orthovanadate, 0.10 mM each) in the manufacturer-provided buffer for PKA (1×, NEB) for 2 hours at 30 °C. The final volume of the reactions was 25 μL. The products were analyzed by SDS-PAGE with subsequent gel imaging, as described above. The percentage of phosphoproteins or biotinylated phosphoproteins observed after inhibitor treatment was calculated by dividing the protein signal after treatment to the protein signal before treatment.
2-D gel analysis
HeLa cell protein sample (100 μg) was mixed with 125 μL of manufacturer provide rehydration/sample buffer (1×, Bio-Rad). Then the mixture was applied to an IPG strip (ReadyStrip IPG strip, pH 3-10, Bio-Rad) and maximum sample absorption was achieved after 12-16 hours. Focusing was performed at 250 V for 20 min, 4000 V for 2 hours and 4000 V for 10000 Vhours with 50 μA current per strip at 20°C. Then the strip was equilibrated for 10 minutes in manufacturer-provided Buffer 1 (0.375 M Tris-HCl (pH 8.8), 6 M urea, 30% (v/v) glycerol, 2% (w/v) SDS and 2% (w/v) DTT) followed by manufacturer-provided Buffer 2 (0.375 M Tris-HCl (pH 8.8), 6 M urea, 30% (v/v) glycerol, 2% (w/v) SDS and 2.5% (w/v) iodoacetamide). The IPG strip was placed on a 12% SDS-PAGE gel and electrophoresis was carried out at a constant 200 V for 45 minutes.
Supplementary Material
Acknowledgments
We thank the NIH (GM067657) and Wayne State University for funding, Q. Yang and H. Hachem for technical assistance, and T. Anthony, P. Dedigama, M. Embogama, T. Faner, A. Fouda, S. Garre for comments on the manuscript.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Hunter T. Cell (Cambridge, Massachusetts) 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- 2.Adams JA. Chemical Reviews (Washington, D C) 2001;101:2271–2290. doi: 10.1021/cr000230w. [DOI] [PubMed] [Google Scholar]
- 3.a) Parang K, Till JH, Ablooglu AJ, Kohanski RA, Hubbard SR, Cole PA. Nature Structural Biology. 2001;8:37–41. doi: 10.1038/83028. [DOI] [PubMed] [Google Scholar]; b) Lawrence DS. Accounts of Chemical Research. 2003;36:401–409. doi: 10.1021/ar020132s. [DOI] [PubMed] [Google Scholar]
- 4.Johnson LN, Barford D. Annual review of biophysics and biomolecular structure. 1993;22:199–232. doi: 10.1146/annurev.bb.22.060193.001215. [DOI] [PubMed] [Google Scholar]
- 5.Cohen P. Nature Reviews Drug Discovery. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 6.Denu JM, Stuckey JA, Saper MA, Dixon JE. Cell. 1996;87:361–364. doi: 10.1016/s0092-8674(00)81356-2. [DOI] [PubMed] [Google Scholar]
- 7.a) Ubersax JA, Woodbury EL, Quang PN, Paraz M, Blethrow JD, Shah K, Shokat KM, Morgan DO. Nature. 2003;425:859–864. doi: 10.1038/nature02062. [DOI] [PubMed] [Google Scholar]; b) Garcia BA, Shabanowitz J, Hunt DF. Methods. 2005;35:256–264. doi: 10.1016/j.ymeth.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 8.a) Muszynska G, Andersson L, Porath J. Biochemistry. 1986;25:6850–6853. doi: 10.1021/bi00370a018. [DOI] [PubMed] [Google Scholar]; b) Posewitz MC, Tempst P. Anal Chem. 1999;71:2883–2892. doi: 10.1021/ac981409y. [DOI] [PubMed] [Google Scholar]; c) Pinkse MW, Uitto PM, Hilhorst MJ, Ooms B, Heck AJ. Anal Chem. 2004;76:3935–3943. doi: 10.1021/ac0498617. [DOI] [PubMed] [Google Scholar]; d) Wolschin F, Wienkoop S, Weckwerth W. PROTEOMICS. 2005;5:4389–4397. doi: 10.1002/pmic.200402049. [DOI] [PubMed] [Google Scholar]; e) Kweon HK, Hakansson K. Anal Chem. 2006;78:1743–1749. doi: 10.1021/ac0522355. [DOI] [PubMed] [Google Scholar]
