

Abstract
DNA double-strand breaks (DSBs) are involved in many cellular mechanisms, including replication, transcription, and genome rearrangements. The recent observation that hot spots of DSBs in human chromosomes delimit DNA domains that possess coordinately expressed genes suggests a strong relationship between the organization of transcription patterns and hot spots of DSBs. In this study, we performed mapping of hot spots of DSBs in a human 43-kb ribosomal DNA (rDNA) repeated unit. We observed that rDNA units corresponded to the most fragile sites in human chromosomes and that these units possessed at least nine specific regions containing clusters of extremely frequently occurring DSBs, which were located exclusively in non-coding intergenic spacer (IGS) regions. The hot spots of DSBs corresponded to only a specific subset of DNase-hypersensitive sites, and coincided with CTCF, PARP1, and HNRNPA2B1 binding sites, and H3K4me3 marks. Our rDNA-4C data indicate that the regions of IGS containing the hot spots of DSBs often form contacts with specific regions in different chromosomes, including the pericentromeric regions, as well as regions that are characterized by H3K27ac and H3K4me3 marks, CTCF binding sites, ChIA-PET and RIP signals, and high levels of DSBs. The data suggest a strong link between chromosome breakage and several different mechanisms of epigenetic regulation of gene expression.
Keywords: double-strand breaks, fragile sites, rDNA, IGS, PARP1, HNRNPA2B1, 4C
Introduction
The most actively transcribed genes in eukaryotes are ribosomal RNA (rRNA) genes. They are organized in clusters of tandemly repeated transcriptional units abutting each other in a head-to-tail orientation, and are devoid of intervening non-ribosomal DNA sequences (Little and Braaten, 1989). Ribosomal RNA accounts for up to 80% of all cellular RNA production (Moss et al., 2007). In human genomic ribosomal DNA (rDNA), 42999-bp tandemly arrayed units are located in the middle of the short (p) arms of the five acrocentric chromosomes 13, 14, 15, 21, and 22 (Worton et al., 1988; Reddy and Sulcova, 1998). Analysis of the patterns of rDNA inheritance revealed that there is high variability between and within human individuals (Stults et al., 2008). The length of the cluster can vary from 50 kb to >6 Mb, thus providing each person with a unique rDNA pattern. Each unit on each chromosome consists of a 13-kb coding region containing 18S, 5.8S, and 28S RNA genes. However, the major part of rDNA is occupied by a non-coding intergenic spacer (IGS) region that possesses, at its 3′ end, a region crucial for regulation of rRNA transcription elements, i.e. an enhancer, spacer promoter, and the core promoter of the adjoining rDNA repeat (McStay and Grummt, 2008).
The IGS was considered for a long time as transcriptionally silent chromatin (Grummt and Pikaard, 2003; Santoro, 2005), but it was recently shown that the IGS is transcribed at a very low level (Zentner et al., 2011). More prominent IGS transcription produces small 150–300 nucleotide non-coding RNAs (ncRNAs) that are complementary to the rDNA promoter, and are required for both establishing and maintaining a specific heterochromatin structure at the promoter of a subset of rDNA arrays (Mayer et al., 2006). The nucleolar remodeling complex (NoRC) is a member of the ATP-dependent chromatin remodeling complexes, and is involved in establishing the heterochromatic state at the rDNA promoter by recruitment of proteins possessing histone-modifying or DNA-methylating activities (Santoro et al., 2002; Zhou et al., 2002; Santoro and Grummt, 2005 ). The IGS contains one or more PolI promoters (Moss et al., 1980) that are targeted by ncRNAs covering the rDNA promoter(s) in association with NoRC.
Although the human genome-sequencing project is largely complete, the most highly repetitive regions of the genome have still not been assembled, including rDNA, which is not included in the reference genome assemblies. Nevertheless, recently it was demonstrated that short-sequence reads generated from ChIP-Seq experiments can be accurately aligned to specially designed genome assemblies containing rDNA (Zentner et al., 2011). This approach allows both the epigenetic marks and RNA-seq data to be mapped in rDNA units.
In humans, nucleolar organizer regions (NOR) bearing chromosomes undergo translocations more frequently than other chromosomes (Therman et al., 1989; Denison et al., 2002). The data might suggest, on one hand, the presence of hot spots of double-strand breaks (DSBs) inside rDNA, and, on the other hand, the close proximity of rDNA units to chromatin regions possessing DSBs in different chromosomes (Misteli, 2010). However, to date, neither the hot spots of DSBs in rDNA units nor the chromosomal contacts of rDNA units have been mapped.
Recently, we developed a method for the precise mapping of DNA DSBs in eukaryotic chromosomes by amplification and deep sequencing of short DNA fragments delimited by sites of DSBs ligated to a specific oligonucleotide and Sau3A restriction sites (Tchurikov et al., 2011, 2013). The hot spots of DSBs were found to flank the coordinately expressed large domains in Drosophila and human chromosomes that are transcribed by RNA polymerase II. FISH experiments with the amplified DNA on human chromosomes suggest that the rDNA units possess hot spots of DSBs. In Drosophila polytene chromosomes, the hot spots of DSBs mainly correspond to the small islands of heterochromatin scattered among euchromatic regions that often form ectopic contacts. Here, we report the results of the profiling of hot spots of DSBs in human rDNA in cultured HEK293T cells. Our data demonstrate that hot spots of DSBs are non-randomly distributed inside rDNA repeats and are exclusively located in nine specific regions of the IGS. Our data suggest that rDNA units correspond to the most fragile regions in human chromosomes and that the mapped hot spots of DSBs in the IGS coincide with the regions possessing the major CTCF binding sites and H3K4me3 marks, and do not correspond to DNase-hypersensitive sites. Our 4C (circular chromosome conformation capture) data indicate that these IGS regions are often located in close proximity to a set of chromosomal regions that possess specific epigenetic marks, including pericentromeric regions in different chromosomes, where the hot spots of DSBs also occur. Taken together, these data suggest that chromosomal breakage is connected with different mechanisms of epigenetic regulation and chromosomal 3D architecture.
Results
Mapping of hot spots of DSBs in rDNA units
For mapping of DSBs inside rDNA units, we amplified short DNA stretches delimited by nucleotides at DSBs and Sau3A sites (Figure 1A). Thus, the amplified samples contained the whole-genome collection of 50–300 bp DNA fragments at DSBs. We used deep sequencing of amplified DNA to produce long (up to 1232 nt) sequences and mapped the reads in rDNA units. The reads were processed as described in the Supplementary material and in the databases (GEO accession numbers GSE35065 and GSE49302 for 454-sequencing and Illumina reads, respectively). Figure 1B shows the rDNA mapping results of 454-sequencing and Illumina reads generated from, in total, ∼0.6 million and 32 million high-quality reads, respectively. About 12.7% of mapped Illumina reads that represented amplified sites of DSBs in both the human genome (assembly GRCh37/hg19) and rDNA units corresponded to rDNA. However, even if we assume that 300 copies of rDNA are present per genome (Stults et al., 2009), the portion of rDNA should be no more than 0.5% of the genome length. We conclude that rDNA is significantly enriched with DSBs. More detailed statistical analysis revealed that the average DSBs density in the rDNA unit was higher than the average DSBs density of any chromosome in the sequenced portion of the human genome (Supplementary Table S1). This conclusion is consistent with experimental data from FISH experiments, which showed that amplified DNA produces the brightest signals in the regions where rDNA is located, i.e. at the ends of all acrocentric chromosomes bearing clusters of rDNA (Tchurikov et al., 2013). We observed similar data for the mapping of hot spots of DSBs in rDNA units using different deep-sequencing approaches (Figure 1B). The mapping results of both deep-sequencing approaches are generally consistent, but some differences are also observed. The differences refer mainly to the relative abundance of reads in different regions. For example, R7 is the most prominent in Illumina reads, while R5 is more significant in 454-sequencing reads. We used the Pfu enzyme for preparation of the DNA probe for 454-sequencing, and Taq polymerase for Illumina sequencing. The data indicate that the aliquots taken from the same DNA sample, but amplified by different polymerases, could give different results due to PCR biases, which is in agreement with an earlier analysis of deep-sequencing data (Treangen and Salzberg, 2011). We assume that the bias would be more significant for unique amplified sequences than for repetitive ones, including the rDNA sequences.
Figure 1.

Mapping of hot spots of DSBs inside rDNA. (A) Scheme showing the procedure used for amplification of short DNA fragments at DSBs. (B) Mapping results of 454-sequencing and Illumina reads along human rDNA units (GEO accession numbers GSM438363 and GSE49302, respectively). The functional map of human rDNA units is presented at the bottom. rDNA numberings are according to the human ribosomal DNA complete repeating unit sequence (Accession number U13369).
