

Abstract
Nine fluorine-containing vesicular acetylcholine transporter (VAChT) inhibitors were synthesized and screened as potential PET tracers for imaging the VAChT. Compound 18a was one of the most promising carbonyl-containing benzovesamicol analogues; the minus enantiomer, (-)-18a displayed high potency (VAChT Ki = 0.59 ± 0.06 nM) and high selectivity for VAChT versus receptors (> 10,000-fold). The radiosynthesis of (-)-[18F]18a was accomplished by a two-step procedure with 30 – 40% radiochemical yield. Preliminary biodistribution studies of (-)-[18F]18a in adult male Sprague–Dawley rats at 5, 30, 60 and 120 min post-injection (p.i.) were promising. The total brain uptake of (-)-[18F]18a was 0.684 ID%/g at 5 min p.i. and by 120 min p.i. slowly washed out to 0.409 %ID/g.; evaluation of regional brain uptake showed stable levels of ~0.800 %ID/g from 5 to 120 min p.i in the VAChT-enriched striatal tissue of rats, indicating the tracer had crossed the blood brain barrier and was retained in the striatum. Subsequent microPET brain imaging studies of (-)-[18F]18a in nonhuman primates (NHPs) showed high striatal accumulation in the NHP brain; the standardized uptake value (SUV) for striatum reached a maximum value of 5.1 at 15 min p.i. The time-activity curve for the target striatal region displayed a slow and gradual decreasing trend 15 min after injection, while clearance of the radioactivity from the cerebellar reference region was much more rapid. Pretreatment of NHPs with 0.25 mg/kg of the VAChT inhibitor (-)-vesamicol resulted in a ~90% decrease of striatal uptake compared to baseline studies. HPLC metabolite analysis of NHP plasma revealed that (-)-[18F]18a had a good in vivo stability. Together, these preliminary results suggest (-)-[18F]18a is a promising PET tracer candidate for imaging VAChT in the brain of living subjects.
Keywords: VAChT, PET tracer, Vesamicol, F-18, Striatum
Graphical Abstract

1. Introduction
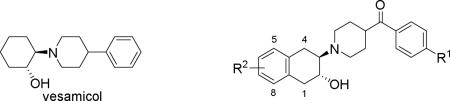
The loss or decline in memory and other cognitive abilities is a clinical syndrome characteristic of dementia which is common seen in Alzheimer disease (AD), Parkinson Disease (PD), and other neurodegenerative diseases.[1-6] The severity of dementia is linked to loss of cholinergic neurons and synapses in the central nervous system (CNS). Identifying a reliable biomarker that is able to assess the loss of the cholinergic neuron is imperative. The vesicular acetylcholine transporter (VAChT) is an important transporter protein in the neuronal cholinergic system and has been accepted as a reliable cholinergic marker for more than 25 years.[7-10] Positron emission tomography (PET) is a non-invasive imaging modality and able to provide functional information for molecular and cellular processes in living subjects. A PET radioligand having high affinity and selectivity for VAChT, and suitable radiopharmaceutical properties would provide a unique tool to measure the loss of cholinergic neurons in the brain of subjects with dementia by assessing changes in VAChT level. The identification of a potent and selective PET ligand with minimal pharmacological effects has posed significant difficulties. Vesamicol (1, initially named AH 5183, Figure 1) was first reported in 1969 [11, 12] as a potential local anesthetic. Bahr and Parsons reported that L-isomer and D-isomer of vesamicol had a different binding activity in Torpedo synaptic vesicles in 1986; vesamicol was later discovered to be a stereoselective inhibitor of VAChT.[13-15] Although tritiated vesamicol has been widely used for in vitro studies of cholinergic loss, vesamicol and many analogues have significant binding affinity for receptors that limits their use as VAChT imaging agents because receptors are highly expressed throughout the brain.[16-18] To overcome this challenge, investigators have put tremendous efforts into exploring vesamicol derivatives to improve the imaging of the VAChT binding and to reduce the σ receptor binding affinity. Identification of benzovesamicol (2, BVM, Figure 1) was a significant achievement, as this structural modification not only retained the high VAChT affinity, but also offered position(s) on the aromatic ring in the tetralin fragment for introducing substitution group(s).[19-22] Among the benzovesamicol analogues shown in Figure 1, 5-(N-methyl-amino)benzovesamicol (3, MABV) and N-ethyl-fluoroacetamidobenzovesamicol (4, NEFA) are more potent inhibitors and more selective for VAChT versus σ receptors than vesamicol.[23] Furthermore, the single-photon emission computerized tomography (SPECT) radiotracer (-)-5-[123I]iodo-benzovesamicol ([123I]5, [123I]IBVM) was used for mapping cholinergic terminals in the human brain [7-9, 24-27] although quantification of peak uptake in the target regions required at least 6 hr post-injection (p.i.). Although SPECT is a useful tool for studies of ligands with slow kinetic processes,[28] PET offers higher spatial resolution and sensitivity for clinical studies of ligands with more rapid pharmacokinetics. [18F]fluoroethoxy-benzovesamicol ([18F]6, [18F]FEOBV) was recently approved for the clinical studies in human subjects as a PET ligand for assessing the VAChT levels in the brain as a biomarker of cholinergic function.[29-32] Although equilibrium kinetics in the brain of both NHP and human subjects show delayed equilibrium of > 360 min p.i.,[32] [18F]FEOBV offers advantages over SPECT ligands for both preclinical and clinical imaging of cholinergic loss. Studies of FEOBV in a rat model of cholinergic dysfunction showed a good correlation between PET measures of [18F]FEOBV uptake and ex vivo autoradiography of the same animals post PET; ex vivo brain autoradiography measures of [18F]FEOBV binding in human AD cases and age-matched controls also showed differences consistent with cholinergic loss.[33, 34]
Figure 1.

Vesamicol, BVM and their derivatives.
We previously reported a new class of VAChT analogues in which a carbonyl group is interposed between the phenyl and piperidine ring of the benzovesamicol structure; several lead candidates displayed high in vitro binding affinity and high selectivity for VAChT versus other CNS targets.[35-40] Evaluation of these 11C- and 18F-labeled radiotracers in rodents and microPET imaging studies in NHPs suggested that the radiolabeled version of several ligands (7-10, Figure 1) bound in vivo to the VAChT enriched striatal regions.[38-41] The half-life of 18F (T1/2 = 109.8 min) permits longer scan sessions that generate higher target-to-reference ratios; 18F PET tracers also place fewer time constraints on tracer production. Here we further explore carbonyl-containing benzovesamicol analogues using the following strategies: 1) Incorporating PEGylated groups on the phenyl ring linked to the piperidinyl ring by the carbonyl group (Figure 1), which can improve clearance kinetics by decreasing lipophilicity;[42-44] 2) Introducing a fluoroethoxy group on the tetralin moiety to further improve affinity and selectivity for VAChT versus the σ receptors; 3) Incorporating a fluorine atom to provide position(s) for 18F radiolabeling. In this manuscript, we report the synthesis and in vitro and in vivo biological evaluation of the new fluorinated carbonyl-containing analogues to determine their suitability as 18F labeled ligands for imaging VAChT in vivo.
2. Results and Discussion
2.1 Chemistry
The syntheses of the PEGylated compounds 12 – 14 are shown in Scheme 1. Intermediate 11 was synthesized as previously reported.[40] Fluoroethoxy-containing 12 was obtained by O-alkylation of 11 with 2-bromo-1-fluoroethane. PEGylated analogues 13 and 14 were obtained by treating 11 with the corresponding fluorinated PEGylated tosylates. The syntheses of benzovesamicol analogues containing a 5-substituted carbonyl group are shown in Scheme 2. The epoxide compound 15 was synthesized as previously described.[36, 38] Commercially available (4-fluorophenyl)(piperidin-4-yl)methanone (16) was reacted with 15 to afford the phenol regioisomers 17a and 17b. The desired ligands 18a and 18b were obtained via O-alkylation of 17a and 17b respectively with -2-fluoroethyl tosylate.
Scheme 1.

Syntheses of compounds 12 - 14
Scheme 2.

Syntheses of compounds 18a and 18b
Since VAChT inhibitors demonstrate stereoselective binding, we initially attempted to resolve the potent racemic compound (±)-18a directly using a Chiralcel OD column without success. To overcome this challenge, racemic compound (±)-18a was first converted to the acetate ester (±)-19a, which was easily resolved on Chiralcel OD column using a mobile phase of 3% 2-propanol in hexanes, as shown in Scheme 3. Using a semi-preparative Chiralcel OD column, the minus isomer (-)-19a eluted at 25-29 min and the plus isomer (+)-19a eluted at 30-38 min. Hydrolysis of the enantiomers (-)-19a and (+)-19a using saturated sodium carbonate aqueous solution afforded the desired enantiomers (-)-18a and (+)-18a.
Scheme 3.

