

Abstract
Overexpression of survivin is observed in various hematological malignancies, including acute myeloid leukemia (AML). Studies show that elevated expression of survivin correlates with a worse clinic outcome in AML patients. It remains unclear whether inhibition of survivin may alter the efficacy of chemotherapy against AML. Here, we evaluate the effects of specific knockdown of survivin on AML cells’ sensitivity to chemotherapy, and investigate the therapeutic potential of the transcription inhibitor of survivin YM155 either alone or in combination with chemotherapeutic agents. We found Kasumi-1 and HL-60 cells had relatively higher expression levels of survivin among all AML cell lines tested. Specific knockdown of survivin in Kasumi-1 and HL-60 cells resulted in: inhibition of cell proliferation; cell cycle G2/M arrest; induction of DNA damage response and apoptosis. Downregulation of survivin enhanced etoposide- or doxorubicin-induced anti-proliferative/anti-survival activity in AML cells. The small molecule inhibitor YM155 reduced survivin in a dose- and time-dependent manner and trigged apoptosis in Kasumi-1 and HL-60 cells. The combinatorial effects of YM155 and chemotherapeutics were either synergetic or antagonistic, depending upon the drugs used for combination and the type of AML cells being treated. Collectively, our data demonstrate that survivin plays an important role in the maintenance and proliferation of AML cells. While specific knockdown of survivin enhances chemosensitivity, the combinations of YM155 and chemotherapeutic agents exhibit synergetic or antagonistic effects on AML cells. Our findings provide a rationale for further assessment of survivin-targeted therapy in the treatment of patients with AML.
Keywords: survivin, targeted therapy, YM155, chemotherapy, acute myeloid leukemia
1. Introduction
Survivin (coding gene: BIRC5), the smallest mammalian member of IAP (inhibitor of apoptosis) family, is a dual functional protein acting as a critical apoptosis inhibitor and key cell cycle regulator [1]. The anti-apoptotic function of survivin is mainly via inhibition of caspase-9, with help of a cellular protein named hepatitis B X - interacting protein (HBXIP) [2]. It also forms a complex with XIAP to stabilize XIAP from ubiquitination/proteasomal destruction [3]. As a cell cycle regulator, expression of survivin peaks at G2/M phase, regulating mitosis within a multi-protein complex called chromosomal passenger complex (CPC), ensuring accurate segregation of sister chromatids[4; 5]. Survivin is usually expressed in embryonic tissues during development and undetectable in most terminally differentiated tissues. It has also been detected in some normal proliferating adult tissues and cells, such as hematopoietic stem/progenitor cells, vascular endothelial cells, T cells, erythroid cells, ovary, testes, and liver [6]. Overexpression of survivin is observed in a wide variety of human cancers and the increased survivin is correlated with a poor clinic outcome, tumor recurrence, and therapeutic resistance [7; 8; 9]. For instance, we have shown that overexpression of erbB3 receptor confers paclitaxel resistance in erbB2-overexpressing breast cancer cells via PI-3K/Akt-dependent upregulation of survivin [10]; and targeting of erbB3 with a therapeutic antibody (Ab) re-sensitizes the breast cancer cells to paclitaxel treatment mainly through downregualtion of survivin [11].
Elevated expression of survivin and its correlation with adverse clinical outcome has also been found in hematological malignancies, including diffuse large B-cell lymphoma (DLBCL), childhood B-cell precursor acute lymphoblastic leukemia, multiple myeloma (MM), BCR-ABL positive chronic myeloid leukemia (CML), adult T-cell leukemia/lymphoma (ATL), anaplastic large-cell lymphoma (ALCL), and acute myeloid leukemia (AML) [12]. It has been shown that survivin was upregulated in AML cells by hematopoietic cytokines, like GM-CSF, G-CSF and SCF, via MAPK and PI-3K signaling pathways [13]. Survivin-specific antisense oligonucleotides induced growth inhibition and cell death in HL-60 cells [14]. In another study, the expression of survivin was detected by a validated reverse-phase protein array in samples from 511 newly diagnosed AML patients. Detailed analysis discovered significantly higher levels of survivin in CD34+38−AML stem/progenitor cells than in bulk blasts and total CD34+ AML cells; and the higher levels of survivin associated with a shorter overall and event-free survival [15]. Furthermore, the expression of survivin correlated with the expression of multiple proteins involving in proliferation and cell survival, supporting the role of survivin as a prognostic marker and a potential therapeutic target in AML [15].
YM155 (Astellas Pharma US, Inc., Northbrook, IL) is a small molecule suppressant of survivin transcription. It competes the binding sites of Sp1 on survivin promoter [16] and thereby disrupts the ILF3 transcriptional complex [17; 18]. Clinical trials of YM155 used as a single agent or in combination with other drugs, have been carried out in various solid tumors, including non-small cell lung cancer [19; 20], prostate cancer [21] and melanoma [22]. In hematological malignancies, phase II clinical trials of YM155 as monotherapy in DLBCL showed weak efficacy [23]. Recent studies with mouse models of human B-Cell non-Hodgkin lymphoma or adult T-cell leukemia revealed that YM155 in combination with monoclonal antibody Rituximab (anti-CD20) [24] or alemtuzumab/Campath-1H (anti-CD25) [25], respectively, showed significant improvements in tumor regression and survival. Thus, corresponding clinical trials are ongoing in CD20-positive B cell non-Hodgkin's lymphoma patients (NCT01007292) and ATL patients (NCT00061048). To date, no clinical studies initiated to test the therapeutic potential of YM155 in AML patients. In current report, we evaluated the influence of survivin silencing on AML cell proliferation and chemosensitivity. We also investigated the inhibitory effects of YM155 alone or in combination with chemotherapeutic agents on AML cells.
2. Materials and methods
2.1 Reagents and antibodies
YM155 was purchased from Selleck Chemicals (Houston, TX, USA). Doxorubicin was purchased from ALEXIS Biochemicals (San Diego, CA, USA). Cytarabine (Pfizer Inc., New York, NY, USA.) and etoposide (Hengrui Medicine CO. LTD, Jiangsu, China) were obtained from pharmacy of Institute of Hematology and Blood Disease Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Tianjin, China. YM155 and etoposide were dissolved in dimethyl sulfoxide (DMSO) to make a stock solution at 20mmol/L. Cytarabine and doxorubicin were dissolved in water to make a stock solution at 20mmol/L and 10mmol/L, respectively. All the stock solutions were stored at −80 °C.
MISSION Non-target shRNA, which does not target human and mouse genes, control vector (pLKO.1-ConshRNA), pLKO.1 containing human survivin shRNA (pLKO.1-SurshRNA) were purchased from Sigma (St. Louis, MO, USA). Clone IDs of survivin targeting shRNAs used in our studies were: TRCN0000073720S (S3), TRCN0000073721 (S4) and TRCN0000222542 (S5). The packaging plasmids psPAX2 and pMD2.G for lentiviral expression vector were from Addgene (Cambridge, MA, USA).
