

Abstract
Cellular FLICE-like inhibitory protein (c-FLIP) has been identified as a protease-dead, procaspase-8-like regulator of death ligand-induced apoptosis, based on observations that c-FLIP impedes tumor necrosis factor-α (TNF-α), Fas-L, and TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by binding to FADD and/or caspase-8 or -10 in a ligand-dependent fashion, which in turn prevents death-inducing signaling complex (DISC) formation and subsequent activation of the caspase cascade. c-FLIP is a family of al ternatively spliced variants, and primarily exists as long (c-FLIPL) and short (c-FLIPS) splice variants in human cells. Although c-FLIP has apoptogenic activity in some cell contexts, which is currently attributed to heterodimerization with caspase-8 at the DISC, accumulat ing evidence indicates an anti-apoptotic role for c-FLIP in various types of human cancers. For example, small interfering RNAs (siRNAs) that specifically knocked down expression of c-FLIPL in diverse human cancer cell lines, e.g., lung and cervical cancer cells, augmented TRAIL-induced DISC recruitment, and thereby enhanced effector caspase stimulation and apoptosis. Therefore, the outlook for the therapeutic index of c-FLIP-targeted drugs appears excellent, not only from the efficacy observed in experimental models of cancer therapy, but also because the current understanding of dual c-FLIP action in normal tissues supports the notion that c-FLIP-targeted cancer therapy will be well tolerated. Interestingly, Taxol, TRAIL, as well as several classes of small molecules induce c-FLIP downregulation in neoplastic cells. Efforts are underway to develop small-molecule drugs that induce c-FLIP downregulation and other c-FLIP-targeted cancer therapies. In this review, we assess the outlook for improving cancer therapy through c-FLIP-targeted therapeutics.
Keywords: Taxol, apoptosis, caspase-8, caspase-10, c-FLIP, leukemia, death receptors
INTRODUCTION
Chemotherapeutic drug resistance is a major clinical problem and an important cause of treatment failure in cancer. One of the important goals of molecular oncology is to identify the underlying drug resistance mechanisms, with the prospect that more effective and novel therapies for cancer can be developed. Several mechanisms have been found to cause resistance to chemotherapeutic agents in cancer cells in vitro [1-5]. Identifying novel mechanisms of resistance to chemotherapeutic agents will assist in the design of more effective strategies to overcome resistance in cancer cells. Defects in apoptotic signaling in malignant cells contribute to the drug resistance in various cancer types [6]. Furthermore, death receptor-mediated apoptosis is deficient in some drug resistant cancer cells. Therefore, strategies to lower the thresholds for triggering apoptosis in various cancers may lead to new and more effective therapeutic regimens. The death-inducing cytokine tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) holds enormous promise as a cancer therapeutic due to its highly selective apoptosis-inducing action on neoplastic versus normal cells [7-10]. However, to exploit this opportunity, the problems of TRAIL resistance in cancer must first be overcome [11-15]. Cellular FLICE-like inhibitory protein (c-FLIP), a catalytically inactive caspase-8/-10 homologue, is involved in TRAIL and chemotherapeutic drug resistance in a wide range of human malignancies [11, 13, 16-20]. Substantial levels of c-FLIP are expressed in deadly human cancers such as ovarian, colon, glioblastoma, breast, colorectal, and prostate cancers, and it is implicated in the TRAIL resistance owing to its overexpression in a substantial proportion of these malignancies [21-24]. Furthermore, interference with c-FLIP expression sensitizes these tumor cells to TRAIL and other tumor necrosis factor-related death ligands, such as FAS ligand, in experimental models [17, 20, 25, 26]. c-FLIP is an important modulator of the initiator procaspases-8 and -10 and thereby regulates life and death in normal cells and tissues, and renders resistance to death receptor-mediated apoptosis in various cancer cells. In addition to its function as an apoptosis modulator, c-FLIP exerts other cellular functions including increased cell proliferation and tumorigenesis [27]. Moreover, dysregulation of c-FLIP expression has been associated with diseases such as cancer and autoimmune diseases [28, 29]. Therefore, c-FLIP is a critical target for therapeutic intervention. In this review, we assess the outlook for improving the outcome of cancer therapy by targeting c-FLIP and exploring the possibility of its degradation and/or decreasing its expression in order to provide a potentially safe approach to the treatment of cancer. The possibility of developing novel modalities of cancer therapy that improve the efficacy and lessen the toxicity of cancer chemotherapy by targeting specific c-FLIP isoforms is discussed.