Nine regions spanning from 242 to 796 bp and possessing hot spots of DSBs were selected. We selected the regions where >700 Illumina reads were mapped. Reads with the mapped simple sequences were discarded (e.g. the regions starting at 15768 and 15931 bp on the consensus sequence, i.e. HSU13369). The selected regions correspond to the DNA stretches aligning only to rDNA. Although a number of Alu elements are present in the IGS, it was possible to map the corresponding reads unambiguously, as these molecular fossils possess a high (20%–27%) divergence among members, each possessing a unique signature of diverged nucleotide positions, which make them characteristic to specific regions in the rDNA units (Gonzalez et al., 1989, 1993).
By visual inspection, all nine selected hot spots of DSBs were non-randomly distributed inside the rDNA units in specific regions of the non-coding IGS. Nevertheless, we tested the hypothesis of homogeneity of DSBs distribution in both the entire rDNA unit and the IGS using the z-ratio criterion for an entropy-like function (Chechetkin, 2013). The resulting values were: z = 2.5903 (P-value <0.01) for rDNA, and z = 2.7025 (P-value <0.01) for IGS, and thus, we conclude that the distribution of hot spots of DSBs in both rDNA and the IGS is non-random.
Supplementary Figures S1–S5 schematically show the multiple sequence alignments of reads in the regions R1–R9 as visualized by UGENE, http://ugene.unipro.ru/ (Okonechnikov et al., 2012). We did not observe any consensus sequence or particular sequence motifs at the sites of hot spots of DSBs inside rDNA. Currently, to answer the question of which enzyme(s) are responsible for DNA cleavage at hot spots, we are studying the cut sites and sequences around them in rDNA and in the whole genome in more detail.
The hot spots of DSBs in rDNA coincide with regions possessing active chromatin marks
The data on the specific distribution of the hot spots of DSBs inside rDNA units prompted us to study the chromatin features in these regions. It is known that CTCF is associated with diverse regulatory events in the human genome. It can act as a transcriptional activator, repressor, and insulator, and can form chromatin loops (Holwerda and de Laat, 2013; Ong and Corces, 2014). The available raw data on CTCF binding sites in HEK293T cells were used for mapping inside rDNA. Figure 2 shows the profile of CTCF binding sites in rDNA. Most CTCF binding sites occurred inside the IGS. We observed that the major R4–R9 hot spots of DSBs coincided with the major peaks of CTCF binding sites. Moreover, there is a clear correlation between the frequencies of observed DSBs and the levels of CTCF binding inside the IGS. Nevertheless, there are CTCF binding sites in the regions where hot spots of DSBs were not detected, e.g. the regions around the coordinates 6500, 17000, and 42000 bp. The latter region corresponds to the upstream control element (UCE) and core promoter (CP) sequences (McStay and Grummt, 2008). The data indicate that, in rDNA units, CTCF recognizes not only the specific sites at DSBs, but some others as well. It might be that some peaks of CTCF binding originate from active rDNA units, while others originate from silenced units. The functional role of CTCF in different hot spots of DSBs is hard to predict. Nevertheless, the mapping data indicate a strong link between the regions that are important for regulation via CTCF and chromosome breakage at NORs. This analysis cannot discriminate the signals coming from active or inactive rDNA units.
Figure 2.
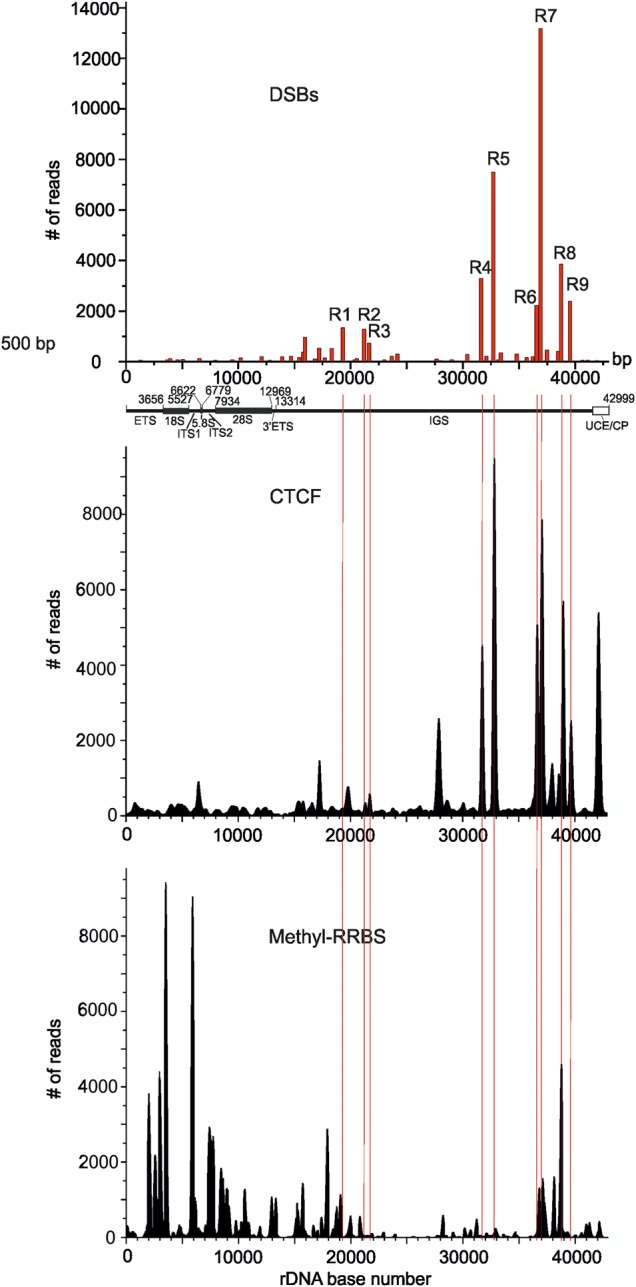
Comparison of CTCF binding and DNA methylation profiles with the observed pattern of hot spots of DSBs inside rDNA. At the top, the position of nine hot spots of DSBs obtained from Illumina reads is presented. Thin red lines show the position of nine hot spots of DSBs inside the IGS. The raw data on CTCF binding and on DNA methylation in HEK293 cells were used for the mapping inside the rDNA unit (Accession numbers wgEncodeEH000396 and GSE27584, respectively). UCE/CP: upstream promoter element and core promoter.
In contrast, the major peaks of the DNA methylation profile inside rDNA are located inside the coding region (Figure 2). Interestingly, only R6–R9 hot spots of DSBs precisely coincide with the corresponding peaks of the DNA methylation profile, suggesting that modulation of expression by DNA methylation is characteristic of only a specific subset of hot spots of DSBs inside rDNA units.
Hot spots of DSBs in rDNA units correspond to H3K4me3 sites, and only to a specific subset of DNase-I-hypersensitive sites
To investigate whether some of the observed hot spots of DSBs inside rDNA correspond to active units, we performed mapping of the active chromatin mark (H3K4me3) along the rDNA repeat. This prominent histone mark is a promoter-specific histone modification that is associated with active transcription and with active genes (Lauberth et al., 2013). Figure 3A shows the profile of H3K4me3. Clearly, the profiles of all almost all the hot spots of DSBs and the H3K4me3 mark exhibit a striking consistency: both the positions and the spans of the peaks demonstrate similarity. The result was unexpected, because the promoter sequences inside rDNA units were described previously only within UCE/CP sequences. The result again argues in favor of a strong connection between the hot spots of DSBs and the regulation of transcription inside rDNA units.
Figure 3.

Relationship between distribution of H3K4me3 marks, chromatin accessibility for DNase I, and hot spots of DSBs inside rDNA. (A) At the top, the position of nine hot spots of DSB obtained from Illumina deep sequencing is presented. The raw data on distribution of H3K4me3 marks in HEK293 cells and on DNase I hypersensitivity in HEK293T cells were used for the mapping in the rDNA unit (GEO accession numbers GSM945288 and GSM1008573, respectively). Thin red lines show the position of nine hot spots of DSBs inside IGS. UCE/CP: upstream promoter element and core promoter. (B) Correlation heatmap of pairwise comparisons between median signals for DSBs, H3K4me3, CTCF, DNase-I-hypersensitive sites (DNaseI-HSS), and DNA methylation sites (Methyl-RRBS; Meissner et al., 2005) inside the entire rDNA. (C) Correlation heatmap of pairwise comparisons between median signals for DSBs, H3K4me3, CTCF, DNase-I-hypersensitive sites (DNaseI-HSS), and DNA methylation sites (Methyl-RRBS) inside the IGS.
The nature of spontaneous DSBs observed is not yet known (Tchurikov et al., 2013). However, it is reasonable to suppose that the regions of hot spots of DSBs should possess free DNA, i.e. DNA not protected by very tight packaging with proteins. To investigate this, we decided to compare the pattern of the observed DSBs and the profile of DNase-seq data presenting hypersensitivity. For the mapping, we used DNase-seq from HEK293T cells obtained from the ENCODE. Figure 3A shows that the majority of DNase-hypersensitive sites are located in the coding region. At face value, it seems that there is an overlap between the sites of DSBs and the DNase I sites. These DNase-seq data were obtained using the well-known Duke's protocol (http://genome.ucsc.edu/ENCODE/protocols/general/Duke_DNase_protocol.pdf) that uses the isolation of nuclei and subsequent DNase I digestion of DNA. Comparison with our straightforward and more rapid procedure for the isolation of DNA samples for amplification of DNA termini at DSBs suggests that ‘our DSBs’, either preexisting in vivo or introduced during incubation of cells inside of the agarose plugs, should already be present in the nuclei preparation before DNase I treatment. That is why we expected that the hot spots of DSBs described in this study should be present among the mapped DNase-hypersensitive sites.