- 9.a) Kawada N. Journal of Biological Chemistry. 2001;276:25318–25323. doi: 10.1074/jbc.M102630200. [DOI] [PubMed] [Google Scholar]; b) Oh J, Pyo JH, Jo EH, Hwang SI, Kang SC, Jung JH, Park EK, Kim SY, Choi JY, Lim J. PROTEOMICS. 2004;4:3485–3497. doi: 10.1002/pmic.200401018. [DOI] [PubMed] [Google Scholar]; c) Kim YM, Song EJ, Seo J, Kim HJ, Lee KJ. J Proteome Res. 2007;6:593–601. doi: 10.1021/pr060326s. [DOI] [PubMed] [Google Scholar]
- 10.a) Khan A, Packer NH. Journal of proteome research. 2006;5:2824–2838. doi: 10.1021/pr060305y. [DOI] [PubMed] [Google Scholar]; b) Lee RS, Monigatti F, Briscoe AC, Waldon Z, Freeman MR, Steen H. Journal of proteome research. 2008;7:4022–4030. doi: 10.1021/pr800301h. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kim KH, Moon MH. Journal of proteome research. 2009;8:4272–4278. doi: 10.1021/pr900363s. [DOI] [PubMed] [Google Scholar]
- 11.a) Steinberg TH, Agnew BJ, Gee KR, Leung WY, Goodman T, Schulenberg B, Hendrickson J, Beechem JM, Haugland RP, Patton WF. Proteomics. 2003;3:1128–1144. doi: 10.1002/pmic.200300434. [DOI] [PubMed] [Google Scholar]; b) Reinders J, Sickmann A. Proteomics. 2005;5:4052–4061. doi: 10.1002/pmic.200401289. [DOI] [PubMed] [Google Scholar]
- 12.a) Marcus K, Immler D, Sternberger J, Meyer HE. Electrophoresis. 2000;21:2622–2636. doi: 10.1002/1522-2683(20000701)21:13<2622::AID-ELPS2622>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]; b) Sun T, Campbell M, Gordon W, Arlinghaus RB. Biopolymers. 2001;60:61–75. doi: 10.1002/1097-0282(2001)60:1<61::AID-BIP1004>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 13.Bodenmiller B, Mueller LN, Mueller M, Domon B, Aebersold R. Nat Meth. 2007;4:231–237. doi: 10.1038/nmeth1005. [DOI] [PubMed] [Google Scholar]
- 14.a) Green KD, Pflum MKH. Journal of the American Chemical Society. 2007;129:10–11. doi: 10.1021/ja066828o. [DOI] [PubMed] [Google Scholar]; b) Green KD, Pflum MKH. ChemBioChem. 2009;10:234–237. doi: 10.1002/cbic.200800393. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Suwal S, Pflum MKH. Angewandte Chemie, International Edition. 2010;49:1627–1630. S1627/1621–S1627/1613. doi: 10.1002/anie.200905244. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Senevirathne C, Green KD, Pflum MKH. Current Protocols in Chemical Biology. John Wiley & Sons, Inc.; 2009. [Google Scholar]
- 15.a) Hiriyanna KT, Baedke D, Baek KH, Forney BA, Kordiyak G, Ingebritsen TS. Anal Biochem. 1994;223:51–58. doi: 10.1006/abio.1994.1545. [DOI] [PubMed] [Google Scholar]; b) Cassel D, Glaser L. Proceedings of the National Academy of Sciences of the United States of America. 1982;79:2231–2235. doi: 10.1073/pnas.79.7.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Endo S, Critz SD, Byrne JH, Shenolikar S. J Neurochem. 1995;64:1833–1840. doi: 10.1046/j.1471-4159.1995.64041833.x. [DOI] [PubMed] [Google Scholar]