APOPTOSIS SIGNALING PATHWAYS
Two well-studied pathways are involved in apoptosis, the mitochondrion-initiated pathway (Fig. 1) and the cell surface death receptors pathway (Fig. 2) [30-32]. In the mitochondrial pathway, cytochrome c, certain caspases, apoptosis-inducing factor, Smac/DIABLO, an inhibitor of apoptosis protein (IAP)-binding protein, and other apoptosis-inducing factors are released from the mitochondrial intramembrane space to the cytosol [33]. Once released, cytochrome c and dATP bind to apoptotic proteinase-activating factor-1 (Apaf-1), and this complex along with adenine nucleotides promotes procaspase-9 autoactivation [34], which in turn activates caspases-2, -3, -6, -7, -8, and -10. In the death receptor-mediated apoptosis pathway (e.g. Fas/Fas ligand interaction and cell death), the initiator caspases-8 and -10 activate the downstream caspases including caspase-3. Active caspases-8 and -10 are known to cleave a pro-apoptotic Bcl-2 family member, Bid, and the truncated Bid induces mitochondrial cytochrome c release [32-35], thereby linking the two pathways. After activation, both caspases-8 and -9 activate caspase-3, which in turn cleaves other caspases and many cellular proteins including fodrin, protein kinase C, poly(ADP-ribose) polymerase, gelsolin, and DNA fragmentation factor-45 (DFF45) [32, 36, 37]. A third pathway also has been recently identified [38]. In this pathway, Bid is cleaved downstream of the point of Bcl-2 action, catalyzed by caspase-3, which occurs upstream of caspase-8 activation, thereby acting as a potential feedback loop for amplifying the apoptosis-associated release of cytochrome c from the mitochondria [38].
Fig. (1). Mitochondrial death pathway.

The mitochondrial pathway of apoptosis is usually caused by disruption of mitochondrial membrane potential (ΔΨm), release of cytochrome c, activation of caspase-9, and subsequent activation of downstream caspases-2, -3, -6, -7, -8 and -10, which eventually leads to apopto sis. On the other hand, caspase-dependent apoptosis induced by death receptor ligation (e.g., Fas ligand/Fas or TNF-α/TNF- α receptor) can function independently of the mitochondria by caspase-8 activation, which directly activates caspase-3, -6 and -7, or by causing proteolytic cleavage of the pro-apoptotic protein Bid to a truncated form (tBid), which in turn is inserted in the mitochondrial membrane and induces release of cytochrome c. Release of the apoptosis inducing factor (AIF) from mitochondria causes caspase-independent apoptosis.
Fig. (2). The cell death receptor pathway.

FAS-ligand, TNF-α, and TRAIL bind to their respective receptors leading to the recruitment of FADD via a death domain (DD) interaction. After FADD binding, the death effector domain (DED) of FADD interacts with the DEDs of procaspases-8 and -10 forming the death-inducing signaling compex (DISC). Following DISC formation, caspases-8 and -10 are activated, disassociate from the DISC, and activate downstream effector caspases. c-FLIP binds to procaspases-8, -10, and FADD, and inhibits DISC formation.
c-FLIP AND THE DEATH RECEPTOR APOPTOSIS PATHWAY
The TNF receptor (TNFR) superfamily consists of cell surface receptors that are activated by cognate ligands and signal proliferation, differentiation, and apoptosis (Fig. 2). Death receptors in the TNFR superfamily include the TNF-α receptor, Fas (APO-1/CD95), and TRAIL receptors DR4 (also known as TRAIL receptor 1) and DR5 (also known as TRAIL receptor 2). Other TNFR superfamily members protect against apoptosis by serving as cell surface decoy receptors that compete for death ligand binding but cannot induce apoptosis [39, 40]. The death receptors contain an intracellular death domain (DD) which is essential for inducing apoptosis upon receptor ligation; the decoy receptors cannot signal apoptosis for lack of a functional DD [40]. The DD is a canonical 80 amino acid cytoplasmic docking surface in death receptors, that upon ligand binding to the receptor, homotypically binds a similar domain in the death adaptor protein FADD or TRADD [41]. Death adaptor proteins also have a death effector domain (DED) that forms a distinct docking surface for homotypic binding of initiator (apical) caspases. Once the initiator caspases are recruited to death receptor-tethered FADD, the DISC (death-inducing signaling complex) is formed, whereupon the caspases are trans-proteolyzed, dimerized, and rendered active; executioner caspase activation and apoptosis ensues. An important regulator of death receptor-induced death is c-FLIP, which binds to procaspases-8, -10 and FADD, and when overexpressed, can protect tumor cells from apoptosis. This supports the notion that cancer therapeutics that target apical proteins in the death receptor pathway, e.g., caspase-8 and -10 isoforms and c-FLIP variants (see below), may have a favorable therapeutic index. In the following sections we will discuss apoptosis induced by TRAIL and other chemotherapeutic agents such as Taxol (paclitaxel) and doxorubicin, through downregulation of c-FLIP variants, and address the possibilities for using c-FLIP as a therapeutic target.