The nature of the major portion of the DNase I sites is clearly different from the hot spots of DSBs described here without usage of any exogenous enzyme. First of all, DNase-sensitive sites are present in both the coding and non-coding regions, while the hot spots of DSBs reside only in the IGS. Detailed analysis reveals many differences in the profiles, e.g. there is a major peak of the DNase I site at coordinate 29000 bp, while there is no corresponding hot spot of DSBs; there are DNase I sites at both a UCE/CP, as well as at the external transcribed spacer (ETS), where DSBs were not detected. Taken together, the data suggest that only a specific subset of DNase-I-hypersensitive sites corresponds to the hot spots of DSBs. This discrepancy strongly indicates that the nature of DNase-sensitive sites and the hot spots of DSBs that we analyzed are different.
The conclusions based on visual inspections of the data regarding correspondence between DSBs, CTCF binding sites, DNA methylation, H3K4me3 marks, and DNase-I-hypersensitive sites were also confirmed by the calculated correlation scores. The corresponding mapping data inside the entire rDNA or only inside the IGS were median-smoothed in 100-bp windows, and the correlation scores were clustered and plotted in a heatmap (Figure 3B and C). These data strongly indicate that the hot spots of DSBs, CTCF binding sites, and H3K4me3 marks are distributed similarly along rDNA units and show very high correlation scores with one another. DNase-I-hypersensitive sites demonstrate much lower (but still meaningful) correlations (especially inside the IGS) with the group consisting of DSBs, CTCF binding sites, and H3K4me3 marks, while the DNA methylation pattern is not correlated with the group either inside the entire rDNA or inside the IGS. The conclusion was independently confirmed by the statistical analysis using GenometriCorr package (Favorov et al., 2012). The data are shown in Supplementary Table S2.
DNA breakage inside rDNA is enhanced by replication stress or heat-shock treatment
Based on our results, we hypothesized that the DNA breakage at the detected hot spots of DSBs in rDNA units could be functionally modulated by treatments that affect replication or transcription, and this could be monitored by real-time PCR. Initially, we compared the level of breakage that could be introduced by the procedure used for isolation of the original DNA samples that were used for mapping of DSBs. The procedure includes a short heat-shock treatment of cells when introducing them into low-melt agarose, which involves a short (2–3 min) incubation at 42°C (see the Materials and methods section), which is why we isolated ‘intact’ and ‘damaged’ DNA samples as described previously (the details are described in the Supplementary Methods). In these experiments, we also used DNA samples from HEK293T cells that were treated with hydroxyurea (to induce replication stress) or heat-shock-treated cells (to induce changes in transcription patterns). In both cases, the DNA samples were isolated immediately after precipitation of cells.
For monitoring of DNA breakage, we selected the R5 region, which was one of the most prominent hot spots of DSBs in rDNA. We mapped 7503 Illumina reads at this region (Figure 1B). Figure 4A shows the position-specific primers used in PCR across R5, indicated by the mapped Illumina reads in this region. We amplified the entire 768-bp DNA fragment containing the 311-bp R5 and the flanking regions (192-bp left side and 204-bp right side). This 768-bp region does not possess Alu sequences. The data shown in Figure 4B demonstrate that ∼50% of DNA molecules at R5 are damaged. If we assume that each of five rDNA clusters has ∼60 units, it follows that ∼30 units per cluster are damaged only at R5. This estimation is in agreement with the statistical analysis of DSBs density inside the 43-kb rDNA unit and in the whole human genome, strongly suggesting that rDNA arrays are one of the most fragile sites in the human genome. We also observed DNA damage around R5 on both flanks, where only a small number of reads were mapped. In the flank regions, ∼10%–15% of the DNA molecules were damaged. In all cases, hydroxyurea-induced replication stress or heat-shock treatment slightly increased the damage, strongly suggesting in vivo DNA breakage at R5 during these treatments. Previously, these effects were demonstrated in several different genomic regions, including a hot spot of DSBs at the WWOX gene (Tchurikov et al., 2013). Here, we show that the same is true for rDNA, in which spontaneous DNA breakage is also increased in vivo by both replication stress and by changes in transcription inside the rDNA units in heat-shocked cells (Parker and Bond, 1989; Waters and Schaal, 1996).
Figure 4.

Analysis of hot spots of DSBs by quantitative PCR across R5 and ChIP experiments. (A) Distribution of Illumina reads at R5. Only the top 610 reads from 3767 mapped reads are shown schematically using UGENE software (http://ugene.unipro.ru/). Red and blue arrows indicate the positions of primers used in PCR experiments designed for amplification of the entire 768-bp region or its flanks. (B) PCR experiments using untreated HEK293T cells or cells incubated in the presence of 0.2 mM hydroxyurea for 18 h or heat-shock-treated cells. For details, see Supplementary Methods. The results of four independent experiments are shown. RQ: relative quantities compared with undamaged DNA. The ribosomal 5.8S gene was used as a control region that does not possess hot spots of DSBs. (C) ChIP experiments using antibodies to PARP1 or to HNRNPA2B1. Results of PCR across the entire R5 or its flanks using immunoprecipitated DNA are presented. Percentage of input DNA is indicated. The results of four independent experiments are shown. PCR across the ribosomal 5.8S gene using immunoprecipitated DNA was used as a control for a region that does not possess hot spots of DSBs (Tchurikov et al., 2013). *P≤ 0.01.
Binding of PARP1 and HNRNPA2B1 at R5
Recently, it was shown that PARP1 and HNRNPA2B1 bind at hot spots of DSBs in human cells (Tchurikov et al., 2013). It was supposed that PARP1 binding might be connected with the suggested mechanism of coordinated expression of genes located in domains delimited by hot spots of DSBs. It is known that PARP1 behaves as a strong regulator of chromatin structure and transcription (Kraus and Lis, 2003; Krishnakumar and Kraus, 2010; Tchurikov et al., 2013) and is even capable of somatic cell reprogramming (Doege et al., 2012). To elucidate whether PARP1 and HNRNPA2B1 could bind at hot spots of DSBs in rDNA, we used a ChIP assay using the same set of primers for amplification of the entire R5 area and the flanks (Figure 4A). The results of real-time PCR experiments with DNA samples isolated from immunoprecipitated chromatin indicate that both proteins bind at R5 and around it (Figure 4C). We used the DNA in chromatin samples that were sonicated to 150–500 bp. We assume that the binding of these proteins at the flanks of the hot spot of DSBs is due to the use of rather long fragments of sonicated DNA. Alternatively, there could be a long area around the R5 region where the proteins bind. We hope that the ChIP-Seq experiments that we are currently performing will resolve this question and will provide a detailed profile of the binding of both proteins along the rDNA units. In any case, the current results indicate that PARP1 and HNRNPA2B1 bind in vivo at this hot spot of spontaneous DSBs in rDNA.
Whole-genomic interactions of IGS areas possessing R4 and R5 hot spots
We observed that R1–R9 islands of low nucleosome occupancy merge into heterochromatic regions of the IGS. Heterochromatin regions are prone to forming both intra- and inter-chromosomal contacts, and human NORs interact with one another, as well as with the non-NORs (Manuelidis and Borden, 1988). Therefore, we decided to detect all the contacts in the entire genome of a 2.2-kb IGS fragment containing R4 and R5 hot spots using a 4C approach. Figure 5A shows schematically the procedure used for amplification of different genomic EcoRI-FaeI fragments ligated in cross-linked chromatin preparations with an EcoRI site at coordinate 30487 of the rDNA unit. R5 and R4 are located at distances of 2218 and 1136 bp from this coordinate, respectively.
Figure 5.

The major contacts of rDNA units in the sequenced portion of the human genome. (A) Design of the 4C-rDNA experiment for analysis of genome-wide contacts of rDNA units at R4 and R5. A physical map of the 12-kb region containing R4 and R5 and the positions of EcoRI sites (E) and only the nearest FaeI site (F) to the EcoRI site at the coordinate 30487 are shown. The primers selected inside the 415-bp EcoRI-FaeI fragment are shown. The major steps of the 4C procedure are illustrated by a scheme. rDNA fragments are not shown to scale. Details of the procedure are described in Supplementary Methods. (B) Circos presentation of rDNA contacts representing at least 1000 mapped 4C reads. Only one rDNA unit was included at the tip of chr14. rDNA contacts with particular regions inside 16 chromosomes are presented. (C) The major sites of contacts of rDNA units are shown along cytobands in chr1, chr2, chr3, chr4, chr9, and chr21 as visualized in the Integrated Genome Browser (Affymetrix) (http://bioviz.org/igb/). The mapping was performed in the human genome assembly of February 2009 (GRC37/hg19).