- 16.a) Parker LL, Schilling AB, Kron SJ, Kent SB. J Proteome Res. 2005;4:1863–1866. doi: 10.1021/pr050150e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jeong S, Nikiforov TT. BioTechniques. 1999;27:1232–1238. doi: 10.2144/99276rr01. [DOI] [PubMed] [Google Scholar]; c) Kwon SW, Kim SC, Jaunbergs J, Falck JR, Zhao Y. Mol Cell Proteomics. 2003;2:242–247. doi: 10.1074/mcp.M300039-MCP200. [DOI] [PubMed] [Google Scholar]; d) Sun IY, Allfrey VG. The Journal of biological chemistry. 1982;257:1347–1353. [PubMed] [Google Scholar]; e) Allen JJ, Lazerwith SE, Shokat KM. J Am Chem Soc. 2005;127:5288–5289. doi: 10.1021/ja050727t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suwal S, Pflum MH. Angew Chem Int Ed Engl. 2010;49:1627–1630. doi: 10.1002/anie.200905244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Green KD, Pflum MH. J Am Chem Soc. 2007;129:10–11. doi: 10.1021/ja066828o. [DOI] [PubMed] [Google Scholar]
- 19.Barford D. Curr Opin Struct Biol. 1995;5:728–734. doi: 10.1016/0959-440x(95)80004-2. [DOI] [PubMed] [Google Scholar]
- 20.Cohen P. Methods Enzymol. 1991;201:389–398. doi: 10.1016/0076-6879(91)01035-z. [DOI] [PubMed] [Google Scholar]
- 21.Denu JM, Dixon JE. Curr Opin Chem Biol. 1998;2:633–641. doi: 10.1016/s1367-5931(98)80095-1. [DOI] [PubMed] [Google Scholar]
- 22.a) Villafranca JE, Kissinger CR, Parge HE. Curr Opin Biotechnol. 1996;7:397–402. doi: 10.1016/s0958-1669(96)80114-5. [DOI] [PubMed] [Google Scholar]; b) Barford D. Trends in biochemical sciences. 1996;21:407–412. doi: 10.1016/s0968-0004(96)10060-8. [DOI] [PubMed] [Google Scholar]; c) Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Cell. 1998;92:441–450. doi: 10.1016/s0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]; d) Songyang Z, Carraway KL, 3rd, Eck MJ, Harrison SC, Feldman RA, Mohammadi M, Schlessinger J, Hubbard SR, Smith DP, Eng C, et al. Nature. 1995;373:536–539. doi: 10.1038/373536a0. [DOI] [PubMed] [Google Scholar]; e) Goldberg J, Huang HB, Kwon YG, Greengard P, Nairn AC, Kuriyan J. Nature. 1995;376:745–753. doi: 10.1038/376745a0. [DOI] [PubMed] [Google Scholar]
- 23.a) Egloff MP, Cohen PT, Reinemer P, Barford D. J Mol Biol. 1995;254:942–959. doi: 10.1006/jmbi.1995.0667. [DOI] [PubMed] [Google Scholar]; b) Griffith JP, Kim JL, Kim EE, Sintchak MD, Thomson JA, Fitzgibbon MJ, Fleming MA, Caron PR, Hsiao K, Navia MA. Cell. 1995;82:507–522. doi: 10.1016/0092-8674(95)90439-5. [DOI] [PubMed] [Google Scholar]; c) Kissinger CR, Parge HE, Knighton DR, Lewis CT, Pelletier LA, Tempczyk A, Kalish VJ, Tucker KD, Showalter RE, Moomaw EW, et al. Nature. 1995;378:641–644. doi: 10.1038/378641a0. [DOI] [PubMed] [Google Scholar]
- 24.Mueller EG, Crowder MW, Averill BA, Knowles JR. Journal of the American Chemical Society. 1993;115:2974–2975. [Google Scholar]