STRUCTURE OF c-FLIP
Initially, viral FLICE-inhibitory proteins (v-FLIPs) were identified by a bioinformatic search for novel virus-encoded apoptotic regulatory molecules containing a DED [42-44]. Following the characterization of v-FLIPs, the mammalian cellular homologue was identified and called c-FLIP [45]. Animal models have revealed that c-FLIP plays an important role in T cell proliferation and heart development. Moreover, abnormal c-FLIP expression has been found in various diseases such as cancers, multiple sclerosis, Alzheimer’s disease, diabetes mellitus, and rheumatoid arthritis. So far, 11 distinct c-FLIP splice variants have been reported, three of which are expressed as proteins: the 26 kDa short form (c-FLIPS), the 24 kDa form of c-FLIP (c-FLIPR) and the 55 kDa c-FLIPL [46, 47] (Fig. 3). The structures of c-FLIPS and the v-FLIPs are similar, except that the two DEDs of c-FLIPS are followed by 20 amino acids that appear to be crucial for its ubiquitylation and targeting for proteasomal degradation [48]. c-FLIPR also contains two DEDs but lacks the additional carboxy (C)-terminal amino acids that are present in c-FLIPS. The C-terminus of c-FLIPL is longer than that of c-FLIPS and closely resembles the structure of caspases-8 and -10 [47], but this region of c-FLIPL does not contain a functional caspase domain. This lack of caspase activity is the result of the substitution of several amino acids, particularly the crucial cysteine residue in the catalytic domain which is necessary for the catalytic activity of caspases [49]. Additionally, c-FLIPL harbors a caspase-8 cleavage site at position Asp-376 (LEVD); c-FLIPL cleavage at this site produces the proteolytic fragment variant p43c-FLIP [50, 51].
Fig. (3). Structures of c-FLIP variants.
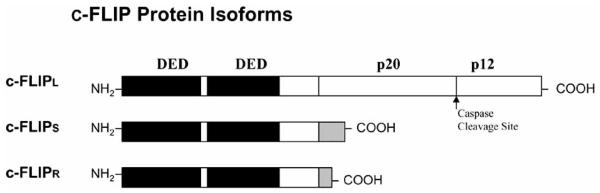
Three c-FLIP variants, c-FLIPL, c-FLIPS, and c-FLIPR, contain two death effector domains (DEDs) at their N-termini. In addition to two DEDs, c-FLIPL contains a large (p20) and a small (p12) caspase-like domain without catalytic activity. c-FLIPS and c-FLIPR consist of two DEDs and a small C-terminus (adapted from [46]).
c-FLIP FUNCTION
As discussed above, c-FLIP has been identified as a protease-dead, procaspase-8-like regulator of death ligand-induced apoptosis [27, 45, 52, 53]. It has been shown that c-FLIP interferes with TNF-α, Fas-L, and TRAIL-induced signaling pathways by binding to FADD and/or caspase-8/10 in a ligand-dependent fashion, which in turn prevents DISC formation and subsequent activation of the caspase cascade [27, 53, 54]. However, studies using c-FLIP-deficient mice support a dual function for c-FLIP by confirming a role for c-FLIP in Fas L- and TNF-α-induced apoptosis, and revealing that c-FLIP has a similar function to caspase-8 in heart development [55]. Nonetheless, a now extensive literature encompassing diverse types of human cancer cells indicates that the action of c-FLIP is generally anti-apoptotic in cancer cells.
While the precise mechanism of c-FLIP variant regulation of apoptosis remains elusive, the profound structural differences between human c-FLIP variants clearly indicate distinct regulatory roles for c-FLIPL and c-FLIPS in apoptosis. In fact, c-FLIPS inhibits TRAIL-induced DISC formation and apoptosis [56], while c-FLIPL is responsible for the above described dual functions whereby it inhibits Fas-induced caspase-8 activation when expressed at high levels, but enhances caspase-8 activation when its expression level is low [53-59]. These opposing c-FLIPL functions may reflect observations that c-FLIPL activates caspases-8 and -10 in vitro by forming heterodimeric enzyme molecules with a substrate specificity and catalytic activity indistinguishable from caspase-8 homodimers, despite the fact that c-FLIPL is protease dead [60]. In addition, accumulating evidence has revealed that c-FLIP may be an effector that contributes to survival signaling by the Akt, ERK, and NF-κB signaling pathways [41, 57, 61, 62] (Fig. 4).
Fig. (4). Model depicting the multifunctional roles of c-FLIP on various signaling pathways.

As discussed in the text, in addition to its functional role in inhibiting apoptosis by binding to procaspases-8 and -10 and inhibiting their activation, c-FLIP also has the following functions: (1) initiating cell proliferation by recruiting and activating (a) downstream signaling proteins including TRAF, RIP, IKK, and NFkB, or (b) RAF and extracellular signal-regulated kinase (ERK), (2) controlling cell survival by activating Akt, JNK, and Wnt signaling pathways, and (3) participating in carcinogenesis.