Chiral separation of (±)-18a
2.2 In vitro binding affinity studies
The newly synthesized analogues were screened using our standard in vitro binding assays [36, 37, 40, 45] to measure their affinities to VAChT and σ receptors (Table 1). [3H]vesamicol was used as the radioligand for the VAChT competitive binding assay; [3H]pentazocine and [3H]ditolylguanidine([3H]DTG) in the presence of 1 μM (+)-pentazocine were used for σ1 and σ2 receptor binding assays respectively. PEGylated compounds 12-14 displayed similar binding affinities for VAChT (0.87 to 1.74 nM), while the selectivity for VAChT over both σ1 and σ2 receptors was greatly increased by extending the PEG chain: Ki-σ1 values were 13.23 ± 0.20, 1,580 ± 46, and > 10,000 nM for compounds 12, 13, and 14 respectively. In comparison of compounds 17a to 17b, and 18a to 18b, the hydroxyl or fluoroethoxy substitution on the 8-position resulted in > 10-fold decrease of VAChT affinity with the Ki values of 4.64 ± 0.32, 56.20 ± 4.08 nM for 17a and 17b, 1.55 ± 0.18, 92.80 ± 22.20 nM for 18a and 18b respectively; the observed stereoselectivity for VAChT is consistent with previous reports.[46, 47] Enantiopure (-)-18a was more potent than (+)-18a with Ki values of 0.59 ± 0.06, 13.0 ± 2.20 nM for (-)-18a and (+)-18a respectively. The results of these in vitro binding studies suggested that: 1) PEGylation not only reduces the lipophilicity, but also improves selectivity for VAChT versus σ receptors; 2) the 5-substitution of the tetralin moiety provides higher binding affinity for VAChT than the non-substituted compound 10 or the 8-substituted counterpart; the observation that 5-position substituted compounds demonstrated high potency is consistent with literature reports;[46, 47] 3) VAChT binding has stereoselectivity. For racemic compound 18a, the minus isomer (-)-18a is more potent than its plus isomer (+)-18a. Enantiopure (-)-18a displayed high VAChT binding affinity (0.59 ± 0.06 nM) and high selectivity versus σ receptors (>10,000-fold), and acceptable lipophilicity, with calculated LogP value of 3.45. Compound (-)-18a was chosen for radiolabeling with 18F and for further evaluation in rodents and nonhuman primates.
Table 1.
Binding affinities of the fluoro-containing VAChT inhibitors
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Ligands | R1 | R2 | Ki-VAChT | Ki-σ1 | Ki-σ2 | VAChT/ σ1 | VAChT/ σ2 | Log P*a |
| Vesamicol*b | 20.00 | 73.80 | 346.0 | 3.69 | 17.30 | 1.40 | ||
| FBBV*c | H | H | 2.70 ± 0.40 | 191.1 ± 57.7 | 251.3 ± 39.2 | 71 | 93 | 2.99 |
| 12 | FCH2CH2O | H | 1.74 ± 0.32 | 13.23 ± 0.20 | 1,885 ± 239 | 7.60 | 1083 | 3.15 |
| 13 | F(CH2CH2O)2 | H | 0.87 ± 0.09 | 1,580 ± 46 | 8,200 ± 97 | 1800 | 9300 | 2.79 |
| 14 | F(CH2CH2O)3 | H | 1.23 ± 0.09 | >10,000 | >10,000 | >8,000 | >9,000 | 2.43 |
| 17a | F | 5-OH | 4.64 ± 0.32 | 8,640 ± 974 | 1,851 ± 408 | 1,862 | 398.0 | 2.63 |
| 17b | F | 8-OH | 56.20 ± 4.08 | 28.70 ± 4.10 | 2,600 ± 178 | 0.51 | 46.30 | 2.59 |
| 18a | F | 5-OCH2CH2F | 1.55 ± 0.18 | 821 ± 34 | >10,000 | 529 | >6000 | 3.45 |
| (+)-18a | F | 5-OCH2CH2F | 13.00 ± 2.19 | >10,000 | 7,800 ± 882 | 1022 | 600.0 | 3.45 |
| (−)-18a | F | 5-OCH2CH2F | 0.59 ± 0.06 | >10,000 | >10,000 | >100,00 | >10,000 | 3.45 |
| 18b | F | 8-OCH2CH2F | 92.80 ± 22.20 | >10,000 | 7,145 ± 549 | 667.0 | 77.00 | 3.47 |
Calculated value at pH = 7.4 by ACD/I-Lab ver. 7.0 (Advanced Chemistry Development, Inc., Canada).
Reference 37.
Reference 36.
2.3 Radiochemistry
The radiosynthesis of (-)-[18F]18a was accomplished by a two-step 18F labeling strategy starting with (-)-17a as shown in Scheme 4. Ethylene ditosylate was first reacted with [18F]KF/Kryptofix 2.2.2 in acetonitrile and then purified on a reversed phase HPLC system to afford [18F]fluoroethyl tosylate ([18F]20) with a 60-75% radiochemical yield. Nucleophilic substitution of (-)-17a with [18F]20 in dimethyl sulfoxide (DMSO) followed by HPLC purification afforded (-)-[18F]18a in 50-60% radiochemical yield (decay corrected to end of synthesis, n > 20). The radioactive (-)-[18F]18a was authenticated by co-injection with the nonradiolabeled standard (-)-18a on an analytical HPLC system. The two-step radiosynthesis of (-)-[18F]18a took ~ 2.0 hr with overall radiochemical yield of 30 to 40%, specific activity > 74 GBq/μmol (decay corrected to EOS) and radiochemical purity > 98% (n > 20).
Scheme 4.

Radiosynthesis of (-)-[18F]18a
2.4 Ex vivo autoradiography study in rats
To confirm the target binding selectivity of (-)-[18F]18a in the rat brain, ex vivo autoradiography was performed. An adult male Sprague-Dawley (SD) rat was euthanized 60 min p.i. The brain was quickly removed and snap-frozen then sectioned at 100 μm and mounted on glass slides. The brain sections were exposed to the film in an imaging cassette for 12 hr; the distribution of radioactivity is shown in Figure 2 with the outlined regions of interest based on standard anatomical guides.[48] The results show (-)-[18F]18a has the highest accumulation in the VAChT enriched striatum, which is consistent with the literature reports.[49] The uptake ratio of radioactivity in striatum versus cerebellum reached 4.19 ± 0.37 at 60 min p.i.
Figure 2.

Autoradiography of (-)-[18F]18a in the male Sprague-Dawley (SD) rat brain. A). The representative horizontal slice (100 μm thick) of the SD rat brain shows the highest accumulation of radioactivity in the VAChT enriched striatal regions. B). Quantification of autoradiography from entire 30 brain slices indicates a striatum-to-cerebellum ratio of 4.19 ± 0.37 at 60 min post-injection. Histograms are the average of relative radioactivity of 30 brain slices. Error bars are the standard derivations.
2.5 Biodistribution studies in rats
Biodistribution and regional brain uptake studies of (-)-[18F]18a were performed in adult male SD rats at 5, 30, 60, and 120 min p.i.; the results are shown in Table 2. The initial uptake (%ID/g) at 5 min p.i. in blood, heart, lung, muscle, fat, pancreas, spleen, kidney, liver, and brain was 0.209, 0.706, 2.255, 0.171, 0.144, 1.202, 1.265, 2.363, 1.065 and 0.684 respectively. The clearance of the radioactivity was rapid from the heart, lung, spleen, kidney, and liver; the uptake (ID%/g) decreased to 0.390, 0.973, 0.685, 0.883, and 0.523, respectively, at 30 min and further decreased at 60 and 120 min p.i. The high initial kidney uptake suggests the radiotracer may be initially excreted through the urinary system. From 5 to 120 min, the bone uptake of the radioactivity remained stable, suggesting that there is no significant metabolic defluorination in vivo. For brain regions of interest, the uptake (ID%/g) of the cerebellum, brain stem, cortex, striatum, thalamus, and hippocampus was 0.593, 0.683, 0.777, 0.782, 0.691, and 0.645 respectively at 5 min post injection of (-)-[18F]18a; the initial uptake of the striatal region was a little higher than other brain regions but no significant difference was observed, as shown in Table 2 and Figure 3. However, the striatal region retained the highest levels (%ID/g) at 0.886, 0.819, and 0.761 at 30, 60 and 120 min, respectively, in contrast with other brain regions. The uptake ratios for target to non-target brain regions (striatum-to-cerebellum) increased from 1.3-fold at 5 min to 3.0-fold at 30 min and remained steady up to 120 min p.i. as shown in Figure 3.
Table 2.
Biodistribution of (−)-[18F]18a in male Sprague-Dawley ratsa
| Organs | 5 min | 30 min | 60 min | 120 min |
|---|---|---|---|---|
| Blood | 0.209 ± 0.029 | 0.270 ± 0.033 | 0.338 ± 0.088 | 0.356 ± 0.019 |
| Heart | 0.706 ± 0.170 | 0.390 ± 0.031 | 0.357 ± 0.068 | 0.336 ± 0.016 |
| Lung | 2.255 ± 0.415 | 0.973 ± 0.107 | 0.738 ± 0.087 | 0.551 ± 0.070 |
| Muscle | 0.171 ± 0.055 | 0.258 ± 0.025 | 0.253 ± 0.044 | 0.235 ± 0.010 |
| Fat | 0.144 ± 0.054 | 0.324 ± 0.071 | 0.548 ± 0.354 | 0.560 ± 0.135 |
| Pancreas | 1.202 ± 0.323 | 1.200 ± 0.347 | 1.324 ± 0.346 | 0.819 ± 0.367 |
| Spleen | 1.265 ± 0.452 | 0.685 ± 0.082 | 0.505 ± 0.069 | 0.376 ± 0.034 |
| Kidney | 2.363 ± 0.520 | 0.883 ± 0.068 | 0.604 ± 0.092 | 0.489 ± 0.052 |
| Liver | 1.065 ± 0.121 | 0.523 ± 0.044 | 0.456 ± 0.122 | 0.382 ± 0.029 |
| Bone | 0.426 ± 0.086 | 0.369 ± 0.020 | 0.403 ± 0.095 | 0.537 ± 0.035 |
| Total brain | 0.684 ± 0.079 | 0.482 ± 0.037 | 0.425 ± 0.075 | 0.409 ± 0.016 |
%ID/g values (mean ± SD) with 4 rats per group
Figure 3.