Figure 5B shows a Circos presentation of the 4C data for the most frequent chromosomal contacts of rDNA units inside sixteen chromosomes. Each contact is represented by at least 1000 reads. As the current assemblies of the human genome do not contain rDNA, we added one rDNA unit to the proximal tip of chr14, on which rDNA is endogenously located, so that self-rDNA contacts could also be observed in the Circos presentation. The overviews of the contacts in chr1–4, chr9, and chr21 are shown in Figure 5C. Supplementary Figures S6 and S7 present the distribution of such regions in the other frequently contacted chromosomes along their cytobands. These contacts of rDNA units are located close to specific regions in sixteen chromosomes. Each chromosome indicated in Figure 5C and in Supplementary Figures S6 and S7 has from one to three frequent contacts. These contacts usually correspond to pericentric heterochromatin (PC-HC) regions, although some prominent contacts were found in euchromatic regions and at telomeres. The most prominent rDNA contacts were observed in pericentromeric regions inside chr16 and Y (Supplementary Figure S8A, B, Tables S3 and S4).
To characterize the features of the contact regions of rDNA, we used the UCSC Genome Browser on the GRCh37/hg19 assembly. We observed that the peaks of rDNA contacts often overlapped with the prominent sites of the layered H3K27Ac mark (Figure 6A, Supplementary Figures S9–S11), which is often found near active regulatory elements of the seven cell lines used in ENCODE, and which may distinguish active enhancers and promoters from their inactive counterparts (The ENCODE Project Consortium, 2012). In some cases, the contact regions were also characterized by sites with high chromatin interaction (ChIA-PET) signals and/or by RIP-Seq signals. The latter shows the profile of mRNAs that co-precipitate with RNA-binding proteins (Supplementary Figure S10). The contact regions also possess higher nucleosomal densities and are often recognized as repeats. However, the observed pattern of the contact regions is not simply a result of mapping inside genomic repeats as we did not observe 4C-rDNA peaks inside a broad region of the repeated sequences shown in Figure 6A. The contact regions that are located in PC-HC often corresponded to the chromosomal stretches that are immediately adjacent to regions that are still absent in the current assemblies of the human genome (Supplementary Figures S9–S11). The data could suggest that many more rDNA contact regions could be found in these HC regions when they are sequenced and mapped in the future.
Figure 6.

Features of the chromosomal regions where rDNA contacts were observed. The UCSC Genome Browser or Integrated Genome Browser was used with the human genome assembly of Feb. 2009 (GRCh37/hg19). The distribution of layered H3K27ac marks, ChIA-PET signals, nucleosome position, and RepeatMasker data are shown. (A) Region of chr10 possesses overlapping profiles of rDNA contacts and H3K27ac marks. Pericentromeric region exhibits overlapping profiles of rDNA contacts, H3K27ac marks, ChIA-PET signals (ENCODE/GIS-Ruan, the protein factors displayed in the track include RNA polymerase II, and CCCTC-binding factor (CTCF)), and regions of higher nucleosome density (MNase-Seq from ENCODE/Stanford/BYU). (B) Region of chr21 illustrates the overlapping profiles of DSBs and rDNA contacts. (C) The fragment of chr21 is shown in more detail. EcoRI sites are shown at the bottom. Thin lines indicated precise correspondence between rDNA contact sites and DSBs inside the chromosome. (D) Statistical analysis of spatial correlations between rDNA contact regions, H3K27ac marks, and DSBs obtained by the GenometriCorr package. Summary results for all chromosomes together are displayed in two panels. Color-coded density plots in each panel represent deviation from the expected uniform distribution. Red color indicates positive deviation from the expected uniform distribution; blue indicates negative deviation from the expected uniform distribution. The overlay line indicates the density of the data at each absolute or relative distance. Query corresponds to a set of intervals corresponding to the genomic contacts of rDNA units (4C-rDNA data).
Contacts of R4 and R5 regions of rDNA units with PC-HC regions possessing hot spots of DSBs
Our 4C-rDNA data reveal that the contacts of EcoRI fragments that possess R4 and R5 clusters of DSBs inside rDNA units in PC-HC regions in different chromosomes often correspond to the sites also possessing hot spots of DSBs. Figure 6B shows such an example in the centric region located close to p11.2 in chr21. More detailed analysis (Figure 6C) strongly suggests that these regions of DSBs in rDNA units are located in chromatin in close proximity with the region of chr21 located between coordinates 11140000 and 11170000. The distribution of EcoRI sites along this 30-kb stretch in chr21 corresponds to the distribution of contact sites revealed by 4C analysis. The data also suggest the suitability of the method used. Supplementary Figures S12–S14 show the profiles of DSBs along sixteen chromosomes in which the most frequent contacts of rDNA units were detected (the regions shown by the red stars indicate mapping of both the sites of DSBs and rDNA contacts). Some additional examples of good correspondence between the hot spots of DSBs in the centric regions of several chromosomes (chr4, 10, 17, and 20) and the 4C-rDNA contacts are also shown in Supplementary Figure S15 with an accuracy of a few kb, depending on the distance between the site of cross-linking and the EcoRI site involved in 4C analysis. The data argue in favor of the conclusion that the IGS regions possessing hot spots of DSBs in rDNA units are often located in physical proximity to the hot spot sites of DSBs located in different chromosomes.
Figure 6A–C presents the data concerning the properties of genomic regions that contact rDNA units as seen by eye in the UCSC Genome Browser. For statistical testing and for fully informed and thorough data exploration, we performed a genome-wide analysis to study whether the set of intervals corresponding to genomic contacts of rDNA units (4C-rDNA reads) are spatially correlated across the genome with some other set of intervals that describe the distribution of H3K27ac and H3K4me3 marks, DSBs, CTCF binding sites, and ChIA-PET peaks, as well as the locations of SINEs or coding regions. For this study, we used two independent packages, GenometriCorr (Favorov et al., 2012) and Genomic HyperBrowser (Sandve et al., 2010), which led us to the same conclusions. Figure 6D shows the result obtained with the GenometriCorr package. Color-coded density plots in each panel represent the deviation from the expected uniform distribution, generated by Monte-Carlo simulations, and show that in all chromosomes, there is a high positive deviation of rDNA contact regions and the chromosomal sites possessing H3K27ac marks from the expected uniform distribution. The sets of intervals corresponding to the contact regions and H3K27ac marks significantly overlap or are located very close to each other (up to 1.7 kb away). Similarly, regions possessing DSBs often overlap with the contact regions or are located very close to them (up to a distance of 1.5–3 kb).
The data shown in Supplementary Figure S16 also demonstrate that rDNA contact regions overlap with H3K4me3 marks and are often located very close to these marks at around a distance of up to 170 bp. ChIA-PET signals overlap with the contact regions rarely but they are located very close, at a distance not exceeding 2.5–3 kb. In contrast, CTCF binding sites often overlap with the rDNA contact regions, and are also located at a distance not more than 300 bp away. SINEs demonstrate a high correlation with the contact regions only at some intervals of ∼250 bp in length, and then there are periodic regions of high correlation spanning up to 6 kb.
Interesting results were obtained regarding correlation with coding regions. Both approaches suggested that these two sets of intervals (rDNA contacts and coding regions) overlap significantly less than expected by chance. The data on relative distances of the 4C-rDNA set of intervals indicate that there is no overlap of coding regions and rDNA contact regions (Supplementary Figure S16, E, left panel). Nevertheless, there is a region of high positive correlation at a distance of 100 bp (a small region of negative correlation is not seen at the scale shown in the absolute correlation panel). The results obtained by Genomic HyperBrowser are consistent with these results.
It follows that the results initially obtained using the UCSC Genome Browser (Figure 6A–C and Supplementary Figures S9–S11), which suggested that rDNA units often contact chromosomal regions possessing H3K27ac marks, ChIA-PET signals, and DSBs, were clearly confirmed by the genome-wide statistical data.