- 25.a) Jia Z, Barford D, Flint AJ, Tonks NK. Science (New York, NY) 1995;268:1754–1758. doi: 10.1126/science.7540771. [DOI] [PubMed] [Google Scholar]; b) Zhang ZY. The Journal of biological chemistry. 1995;270:11199–11204. doi: 10.1074/jbc.270.19.11199. [DOI] [PubMed] [Google Scholar]
- 26.Oda Y, Nagasu T, Chait BT. Nat Biotechnol. 2001;19:379–382. doi: 10.1038/86783. [DOI] [PubMed] [Google Scholar]
- 27.a) Boeri Erba E, Matthiesen R, Bunkenborg J, Schulze WX, Di Stefano P, Cabodi S, Tarone G, Defilippi P, Jensen ON. J Proteome Res. 2007;6:2768–2785. doi: 10.1021/pr060675m. [DOI] [PubMed] [Google Scholar]; b) Alberts AS, Montminy M, Shenolikar S, Feramisco JR. Molecular and cellular biology. 1994;14:4398–4407. doi: 10.1128/mcb.14.7.4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.a) Subramanian G, Sud M. ACS Medicinal Chemistry Letters. 2010;1:395–399. doi: 10.1021/ml1001097. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jester BW, Gaj A, Shomin CD, Cox KJ, Ghosh I. Journal of medicinal chemistry. 2012;55:1526–1537. doi: 10.1021/jm201265f. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Comeau AB, Critton DA, Page R, Seto CT. Journal of medicinal chemistry. 2010;53:6768–6772. doi: 10.1021/jm100528p. [DOI] [PubMed] [Google Scholar]; d) McCluskey A, Sim AT, Sakoff JA. Journal of medicinal chemistry. 2002;45:1151–1175. doi: 10.1021/jm010066k. [DOI] [PubMed] [Google Scholar]
- 29.a) Klammer M, Kaminski M, Zedler A, Oppermann F, Blencke S, Marx S, Muller S, Tebbe A, Godl K, Schaab C. Mol Cell Proteomics. 2012;11:651–668. doi: 10.1074/mcp.M111.016410. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yan GR, Xiao CL, He GW, Yin XF, Chen NP, Cao Y, He QY. PROTEOMICS. 2010;10:976–986. doi: 10.1002/pmic.200900662. [DOI] [PubMed] [Google Scholar]; c) Imami K, Sugiyama N, Imamura H, Wakabayashi M, Tomita M, Taniguchi M, Ueno T, Toi M, Ishihama Y. Mol Cell Proteomics. 2012 doi: 10.1074/mcp.M112.019919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.a) Anderson NG, Matheson A, Anderson NL. Proteomics. 2001;1:3–12. doi: 10.1002/1615-9861(200101)1:1<3::AID-PROT3>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]; b) Rabilloud T. Proteomics. 2002;2:3–10. [PubMed] [Google Scholar]
- 31.a) Lottspeich F. Angew Chem Int Ed Engl. 1999;38:2476–2492. doi: 10.1002/(sici)1521-3773(19990903)38:17<2476::aid-anie2476>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]; b) Gygi SP, Corthals GL, Zhang Y, Rochon Y, Aebersold R. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:9390–9395. doi: 10.1073/pnas.160270797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mulkey RM, Endo S, Shenolikar S, Malenka RC. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- 33.a) Green KD, Pflum MH. ChemBioChem. 2009;10:234–237. doi: 10.1002/cbic.200800393. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Senevirathne C, Green KD, Pflum MKH. Current Protocols in Chemical Biology. Vol. 4. John Wiley & Sons, Inc.; 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.