Regional brain uptake and striatum/organ ratio of (-)-[18F]18a in male SD rats. Both figures showed the radioactivity retention was the highest in the striatum at 30, 60 and 120 min p.i., while the radioactivity was quickly washed out in other brain regions. CBL: cerebellum; STR, striatum; BSTM, brain stem; CTX, cortex; THA, thalamus.
2.6 MicroPET imaging studies in NHPs
Following the promising data from rat ex vivo autoradiography and biodistribution studies, microPET imaging of (-)-[18F]18a in the brains of male cynomolgus monkeys was used to determine its potential as a PET tracer for imaging the VAChT in vivo. As shown in Figure 4, the microPET imaging results suggested that (-)-[18F]18a was able to enter the NHP brain and had the highest accumulation in the striatal region including both caudate and putamen. This high accumulation of radioactivity in the striatal region further indicates that (-)-[18F]18a binds to VAChT in vivo. The time-activity curves showed that the radioactivity accumulation in striatum reached maximum with a standardized uptake value (SUV) of 5.1 at 15 min p.i. and decreased slowly over 120 min p.i. Compared with [18F]FEOBV which displayed slow striatal kinetics, (-)-[18F]18a demonstrated a much faster equilibrium for striatal binding in NHP. The uptake ratio of the target (striatum) versus non-target (cerebellum) increased gradually and reached ~3.0 at 80 min p.i. To test the specificity of (-)-[18F]18a binding to VAChT in the brain, microPET studies were performed in a monkey pretreated with 0.25 mg/kg (-)-vesamicol, a known VAChT inhibitor, 5 min before (-)-[18F]18a injection. Striatal uptake in this blocking study was reduced about ~90% compared to the striatal uptake in the baseline condition (Figure 4), while the change in the cerebellar region was negligible (Figure 4); this further suggested that (-)-[18F]18a binds to VAChT specifically in the monkey brain.
Figure 4.

Representative microPET images of (-)-[18F]18a in cynomolgus monkey brain. Upper panel: PET image (left) at baseline condition and PET image, pretreated the animals using 0.25 mg/kg (-)-vesamicol 5 min prior to injection of the radiotracer (right). High uptake of (-)-[18F]18a in striatum produced clear visualization of striatum (putamen and caudate) in the baseline condition. The pretreatment using 0.25 mg/kg (-)-vesamicol significantly reduced the uptake of (-)-[18F]18a in the striatum (right). Lower panel: time tissue-activity curves for cerebellum (red open circles), striatum (red open triangles) at baseline condition, cerebellum (blue filled circles), and striatum (blue filled triangles) at the condition that animal was pretreated using 0.25 mg/kg (-)-vesamicol. SUV: standardized uptake value.
2.7 Metabolite studies in NHP plasma
Because the microPET imaging data suggested that (-)-[18F]18a was a promising candidate VAChT-specific PET radiotracer, radioactive metabolite analysis was performed in NHP plasma samples post injection of (-)-[18F]18a. 70 - 82% of the radioactivity was in the plasma after centrifugation to separate the red blood cells. Following solvent extraction and deproteination, 76 - 87% of the radioactivity in plasma was in the supernatant. The solvent extract was then injected onto HPLC to identify the percentage of radioactive parent compound versus any radiolabeled metabolites. The HPLC metabolite analysis of the plasma samples showed three radioactive peaks: the parent compound (-)-[18F]18a, radioactive metabolite 1 and radioactive metabolite 2; the retention times were 9 – 11 min for (-)-[18F]18a, 2 – 4 min for the major hydrophilic radioactive metabolite 1 and 6 – 7 min for the radioactive metabolite 2 which represented < 1.5% of the total radioactivity collected from the HPLC and is considered negligible. The percentage of the remaining parent compound (-)-[18F]18a was 66, 41, 21, and 16% at 15, 30, 60 and 90 min p.i. respectively. The level of radioactive metabolite 1 was 3.0, 30, 59, 78 and 82% at 5, 15, 30, 60 and 90 min respectively as shown in Figure 5. To test if the hydrophilic radioactive metabolite 1 has capability in penetrating the blood brain barrier and thus confound PET measurement, we evaluated the stability of the tracer in rat blood and brain. The radiometabolite analysis of the rat plasma samples revealed that (-)-[18F]18a in rat metabolized much faster than in monkeys, indicating the metabolism of (-)-[18F]18a has a species difference. This limits the utility of rodent data in predicting the metabolism of (-)-[18F]18a in nonhuman primates or human subjects. Future metabolite analysis of the human plasma samples, kinetic modeling of tracer uptake, and clearance in human subjects are necessary to determine if the major radioactive metabolite can enter into the human brain.
Figure 5.

Radiometabolite analysis of NHP arterial plasma samples post injection of (-)-[18F]18a. The black diamonds represent the percentage intact radioactive parent compound at 5, 15, 30, 60, 90, and 120 min p.i.
3. Experimental
3.1 General
All the solvents and reagents were obtained commercially and used as received, unless otherwise stated. All anhydrous reactions were carried out in oven-dried or flame-dried, and nitrogen purged glassware. Anhydrous tetrahydrofuran was dried over sodium and fresh distilled prior to use. Anhydrous dichloromethane was distilled over calcium hydride prior to use. Reactions were monitored by thin layer chromatography (TLC) using silica gel 60 F254 glass plates (EMD Chemicals Inc.). Flash column chromatography was performed over silica gel (32-63 μm), HPLC grade solvents were used for chromatography. 1H NMR were recorded on a Varian Mercury-VX 400 or 300 MHz spectrometer. The chemical shifts are reported as values (ppm) relative to tetramethylsilane (TMS) as an internal reference. The SpectraSystem was used for both analytical and semi-preparative HPLC. A Chiralcel OD normal phase HPLC column was used to resolve enantiomers. The specific optical rotation was determined on an automatic polarimeter (Autopol 111, Rudolph Research, Flanders, NJ). Elemental analyses were determined by Atlantic Microlab, Inc. (Norcross, GA).
[18F]Fluoride was produced from a RDS111 cyclotron (Siemens/CTI Molecular Imaging, Knoxville, TN) by 18O(p, n)18F reaction through proton irradiation of enriched 18O water (95%). [18F]Fluoride was firstly passed through an ion-exchange resin and then eluted using 0.02 M potassium carbonate (K2CO3) solution.
3.1.1 (4-(2-Fluoroethoxy)phenyl)(1-(3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)methanone (12)
To a solution of 11 (30 mg, 0.08 mmol) in acetone (10 mL) was added K2CO3 (12 mg, 0.08 mmol) and 2-bromo-1-fluoroethane (54 mg, 0.2 mmol). The reaction mixture was refluxed overnight until the reaction was completed as determined by TLC. After the solvent was removed, 20 mL of water was added to the flask, and the mixture was extracted with ethyl acetate (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography using ethyl acetate/hexane(1/4, v/v) to give compound 12 (26.1 mg, 77%) as a pale yellow solid. 1HNMR (400 MHz, CDCl3) δ 1.76–1.95 (m, 4 H), 3.88 (dt, J = 2.7, 12.4 Hz, 1 H), 2.75–3.02 (m, 7 H), 3.24-3.35 (m, 2 H), 3.88 (td, J = 5.0, 13.8 Hz, 1 H), 4.05 (t, J = 4.0 Hz, 1 H). 4.33 (t, J = 4.2 Hz, 1 H). 4.71 (t, J = 4.0 Hz, 1 H), 4.87 (t, J = 4.2 Hz, 1 H), 6.99 (d, J = 8.4 Hz, 2 H), 7.09–7.16 (m, 4 H), 7.95 (d, J = 9.0 Hz, 2 H). 13C NMR (75.5 MHz, CD3OD) δ 201.4, 165.9, 162.4, 136.0, 135.0, 131.0, 129.1, 126.2, 116.6, 115.0, 108.6, 82.5 (J = 167.2 Hz), 68.0, 66.3 (d, J = 23.2 Hz), 50.3, 47.0, 43.3, 38.4, 31.2, 29.7, 28.0, 24.3; HRMS (ESI) Calcd for C24H29FNO3 (M+H)+ 398.2131, found: 398.2129. To 26 mg (0.065 mmol) of 12 in a glass vial was added in 2.5 mL of dichloromethane, and 2.94 mg (0.0325 mmol) of oxalic acid in methanol was added. After recrystallization, 31.4 mg of the oxalate salt was obtained, Mp: 196.2 – 197.0 °C.