Interactions inside and between rDNA units
Figure 7 shows the data of a 4C-rDNA experiment for the contacts mapped inside the rDNA unit itself. The mapping results strongly indicate that the reads at three non-anchor EcoRI sites were not the result of partial digestion products or circularization of the 12-kb EcoRI fragment during the 4C procedure. We expected that the results would be distance-dependent and ligation would mainly occur between more closely located EcoRI sites along the rDNA repeats. However, we found that three EcoRI sites contacted the anchor site in an unpredictable fashion. The nearest EcoRI site located inside the promoter region (coordinate 42487 bp) at a distance of 12 kb practically escapes interactions with the anchor site and produced only 95 4C reads. Surprisingly, a more distant EcoRI site located inside 3′ region of the 28S gene at a distance of 18.25 kb from the anchor site produced ∼1 million reads. Finally, the site located at coordinate 5297 inside the 18S gene at 25-kb distance from the anchor produced 21878 reads. The positions of the reads in respect to EcoRI sites suggest the formation of loops inside rDNA units or contacts between them (Figure 7G). The details are described in the legend to Figure 7. In the 18-kb loop, the region at coordinate 30487 (anchor) is located very frequently in close proximity to the terminator region. In the 25-kb loop, the anchor region forms the contact with the 18S gene. A small number of mapped reads suggest the formation of a 12-kb loop that is shaped by the contacts between the anchor region possessing R4 and R5 and the promoter region. At least one interaction site corresponds to the contacts between rDNA units. Reads located to the right of the anchor EcoRI site (coordinate 30487) could originate only from the contacts between different rDNA units at the anchor EcoRI sites. Our 4C data on internal interactions in rDNA units between the region of the IGS enriched with DSBs, coding regions inside 18S and 28S genes, and the promoter region are consistent with the previous 3C data that suggested spatial proximity between the promoter and the coding regions inside the 18S and 28S genes in human rDNA (Denissov et al., 2011).
Figure 7.

Contacts inside and between rDNA units. (A) Vertical bars present the number of mapped 4C-rDNA reads mapped inside rDNA units. Blue bars indicate the regions where reads were mapped in respect to four EcoRI sites inside the rDNA unit. Thin red lines inside IGS show the positions of R1–R9 possessing hot spots of DSBs. The reads at coordinate 5297 were detected only to the left of the EcoRI site suggesting that the contacts involved only the fragment possessing ETS and the 5′ part of 18S gene, but not the region between coordinates 5297 and 12383. We detected only a small number of reads (∼1%) to the left of coordinate 12383, while the rest of them—∼1 million reads—were mapped exclusively to the right of the coordinate, indicating that this particular region at the 3′ end of 28S rDNA gene is very often located in close proximity to the anchor site (shown with a red vertical bar at coordinate 30487). We detected the reads on both sides around the anchor. The reads to the left of coordinate 30487 may result from both the partial digestion products during EcoRI treatment, and from the contacts with the neighboring 18-kb region repairing the site during ligation (see Figure 5A). The reads to the right of coordinate 30487 could appear only from the contacts with the same 12-kb fragment from another rDNA unit. These reads are mapped in two regions coming either from EcoRI and FaeI sites during inverse PCR. We did not observe any reads that potentially could appear after circularization of 12-kb EcoRI fragments possessing the 4C-anchor (to the left of coordinate 42487). The location of a small number of reads only to the right of EcoRI site at coordinate 42487 suggests that a small portion of rDNA units are observed in close proximity to the corresponding region to the anchor site (coordinate 30487). (B) Circos presentation of contacts inside the rDNA unit itself. EcoRI sites are shown by short, green lines. The contacts of the anchor site (shown by the red arrow) were detected inside the 18S and 28S genes, downstream from the anchor site, and in the region of the UCE/CP. (C) Overview of reads at the coordinate 5297. The sizes of EcoRI-FaeI fragments involved in the 4C procedure were 234 bp and 2.8 kb. The asterisk at one FaeI site indicates the diverged sequence. (D) Overview of reads at the coordinate 12383. The sizes of EcoRI-FaeI fragments involved in the 4C procedure were 522 bp and 104 bp. 99% of reads were detected to the right of the EcoRI site. (E) Overview of reads at the anchor EcoRI site (coordinate 30487). A small number of reads located to the left of the EcoRI site correspond to partial digestion products during EcoRI treatment (see Figure 5A). Reads coming from EcoRI and FaeI sites during inverse PCR are seen in two columns (only the top 596 rows are shown). They could originate only from the contacts between the same regions at the anchor EcoRI site from different rDNA units. (F) Overview of reads at the coordinate 42487. The sizes of EcoRI-FaeI fragments involved in the 4C procedure were 1.5 kb and 4.2 kb. Although the size of the latter fragment is rather large, the reads at this coordinate were detected only to the right of the EcoRI site, which strongly indicates that there was practically no circularization of 12-kb fragments (located between coordinates 30487 and 42487) during the 4C procedure. (G) Possible formation of loops inside the rDNA units and between them. Red ovals indicate the cross-linking proteins.
Discussion
Our data strongly indicate that rDNA units belong to the most fragile regions in the human genome and possess at least nine hot spots of DSBs at specific regions of the IGS. The real-time PCR data indicate that up to half of DNA molecules comprising rDNA units are subjected to breakage at a single hot spot in vivo (Figure 4), which means that practically all units should possess the breaks. However, during mapping of DSBs and analysis of epigenetic marks in rDNA (CTCF binding and H3K4me3 marks), we could not discriminate between the signals coming from active and inactive rDNA units, or between the units in the same or different clusters in NORs. Nevertheless, the data on the striking correspondence between CTCF binding sites, H3K4me3 marks, and the pattern of hot spots of DSBs suggest a strong link between the observed fragile sites and transcription regulation in rDNA units.
The data on the high similarity of the CTCF and H3K4me3 profiles of rDNA in additional 12 cell lines, including donor monocytes (Supplementary Figures S17 and S18), suggest a very high consistency of binding sites of insulator binding protein CTCF and of active H3K4me3 marks at rDNA in different cell lines. As in HEK293T cells CTCF and H3K4me3 profiles practically coincide with the profile of DSBs, the data shown in Supplementary Figures S17 and S18 may indicate that similar patterns of DNA breakage at rDNA could also be observed in multiple cell types.
The breakage in rDNA is clearly increased under the replication stress induced by hydroxyurea (Petermann et al., 2010) or by heat-shock treatment, leading to a dramatic inhibition of transcription and termination in active rDNA units (Parker and Bond, 1989). A high level of DSBs in rDNA could also be explained by extensive transcription of rDNA units, and perhaps an incomplete mitotic shutdown of transcription before condensation of the chromosomes. It has been described that adenovirus type 12 infection in human cells induces several fragile sites that may be linked through transcription and that persist into metaphase, thus interfering with chromatin packing and replication, leading to DNA breaks (Li et al., 1998). More detailed analysis revealed that loss of Cockayne syndrome group B protein induces fragility in the same loci suggesting a link between basal transcription apparatus and fragility (Yu et al., 2000).
These facts suggest a mechanistic link between the hot spots of DSBs and transcription and replication in rDNA units, although the particular role of the DNA breaks in these processes remains to be elucidated. It was speculated that hot spots of DSBs reflect the existence of physiological mechanisms for DNA breakage (recombination, transcription, replication, transpositions, and formation of chromosomal structures) (Tchurikov et al., 2013). DSBs could reduce topological stress in DNA caused by binding of protein complexes or RNA molecules and could help formation of open or closed chromatin structures, or particular 3D chromosomal structures, including topological chromatin loops. From this point of view, the existence of specific genomic regions designed for binding of critical regulatory machineries inevitably should lead to the appearance of hot spots of DSBs at these sites or nearby. Our data on the strong link between profiles of insulator protein CTCF-binding sites and H3K4me3 marks with profiles of hot spots of DSBs inside rDNA support this view.
One clue about the possible role of DSBs on the regulation of transcription comes from the data on the binding of PARP1 and HNRNPA2B1 around the hot spots of DSBs. We propose that the breaks could serve as the signals for recruitment of important regulators at particular sites at specific stages of cellular development or the cell cycle. It is known that PARP1 is a potent regulator of gene expression and may play an important role in the establishment of early epigenetic marks during somatic cell reprogramming by regulation of 5-methylcytosine modifications (Doege et al., 2012). Recently, it was demonstrated that PARP1 is involved in the inheritance of silent rDNA chromatin structures by association with TIP5, the large subunit of NoRC, via non-coding pRNA originating from a PolI promoter located 2 kb upstream of the transcription start site, and which binds to the methylated promoter of silent rDNA units and represses transcription (Guetg et al., 2012). Our data indicate that PARP1 is also recruited to sites of DSBs inside the IGS. It is not clear whether PARP1 in these regions is associated with active or inactive rDNA units.
We propose that the H3K4me3 marks of active chromatin in rDNA come from transcribing units located in different rDNA clusters. The hot spots of DSBs are mainly distributed in the regions associated with H3K4me3 and CTCF binding sites. The X-ChIP approach does not allow mapping of the profiles of modified histones more precisely, as cross-linked chromatin DNA is usually sonicated to 300–500 bp to ensure that intra-nucleosomal DNA is not fragmented and that nucleosomes are not lost. Thus, we propose that the hot spots of DSBs are located in inter-nucleosomal DNA at open chromatin regions very close to H3K4me3 marks and CTCF binding sites. PARP1 and HNRNPA2B1, which often bind at hot spots of DSBs (Tchurikov et al., 2013), are probably also located close to the hot spots of DSBs in rDNA units. It is known that PARP1 is important for nucleoli biogenesis in Drosophila, and if PARP1 activity is disrupted, nucleolar proteins that normally colocalize under wild-type conditions disperse into the nucleoplasm and do not show any colocalization (Boamah et al., 2012). Recently, it was shown that, in HEK293T cells, PARP1 represses rRNA transcription via non-coding RNA and is implicated in the formation of silent rDNA chromatin (Guetg et al., 2012). Our data described here and previously (Tchurikov et al., 2013) suggest that this function of PARP1 is mechanistically linked with the hot spots of DSBs located inside the IGS. Independent confirmation of this notion comes from the recent data demonstrating that DNA damage-induced conformation changes underlie the DNA-dependent activation mechanism of human PARP1 (Langelier et al., 2012).