Antibodies were obtained as follows: Survivin (6E4) mouse mAb, Bcl-2 rabbit Ab, Bcl-xL rabbit Ab, caspase-3 rabbit mAb (8G10), caspase-8 mouse mAb (1C12), PARP rabbit mAb, P-Histone H2A.X (Ser139) rabbit Ab, Histone H2A rabbit polyclonal Ab II, P-CHK2 (Thr68) rabbit polyclonal Ab, and CHK2 rabbit polyclonal Ab (Cell Signaling Technology, Inc., Beverly, MA), and β-actin mouse mAb (AC-75) (Sigma Chemical Co.). All other reagents were purchased from Sigma unless otherwise specified.
2.2 Cells and cell culture
Human acute myeloid leukemia (AML) cell lines Kasumi-1, HL60, NB4 and U937 were maintained in our laboratory. All cell lines were maintained in RPMI1640 cell culture medium supplemented with 10% fetal bovine serum (FBS) at a 37 °C humidified atmosphere containing 95% air and 5% CO2 and were split twice a week.
2.3 Quantification of apoptosis
An apoptosis ELISA kit (Roche Diagnositics Corp., Indianapolis, IN) was used to quantitatively measure cytoplasmic histone-associated DNA fragments (mononucleosomes and oligonucleosomes) as previously reported [11; 26].
2.4 Flow cytometric analysis of cell cycle
Flow cytometric analyses were performed to define the cell cycle distribution for treated and untreated cells [26; 27]. In brief, cells grown in 100mm culture dishes were harvested and resuspended in cold PBS, fixed with 9 volumes of 70% ethanol. Cells were then stained for total DNA content with a solution containing 50μg/ml propidium iodide, 50μg/ml RNaseA, 0.1% Triton X-100 and 0.1mmol/L EDTA in PBS for 30 min at 37°C. Cell cycle distribution was analyzed at the Flow Cytometry Core Facility of University of Colorado Cancer Center with a FACScan flow cytometer (BD Biosciences, San Jose, CA).
2.5 Reverse transcription-PCR and quantitative real-time (qRT)-PCR
Total RNA was extracted using a modified chloroform/phenol procedure (TRIZOL®, Invitrogen, Carlsbad, CA). First-strand cDNA was generated using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) following the manufacturer’s instructions. The expression of human survivin mRNA (forward primer: 5’-GCCCAGTGTTTCTTCTGCTT-3’; reverse primer: 5’-TCTCCGCAGTTTCCTCAAAT-3’) was examined by both conventional reverse transcription-PCR and qRT-PCR as we described previously [28]. The qRT-PCR was performed using the Absolute* Blue QPCR Master Mixes (Thermo Fisher Scientific Inc., Waltham, MA) according to the manufacturer’s protocol. The expression of β-actin mRNA (forward primer: 5’-AGAGCTACGAGCTGCCTGAC-3’; reverse primer: 5’-AGCACTGTGTTGGCGTACAG-3’) was used as an internal control. All qRT–PCR reactions were carried out on a 7500 Fast Real-Time PCR system (Applied Biosystems).
2.6 Western blot analysis
Protein expression or activation was determined by Western blot analyses as previously described [11; 26]. The Coomassie Plus protein assay reagent (Thermo Fisher Scientific Inc.) was used to measure the protein concentration of total cell lysates. Equal amounts of cell lysates were boiled in Laemmli SDS-sample buffer, resolved by SDS-PAGE, subjected to western blot analysis with specific primary antibodies as described in the figure legends. After the blots were incubated with horseradish peroxidase-labeled secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA), signals were detected using the enhanced chemiluminescence reagents (GE Healthcare Bio-Sciences Corp., Piscataway, NJ).
2.7 Cell proliferation assays
The CellTiter96 AQ non-radioactive cell proliferation kit (Promega Corp., Madison, WI) was used to evaluate cell viability as previously described [26; 27]. In brief, cells were plated on 96-well plates with 0.1 ml complete medium containing 5% FBS as control, or 0.1 ml of the same medium with either YM155, etoposide, doxorubicin or cytarabine alone, or their combinations, and incubated for 48 hr in a cell culture incubator. After reading all wells at 490nm with a microplate reader, the percentages of surviving cells from each group relative to controls, defined as 100% survival, were determined by reduction of MTS.
2.8 Statistical analysis
Statistical analyses of the experimental data were performed using a two-sided Student’s t test. Significance was set at a P < 0.05. Calculation of IC50, combination index (CI) and evaluation of synergy or antagonism between YM155 and chemotherapeutic agents were performed as described in literature [29] using the CompuSyn software (ComboSyn, Inc., Paramus, NJ. USA).
3. Results
3.1 Different expression of survivin is observed in AML cells and higher levels of survivin correlate with a worse overall survival in AML patients
To strengthen the hypothesis that survivin may be developed as a valuable target in the treatment of AML patients, we first examined the expression levels of survivin in several human AML cell lines. Western blot analyses showed strikingly high levels of survivin in Kasumi-1 and HL-60 cells as compared to U-937 and NB4 cells (Fig. 1A). It appeared that Kasumi-1 cells also expressed high levels of Bcl-2 and Bcl-xL, the other two well-known, functionally related anti-apoptotic proteins, whereas HL-60 cells had highest Bcl-2 levels and the least expression of Bcl-xL among the AML cell lines tested (Fig. 1A). We next performed PCR analyses of survivin mRNA to explore whether the high protein levels of survivin were due to a mechanism involving in gene transcription. Both conventional reverse transcription-PCR (Fig. 1B) and quantitative real-time (qRT)-PCR (Fig. 1C) assays revealed that the expression of survivin mRNA was much higher in Kasumi-1 and HL-60 cells than that in U-937 and NB4 cells, suggesting that the expression of survivin in AML cells might be regulated at transcriptional level. To further assess the significance of survivin expression in AML, we analyzed the TCGA dataset of AML patient samples. By utilizing the University of California Santa Cruz (UCSC) Cancer Genomics Browser (https://genome-cancer.ucsc.edu), we generated Kaplan-Meier survival curves following the description in literature [30] and discovered that the AML patients with higher expression of survivin (red line) had a worse overall survival than those with basal expression of survivin (green line) (Fig. 1D). These data provide a strong rationale to use Kasumi-1 and HL-60 as a valuable cell model to corroborate survivin as a molecular target for AML therapy.
Figure 1. Elevated expression of survivin is observed in AML cell lines, and associated with a worse survival in AML patients.