3.1.2 (4-(2-(2-Fluoroethoxy)ethoxy)phenyl)(1-((2R,3R)-3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)-methanone (13)
To a solution of 11 (40 mg, 0.11 mmol) in 2.0 mL DMF was added 2-(2-fluoroethoxy)ethyl tosylate (60 mg, 0.22 mmol) and K2CO3 (35 mg, 0.25 mmol). The mixture was stirred at room temperature overnight. Ethyl acetate (35 mL) was added into the reaction mixture, and then sequentially washed with saturated NaHCO3 aqueous solution and brine. The combined organic solution was dried over anhydrous sodium sulfate, concentrated under reduced pressure. The crude product was purified on a silica gel column to give compound 13 (38 mg, 78%) as colorless syrup. 1HNMR (400 MHz, CDCl3) δ 7.94 (d, J=8.7 Hz, 2H), 7.12 (s br, 4H), 6.98 (d, J=8.4 Hz, 2H), 4.70-4.65 (m, 1H), 4.55-4.49 (m, 1H), 4.25-4.20 (m, 2H), 4.00-3.75 (m, 5H), 3.40-3.10 (m, 2H), 3.10-2.70 (m, 7H), 2.5-2.30 (m, 1H), 2.00-1.70 (m, 4H). The oxalate salt was obtained as described above. Mp: 203 – 206 °C. Anal. Calcd of C28H34FNO8: C, 63.27; H, 6.45; N, 2.63. Found: C, 63.29; H, 6.49; N, 2.79. HRMS (ESI) Calcd for C26H33FNO4 (M+H)+ 442.2394, found: 442.2392.
3.1.3 (4-(2-(2-(2-Fluoroethoxy)ethoxy)ethoxy)phenyl)(1-((2R,-3R)-3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)methanone (14)
Following the similar procedure for preparing compound 13, compound 14 was afforded as colorless oil (41.6 mg, 41%). 1HNMR (400 MHz, CDCl3) δ 7.93 (d, J=9.0 Hz, 2H), 7.11 (s br, 4H), 6.97 (d, J=9.0 Hz, 2H), 4.70-4.63 (m, 1H), 4.55-4.45 (m, 1H), 4.25-4.18 (m, 3H), 4.00-3.80 (m, 3H), 3.80-3.70 (m, 6H), 3.40-3.20 (m, 2H), 3.10-2.70 (m, 7H), 2.5-2.30 (m, 1H), 2.00-1.70 (m, 4H). The oxalate salt was obtained as described above. Mp: 189.8 – 191.8 °C. Anal. Calcd for C30H38FNO9: C, 62.60; H, 6.65; N, 2.43. Found: C, 62.63; H, 6.62; N, 2.50. HRMS (ESI) Calcd for C26H37FNO5 (M+H)+ 486.2656, found: 486.2655.
3.1.4 (1-(3,5-Dihydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)-piperidin-4-yl)(4-fluorophenyl)methanone (17a) and (1-(3,8-dihydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(4-fluorophenyl)methanone (17b)
Compound 15 (200 mg, 1.2 mmol), (4-fluorophenyl)(piperidin-4-yl)methanone 16 (585 mg, 2.4 mmol) and triethylamine (1 mL) were mixed in ethanol (5 mL) and heated at 60 °C for 2 d. Ethyl acetate (50 mL) was used to dilute the solution and then washed with brine (2 × 10 mL). After concentration, the residue was loaded onto silica gel column and eluted using CH2Cl2/methanol (20/1, v/v). Compound 17a was obtained as the first component, a white solid (109 mg, 25%). Mp 229 – 230 °C. 1H NMR (300 MHz, CDCl3) δ 8.00 – 7.94 (m, 2H), 7.15 – 7.11 (m, 2H), 7.02 (t, J = 7.8 Hz, 1H), 6.66 (dd, J = 25.2 Hz, 7.8 Hz, 2H), 5.01 (br, 1H), 3.92 – 3.83 (m, 1H), 3.32 – 3.23 (m, 2H), 3.07 – 2.97 (m, 2H), 2.89 – 2.77 (m, 4H), 2.64 – 2.45 (m, 2H), 1.97 – 1.89 (m, 3H), 1.88 – 1.76 (m, 1H). 13C NMR (75.5 MHz, CDCl3) δ 201.1, 166.2 (d, J = 301.0 Hz), 153.6, 135.8, 130.9, 130.8, 126.9, 121.8, 121.4, 115.8 (d, J = 21.7 Hz), 112.1, 66.5, 65.3, 51.9, 43.8, 37.8, 29.5, 29.2, 28.5, 20.0. HRMS (ESI) Calcd for C22H25FNO3 (M+H)+ 370.1818, found: 370.1817.
Compound 17b was obtained as the second component, a white solid (120 mg, 27%). Mp 208 – 210 °C. 1H NMR (400 MHz, CDCl3) δ 1.76-1.93 (m, 4H), 2.46-2.64 (m, 2H), 2.76-2.89 (m, 4H), 2.97-3.07 (m, 2H), 3.25-3.32 (m, 2H), 3.88 (s, 1H), 6.61 (d, J = 8.4Hz, 2H), 6.72 (d, J = 7.5Hz, 1H), 7.15 (t, J = 8.1Hz, 2H), 7.98 (t, J = 8.4Hz, 2H). 13C NMR (75.5 MHz, CD3SOCD3) δ 201.5, 165.4 (d, J = 251.6 Hz), 155.2, 137.1, 132.8, 131.6 (d, J = 9.3 Hz), 126.6, 121.6, 119.7, 116.2 (d, J = 21.7 Hz), 112.1, 66.5, 65.7, 50.3, 46.6, 43.6, 32.9, 29.6, 29.4, 27.8. HRMS (ESI) Calcd for C22H25FNO3 (M+H)+ 370.1818, found: 370.1833.
3.1.5 (1-(8-(2-Fluoroethoxy)-3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(4-fluorophenyl)methanone (18a)
Compound 17a (74 mg, 0.2 mmol), 2-fluoroethyl tosylate (52 mg, 0.24 mmol) and Cs2CO3 (98 mg, 0.3 mmol) were mixed in THF (5 mL) and refluxed overnight. The reaction mixture was partitioned between ethyl acetate and water. After drying over anhydrous Na2SO4, the organic phase was concentrated and purified on silica gel column by CH2Cl2/ethyl acetate (1/1, v/v) to get a white solid 18a (65 mg, 78%). Mp 149 – 152 °C. 1H NMR (400 MHz, CDCl3) δ 1.79 -1.95 (m, 4H), 2.47-2.61 (m, 2H), 2.72-2.99 (m, 5H), 3.10 (d, J = 16.5Hz, 1H), 3.24-3.35 (m, 2H), 3.84 (m, 1H), 4.18 (s, 1H), 4.27 (s, 1H), 4.72 (s, 1H), 4.88 (s, 1H), 6.65 (d, J = 8.4Hz, 1H), 6.76 (d, J = 8.4Hz, 1H), 7.08-7.19 (m, 3H), 8.00 (t, J = 8.7Hz, 2H). 13C NMR (75.5 MHz, CD3COCD3) δ 200.5, 165.4 (d, J = 254.2 Hz), 156.3, 135.7, 132.8, 131.2 (d, J = 9.2 Hz), 126.6, 124.0, 121.5, 115.5 (d, J = 22.0 Hz), 108.3, 82.1 (d, J = 168.4 Hz), 67.4 (d, J = 19.6 Hz), 66.5, 65.2, 51.6, 44.6, 43.5, 38.2, 20.1; HRMS (ESI) Calcd for C24H28F2NO3 (M+H)+ 416.2037, found: 416.2041. The free base was converted to oxalate salt as described above.
3.1.6 (1-(5-(2-Fluoroethoxy)-3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(4-fluorophenyl)methanone (18b)
Compound 18b was obtained by using 17b as the starting material, following the procedure for making 18a. Mp 137 – 138 °C. 1H NMR (400 MHz, CDCl3) δ 1.79 -1.92 (m, 4H), 2.36-2.54 (m, 2H), 2.75-3.02 (m, 6H), 3.27 (s, 1H), 3.50(d, J = 16.8Hz, 2H), 3.84 (m, 1H), 4.11 (s, 1H), 4.24 (s, 1H), 4.67 (s, 1H), 4.83 (s, 1H), 6.66 (d, J = 8.1Hz, 1H), 6.74 (d, J = 7.5Hz, 1H), 7.07-7.17(m, 3H), 7.98 (t, J = 8.7Hz, 2H). 13C NMR (75.5 MHz, CD3COCD3) δ 200.4, 165.4 (d, J = 256.4 Hz), 156.3, 136.8, 132.8 (d, J = 3.0 Hz), 131.1 (d, J = 9.2 Hz), 130.4, 123.2, 121.5, 115.6 (d, J = 22.0 Hz), 108.4, 82.1 (d, J = 168.4 Hz), 67.4 (d, J = 19.8 Hz), 66.1, 65.7, 51.4, 44.9, 43.6, 32.3, 26.3. HRMS (ESI) Calcd for C24H28F2NO3 (M+H)+ 416.2032, found: 416.2029.
3.1.7 3-(4-(4-Fluorobenzoyl)piperidin-1-yl)-5-(2-fluoroethoxy)-1,2,3,4-tetrahydronaphthalen-2-yl acetate (19a)
Racemic compound (±)-18a (140 mg, 0.337 mmol), acetic anhydride (200 μL) and pyridine (200 μL) were dissolved in CH2Cl2 (10 mL) and stirred at ambient temperature overnight. The product was extracted using ethyl acetate, and then dried over anhydrous Na2SO4. After solvent was removed under vacuum, the residue was purified on silica gel column by ethyl acetate/hexane (1/5 to 1/3, v/v) to afford (±)-19a as a white solid (148 mg, 96%). Mp 70 - 73 °C. 1H NMR (400 MHz, CDCl3) δ 1.58-1.86 (m, 4H), 2.10 (s, 3H), 2.45-2.76 (m, 3H), 2.80-3.20 (m, 7H), 4.02-4.25 (m, 3H), 4.65 (s, 1H), 4.80 (s, 1H), 6.57-6.74 (m, 2H), 7.01-7.15 (m, 3H), 7.95 (br s, 2H). 13C NMR (75.5 MHz, CD3OD) δ 202.0, 171.0, 156.2, 131.0, 130.9, 126.6, 121.1, 120.8, 115.4 (d, J = 21.9 Hz), 115.2, 108.6, 108.4, 81.9 (d, J = 169.3 Hz), 70.0, 66.2 (d, J = 25.3 Hz), 63.3, 49.5, 43.5, 37.9, 34.7, 29.0, 22.2, 21.1, 20.0. HRMS (ESI) Calcd for C26H30F2NO4 (M+H)+ 458.2143, found: 458.2152.