We found that, very often, rDNA contacts in different chromosomes correspond to the PC-HC regions. The ectopic contacts of nucleoli with specific regions in different chromosomes were described in Drosophila (Ananiev and Barsky, 1985). In human cells, nucleoli were found to be associated with the heterochromatin of non-NOR-bearing chromosomes (Boamah et al., 2012). Moreover, it was described that centromeric recruitment establishes allelic exclusion at the Igh locus in B cells (Roldan et al., 2005). More recently, large fragments of genomic DNA (up to 1 Mb or more), isolated with nucleoli preparations, were extensively studied using 2D-FISH analysis and deep sequencing (Nemeth et al., 2010). It was found that pericentromeric and centromeric repetitive sequences are over-represented in nucleoli-associated DNA and that a large part of chr19 associates with the nucleolus. It was also shown that nucleolar-associated domains (NADs), co-purified with nucleoli, associate with nucleoli in a reproducible and heritable manner, possess specific sequences from most human chromosomes, and are characterized by low gene density and a statistically significant enrichment in transcriptionally repressed genes (van Koningsbruggen et al., 2010). The important data on the contacts of rDNA units with pericentromeric and centromeric repetitive sequences are clearly consistent with our observation (Figure 5C, Supplementary Figures S9–S11). However, the data also show several important differences. First, we did not observe that the major part of chr19 is involved in contacts with rDNA. We found the largest number of contact sites in chr1, chr4, chr10, chr14, and chr21 (Supplementary Figure S8). Secondly, although we found that rDNA contact sites did not overlap with coding regions, we also detected a statistically significant correlation between the contact sites and the coding regions at small distances of ∼100 bp (Supplementary Figure S16, E). Thirdly, we observed that rDNA units very often contact with the regions possessing active chromatin marks (CTCF binding sites, H3K4me3, and H3K27ac). The observed differences are probably due to the great difference in the scales used. We used another approach, 4C, which gives a higher resolution depending on the length of the EcoRI-FaeI DNA fragments. As the latter enzyme is a four-base cutter, the resolution is mostly lower than a few kb. However, in the cited papers, NADs were studied using rather large chromosomal DNA fragments up to 1 Mb in length, which were reproducibly co-purified with nucleoli preparations.
We propose that the observed PC-HC contacts are important for regulation of rDNA units. It is not clear yet whether the contacts are required for establishing or maintaining silent or active states of rDNA units, or both. In non-NOR-bearing chr18, we observed frequent contacts of rDNA with both flanks of the centromeric region, but only the q side possesses the active chromatic marks (Supplementary Figure S10). Among the contacts of rDNA with PC-HC regions, we often observed the active chromatin marks. Heterochromatin regions are associated with the repressed state of DNA, but PC-HC also possesses actively expressed genes. It might be that, in future, many more frequent contacts of rDNA units will be detected in the repressed heterochromatin regions composed of compact chromatin fibers (Gilbert et al., 2004) that are absent in the current assemblies of the human genome. The supposition is based on the fact that many of the ‘good’ 4C reads corresponding to contacts of rDNA cannot be mapped at present. It is known that inactive human NORs are indistinguishable from the surrounding heterochromatin (Stults et al., 2008), which is why we do not propose that the currently mapped contacts of rDNA (Figure 6B, C and Supplementary Figures S9–S11) correspond to contacts of active rDNA units.
We think it likely that the detected set of inter-chromosomal contacts of IGS might indicate the existence of specific nuclear compartment(s) formed by chromosomal 3D structures. In our 4C analysis, we could detect only the individual NOR-contacting regions. However, it cannot be excluded that these regions also contact, forming a specific nuclear compartment(s). The number and size of nucleoli vary in different cell types. However, normal human cells have only one nucleolus. It follows that all five NORs could interact forming a single nucleolus with which all chromosomal-contacting regions are associated. Such a structure should form a specific 3D chromosomal ‘clamp’ or compartment holding together rDNA units from five acrocentric chromosomes and the most frequently attached chromosomal regions. The evidence in favor of this comes from isolated nucleoli that have been reproducibly co-purified with DNA containing a specific set of DNA fragments (Nemeth et al., 2010; van Koningsbruggen et al., 2010). In this study, we more precisely delimited NOR-contacting regions and described their specific features. These non-coding regions are often characterized by H3K27ac marks, ChIA-PET signals (regions involved in chromatin interactions), RIP signals (RNA-binding regions), and hot spots of DSBs. It has been described that PARP1 and HNRPA2B1 specifically bind at hot spots of DSBs (Tchurikov et al., 2013). Taken together, these data suggest that such compartments include interacting domains that are regulated by PARP1 and non-coding RNAs. IGS regions possess hot spots of DSBs, PARP1 and CTCF binding sites, and H3K4me3 marks. It has been described that non-coding RNAs transcribed from IGS regions are important for regulation of rDNA units (Mayer et al., 2006). We propose that these regulatory RNAs arise from the sites where hot spots of DSBs and H3K4me3 marks were detected.
The mapping of 4C reads also revealed rDNA contacts inside a unit itself or between the units corresponding to the same or another cluster. From Hi-C data, it is known that regions tend to be closer in space if they belong to the same compartment (Lieberman-Aiden et al., 2009). Nucleoli provide a unique example of a compartment that brings together active copies of rDNA during interphase. During the metaphase, entire NORs are packaged in a form that is as condensed as the surrounding heterochromatin (Stults et al., 2008). In our experiment, we used unsynchronized HEK293T cells, which is why we cannot discriminate the rDNA-rDNA contacts of active or repressed rDNA units.
The comparison of the mapping results of 4C-rDNA and hot spots of DSBs along the 16 chromosomes most frequently contacting rDNA (Figure 5) revealed that the contact regions mostly correspond to chromosomal sites possessing hot spots of DSBs (Supplementary Figures S12–S14). In Figure 6B and C, the overlapping of profiles of DSBs and 4C-rDNA contacts in the centric region of chr21 is shown. We observed a very good correlation between positions of chromosomal breaks, the rDNA contact sites, and the EcoRI sites in the region. Many such examples in different chromosomes were observed. In 10 chromosomes (chr1, 2, 4, 7, 10, 11, 14, 17, 20, and 21), we found the overlapping hot spots of DSBs and rDNA contacts in centric regions (Supplementary Figures S12–S14). That is why we conclude that there is a potential for translocations between hot spots of DSBs in both arrays of rDNA units (chr13, 14, 15, 21, and 22) and different regions of 10 chromosomes (chr1, 2, 4, 7, 10, 11, 14, 17, 20, and 21). As chr14 and chr21 possess NORs, potentially they could translocate their arms. If we assume that there are 70 copies of rDNA units in a cluster (Sakai et al., 1995; Stults et al., 2008) at least 70× ‘molar excess’ of free DNA ends coming from a cluster should be present at frequently occurring DSB sites in the above-mentioned 14 chromosomes. Therefore, one could expect that during non-homologous end joining (NHEJ), translocations could occur involving the long or short arms of 5 acrocentric chromosomes bearing rDNA clusters (chr13, 14, 15, 21, and 22), and one arm from 14 chromosomes possessing hot spots of DSBs at contact sites of rDNA clusters.
The known examples of translocations are in good agreement with our results. For example, the long arm of chr21 is often attached to chr14 or itself, which leads to trisomy 21 and Down syndrome. These translocations correspond to so-called whole-arm or centric-fusion Robertsonian translocations (ROBs). ROBs in humans occur in five chromosomes bearing NORs. As we show here, rDNA units are the most fragile regions in the human genome. The hot spots of DSBs in arrays of rDNA units in five acrocentric chromosomes might provide the molecular basis for ROB. Currently, we are testing this possibility experimentally using NGS.
rDNA fragility could also lead to rDNA copy-number variation, resulting in striking variability between and within human individuals (Stults et al., 2008) because DSBs are potent inducers of homologous recombination. As rDNA consists of tandemly repeated units, the damage may be repaired by recombination with another copy. In this way, the repeat could lose a number of copies between the damaged site and the template copy for repair. It has been shown that in ∼54% of solid tumors, there are rDNA cluster alterations before the start of the clonal tumor expansion (Stults et al., 2009).