A, Four AML cell lines in logarithmic phase were collected and subjected to (B) western blot analyses with specific antibody directed against survivin, Bcl- 2, Bcl-xL or β-actin. B, Total RNA extracted from the AML cell lines was subjected to first-strand cDNA synthesis using a reverse transcription kit from Applied Biosystems. The partial coding sequence of survivin and β-actin was amplified with specific primers. The PCR products were separated on a 1% agarose gel containing ethidium bromide and visualized under a UV light. Densitometry analyses of the signals were carried out. The arbitrary numbers indicate the ratio of survivin:β-actin for each sample. C, The mRNA levels of survivin were measured by qRT-PCR. Data show the relative expression levels of survivin in each cell line as compared to that in U937, defined as 1. D, Kaplan-Meier plot generated from UCSC Cancer Genomics Browser. The AML patients from TCGA database with higher expression levels of survivin mRNA (red line) have a worse overall survival than those with basal expression levels of survivin mRNA (green line).
3.2 Silencing of survivin induces growth inhibition, cell cycle G2/M arrest, and apoptosis in AML cells
To study the influence of survivin expression on AML cell maintenance and proliferation, we infected Kasumi-1 and HL-60 cells with the lentivirus containing either non-targeted control shRNA or survivin-specific shRNA. As compared to control, all three sequences of survivin-specific shRNA (S3, S4, S5) markedly downregulated survivin expression in Kasumi-1 and HL-60 cells at 48 hr and 72 hr post lentiviral infection (Fig. 2A). The shRNAs we used were specific for survivin, as they had no significant inhibitory effect on Bcl-2 and Bcl-xL in both Kasumi-1 and HL-60 cells. These data are consistent with our previous findings in human breast cancer cells [10]. It seemed that the expression levels of Bcl-xL were slightly increased in Kasumi-1 cells at 72 hr post lentiviral infection (Fig. 2A), which might be a compensatory induction upon survivin downregulation. However, this phenomenon was not seen in HL-60 cells. Importantly, specific knockdown of survivin significantly reduced proliferation of both Kasumi-1 and HL-60 cells (Fig. 2B). Especially in Kasumi-1 cells, the cell survival rate tested on day 5 of survivin knockdown (with shRNA-S3 and S5) was lower than that on day 3, suggesting a possibility of cell death. This observation was supported by an early report showing that decreased expression of survivin with antisense oligonucleotides promoted myeloid leukemia cells undergoing apoptosis through a mitochondrial pathway [14].
Figure 2. Specific knockdown of survivin expression inhibits cell proliferation.

Kasumi-1 and HL-60 cells infected with lentivirus containing either ConshRNA (Con) or SurshRNA (S3, S4, and S5) were subjected to the following experiments. A, After 48 hr or 72 hr lentiviral infection, cells were collected and subjected to Western blot analyses with specific antibody directed against survivin, Bcl-2, Bcl-xL, or β-actin. B, After 48 hr lentiviral infection, cells were plated onto 96-well plates at the density of 8000 cell/well, MTS assays were performed to examine cell growth. All wells were read at 490nM with a microplate reader on Day 1, 3, and 5. Reading of Day 1 was set as control. Curves show the relative proliferation rates of cells.
Since survivin plays a pivotal role in cell cycle regulation [4; 5], we next performed flow cytometry analyses (Fig. 3A & B) to examine whether downregulation of survivn blocked cell cycle progression, and thus contributing to the growth inhibitory effects. As compared to control shRNA, knockdown of survivin with specific shRNAs dramatically increased the cell population at G2/M phase (S3 and S4 in Kasumi-1 cells, S3 and S5 in HL-60 cells). This observation is in agreement with the unique feature of survivin, whose expression is cell cycle-dependent, peaks at G2/M phase, and executes its function as an indispensable regulator of cell mitosis [1; 31]. These data suggest that cell cycle G2/M arrest may contribute to survivn knockdown-induced growth inhibition in AML cells. As perturbed mitotic progress inhibits acute segregation of sister chromatids, which can cause DNA damage and lead to mitotic catastrophe and eventual cell death, we then investigated whether specific knockdown of survivin in AML cells would cause DNA damage and apoptosis. We found that the phosphorylation levels of H2A.X (γH2A.X) and CHK2 (P-CHK2), two well-known DNA damage markers increased upon reduction of survivin in both Kasumi-1 and HL-60 cells (Fig. 3C). In comparison to the changes of P-CHK2, dramatic induction of γH2A.X was observed, with the most profound upregulation caused by the survivin-specific shRNA S3, consistent with the findings that shRNA-S3 was the most effective sequence to downregulate survivin and induce cell cycle G2/M arrest (Fig. 3A & B). Furthermore, specific knockdown of survivin also promoted AML cells undergoing apoptosis, evidenced by PARP cleavage (Fig. 4A), a hallmark of apoptosis and significant increases in DNA fragments (Fig. 4B). The enhanced cleavage of caspase-8 and caspase-3 suggested that survivin knockdown-induced apoptosis in Kasumi-1 and HL-60 cells might be caspase dependent (Fig. 4A). Collectively, our data indicate that silencing of survivin blocks AML cells go through G2-M checkpoint or halt at mitotic phase, and thereby induces cell cycle G2/M arrest and apoptosis.
Figure 3. Specific knockdown of survivin expression induces cell cycle G2/M arrest in AML cells.
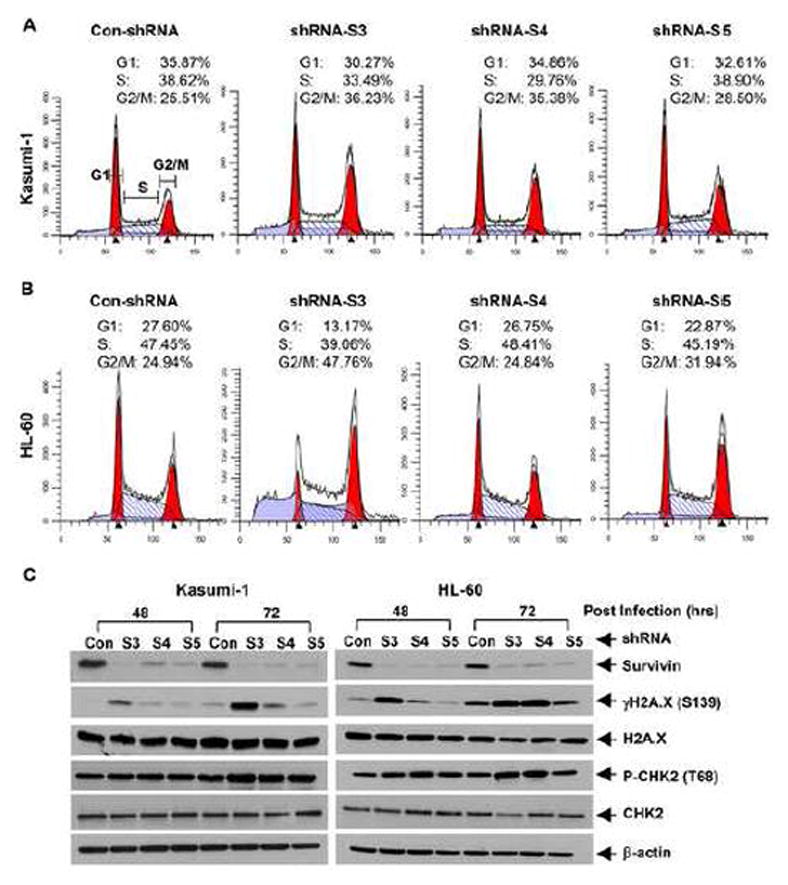
Kasumi-1 and HL-60 cells were infected with lentivirus containing either ConshRNA (Con) or SurshRNA (S3, S4, and S5). A & B, After 48 hr, cells were collected and subjected to flow cytometric analysis of cell cycle distribution. Data show the representative of three independent experiments. C, After 48 or 72 hr, cells were collected and subjected to Western blot analyses with specific antibody directed against survivin, γH2A.X (Ser139), H2A.X, P-CHK2 (Thr68), CHK2, or β-actin.