3.1.8 (-)-(1-(8-(2-Fluoroethoxy)-3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(4-fluorophen-yl)-methanone ((-)-18a)
Enantiomers of (±)-19a (132 mg, 0.29 mmol) were separated by HPLC using a Chiralcel OD column (250 mm × 10 mm), 2-propanol/hexane (3/97, v/v) as mobile phase, flow rate of 4.0 mL/min, and UV wavelength at 254 nm) to give (-)-19a (53 mg, 40%) and (+)-19a (61 mg, 46%) respectively. Each enantiopure compound was mixed with saturated Na2CO3 aqueous solution (3 mL) in EtOH (3 mL) separately and stirred for 72h at room temperature. Water (20 mL) was used to dilute the reaction mixture followed by extraction using CH2Cl2 (20 mL × 3). The combined organic phase was dried over anhydrous Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified on silica gel column using ethyl acetate/hexane (1/1, v/v) to afford (-)-18a as a white solid (40 mg, 88% yield). 1H NMR (400 MHz, CDCl3) δ 8.00 (t, J = 8.0 Hz, 2H), 7.17 – 7.09 (m, 3H), 6.76 (d, J = 8.0 Hz, 1H), 6.65 (d, J = 8.0 Hz, 1H), 4.87 - 4.85 (m, 1H), 4.75 - 4.73 (m, 1H), 4.27 – 4.15 (m, 3H), 3.85 – 3.75 (m, 1H), 3.30 – 3.20 (m, 2H), 3.13 – 3.02 (m, 1H), 2.99 – 2.70 (m, 5H), 2.60 – 2.43 (m, 2H), 1.98 – 1.73 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 200.9, 165.6 (d, J = 253.5 Hz), 156.1, 135.6, 132.3, 130.8, 126.7, 124.0, 122.0, 115.8 (d, J = 21.7 Hz), 108.1, 82.0 (d, J = 169.8 Hz), 67.2 (d, J = 20.2 Hz), 66.4, 65.1, 52.1, 44.3, 43.8, 37.9, 29.6, 29.2, 20.0.The free base of (-)-18a was converted to oxalate salt, Mp: 185.0-185.3 °C. The optical rotation of the oxalate salt was [α]D20 = -37.1° (0.7 mg/mL in 1/1 acetonitrile/H2O). HRMS (ESI) Calcd for C24H28F2NO3 (M+H)+ 416.2032, found: 416.2022.
3.1.9 (+)-(1-(8-(2-Fluoroethoxy)-3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)pipe-ridin-4-yl)(4-fluorophenyl)-methanone ((+)-18a)
Starting with (+)-19a (58 mg), the procedure of making (-)-18a afforded compound (+)-18a as a yellow oil (44.0 mg, 83% yield). 1H NMR (400 MHz, CDCl3) δ 8.00 (t, J = 8.0 Hz, 2H), 7.17 – 7.09 (m, 3H), 6.76 (d, J = 8.0 Hz, 1H), 6.65 (d, J = 8.0 Hz, 1H), 4.87 - 4.85 (m, 1H), 4.75 - 4.73 (m, 1H), 4.27 – 4.15 (m, 3H), 3.85 – 3.75 (m, 1H), 3.30 – 3.20 (m, 2H), 3.13 – 3.02 (m, 1H), 2.99 – 2.70 (m, 5H), 2.60 – 2.43 (m, 2H), 1.98 – 1.73 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 200.9, 165.6 (d, J = 253.4 Hz), 156.1, 135.6, 130.8, 126.7, 124.0, 122.0, 115.8 (d, J = 21.7 Hz), 108.2, 82.0 (d, J = 169.9 Hz), 67.2 (d, J = 20.2 Hz), 66.5, 65.1, 52.1, 44.3, 43.8, 37.9, 29.6, 29.2, 20.0. The free base was converted to oxalic salt, Mp: 191.0 – 192.2 °C. The optical rotation of (+)-18a was [α]D20 = 38.4° (0.65 mg/mL in 1/1 acetonitrile/H2O). HRMS (ESI) Calcd for C24H28F2NO3 (M+H)+ 416.2032, found: 416.2021.
3.2 In vitro biological evaluation
3.2.1 VAChT binding affinity studies
Binding affinities of the new compounds were determined by competing them against 5 nM [3H]vesamicol for binding to human VAChT present in synaptic-like microvesicles in postnuclear supernatant prepared from PC12A123.7 cells.[50] Nonspecific binding was determined from samples that contained 1 μM of nonradioactive (±)-vesamicol. The test compounds were assayed in increments of 10-fold from 0.1 to 10,000 nM concentration. The surfaces of containers were pre-coated with Sigmacote (Sigma-Aldrich, MO). Samples containing 200 μg postnuclear supernatant in 200 μL of 110 mM potassium tartrate, 20 mM HEPES (pH 7.4 with KOH), 1 mM dithiothreitol and 0.02 % sodium azide were incubated at 22 °C for 24 h. A volume of 90 μL was filtered in duplicate through GF/F glass fiber filters coated with polyethylenimine and washed. Filter-bound radioactivity was determined by liquid scintillation spectrometry for 10 min per sample. Averaged data were fitted by regression with a rectangular hyperbola to estimate Ki. All compounds were independently assayed at least two times.
3.2.2 Sigma receptors binding affinity studies
The σ receptor binding affinity studies were performed following our previously reported procedure. [51, 52] The σ1 receptor binding assays were performed in 96-well plates by incubating test compounds with approximately 300 μg protein of guinea pig brain membrane homogenates and 5 nM (+)-[3H]pentazocine (1.30 GBq/μmol, Perkin Elmer, Boston, MA). Non-specific binding was determined from samples containing 10 μM cold haloperidol. The σ2 receptor binding assays were similarly performed by incubating test compound(s) with rat liver membrane homogenates (~300 μg protein) and ~5 nM [3H]DTG (2.15 GBq/μmol, Perkin Elmer, Boston, MA) in the presence of 1 μM (+)-pentazocine to block σ1 sites. Non-specific binding was determined from samples that contained 10 μM of cold haloperidol. Data from the competitive inhibition experiments were modeled using nonlinear regression analysis to determine the concentration that inhibits 50% of the specific binding of the radioligand (IC50 value). Competitive binding curves were best fit to a one-site fit and gave pseudo-Hill coefficients of 0.6 – 1.0. Ki values were calculated using the method of Cheng and Prusoff 36 and are presented as the mean ± SEM. For these calculations, we used a Kd value of 7.89 nM for (+)-[3H]pentazocine in guinea pig brains and a Kd value of 30.73 nM for [3H]ditolylguanidine in rat livers.
3.3 Radiochemistry
3.3.1 Radiosynthesis of 1-[18F]fluoro-2-tosyloxyethane ([18F]20)
A sample of ~5.50 GBq [18F]/fluoride was added to a reaction vessel containing Kryptofix 2.2.2 (6.5 - 7.0 mg). To the mixture, acetonitrile (3 × 1.0 mL) was added to remove azeotropically water using nitrogen gas to bubble though the mixture at ~110 °C. After all the water was removed, 1, 2-ethylene ditosylate (5.0 – 5.5 mg) was dissolved into acetonitrile (200 μL) under vortex, and then the solution was transferred to the reaction vessel containing [18F]fluoride/Kryptofix /K2CO3. The reaction vessel was capped and the reaction mixture was briefly mixed, then heated about 10 min in an oil bath that was preheated to 110 °C. After reaction, the vessel was lifted up from the oil bath, the reaction mixture was diluted using 3.0 mL of HPLC mobile phase composed of 50/50 ratio of acetonitrile/0.1 M aqueous ammonium formate (pH ~6.5), and passed through an alumina Neutral Sep-Pak Plus cartridge. The crude product was then loaded onto an Agilent SB-C18 semi-preparative HPLC column (250 mm × 10 mm) with a UV detector set at 254 nm. The HPLC system was equipped with a 5 mL injection loop. At the flow rate of 4.0 mL/min, the retention time of the product was 9.5-10 min. The retention time of the precursor was 23-24 min. The product eluted from the HPLC was diluted using ~50 mL sterile water and then passed through a C-18 Sep-Pak Plus cartridge to trap [18F]20 on the Sep-Pak. Using ether (2.5 mL) to elute the radioactivity afforded [18F]20 (2.60-3.0 MBq, 60-75% radiochemistry yield, decay corrected to end of synthesis, EOS). The synthesis of [18F]20 took about 40 min.