Materials and methods
Isolation of DNA fragments at DSBs for Illumina sequencing
DNA preparations possessing spontaneous DSBs were isolated from HEK293T cells in agarose plugs as described previously (Tchurikov and Ponomarenko, 1992; Tchurikov et al., 1998, 2000). About 1.5 µg of isolated DNA was treated with the Klenow fragment of E. coli DNA polymerase I and then ligated at the site of spontaneous DSBs with a molar excess of double-stranded biotinylated oligonucleotide (the details are described in Supplementary Methods). The DNA was then digested with Sau3A to shorten the fragments attached to the ligated oligonucleotide to 100–300 bp. The selection of terminal regions possessing DSB sites was performed using SA-PMP (Promega) according to the manufacturer's recommendations. After extensive washing, the DNA preparation was eluted from the SA-PMP and then ligated with a 100-fold molar excess of double-stranded Sau3A adaptor. The final DNA samples were amplified by PCR.
Chromatin immunoprecipitation
The HEK293T cell suspension was treated with 1% formaldehyde at 20°C for 10 min. The nuclei were washed and lysed, and chromatin was sheared to an average length of 600 bp by sonication. X-ChIP was carried out using the OneDay ChIP kit (Diagenode), with 4 mg of antibodies against PARP1 (ActiveMotif) or HNRNPA2B1 (Sigma Aldrich). The negative control was DNA precipitated using 4 mg of non-specific IgG from rabbit serum. The primers used for PCR across the whole R5 stretch were 5′ CAATGTAACTACTACAGCAAAATGAG 3′ (plus primer) and 5′ CTTCATAGTAACTCCGTAAACTGGAAC 3′ (minus primer). For amplification of the left side of R5, another minus primer was used: 5′ CCACAGGGTTATGACTTCAGAATC 3′, and for amplification of the right side of R5, another plus primer was used: 5′ AACGCAATAAATGTCAACGGTGAG 3′.
4C procedure
DNA samples for 4C experiments were performed according to procedures described previously (Dekker et al., 2002; Osborne et al., 2004). Cells were fixed in 1.5% formaldehyde, and nuclei were isolated, followed by digestion with EcoRI enzyme and ligation of extensively diluted DNA to favor intra-molecular ligations. To shorten the ligation products, digestion with FaeI was performed followed by ligation of diluted DNA samples to favor circularization (Figure 5A). The details are described in Supplementary Methods. The final DNA was used for preparation of a DNA library that was subjected to deep sequencing in MiSeq.
Computer treatments
Raw data were obtained using a Genome Analyzer IIx machine (Illumina). Data were then decoded to FASTQ format using Illumina Casava 1.8 software. Quality evaluation was performed by FastQC 0.10.1 software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Further elimination of primer sequences was performed by cutadapt 1.2.1 software (http://code.google.com/p/cutadapt/) with the assumption that a primer should be at either end of a read. We selected only the sequences possessing at least one copy of a primer that had been cut off. All sequences shorter than 30 bp were removed from the dataset. Then we performed the estimation of read quality by FastQC. The final mapping was performed using BWA 0.7.5a (http://bio-bwa.sourceforge.net) and Samtools 0.1.19 (http://samtools.sourceforge.net) with the human rDNA complete repeating unit (GenBank accession number U13369) and Homo sapiens masked genome (assembly GRCh37p10/hg19) as the database (taken in the form of MFA files from ftp://ftp.ncbi.nih.gov/genomes/H_sapiens/Assembled_chromosomes/seq).
The same procedure was performed for 4C-rDNA data obtained using an Illumina MiSeq machine. Both raw data and the final mappings were submitted to GEO database (http://www.ncbi.nlm.nih.gov/geo) with accession numbers GSE49302 and GSE49193. A circular presentation of 4C data was created by the means of Circos 0.64 software http://www.circos.ca). The details are described in Supplementary Methods.
The mapping of CTCF binding sites, methyl-RRBS and H3K4me3 marks, and DNase I hypersensitive sites inside rDNA was performed as described previously (Zentner et al., 2011). Data sets were aligned to rDNA sequence with Bowtie, allowing two mismatches per read. Prior to alignment, non-unique reads were removed from each FASTQ file. During alignment, reads with more than one reportable alignment were discarded using the ‘-m 1’ option. Peaks were detected with F-seq. The fragment size was set to 200 bp for all analyses except DNase I, for which it was 0.
Statistical analysis
To evaluate the correlations of genome-wide datasets between each other, we used the GenometriCorr R package (Favorov et al., 2012), which combines Jaccard and projection tests both in absolute and relative reference/query distances. To double-check the obtained results, we also performed the tests of close proximity in HyperBrowser (Sandve et al., 2010). The tracks for the comparison were constructed in the following way.
A CDs database (coding regions) was created by parsing GBS files of reference Homo sapiens genome hg19 build GRCh37 patch 13 from NCBI ftp site: ftp://ftp.ncbi.nih.gov/genomes/H_sapiens/Assembled_chromosomes/gbs/. SINEs mapping was taken from the DFAM database ver. 1.2 (http://www.dfam.org). Dfam is a database of repetitive DNA based on profile-hidden Markov models (Wheeler et al., 2013).
We used H3K4me3 peaks for correlation from accession [ENCODE: wgEncodeEH000953 HEK293 H3K4me3 Histone Mod narrowPeak]. We converted two experimental sets (rep1, rep2) into one by calculating the intersection between them (bedtools intersectBed).
We used CTCF peaks for correlation from accession [ENCODE: wgEncodeEH000396 HEK293 CTCF TFBS narrowPeak]. We converted two experimental sets (rep1, rep2) into one by calculating the intersection between them (bedtools intersectBed).
The composite track layered H3K27ac was composed from the following accession numbers [ENCODE: wgEncodeEH000030, wgEncodeEH000997, wgEncodeEH000111, wgEncodeEH000055, wgEncodeEH000043, wgEncodeEH000064, wgEncodeEH000097]. We calculated the union of raw signals from all these subtracks using bedtools (bedtools unionBedGraphs), then calculated the average united signal <S> and its standard deviation σ per chromosome, and finally selected for peaks the data that were higher than <S>+3σ and closer than 1 kb. The resulting bed track was used to perform all genome-wide calculations.
The combined track ChIA-PET was composed from the following ENCODE accession numbers: [ENCODE: wgEncodeEH002075 K562 CTCF ChIA-PET, wgEncodeEH001428 K562 Pol2 ChIA-PET, wgEncodeEH001426 HeLa-S3 Pol2 ChIA-PET, wgEncodeEH002076 MCF-7 CTCF ChIA-PET, wgEncodeEH001430 MCF-7 Pol2 ChIA-PET, wgEncodeEH001427 HCT-116 Pol2 ChIA-PET, wgEncodeEH001431 NB4 Pol2 ChIA-PETMark] in the same way as the previous track.
Data access
The mapping result was deposited into the GEO database with the accession numbers GSE35065, GSE49302, and GSE49193. The reads are presented in .gff and .wig files, which are divided by chromosomes for convenience. The data in the .gff and .wig files are the same, and only the format differs.
Supplementary material
Supplementary material is available at Journal of Molecular Cell Biology online.
Funding
This work was supported by a grant from the Molecular and Cellular Biology Program of the Russian Academy of Sciences and by grants from the Russian Foundation for Basic Research (#12-04-01416-a, #12-04-01311-а, #14-04-01638-a, and #15-04-00299-a), and by a President Grant for Government Support of Young Russian Scientists MK-1934.2014.4.
Conflict of interest: none declared.
Supplementary Material
Acknowledgements
We thank Dr M.A. Gorbacheva (Engelhardt Institute of Molecular Biology, Moscow, Russia) for technical assistance and Dr G.E. Zentner (Fred Hutchinson Cancer Research Center, Seattle, USA) for his help.