Figure 4. Specific knockdown of survivin expression promotes AML cells undergoing apoptosis.

Kasumi-1 and HL-60 cells infected with lentivirus containing either ConshRNA (Con) or SurshRNA (S3, S4, and S5) for 48 or 72 hr were collected and subjected to Western blot analyses with specific antibody directed against caspase-8 (Pro-Casp-8, full-length caspase- 8; C-Casp-8, cleaved caspase-8), caspase-3 (Pro-Casp-3, full-length caspase-3; C-Casp-3, cleaved caspase-3), PARP (F-PARP, full-length PARP; C-PARP, cleaved PARP), or β-actin (A), or a specific apoptosis ELISA (B). Bars, SD.
3.3 Downregulation of survivin enhances chemotherapy-induced DNA damage response and apoptosis in AML cells
To further explore survivin as a useful target in AML treatment, we next investigated if knockdown of survivin would enhance chemotherapy-mediated anti-proliferative/anti-survival effects on AML cells. Kasumi-1 and HL-60 cells were infected with the lentivirus containing either control shRNA or survivin-specific shRNAs. After 24 hr, the cells were then treated with etoposide or doxorubicin, the commonly used chemotherapeutics in AML for another 24 hr. As compared to the control cells treated with etoposide, specific knockdown of survivin clearly increased PARP cleavage and activation of caspase-8 and caspase-3-triggered by etoposide (Fig. 4A), and significantly enhanced etoposide-induced DNA fragmentation (Fig. 4B). Western blot assays discovered that downregulation of survivin in combination with etoposide markedly increased P-CHK2 and γH2A.X in Kasumi-1 and HL-60 cells, respectively. However, it only led to a slight induction of P-CHK2 in HL-60 cells and of γH2A.X in Kasumi-1 cells. There were no significant changes on expression of the anti-apoptotic proteins Bcl-2 and Bcl-xL. Similar results of the molecular markers of apoptosis and DNA damage response were also observed in Kasumi-1 cells treated with doxorubicin (Fig. S1). Interestingly, both the levels of P-CHK2 (Thr68) and electrophoretic mobility of CHK2 reduced in Kasumi-1 cells upon doxorubicin treatment (Fig. S1), implying that the phosphorylation of CHK2 might occur on the residues other than Thr68. Previous report regarded Thr68 as the first phosphorylation site of CHK2 after doxorubicin treatment, which was subsequently followed by phosphorylation on Ser33/35 [32]. Phosphorylation on multiple sites of CHK2 would probably decrease its electrophoretic mobility. Our data indicated that specific knockdown of survivin significantly enhanced chemosensitivity in AML cells, providing a theoretical basis to facilitate the development of a novel combinatorial approach to treat patients with AML.
3.4 The transcriptional inhibitor of survivin YM155 induces growth inhibition, DNA damage response, and apoptosis in AML cells
To identify a feasible tool for clinical use in targeting of survivin, we considered YM155, a small molecule inhibitor of survivin transcription. YM155 exhibits potent inhibitory effect on survivin promoter [33]; and it is currently under phase I and II clinical trials in various solid tumors and lymphoma [19; 20; 21; 22; 23]. To date, no clinic study is initiated to test the therapeutic potential of YM155 in AML patients. Thus, we sought to evaluate if YM155 could specifically downregulate survivin and induce apoptosis in AML cells. First, we treated Kasumi-1 and HL-60 cells with different concentrations of YM155, and found that YM155 inhibited proliferation of Kasumi-1 (Fig. 5A) and HL-60 (Fig. 6A) in a dose-dependent manner. The IC50 values of YM155 were 40.6nmol/L and 2.5nmol/L for Kasumi-1 and HL-60 cells, respectively. Next, we examined if YM155 could inhibit survivin expression in the two cell lines. While the expression levels of survivin were downregulated by YM155 in a dose- and time-dependent manner, the expression of Bcl-2 and Bcl-xL was not affected (Fig. 6B and data not shown for HL-60). Third, we studied if YM155 could promote AML cells undergoing apoptosis. Treatment of Kasumi-1 cells with YM155 enhanced PARP cleavage and activation of both caspase-8 and caspase-3 (Fig. 6C), and increased histone-associated DNA fragments-quantified by an apoptotic-specific ELISA (Fig. 6D). Similar results were also observed in HL-60 cells (Figs. 7B & C). These data suggest that YM155-induced apoptosis in AML cells was caspase-dependent. In addition, studies on the molecular markers of DNA damage response showed an increased γH2A.X (Ser139) and P-CHK2 (Thr68) in both Kasumi-1 and HL-60 cells upon YM155 treatment (Figs. 6C & 7B). As DNA damage response was a consequential event of cell cycle G2/M arrest in the studies of survivin knockdown with specific shRNAs (Fig. 3), we wondered if YM155-induced DNA damage response was due to its capability to inhibit survivin. Analysis of cell cycle distribution revealed that YM155 mainly reduced the cells at S phase and slightly increased the cells at G1 and G2/M phases (Fig. S2), implying a major effect on DNA synthesis. Thus, the DNA damage response seemed to be a direct event of YM155 treatment, independent from the reduction of survivin. Collectively, these results indicated that YM155 inhibited survivin expression and induced apoptosis and DNA damage response in AML cells in a dose- and time-dependent manner. It appeared that the apoptotic cell death and DNA damage response triggered by YM155 did not result exclusively from survivin downregulation.
Figure 5. Downregulation of survivin enhances etoposide-induced apoptosis and DNA damage response in AML cells.

Kasumi-1 and HL-60 cells infected with lentivirus containing either ConshRNA (Con) or SurshRNA (S3, S4, and S5) for 24 hr were then untreated or treated with etoposide (Kasumi-1: 2.5μmol/L; HL-60: 0.4μmol/L) for additional 24 hr. Cells were collected and subjected to Western blot analyses of survivin, Bcl-2, Bcl-xL, caspase-8, caspase- 3, PARP, γH2A.X (Ser139), H2A.X, P-CHK2 (Thr68), CHK2, or β-actin (A), or a specific apoptosis ELISA (B). Bars, SD.
Figure 6. YM155 downregulates survivin and induces apoptosis and DNA damage response in Kasumi-1 cells.