3.3.2 Radiosynthesis of (-)-[18F]18a
The upper ether layer of the eluted solution of [18F]20 was transferred into a vial first, and the bottom aqueous phase was extracted with an additional 1 mL of ether. The combined ether solution was passed through a set of two Sep-Pak Plus dry cartridges into a reaction vessel. After the ether was evaporated with a nitrogen stream at 25 °C, the solution of precursor (-)-17a (1.0 - 1.4 mg) in DMSO (200 μL), and Cs2CO3 (1.0 - 1.5 mg) were added to the abovementioned reaction vessel containing [18F]20. The vessel was capped, vortexed, and then heated at 100 °C for 15 min. Subsequently, the residual was diluted with 3 mL HPLC mobile phase (35:65 acetonitrile/0.1 M ammonium formate buffer (pH ~4.5)) and loaded onto a semi-preparative HPLC system for purification. The HPLC system contains a 5 mL injection loop, an Agilent SB-C18 column, a UV detector at 254 nm and a radioactivity detector. With 35:65 acetonitrile/0.1 M ammonium formate buffer (pH ~4.5) as eluent, at 4.0 mL/min flow rate, the retention time of the product was 24 - 26 min, whereas the retention time of (-)-17a was 9 - 10 min. The product collection was diluted using sterile water (~50 mL) and then passed through a C18 Sep-Pak Plus cartridge. The trapped product was eluted using ethanol (0.6 mL), followed by 0.9% saline (5.4 mL). After sterile filtration into a glass vial, the (-)-[18F]18a was ready for quality control (QC) analysis and animal studies. To check the quality of the -(-)-[18F]18a, an aliquot of sample was co-injected with nonradiolabeled standard (-)-18a sample solution onto an analytical HPLC system equipped with an Agilent Zorbax SB-C18 column (250 × 4.6 mm) and UV absorbance at 254 nm; the mobile phase consisted of acetonitrile/0.1 M ammonium formate buffer (52/48, v/v). Under these conditions, the retention time of (-)-[18F]18a was ~ 5.0 min at a flow rate of 1.5 mL/min. For this step, the radiochemical purity was > 98%, the radiochemical yield was 50 - 60% (n > 20, decay corrected to EOS) and the specific activity was > 74 GBq/μmol (decay corrected to EOS). The synthesis of (-)-[18F]18a starting with [18F]20 took about 80 min. The entire two-step procedure of making (-)-[18F]18a starting with K[18F]F took ~2.0 h and the radiochemical yield was 30 – 40%.
3.4 Biodistribution studies in rats
All animal experiments were conducted in compliance with the Guidelines for the Care and Use of Research Animals under protocols approved by Washington University's Animal Studies Committee. For the biodistribution studies, the solution of (-)-[18F]18a (~2.2 MBq/120 μL) in 10% ethanol in saline was injected via the tail vein into adult male SD rats under 2-3% isoflurane/oxygen anesthesia. At 5, 30, 60, and 120 min p.i. (n = 4 for each time point), the rats were euthanized under anesthesia. The whole brain was quickly harvested and dissected into regions of cerebellum, brain stem, thalamus, striatum, cortex and hippocampus; the remainder of the brain was also collected to determine the total brain uptake. Samples of blood, heart, lung, muscle, fat, pancreas, spleen, kidney, liver, and bone were also collected, and all the samples were counted in an automated Beckman Gamma 8000 well counter with a standard dilution of the injectate. Tissues were weighed and the %ID/g was calculated. The striatum/organ ratios were calculated using the striatal uptake (%ID/g) divided by the %ID/g of the different brain regions.
For the ex vivo autoradiography study, an adult male SD rat was injected with (-)-[18F]18a (~63.0 MBq) via the tail vein under anesthesia and euthanized at 60 min p.i. The brain was quickly removed, snap-frozen, and horizontal sections (100 μm) were sliced using a chrome brain matrix (Zivic Instruments Inc., Pittsburg, PA). Frozen slides were directly exposed to film in an imaging cassette (Fuji Photo Film Co., Tokyo, Japan) for 12 hrs at −80 °C in the dark. The distribution of radioactivity was visualized by a Fuji Bio-Imaging Analyzer FLA-7000 (Fuji Photo Film Co., Tokyo, Japan). Photo-stimulated luminescence (PSL) from the striatum and cerebellum was quantified using Multi Gauge v3.0 software (Fuji Photo Film Co., Tokyo, Japan). Data were background-corrected, and expressed as photo-stimulated luminescence signals per square millimeter (PSL/mm2).
3.5 In vivo microPET imaging
Three PET studies were performed on adult male cynomolgus monkeys (6-8 Kg) with a microPET Focus 220 scanner (Concorde/CTI/Siemens Microsystems, Knoxville, TN). The animals were fasted for 12 h prior to each PET study. The animals were initially anesthetized using an intramuscular injection with ketamine (10 - 20 mg/kg) and glycopyrrolate (0.013 - 0.017 mg/kg) and then transported to the PET scanner suit. Upon arrival, the animal was intubated with an endotracheal tube and anesthesia was maintained at 0.75 - 2.0% isoflurane/oxygen throughout the procedure. After intubation, a percutaneous venous catheter was placed for radiotracer injection. Core temperature was kept constant at 37 °C with a heated water blanket. In each microPET scanning session, the head was positioned supine in the adjustable head holder with the brain in the center of the field of view. A 10-min transmission scan was performed to check positioning; once confirmed, a 45 min transmission scan was obtained for attenuation correction. Subsequently, a 2 hr dynamic emission scan was acquired after administration of 296 - 410 MBq of (-)-[18F]18a via the venous catheter. In the blocking studies, (-)-vesamicol (0.25 mg/kg) was administrated to the animal 5 min prior to the (-)-[18F]18a injection via the venous catheter.
3.6 MicroPET image processing and analysis
PET scans were collected from 0 – 120 min with the following time frames: 3×1 min, 4×2 min, 3×3 min and 20×5 min. PET image reconstructed resolution was < 2.0 mm full width half maximum for all 3 dimensions at the center of the field of view. Emission data were corrected using individual attenuation and model-based scatter correction and reconstructed using filtered back projection. The first baseline PET image for each animal acted as the target image with the MPRAGE and subsequent PETs co-registered using automated image registration program AIR. All MPRAGE-based volume of interest (VOI) analyses were accomplished by experienced investigators. For quantitative analyses, three-dimensional ROIs (cerebellum and striatum) were transformed to the baseline PET space and then overlaid on all reconstructed PET images to obtain time-activity curves. Activity measures were standardized to body weight and dose of radioactivity injected to yield standardized uptake value (SUV).
3.7 Metabolite studies on NHP plasma
Arterial blood samples (1.2 - 1.5 mL) were collected at 5, 15 30, 60, 90, and 120 min post-injection of the radiotracer in a heparinized syringe.[41, 53] Aliquots of whole blood (1 mL) were counted in a well counter, then centrifuged to separate red blood cells from plasma. Aliquots of plasma (400 μL) were solvent deproteinated using 0.92 mL ice-cold methanol and separated by centrifugation. The supernatant was mixed with water (1/1, v/v) and the mixture was injected to a HPLC system for radioactive metabolite determination. The HPLC system consisted of an Agilent SB C-18 analytic HPLC column (250 mm × 4.6 mm, 5 μA) and an UV detector with wavelength set at 254 nm. The mobile phase was acetonitrile/0.1 M ammonium formate buffer (pH, ~ 4.5, 52/48, v/v), and the flow rate was 1.5 mL/min. The HPLC fractions were collected at 1 min intervals for 16 min; each fraction was counted by a well counter to determine the radioactivity of each collection. The results were corrected for background radiation and physical decay.
To confirm if the major radioactive metabolite is able to enter into the brain, metabolite analysis was performed for the rat plasma samples post injection of (-)-[18F] 18a. 15 – 106 MBq of (-)-[18F] 18a was injected into male SD rats which were euthanized 5, 15, 30 and 60 min p.i. The rat plasma samples were prepared as described above. The rat brain tissue homogenate samples were prepared by following our published procedure,[41, 48] briefly, the whole brain was harvested and the excess blood was blotted; the brain tissue was homogenized on ice; acetonitrile (1.2 mL) was then added to extract the radioactivity and homogenize further. A 1.0 mL aliquot of this brain homogenate was centrifuged to remove the debris; 0.2 mL of the clear supernatant was diluted with 0.2 mL of distilled water to form the HPLC injection sample. The rat plasma and rat brain tissue samples were used for HPLC analysis following the procedure described above.
4. Conclusions
In this study, we successfully synthesized nine new carbonyl-containing fluorinated VAChT ligands. In vitro binding studies suggested that (-)-18a is a potent and selective candidate PET ligand. Ex vivo autoradiography studies and biodistribution studies in rodents and microPET studies in nonhuman primates suggest that (-)-[18F]18a specifically accumulates in the striatum, the VAChT enriched region of the brain. Striatal uptake of (-)-[18F]18a measured in monkeys using PET reached maximum uptake (SUV = 5.1) at 15 min p.i. More importantly, the striatal activity levels gradually decreased over time. Washout of the radioactivity from the nonspecific binding reference region, cerebellum, was much faster than in the target striatal regions. A blocking study using vesamicol strongly suggested that (-)-[18F]18a specifically binds to VAChT in the striatal regions. Over all, (-)-[18F]18a is a promising PET radiotracer candidate for quantifying VAChT levels in vivo. Currently an exploratory Investigational New Drug (IND) application has been approved by the Food and Drug Administration. The translational clinical investigation of (-)-[18F]18a in normal control subjects and patients is in progress.