References
- Ananiev E.V., Barsky V.E. (1985). Electron Microscopic Map of the Polytene Chromosomes of Drosophila Melanogaster Salivary Glands. Moscow: Nauka. [Google Scholar]
- Boamah E.K., Kotova E., Garabedian M., et al. (2012). Poly(ADP-Ribose) Polymerase 1 (PARP-1) regulates ribosomal biogenesis in Drosophila nucleoli. PLoS Genet. 8, e1002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chechetkin V.R. (2013). Statistics of genome architecture. Phys. Lett. A 377, 3312–3316. [Google Scholar]
- Dekker J., Rippe K., Dekker M., et al. (2002). Capturing chromosome conformation. Science 95, 1306–1311. [DOI] [PubMed] [Google Scholar]
- Denison S.R., Multani A.S., Pathak S., et al. (2002). Fragility in the 14q21q translocation region. Genet. Mol. Biol. 25, 271–276. [Google Scholar]
- Denissov S., Lessard F., Mayer C., et al. (2011). A model for the topology of active ribosomal RNA genes. EMBO Rep. 12, 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doege C.A., Inoue K., Yamashita T., et al. (2012). Early-stage epigenetic modification during somatic cell reprogramming by Parp1 and Tet2. Nature 488, 652–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favorov A., Mularoni L., Cope L.M., et al. (2012). Exploring massive, genome scale datasets with the GenometriCorr package. PLoS Comput. Biol. 8, e1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert N., Boyle S., Fiegler H., et al. (2004). Chromatin architecture of the human genome: gene-rich domains are enriched in open chromatin fibers. Cell 118, 555–566. [DOI] [PubMed] [Google Scholar]
- Gonzalez I.L., Petersen R., Sylvester J.E. (1989). Independent insertion of Alu elements in the human ribosomal spacer and their concerted evolution. Mol. Biol. Evol. 6, 413–423. [DOI] [PubMed] [Google Scholar]
- Gonzalez I.L., Tugendreich S., Hieter P., et al. (1993). Fixation times of retroposons in the ribosomal DNA spacer of human and other primates. Genomics 8, 29–36. [DOI] [PubMed] [Google Scholar]
- Grummt I., Pikaard C.S. (2003). Epigenetic silencing of RNA polymerase I transcription. Nat. Rev. Mol. Cell Biol. 4, 641–649. [DOI] [PubMed] [Google Scholar]
- Guetg C., Scheifele F., Rosenthal F., et al. (2012). Inheritance of silent rDNA chromatin is mediated by PARP1 via noncoding RNA. Mol. Cell 45, 790–800. [DOI] [PubMed] [Google Scholar]
- Holwerda S.J., de Laat W. (2013). CTCF: the protein, the binding partners, the binding sites and their chromatin loops. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20120369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus W.L., Lis J.T. (2003). PARP goes transcription. Cell 113, 677–683. [DOI] [PubMed] [Google Scholar]
- Krishnakumar R., Kraus W.L. (2010). PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol. Cell 39, 736–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langelier M.F., Planck J.L., Roy S., et al. (2012). Structural basis for DNA damage–dependent poly(ADP-ribosyl)ation by human PARP-1. Science 336, 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauberth S.M., Nakayama T., Wu X., et al. (2013). H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell 152, 1021–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Yu A., Weiner A.M. (1998). Adenovirus type 12-induced fragility of the human RNU2 locus requires p53 function. J. Virol. 72, 4183–4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E., van Berkum N.L., Williams L., et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little R.D., Braaten D.C. (1989). Genomic organization of human 5S rDNA and sequence of one tandem repeat. Genomics 4, 376–383. [DOI] [PubMed] [Google Scholar]
- Manuelidis L., Borden J. (1988). Reproducible compartmentalization of individual chromosome domains in human CNS cells revealed by in situ hybridization and three-dimensional reconstruction. Chromosoma 96, 397–410. [DOI] [PubMed] [Google Scholar]
- Mayer C., Schmitz K.M., Li J., et al. (2006). Intergenic transcripts regulate the epigenetic state of rRNA genes. Mol. Cell 22, 351–361. [DOI] [PubMed] [Google Scholar]
- McStay B., Grummt I. (2008). The epigenetics of rRNA genes: from molecular to chromosome biology. Annu. Rev. Cell Dev. Biol. 24, 131–157. [DOI] [PubMed] [Google Scholar]
- Meissner A., Gnirke A., Bell G.W., et al. (2005). Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 33, 5868–5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T. (2010). Higher-order genome organization in human disease. Cold Spring Harb. Perspect. Biol. 2, a000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss T., Boseley P.G., Birnstiel M.L. (1980). More ribosomal spacer sequences from Xenopus laevis. Nucleic Acids Res. 8, 467–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss T., Langlois F., Gagnon-Kugler T., et al. (2007). A housekeeper with power of attorney: the rRNA genes in ribosome biogenesis. Cell. Mol. Life Sci. 64, 29–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth A., Conesa A., Santoyo-Lopez J., et al. (2010). Initial genomics of the human nucleolus. PLoS Genet. 6, e1000889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonechnikov K., Golosova O., Fursov M., et al. (2012). Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28, 1166–1167. [DOI] [PubMed] [Google Scholar]
- Ong C.T., Corces V.G. (2014). CTCF: an architectural protein bridging genome topology and function. Nat. Rev. Gen. 15, 234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne C.S., Chakalova L., Brown K.E., et al. (2004). Active genes dynamically colocalize to shared sites of ongoing transcription. Nat. Genet. 36, 1065–1071. [DOI] [PubMed] [Google Scholar]
- Parker K.A., Bond U. (1989). Analysis of pre-rRNAs in heat-shocked HeLa cells allows identification of the upstream termination site of human polymerase I transcription. Mol. Cell. Biol. 9, 2500–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E., Orta M.L., Issaeva N., et al. (2010). Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell 37, 492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy K.S., Sulcova V. (1998). The mobile nature of acrocentric elements illustrated by three unusual chromosome variants. Hum. Genet. 102, 653–662. [DOI] [PubMed] [Google Scholar]
- Roldan E., Fuxa M., Chong W., et al. (2005). Locus ‘decontraction’ and centromeric recruitment contribute to allelic exclusion of the immunoglobulin heavy-chain gene. Nat. Immunol. 6, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K., Ohta T., Minoshima S., et al. (1995). Human ribosomal RNA gene cluster: identification of the proximal end containing a novel tandem repeat sequence. Genomics 26, 521–526. [DOI] [PubMed] [Google Scholar]
- Sandve G.K., Gundersen S., Rydbeck H., et al. (2010). The Genomic HyperBrowser: inferential genomics at the sequence level. Genome Biol. 11, R121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro R. (2005). The silence of the ribosomal RNA genes. Cell. Mol. Life Sci. 62, 2067–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro R., Grummt I. (2005). Epigenetic mechanisms of rRNA silencing: temporal order of NoRC recruitment, histone modifications, chromatin remodeling and DNA methylation. Mol. Cell. Biol. 25, 2539–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro R., Li J., Grummt I. (2002). The nucleolar remodeling complex NoRC mediates heterochromatin formation and silencing of ribosomal gene transcription. Nat. Genet. 32, 393–396. [DOI] [PubMed] [Google Scholar]
- Stults D.M., Killen M.W., Pierce H.H., et al. (2008). Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res. 18, 13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stults D.M., Killen M.W., Williamson E.P., et al. (2009). Human rRNA gene clusters are recombinational hotspots in cancer. Cancer Res. 69, 9096–9104. [DOI] [PubMed] [Google Scholar]
- Tchurikov N.A., Ponomarenko N.A. (1992). Detection of DNA domains in Drosophila, human and plant chromosomes possessing mainly 50- to 150-kilobase stretches of DNA. Proc. Natl Acad. Sci. USA 89, 6751–6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchurikov N.A., Krasnov A.N., Ponomarenko N.A., et al. (1998). Forum domain in Drosophila melanogaster cut locus possesses looped domains inside. Nucleic Acids Res. 26, 3221–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchurikov N.A., Kretova O.V., Chernov B.K., et al. (2000). SuUR protein binds to the boundary regions separating forum domains in Drosophila melanogaster J. Biol. Chem. 279, 11705–11710. [DOI] [PubMed] [Google Scholar]
- Tchurikov N.A., Kretova O.V., Sosin D.V., et al. (2011). Genome-wide profiling of forum domains in Drosophila melanogaster. Nucleic Acids Res. 39, 3667–3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchurikov N.A., Kretova O.V., Fedoseeva D.M., et al. (2013). DNA double-strand breaks coupled with PARP1 and HNRNPA2B1 binding sites flank coordinately expressed domains in human chromosomes. PLoS Genet. 9, e1003429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therman E., Susman B., Denniston C. (1989). The nonrandom participation of human acrocentric chromosomes in Robertsonian translocations. Ann. Hum. Genet. 53, 49–65. [DOI] [PubMed] [Google Scholar]
- Treangen T.J., Salzberg S.L. (2011). Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 13, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Koningsbruggen S., Gierlinski M., Schofield P., et al. (2010). High-resolution whole-genome sequencing reveals that specific chromatin domains from most human chromosomes associate with nucleoli. Mol. Biol. Cell 21, 3735–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters E.R., Schaal B.A. (1996). Heat shock induces a loss of rRNA-encoding DNA repeats in Brassica nigra. Proc. Natl Acad. Sci. USA 93, 1449–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler T.J., Clements J., Eddy S.R., et al. (2013). Dfam: a database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Res. 41, D70–D82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worton R.G., Sutherland J., Sylvester J.E., et al. (1988). Human ribosomal RNA genes: orientation of the tandem array and conservation of the 5′ end. Science 239, 64–68. [DOI] [PubMed] [Google Scholar]
- Yu A., Fan H.Y., Liao D., et al. (2000). Activation of p53 or loss of the Cockayne syndrome group B repair protein causes metaphase fragility of human U1, U2, and 5S genes. Mol. Cell 5, 801–810. [DOI] [PubMed] [Google Scholar]
- Zentner G.E., Saiakhova A., Manaenkov P., et al. (2011). Integrative genomic analysis of human ribosomal DNA. Nucleic Acids Res. 39, 4949–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Santoro R., Grummt I. (2002). The chromatin remodeling complex NoRC targets HDAC1 to the ribosomal gene promoter and represses RNA polymerase I transcription. EMBO J. 21, 4632–4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.