A, Kasumi-1 cells were plated onto 96-well plates with 0.1 ml 1640 medium containing 5% FBS as control, or 0.1 ml of the same medium with indicated concentrations of YM155, and incubated for 48 hr in a cell culture incubator. After reading all wells at 490nM with a microplate reader, the percentages of surviving cells from each group relative to control, defined as 100% survival, were determined by reduction of MTS. B-D, Kasumi-1 cells were cultured in 1640 medium with 5% FBS containing 0, 20, 40, or 80 nmol/L of YM155 for 24 hr, or in 1640 medium with 5% FBS containing 40 nmol/L of YM155 for 0, 16, or 24 hr. Cells were collected and subjected to Western blot analyses of survivin, Bcl-2, Bcl-xL, or β-actin (B), or Western blot analyses of caspase-8, caspase-3, PARP, γH2A.X (Ser139), H2A.X, P-CHK2 (Thr68), CHK2 or β-actin (C), or a specific apoptosis ELISA (D). Bars. SD.
Figure 7. YM155 downregulates survivin and induces apoptosis and DNA damage response in HL-60 cells.

A, The response curve of HL-60 cells to YM155 was generated by MTS assays. B & C, HL-60 cells were cultured in 1640 medium with 5% FBS containing 0, 2 or 4 nmol/L of YM155 for 24 hr, or cultured in 1640 medium with 5% FBS containing 2.5 nmol/L of YM155 for 0, 24, or 48 hr. Cells were collected and subjected to Western blot analyses of survivin, caspase-8, caspase-3, PARP, γH2A.X (Ser139), H2A.X, P-CHK2 (Thr68), CHK2, or β-actin (B) or a specific apoptosis ELISA (C). Bars, SD.
3.5 YM155 in combination with chemotherapeutics exhibits synergistic or antagonistic effects on AML cells
As aforementioned, specific downregulation of survivin enhanced chemosensitivity and YM155 inhibited survivin expression in AML cells. We next explored the anti-proliferative/anti-survival effects of YM155 in combination with chemotherapeutic agents on AML cells. Etoposide, doxorubicin, and cytarabine were used either alone or combined with a fixed ratio with YM155 to treat Kasumi-1 cells for 48 hr. A significant growth inhibition was observed upon treatments with etoposide plus YM155 or doxorubicin plus YM155, as compared to single agent (Fig. 8A left). The potential outcomes for combining two agents may be synergistic, additive, or antagonistic, which can be determined by the values of combination index (CI) [34]. The values of CI quantitatively depict synergism (CI < 1), additive effect (CI = 1), or antagonism (CI > 1). To determine if YM155 in combination with etoposide or doxorubicin may have synergistic effects on Kasumi-1 cells, we performed CI analyses using the CompuSyn software according to the Chou-Talalay equation [29]. YM155 plus etoposide showed a synergistic effect over a wide range of concentrations. YM155 in combination with doxorubicin exhibited a potent synergistic activity with CI well below 1.0 (Fig. 8A right). The combinatorial effect of YM155 and cytarabine was slightly better than either agent alone under certain concentrations (Fig. S3). CI analysis found that YM155 and cytarabine exhibited a synergistic activity over a limited range of concentrations (CI<1). Their antagonistic activity, however, covered a wide range of concentrations (CI>1) (Fig. S3). Similar studies were also performed in HL-60 cells. It seemed that HL-60 cells were very sensitive to the treatments of YM155 alone, whereas the combinatorial treatments did not do significantly better than YM155 alone (Fig. 8B left). Although all the combinations appeared to be more effective than single agent to inhibit proliferation of HL-60 cells, the CI analyses discovered antagonistic effects for the combination of YM155 and etoposide. YM155 in combination with doxorubicin showed antagonistic effects over a wide range of concentrations, and it had addictive or weak synergetic effects in a very limited range of concentrations (Fig. 8B right). Taken together, our data demonstrated that the combinatorial effects of YM155 and chemotherapeutic agents could be synergetic or antagonistic, depending upon the drugs used and the AML cells tested. The concentrations of the drugs also influenced the CI index. Further investigation is warranted to seek optimal regimens containing YM155 for AML therapy. When applying YM155 into clinic practice, the subtypes of AML need to be taken into consideration for selecting the most effective treatments.
Figure 8. YM155 in combination with chemotherapeutic agents exhibits synergistic or antagonistic effects on growth inhibition of AML cells.

Kasumi-1 (A) or HL-60 (B) cells were plated onto 96-well plates with fresh medium (5% FBS) or the same medium containing indicated concentrations of YM155, etoposide, or doxorubicin alone, or the combinations of etoposide and YM155, or doxorubicin and YM155 with a fixed ratio for 48 hr. The percentages of surviving cells as compared to controls, defined as 100% survival, were determined by reduction of MTS. The combination index (CI) curves were calculated using CompuSyn software according to the Chou-Talalay equation.
4. Discussion
Survivin is selectively expressed in tumor, but not normal tissues [1; 31]. Increased survivin associates with a poor clinic outcome, tumor recurrence, and drug resistance in cancer patients [7; 8; 9]. It is believed that inhibition/downregulation of survivin should be a valuable strategy for cancer treatment. Indeed, several approaches targeting of survivin, including the transcriptional inhibitors, antisense oligonucleotide (ASO), immunotherapy, and gene therapy have been developed [7; 8; 35], and some of them are under clinical trials in various human cancers (http://www.clinicaltrials.gov). It is worth mentioning that survivin does exist in some, although not many normal tissues. In hematopoietic system, expression of survivin has been found in CD34+ stem and progenitor cells [36; 37; 38], T lymphocytes [39; 40], polymorphonuclear neutrophils [41], and vascular endothelial cells [42]. In adult, survivin is indispensable for the proliferation and maintenance of hematopoietic stem and progenitor cells [38]. Survivin also plays a vital role in the development of megakaryocyte/erythroid progenitors [43]. During the differentiation process of megakaryocyte/erythroid progenitors, the expression levels of survivin vary according to the destination of cell. A relatively higher level of survivin is critical for proper erythroid differentiation, whereas downregulation of survivin is required for terminal differentiation of megakaryocytes [38; 43]. Thus, when choosing a survivin-targeted therapy for hematological malignancies, its influence on normal hematopoiesis needs to be taken into consideration. LY2181308 (Eli Lilly and Co., Indianapolis, IN) and Terameprocol/EM-1421 (Erimos Pharmaceuticals, Houston, TX) are survivin-targeted agents under clinical investigation in human cancers.