Acknowledgements
This work was supported by NIH grants NS061025, NS075527 and MH092797, grants from the National Institute of General Medical Sciences (8 P41 GM103422-35). The authors thank William H. Margenau, and Robert Dennett in the Cyclotron Facility for 18F radioisotope production. Optical rotation was determined in the laboratory of Dr. Douglas F. Covey in the Department of Molecular Biology and Pharmacology of Washington University. The authors thank John Hood, Christina Zukas and Darryl Craig for their assistance with the nonhuman primate microPET studies. The authors acknowledge NMR core facility and microPET facility of Washington University St. Louis for research assistance.
Abbreviations
- Ac2O
acetic anhydride
- AD
Alzheimer disease
- BVM
benzovesamicol
- calcd
calculated
- CH2Cl2
dichloromethane
- CNS
central nervous system
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- [3H]DTG
[3H]ditolylguanidine
- EOB
end of bombardment
- EtOH
ethanol
- [18F]FEOBV
[18F]fluoroethoxybenzovesamicol
- HPLC
high performance liquid chromatography
- HRMS
high resolution mass spectrometry
- [123I]IBVM
(-)-5-[123I]iodobenzovesamicol
- IC50
half maximum inhibitory constant
- MABV
5-(N-methyl-amino)benzo-vesamicol
- NEFA
N-ethyl-fluoroacetamidobenzo-vesamicol
- NHPs
nonhuman primates
- PD
Parkinson Disease
- PET
positron emission tomography
- p.i.
post-injection
- PSL
photo-stimulated luminescence
- QC
quality control
- SD
Sprague-Dawley
- SPECT
single-photon emission computerized tomography
- SUV
standardized uptake value
- THF
tetrahydrofuran
- TLC
thin layer chromatography
- VAChT
Vesicular Acetylcholine Transporter
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Bohnen NI, Albin RL. The cholinergic system and Parkinson disease. Behav Brain Res. 2011;221:564–573. doi: 10.1016/j.bbr.2009.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ransome MI, Hannan AJ. Behavioural state differentially engages septohippocampal cholinergic and GABAergic neurons in R6/1 Huntington's disease mice. Neurobiol Learn Mem. 2012;97:261–270. doi: 10.1016/j.nlm.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Bohnen NI, Kaufer DI, Hendrickson R, Ivanco LS, Lopresti B, Davis JG, Constantine G, Mathis CA, Moore RY, DeKosky ST. Cognitive correlates of alterations in acetylcholinesterase in Alzheimer's disease. Neurosci Lett. 2005;380:127–132. doi: 10.1016/j.neulet.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 4.Schliebs R, Arendt T. The cholinergic system in aging and neuronal degeneration. Behav Brain Res. 2011;221:555–563. doi: 10.1016/j.bbr.2010.11.058. [DOI] [PubMed] [Google Scholar]
- 5.Muller ML, Bohnen NI. Cholinergic dysfunction in Parkinson's disease. Curr Neurol Neurosci Rep. 2013;13:377. doi: 10.1007/s11910-013-0377-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith R, Chung H, Rundquist S, Maat-Schieman ML, Colgan L, Englund E, Liu YJ, Roos RA, Faull RL, Brundin P, Li JY. Cholinergic neuronal defect without cell loss in Huntington's disease. Hum Mol Genet. 2006;15:3119–3131. doi: 10.1093/hmg/ddl252. [DOI] [PubMed] [Google Scholar]
- 7.Jung YW, Van Dort ME, Gildersleeve DL, Wieland DM. A radiotracer for mapping cholinergic neurons of the brain. J Med Chem. 1990;33:2065–2068. doi: 10.1021/jm00170a002. [DOI] [PubMed] [Google Scholar]
- 8.Kuhl DE, Minoshima S, Fessler JA, Frey KA, Foster NL, Ficaro EP, Wieland DM, Koeppe RA. In vivo mapping of cholinergic terminals in normal aging, Alzheimer's disease, and Parkinson's disease. Ann Neurol. 1996;40:399–410. doi: 10.1002/ana.410400309. [DOI] [PubMed] [Google Scholar]
- 9.Efange SM. In vivo imaging of the vesicular acetylcholine transporter and the vesicular monoamine transporter. FASEB J. 2000;14:2401–2413. doi: 10.1096/fj.00-0204rev. [DOI] [PubMed] [Google Scholar]
- 10.Usdin TB, Eiden LE, Bonner TI, Erickson JD. Molecular biology of the vesicular ACh transporter. Trends Neurosci. 1995;18:218–224. doi: 10.1016/0166-2236(95)93906-e. [DOI] [PubMed] [Google Scholar]
- 11.Brittain RT, Levy GP, Tyers MB. The neuromuscular blocking action of 2-(4-phenylpiperidino) cyclohexanol (AH 5183) Eur J Pharmacol. 1969;8:93–99. doi: 10.1016/0014-2999(69)90133-2. [DOI] [PubMed] [Google Scholar]
- 12.Brittain RT, Levy GP, Tyers MB. Observations on the neuromuscular blocking action of 2-(4-phenylpiperidino)-cyclohexanol (AH 5183) Br J Pharmacol. 1969;36:173p–174p. [PMC free article] [PubMed] [Google Scholar]
- 13.Bahr BA, Parsons SM. Acetylcholine transport and drug inhibition kinetics in Torpedo synaptic vesicles. J Neurochem. 1986;46:1214–1218. doi: 10.1111/j.1471-4159.1986.tb00640.x. [DOI] [PubMed] [Google Scholar]
- 14.Bahr BA, Parsons SM. Demonstration of a receptor in Torpedo synaptic vesicles for the acetylcholine storage blocker L-trans-2-(4-phenyl[3,4-3H]-piperidino)cyclohexanol. Proc Natl Acad Sci U S A. 1986;83:2267–2270. doi: 10.1073/pnas.83.7.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaufman R, Rogers GA, Fehlmann C, Parsons SM. Fractional vesamicol receptor occupancy and acetylcholine active transport inhibition in synaptic vesicles. Mol Pharmacol. 1989;36:452–458. [PubMed] [Google Scholar]
- 16.Efange SM, Mach RH, Smith CR, Khare AB, Foulon C, Akella SK, Childers SR, Parsons SM. Vesamicol analogues as sigma ligands: Molecular determinants of selectivity at the vesamicol receptor. Biochem Pharmacol. 1995;49:791–797. doi: 10.1016/0006-2952(94)00541-s. [DOI] [PubMed] [Google Scholar]
- 17.Shiba K, Ogawa K, Ishiwata K, Yajima K, Mori H. Synthesis and binding affinities of methylvesamicol analogs for the acetylcholine transporter and sigma receptor. Bioorg Med Chem. 2006;14:2620–2626. doi: 10.1016/j.bmc.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 18.Custers FG, Leysen JE, Stoof JC, Herscheid JD. Vesamicol and some of its derivatives: Questionable ligands for selectively labelling acetylcholine transporters in rat brain. Eur J Pharmacol. 1997;338:177–183. doi: 10.1016/s0014-2999(97)81946-2. [DOI] [PubMed] [Google Scholar]
- 19.Haigh JR, Noremberg K, Parsons SM. Acetylcholine active transport by rat brain synaptic vesicles. Neuroreport. 1994;5:773–776. doi: 10.1097/00001756-199403000-00009. [DOI] [PubMed] [Google Scholar]
- 20.Giboureau N, Som IM, Boucher-Arnold A, Guilloteau D, Kassiou M. PET radioligands for the vesicular acetylcholine transporter (VAChT) Curr Top Med Chem. 2010;10:1569–1583. doi: 10.2174/156802610793176846. [DOI] [PubMed] [Google Scholar]
- 21.Rogers GA, Parsons SM, Anderson DC, Nilsson LM, Bahr BA, Kornreich WD, Kaufman R, Jacobs RS, Kirtman B. Synthesis, in vitro acetylcholine-storage-blocking activities, and biological properties of derivatives and analogues of trans-2-(4-phenylpiperidino)cyclohexanol (vesamicol) J Med Chem. 1989;32:1217–1230. doi: 10.1021/jm00126a013. [DOI] [PubMed] [Google Scholar]
- 22.Rogers GA, Parsons SM. Photoaffinity labeling of the vesamicol receptor of cholinergic synaptic vesicles. Biochemistry. 1993;32:8596–8601. doi: 10.1021/bi00084a029. [DOI] [PubMed] [Google Scholar]
- 23.Rogers GA, Kornreich WD, Hand K, Parsons SM. Kinetic and equilibrium characterization of vesamicol receptor-ligand complexes with picomolar dissociation constants. Mol Pharmacol. 1993;44:633–641. [PubMed] [Google Scholar]
- 24.Kuhl DE, Koeppe RA, Fessler JA, Minoshima S, Ackermann RJ, Carey JE, Gildersleeve DL, Frey KA, Wieland DM. In vivo mapping of cholinergic neurons in the human brain using SPECT and IBVM. Journal of Nuclear Medicine. 1994;35:405–410. [PubMed] [Google Scholar]
- 25.Jung YW, Frey KA, Mulholland GK, del Rosario R, Sherman PS, Raffel DM, Van Dort ME, Kuhl DE, Gildersleeve DL, Wieland DM. Vesamicol receptor mapping of brain cholinergic neurons with radioiodine-labeled positional isomers of benzovesamicol. J Med Chem. 1996;39:3331–3342. doi: 10.1021/jm9507486. [DOI] [PubMed] [Google Scholar]
- 26.Lamare F, Mazere J, Attila M, Mayo W, De Clermont-Gallerande H, Meissner W, Fernandez P, Allard M. Improvement of in vivo quantification of [123I]-iodobenzovesamicol in single-photon emission computed tomography/computed tomography using anatomic image to brain atlas nonrigid registration. Mol Imaging. 2013;12:288–299. [PubMed] [Google Scholar]
- 27.Mazere J, Meissner WG, Sibon I, Lamare F, Tison F, Allard M, Mayo W. [123I]-IBVM SPECT imaging of cholinergic systems in multiple system atrophy: A specific alteration of the ponto-thalamic cholinergic pathways (Ch5-Ch6) Neuroimage Clin. 2013;3:212–217. doi: 10.1016/j.nicl.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meikle SR, Kench P, Kassiou M, Banati RB. Small animal SPECT and its place in the matrix of molecular imaging technologies. Phys Med Biol. 2005;50:R45–61. doi: 10.1088/0031-9155/50/22/R01. [DOI] [PubMed] [Google Scholar]
- 29.Mulholland GK, Jung Y-W, Wieland DM, Kilbourn MR, Kuhl DE. Synthesis of [18F]fluoroethoxy-benzovesamicol, a radiotracer for cholinergic neurons. J Labelled Comp Radiopharm. 1993;33:583–591. [Google Scholar]
- 30.Mulholland GK, Wieland DM, Kilbourn MR, Frey KA, Sherman PS, Carey JE, Kuhl DE. [18F]Fluoroethoxy-benzovesamicol, a PET radiotracer for the vesicular acetylcholine transporter and cholinergic synapses. Synapse. 1998;30:263–274. doi: 10.1002/(SICI)1098-2396(199811)30:3<263::AID-SYN4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 31.Kilbourn MR, Hockley B, Lee L, Sherman P, Quesada C, Frey KA, Koeppe RA. Positron emission tomography imaging of (2R,3R)-5-[18F]fluoroethoxybenzovesamicol in rat and monkey brain: A radioligand for the vesicular acetylcholine transporter. Nuc Med Biol. 2009;36:489–493. doi: 10.1016/j.nucmedbio.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petrou M, Frey KA, Kilbourn MR, Scott PJ, Raffel DM, Bohnen NI, Muller ML, Albin RL, Koeppe RA. In vivo imaging of human cholinergic nerve terminals with (−)-5-18F-fluoroethoxybenzovesamicol: Biodistribution, dosimetry, and tracer kinetic analyses. J Nuc Med. 2014;55:396–404. doi: 10.2967/jnumed.113.124792. [DOI] [PubMed] [Google Scholar]
- 33.Parent MJ, Bedard MA, Aliaga A, Minuzzi L, Mechawar N, Soucy JP, Schirrmacher E, Kostikov A, Gauthier SG, Rosa-Neto P. Cholinergic depletion in Alzheimer's disease shown by [18F]FEOBV autoradiography. Int J Mol Imaging. 2013;2013:205045. doi: 10.1155/2013/205045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parent MJ, Cyr M, Aliaga A, Kostikov A, Schirrmacher E, Soucy JP, Mechawar N, Rosa-Neto P, Bedard MA. Concordance between in vivo and postmortem measurements of cholinergic denervation in rats using PET with [18F]FEOBV and choline acetyltransferase immunochemistry. EJNMMI Res. 2013;3:70. doi: 10.1186/2191-219X-3-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Efange SM, Khare AB, von Hohenberg K, Mach RH, Parsons SM, Tu Z. Synthesis and in vitro biological evaluation of carbonyl group-containing inhibitors of vesicular acetylcholine transporter. J Med Chem. 2010;53:2825–2835. doi: 10.1021/jm9017916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tu Z, Efange SM, Xu J, Li S, Jones LA, Parsons SM, Mach RH. Synthesis and in vitro and in vivo evaluation of 18F-labeled positron emission tomography (PET) ligands for imaging the vesicular acetylcholine transporter. J Med Chem. 2009;52:1358–1369. doi: 10.1021/jm8012344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tu Z, Wang W, Cui J, Zhang X, Lu X, Xu J, Parsons SM. Synthesis and evaluation of in vitro bioactivity for vesicular acetylcholine transporter inhibitors containing two carbonyl groups. Bioorg Med Chem. 2012;20:4422–4429. doi: 10.1016/j.bmc.2012.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J, Zhang X, Zhang Z, Padakanti PK, Jin H, Cui J, Li A, Zeng D, Rath NP, Flores H, Perlmutter JS, Parsons SM, Tu Z. Heteroaromatic and aniline derivatives of piperidines as potent ligands for vesicular acetylcholine transporter. J Med Chem. 2013;56:6216–6233. doi: 10.1021/jm400664x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu H, Jin H, Li J, Zhang X, Kaneshige K, Parsons SM, Perlmutter JS, Tu Z. In vitro and ex vivo characterization of (−)-TZ659 as a ligand for imaging the vesicular acetylcholine transporter. Eur J Pharmacol. 2015;752:18–25. doi: 10.1016/j.ejphar.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Padakanti PK, Zhang X, Li J, Parsons SM, Perlmutter JS, Tu Z. Syntheses and radiosyntheses of two C-11 labeled potent and selective radioligands for imaging vesicular acetylcholine transporter. Mol Imaging Biol. 2014:1–8. doi: 10.1007/s11307-014-0748-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Padakanti PK, Zhang X, Jin H, Cui J, Wang R, Li J, Flores HP, Parsons SM, Perlmutter JS, Tu Z. In vitro and in vivo characterization of two C-11-labeled PET tracers for vesicular acetylcholine transporter. Mol Imaging Biol. 2014 doi: 10.1007/s11307-014-0749-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roberts MJ, Bentley MD, Harris JM. Chemistry for peptide and protein PEGylation. Adv Drug Deliv Rev. 2002;54:459–476. doi: 10.1016/s0169-409x(02)00022-4. [DOI] [PubMed] [Google Scholar]
- 43.Harris JM, Chess RB. Effect of pegylation on pharmaceuticals. Nat Rev Drug Discov. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 44.Stephenson KA, Chandra R, Zhuang ZP, Hou C, Oya S, Kung MP, Kung HF. Fluoro-pegylated (FPEG) imaging agents targeting A aggregates. Bioconjug Chem. 2007;18:238–246. doi: 10.1021/bc060239q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang W, Cui J, Lu X, Padakanti PK, Xu J, Parsons SM, Luedtke RR, Rath NP, Tu Z. Synthesis and in vitro biological evaluation of carbonyl group-containing analogues for 1 receptors. J Med Chem. 2011;54:5362–5372. doi: 10.1021/jm200203f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wenzel B, Sorger D, Heinitz K, Scheunemann M, Schliebs R, Steinbach J, Sabri O. Structural changes of benzylether derivatives of vesamicol and their influence on the binding selectivity to the vesicular acetylcholine transporter. Eur J Med Chem. 2005;40:1197–1205. doi: 10.1016/j.ejmech.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 47.Scheunemann M, Sorger D, Wenzel B, Heinitz K, Schliebs R, Klingner M, Sabri O, Steinbach J. Synthesis of novel 4- and 5-substituted benzyl ether derivatives of vesamicol and in vitro evaluation of their binding properties to the vesicular acetylcholine transporter site. Bioorg Med Chem. 2004;12:1459–1465. doi: 10.1016/j.bmc.2003.12.035. [DOI] [PubMed] [Google Scholar]
- 48.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 4th ed. Academic Press, Inc.; San Diego, CA: 1998. [Google Scholar]
- 49.Gilmor ML, Nash NR, Roghani A, Edwards RH, Yi H, Hersch SM, Levey AI. Expression of the putative vesicular acetylcholine transporter in rat brain and localization in cholinergic synaptic vesicles. J Neurosci. 1996;16:2179–2190. doi: 10.1523/JNEUROSCI.16-07-02179.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zea-Ponce Y, Mavel S, Assaad T, Kruse SE, Parsons SM, Emond P, Chalon S, Giboureau N, Kassiou M, Guilloteau D. Synthesis and in vitro evaluation of new benzovesamicol analogues as potential imaging probes for the vesicular acetylcholine transporter. Bioorg Med Chem. 2005;13:745–753. doi: 10.1016/j.bmc.2004.10.043. [DOI] [PubMed] [Google Scholar]
- 51.Tu Z, Xu J, Jones LA, Li S, Dumstorff C, Vangveravong S, Chen DL, Wheeler KT, Welch MJ, Mach RH. Fluorine-18-labeled benzamide analogues for imaging the 2 receptor status of solid tumors with positron emission tomography. J Med Chem. 2007;50:3194–3204. doi: 10.1021/jm0614883. [DOI] [PubMed] [Google Scholar]
- 52.Xu J, Tu Z, Jones LA, Vangveravong S, Wheeler KT, Mach RH. [3H]N-[4-(3,4-dihydro-6,7-dimethoxyisoquinolin-2(1H)-yl)butyl]-2-methoxy-5-methylbenzamide: A novel sigma-2 receptor probe. Eur J Pharmacol. 2005;525:8–17. doi: 10.1016/j.ejphar.2005.09.063. [DOI] [PubMed] [Google Scholar]
- 53.Tu Z, Fan J, Li S, Jones LA, Cui J, Padakanti PK, Xu J, Zeng D, Shoghi KI, Perlmutter JS, Mach RH. Radiosynthesis and in vivo evaluation of [11C]MP-10 as a PET probe for imaging PDE10A in rodent and non-human primate brain. Bioorg Med Chem. 2011;19:1666–1673. doi: 10.1016/j.bmc.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]