LY2181308 is a 2'-O-methoxymethyl modified ASO targeting survivin mRNA. Its clinical activity as monotherapy or in combination with idarubicin or cytarabine has been tested in refractory/relapsed AML patients. LY2181308 was well tolerated as single agent, and showed some clinical benefits and no additional toxicity in combination with chemotherapy [44]. Terameprocol is a transcriptional repressor of survivin promoter. Its suppressive effect on survivin is based upon inhibition of the transcription factor Sp1 [45]. While phase I clinical trials of Terameprocol in refractory/relapsed AML patients has been completed, phase II studies are ongoing [46]. To date, targeted therapy of AML has only been used for PML-RARα positive acute promyelocytic leukemia (APL) with all-trans retinoic acid (ATRA) in the clinic. Chemotherapy remains the first line treatments for majority of the subtypes of AML [47]. In the current study, we not only demonstrated that survivin was essential for the maintenance and proliferation of AML cells, but also showed that targeting of survivin with either specific shRNAs or a transcriptional suppressant YM155 markedly enhanced chemosensitivity against AML. YM155 downregulated survivin and promoted AML cells undergoing apoptosis in a dose- and time-dependent manner. The combinatorial effects of YM155 and chemotherapeutic agents can be synergetic or antagonistic, depending on the drugs used for combination and the type of AML cells. As a transcriptional inhibitor of survivin, YM155 shows potent anticancer ability in vitro, with a profound induction of cell death in various cancer cells at nanomolar levels [33; 48]. In a phase II study of YM155 as single agent in 41 patients with refractory DLBCL, only 1 patient had a complete remission and 2 patients responded with a median progression-free survival of 58 days, indicating a very limited effect in DLBCL [23]. Phase II studies with YM155 as monotherapy had also been carried out in other solid tumors. Adverse events of YM155 as single therapy was well tolerated, however, its antitumor activities were modest [19; 20; 21; 22]. Our data suggest that survivin as a promising target, YM155 may be best used in combination with the drugs showing synergetic effects for the treatment of AML.
Meanwhile, the specificity of YM155 has been challenged [35]. Glaros and colleagues first reported that YM155 was a DNA damaging agent rather than a transcriptional suppressor of survivin, because the concentrations of YM155 required to induce γH2AX and P-KAP1, two hallmarkers of DNA damage response, were much lower than that needed to inhibit survivin [49]. YM155-induced downregulation of survivin was likely a secondary event following DNA damage. It was shown that YM155 did not induce cell cycle G2/M arrest, but reduced early DNA synthesis in a human Merkel cell carcinoma cell line MKL-1. In addition, YM155 only decreased the growth rate of MKL-1 xenografts and the tumors grew back when YM155 was withdrawn [50]. In our studies, we found that YM155, unlike specific knockdown of survivin with shRNAs , did not arrest Kasumi-1 cells at G2/M, rather decreased cell population in S phase (Fig. S2). Our data support YM155 as a DNA damaging agent to inhibit DNA synthesis. Although U-937 and NB4 cells express much lower levels of survivin than Kasumi-1 and HL-60 cells (Fig. 1), we performed the same MTS assays to examine the inhibitory effects of YM155 on U-937 and NB4 cells and found that they exhibited a similar sensitivity to YM155 as compared to Kasumi-1 and HL-60 cells (Data not shown). These results may partially attribute to the non-specificity of YM155. It is likely that survivin is not the only factor predicting for YM155 sensitivity. Other genetic/epigenetic alterations may also contribute to the anti-proliferative effects of YM155 on AML cells. Thus, it is understandable that the mechanisms of action, the specificity and the potency of YM155 in human cancers are under intensive interrogation. More detailed investigations are warranted to test the clinic activity of YM155 in AML as monotherapy or in combination with other drugs. Although YM155 may function with “off-target” effects, if AML patients could benefit from YM155 treatment, it would still be a valuable therapeutic option for AML patients.
In summary, we show that survivin plays a vital role for the maintenance and proliferation of AML cells. Therapeutic targeting of survivin is a useful strategy against AML. YM155, the transcriptional inhibitor of survivin, in combination with chemotherapeutic agents exhibits either synergetic or antagonistic effects dependent upon the drugs or AML cells used. Our findings provide a strong rationale for in vivo testing and clinical assessment of survivin-targeted therapy in the treatment of patients with AML.
Supplementary Material
Highlights.
Survivin plays an important role in the maintenance and proliferation of AML cells.
Specific knockdown of survivin expression enhances chemotherapy-mediated anti-proliferative/anti-survival effects on AML cells.
The combinations of YM155 and chemotherapeutic agents exhibit synergetic or antagonistic activity depending upon the drugs used for combination and the type of AML cells being tested.
Acknowledgments
The authors are grateful to Ms. Lisa Litzenberger for her excellent assistance in arts preparation. This work was supported in part by the NIH/NCI (1R03CA181918-01) and the National Natural Science Foundation of China (NSFC grant No. 81472763) (BL).
Abbreviations
- AML
acute myeloid leukemia
- ELISA
enzyme-linked immunosorbent assay
- PARP
poly (ADP-ribose) polymerase
- CI
combination index
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- G-CSF
granulocyte-colony stimulating factor
- SCF
stem Cell Factor
- MAPK
mitogen-activated protein kinase
- PI3K
phosphoinositide 3-kinase
- XIAP
X-linked inhibitor of apoptosis protein
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium,inner salt
Footnotes
Conflicts of interest
The authors declare that they have no conflict of interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer. 2008;8:61–70. doi: 10.1038/nrc2293. [DOI] [PubMed] [Google Scholar]
- 2.Marusawa H, Matsuzawa S, Welsh K, Zou H, Armstrong R, Tamm I, Reed JC. HBXIP functions as a cofactor of survivin in apoptosis suppression. EMBO J. 2003;22:2729–2740. doi: 10.1093/emboj/cdg263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dohi T, Okada K, Xia F, Wilford CE, Samuel T, Welsh K, Marusawa H, Zou H, Armstrong R, Matsuzawa S, Salvesen GS, Reed JC, Altieri DC. An IAP-IAP complex inhibits apoptosis. J Biol Chem. 2004;279:34087–34090. doi: 10.1074/jbc.C400236200. [DOI] [PubMed] [Google Scholar]
- 4.Lens SM, Vader G, Medema RH. The case for Survivin as mitotic regulator. Curr Opin Cell Biol. 2006;18:616–622. doi: 10.1016/j.ceb.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 5.Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, Altieri DC. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:580–584. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 6.Fukuda S, Pelus LM. Survivin, a cancer target with an emerging role in normal adult tissues. Mol Cancer Ther. 2006;5:1087–1098. doi: 10.1158/1535-7163.MCT-05-0375. [DOI] [PubMed] [Google Scholar]
- 7.Coumar MS, Tsai FY, Kanwar JR, Sarvagalla S, Cheung CH. Treat cancers by targeting survivin: just a dream or future reality? Cancer Treat Rev. 2013;39:802–811. doi: 10.1016/j.ctrv.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 8.Kelly RJ, Lopez-Chavez A, Citrin D, Janik JE, Morris JC. Impacting tumor cell-fate by targeting the inhibitor of apoptosis protein survivin. Mol Cancer. 2011;10:35. doi: 10.1186/1476-4598-10-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zaffaroni N, Daidone MG. Survivin expression and resistance to anticancer treatments: perspectives for new therapeutic interventions. Drug Resist Updat. 2002;5:65–72. doi: 10.1016/s1368-7646(02)00049-3. [DOI] [PubMed] [Google Scholar]
- 10.Wang S, Huang X, Lee CK, Liu B. Elevated expression of erbB3 confers paclitaxel resistance in erbB2-overexpressing breast cancer cells via upregulation of Survivin. Oncogene. 2010;29:4225–4236. doi: 10.1038/onc.2010.180. [DOI] [PubMed] [Google Scholar]
- 11.Wang S, Huang J, Lyu H, Cai B, Yang X, Li F, Tan J, Edgerton SM, Thor AD, Lee CK, Liu B. Therapeutic targeting of erbB3 with MM-121/SAR256212 enhances antitumor activity of paclitaxel against erbB2-overexpressing breast cancer. Breast Cancer Res. 2013;15:R101. doi: 10.1186/bcr3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fulda S. Inhibitor of apoptosis proteins in hematological malignancies. Leukemia. 2009;23:467–476. doi: 10.1038/leu.2008.329. [DOI] [PubMed] [Google Scholar]
- 13.Carter BZ, Milella M, Altieri DC, Andreeff M. Cytokine-regulated expression of survivin in myeloid leukemia. Blood. 2001;97:2784–2790. doi: 10.1182/blood.v97.9.2784. [DOI] [PubMed] [Google Scholar]
- 14.Carter BZ, Wang RY, Schober WD, Milella M, Chism D, Andreeff M. Targeting Survivin expression induces cell proliferation defect and subsequent cell death involving mitochondrial pathway in myeloid leukemic cells. Cell Cycle. 2003;2:488–493. [PubMed] [Google Scholar]
- 15.Carter BZ, Qiu Y, Huang X, Diao L, Zhang N, Coombes KR, Mak DH, Konopleva M, Cortes J, Kantarjian HM, Mills GB, Andreeff M, Kornblau SM. Survivin is highly expressed in CD34+38- leukemic stem/progenitor cells and predicts poor clinical outcomes in AML. Blood. 2012;120:173–180. doi: 10.1182/blood-2012-02-409888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng Q, Ling X, Haller A, Nakahara T, Yamanaka K, Kita A, Koutoku H, Takeuchi M, Brattain MG, Li F. Suppression of survivin promoter activity by YM155 involves disruption of Sp1-DNA interaction in the survivin core promoter. Int J Biochem Mol Biol. 2012;3:179–197. [PMC free article] [PubMed] [Google Scholar]
- 17.Nakamura N, Yamauchi T, Hiramoto M, Yuri M, Naito M, Takeuchi M, Yamanaka K, Kita A, Nakahara T, Kinoyama I, Matsuhisa A, Kaneko N, Koutoku H, Sasamata M, Yokota H, Kawabata S, Furuichi K. Interleukin enhancer-binding factor 3/NF110 is a target of YM155, a suppressant of survivin. Mol Cell Proteomics. 2012;11:M111 013243. doi: 10.1074/mcp.M111.013243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamauchi T, Nakamura N, Hiramoto M, Yuri M, Yokota H, Naitou M, Takeuchi M, Yamanaka K, Kita A, Nakahara T, Kinoyama I, Matsuhisa A, Kaneko N, Koutoku H, Sasamata M, Kobori M, Katou M, Tawara S, Kawabata S, Furuichi K. Sepantronium bromide (YM155) induces disruption of the ILF3/p54(nrb) complex, which is required for survivin expression. Biochem Biophys Res Commun. 2012;425:711–716. doi: 10.1016/j.bbrc.2012.07.103. [DOI] [PubMed] [Google Scholar]
- 19.Giaccone G, Zatloukal P, Roubec J, Floor K, Musil J, Kuta M, van Klaveren RJ, Chaudhary S, Gunther A, Shamsili S. Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J Clin Oncol. 2009;27:4481–4486. doi: 10.1200/JCO.2008.21.1862. [DOI] [PubMed] [Google Scholar]
- 20.Kelly RJ, Thomas A, Rajan A, Chun G, Lopez-Chavez A, Szabo E, Spencer S, Carter CA, Guha U, Khozin S, Poondru S, Van Sant C, Keating A, Steinberg SM, Figg W, Giaccone G. A phase I/II study of sepantronium bromide (YM155, survivin suppressor) with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer. Ann Oncol. 2013;24:2601–2606. doi: 10.1093/annonc/mdt249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tolcher AW, Quinn DI, Ferrari A, Ahmann F, Giaccone G, Drake T, Keating A, de Bono JS. A phase II study of YM155, a novel small-molecule suppressor of survivin, in castration-resistant taxane-pretreated prostate cancer. Ann Oncol. 2012;23:968–973. doi: 10.1093/annonc/mdr353. [DOI] [PubMed] [Google Scholar]
- 22.Lewis KD, Samlowski W, Ward J, Catlett J, Cranmer L, Kirkwood J, Lawson D, Whitman E, Gonzalez R. A multi-center phase II evaluation of the small molecule survivin suppressor YM155 in patients with unresectable stage III or IV melanoma. Invest New Drugs. 2011;29:161–166. doi: 10.1007/s10637-009-9333-6. [DOI] [PubMed] [Google Scholar]
- 23.Cheson BD, Bartlett NL, Vose JM, Lopez-Hernandez A, Seiz AL, Keating AT, Shamsili S, Papadopoulos KP. A phase II study of the survivin suppressant YM155 in patients with refractory diffuse large B-cell lymphoma. Cancer. 2012;118:3128–3134. doi: 10.1002/cncr.26510. [DOI] [PubMed] [Google Scholar]
- 24.Kita A, Mitsuoka K, Kaneko N, Nakata M, Yamanaka K, Jitsuoka M, Miyoshi S, Noda A, Mori M, Nakahara T, Sasamata M. Sepantronium bromide (YM155) enhances response of human B-cell non-Hodgkin lymphoma to rituximab. J Pharmacol Exp Ther. 2012;343:178–183. doi: 10.1124/jpet.112.195925. [DOI] [PubMed] [Google Scholar]
- 25.Chen J, Pise-Masison CA, Shih JH, Morris JC, Janik JE, Conlon KC, Keating A, Waldmann TA. Markedly additive antitumor activity with the combination of a selective survivin suppressant YM155 and alemtuzumab in adult T-cell leukemia. Blood. 2013;121:2029–2037. doi: 10.1182/blood-2012-05-427773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai B, Lyu H, Huang J, Wang S, Lee CK, Gao C, Liu B. Combination of bendamustine and entinostat synergistically inhibits proliferation of multiple myeloma cells via induction of apoptosis and DNA damage response. Cancer Lett. 2013;335:343–350. doi: 10.1016/j.canlet.2013.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang J, Wang S, Lyu H, Cai B, Yang X, Wang J, Liu B. The anti-erbB3 antibody MM-121/SAR256212 in combination with trastuzumab exerts potent antitumor activity against trastuzumab-resistant breast cancer cells. Mol Cancer. 2013;12:134. doi: 10.1186/1476-4598-12-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang S, Huang J, Lyu H, Lee CK, Tan J, Wang J, Liu B. Functional cooperation of miR-125a, miR-125b, and miR-205 in entinostat-induced downregulation of erbB2/erbB3 and apoptosis in breast cancer cells. Cell Death Dis. 2013;4:e556. doi: 10.1038/cddis.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chou T-C, Martin N. CompuSyn Software for Drug Combinations and for General Dose-Effect Analysis, and User's Guide. ComboSyn, Inc; Paramus, NJ: 2007. [ www.combosyn.com] (2007) [Google Scholar]
- 30.Cline MS, Craft B, Swatloski T, Goldman M, Ma S, Haussler D, Zhu J. Exploring TCGA Pan-Cancer data at the UCSC Cancer Genomics Browser. Sci Rep. 2013;3:2652. doi: 10.1038/srep02652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanwar JR, Kamalapuram SK, Kanwar RK. Survivin signaling in clinical oncology: a multifaceted dragon. Med Res Rev. 2013;33:765–789. doi: 10.1002/med.21264. [DOI] [PubMed] [Google Scholar]
- 32.Ouchi M, Ouchi T. Distinct DNA damage determines differential phosphorylation of Chk2. Cancer Biol Ther. 2014;15:1700–1704. doi: 10.4161/15384047.2014.972823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakahara T, Kita A, Yamanaka K, Mori M, Amino N, Takeuchi M, Tominaga F, Hatakeyama S, Kinoyama I, Matsuhisa A, Kudoh M, Sasamata M. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007;67:8014–8021. doi: 10.1158/0008-5472.CAN-07-1343. [DOI] [PubMed] [Google Scholar]
- 34.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 35.Rauch A, Hennig D, Schafer C, Wirth M, Marx C, Heinzel T, Schneider G, Kramer OH. Survivin and YM155: how faithful is the liaison? Biochimica et biophysica acta. 2014;1845:202–220. doi: 10.1016/j.bbcan.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 36.Fukuda S, Mantel CR, Pelus LM. Survivin regulates hematopoietic progenitor cell proliferation through p21WAF1/Cip1-dependent and -independent pathways. Blood. 2004;103:120–127. doi: 10.1182/blood-2003-05-1756. [DOI] [PubMed] [Google Scholar]
- 37.Fukuda S, Pelus LM. Regulation of the inhibitor-of-apoptosis family member survivin in normal cord blood and bone marrow CD34(+) cells by hematopoietic growth factors: implication of survivin expression in normal hematopoiesis. Blood. 2001;98:2091–2100. doi: 10.1182/blood.v98.7.2091. [DOI] [PubMed] [Google Scholar]
- 38.Leung CG, Xu Y, Mularski B, Liu H, Gurbuxani S, Crispino JD. Requirements for survivin in terminal differentiation of erythroid cells and maintenance of hematopoietic stem and progenitor cells. J Exp Med. 2007;204:1603–1611. doi: 10.1084/jem.20062395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xing Z, Conway EM, Kang C, Winoto A. Essential role of survivin, an inhibitor of apoptosis protein, in T cell development, maturation, and homeostasis. J Exp Med. 2004;199:69–80. doi: 10.1084/jem.20031588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okada H, Bakal C, Shahinian A, Elia A, Wakeham A, Suh WK, Duncan GS, Ciofani M, Rottapel R, Zuniga-Pflucker JC, Mak TW. Survivin loss in thymocytes triggers p53-mediated growth arrest and p53-independent cell death. J Exp Med. 2004;199:399–410. doi: 10.1084/jem.20032092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altznauer F, Martinelli S, Yousefi S, Thurig C, Schmid I, Conway EM, Schoni MH, Vogt P, Mueller C, Fey MF, Zangemeister-Wittke U, Simon HU. Inflammation-associated cell cycle-independent block of apoptosis by survivin in terminally differentiated neutrophils. J Exp Med. 2004;199:1343–1354. doi: 10.1084/jem.20032033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zwerts F, Lupu F, De Vriese A, Pollefeyt S, Moons L, Altura RA, Jiang Y, Maxwell PH, Hill P, Oh H, Rieker C, Collen D, Conway SJ, Conway EM. Lack of endothelial cell survivin causes embryonic defects in angiogenesis, cardiogenesis, and neural tube closure. Blood. 2007;109:4742–4752. doi: 10.1182/blood-2006-06-028068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gurbuxani S, Xu Y, Keerthivasan G, Wickrema A, Crispino JD. Differential requirements for survivin in hematopoietic cell development. Proc Natl Acad Sci U S A. 2005;102:11480–11485. doi: 10.1073/pnas.0500303102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erba HP, Sayar H, Juckett M, Lahn M, Andre V, Callies S, Schmidt S, Kadam S, Brandt JT, Van Bockstaele D, Andreeff M. Safety and pharmacokinetics of the antisense oligonucleotide (ASO) LY2181308 as a single-agent or in combination with idarubicin and cytarabine in patients with refractory or relapsed acute myeloid leukemia (AML) Invest New Drugs. 2013;31:1023–1034. doi: 10.1007/s10637-013-9935-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smolewski P. Terameprocol, a novel site-specific transcription inhibitor with anticancer activity. IDrugs. 2008;11:204–214. [PubMed] [Google Scholar]
- 46.Zhu X, Ma Y, Liu D. Novel agents and regimens for acute myeloid leukemia: 2009 ASH annual meeting highlights. J Hematol Oncol. 2010;3:17. doi: 10.1186/1756-8722-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.NCCN. National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology-Acute Myeloid Leukemia. NCCN Guidelines(NCCN.org) 2014;2014 [Google Scholar]
- 48.Nakahara T, Kita A, Yamanaka K, Mori M, Amino N, Takeuchi M, Tominaga F, Kinoyama I, Matsuhisa A, Kudou M, Sasamata M. Broad spectrum and potent antitumor activities of YM155, a novel small-molecule survivin suppressant, in a wide variety of human cancer cell lines and xenograft models. Cancer Sci. 2011;102:614–621. doi: 10.1111/j.1349-7006.2010.01834.x. [DOI] [PubMed] [Google Scholar]
- 49.Glaros TG, Stockwin LH, Mullendore ME, Smith B, Morrison BL, Newton DL. The "survivin suppressants" NSC 80467 and YM155 induce a DNA damage response. Cancer Chemother Pharmacol. 2012;70:207–212. doi: 10.1007/s00280-012-1868-0. [DOI] [PubMed] [Google Scholar]
- 50.Arora R, Shuda M, Guastafierro A, Feng H, Toptan T, Tolstov Y, Normolle D, Vollmer LL, Vogt A, Domling A, Brodsky JL, Chang Y, Moore PS. Survivin is a therapeutic target in merkel cell carcinoma. Sci Transl Med. 2012;4:133ra156. doi: 10.1126/scitranslmed.3003713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.