It has been demonstrated that overexpression of c-FLIPL activates NF-κB and ERK signaling by binding to adaptor proteins in each pathway, such as TNFR-associated factors 1 (TRAF1) and 2 (TRAF2), receptor-interacting protein 1 (RIP), and Raf-1 [63, 64] (Fig. 4). The caspase-8 processed N-terminal fragment of c-FLIPL (p43cFLIP) is more efficient than c-FLIPL at recruiting TRAF2 and RIP1, leading to more robust NF-κB activation [51, 62-65]. Golks et al. [66] recently showed that in nonapoptotic cells, c-FLIP and the procaspase-8 heterodimer result in a novel NH2-terminal fragment of c-FLIP (p22-FLIP) which is the key mediator of NF-κB activation by directly binding to the IKK complex. These results provide a new mechanism of c-FLIP-mediated NF-κB activation. Recently, Chang et al. [67] demonstrated that TNF-α-mediated JNK activation increases turnover of the NF-κB-induced c-FLIP. This is not the result of direct c-FLIP phosphorylation, but rather depends on JNK-mediated phosphorylation and activation of the E3 ubiquitin ligase Itch which specifically ubiquitinates c-FLIP and induces its proteasomal degradation. Thus, JNK antagonizes NF-κB during TNF-α signaling by promoting the proteasomal elimination of c-FLIPL.
In addition to inhibiting caspase-8 and -10 activation [68], c-FLIPL interacts with Daxx (a death domain-associated protein that has been implicated in proapoptosis and transcriptional regulation) and prevents Fas-induced JNK activation [69]. Thus, c-FLIPL acting on both the FADD- and Daxx-mediated signaling pathways may be involved in completely inhibiting Fas-induced cell death. Furthermore, Nakajima et al. [70] demonstrated that c-FLIPL directly interacts with a JNK activator, MAP kinase kinase 7 (MKK7), in a TNF-α-dependent manner and inhibits the interactions of MKK7 with MAP/ERK kinase kinase 1, apoptosis-signal-regulating kinase 1, and TGF-β-activated kinase 1. This interaction of c-FLIPL with MKK7 might selectively suppress JNK activation (Fig. 4).
Recent results also indicate that the calcium/calmodulin-dependent protein kinase II (CaMK II)-mediates the upregulation of c-FLIP, thereby protecting melanoma cells from TRAIL-induced apoptosis. Treating resistant cells with the CaMK II inhibitor KN-93 inhibited CaMK II activity, reduced c-FLIP expression, inhibited c-FLIP phosphorylation, and rescued Fas agonistic antibody (CH-11) sensitivity [19, 71]. Targeting this pathway may provide novel therapeutic strategies in treating cancers with upregulated CaMK II. Interestingly, phosphorylation of c-FLIP variants by CaMK II appears to promote c-FLIPL recruitment to the DISC and inhibit TRAIL-induced apoptosis [19, 71], but phosphorylation of c-FLIPL by protein kinase C or the bile acid glycochenodeoxycholate results in decreased c-FLIPL recruitment to the DISC and increased sensitivity of hepatocellular carcinoma cells to TRAIL-triggered apoptosis [72]. Thus, the particular site of phosphorylation in c-FLIPL appears to influence the functional outcome of this protein on apoptosis.
c-FLIPS also suppress apoptosis by inhibiting caspase-8 activation, although at different levels of procaspase-8 processing [73, 74]. c-FLIPL induces a conformation of procaspase-8 that triggers partial but not complete proteolytic processing, while in contrast c-FLIPS even prevents partial procaspase-8 activation at the DISC [74]. Using an in vitro induced proximity assay, Boatright et al. [60] provide evidence that c-FLIPL is an activator of caspase-8/-10 and demonstrate that the resulting heterodimer is enzymatically active with a substrate specificity identical to that of the caspase-8 homodimer.
Increased expression of c-FLIP can alter cell cycle progression and enhance cell proliferation and carcinogenesis. Overexpression of c-FLIPL inhibited the ubiquitinylation and proteasomal degradation of -β-catenin, resulting in an increase of the target gene cyclin D1, colony formation, and invasive activity in prostate cancer cells. The FLIP/β-catenin/cyclin D1 signals contributing to colony formation and invasion were reversed by selective silencing of c-FLIP expression [41]. Similarly, c-FLIPL, in cooperation with FADD, enhances canonical Wnt signaling by inhibiting proteasomal degradation of β-catenin, thus suggesting a new mechanism involved with tumorigenesis, in addition to inhibiting Fas signaling [75] (Fig. 4). Recently, Wang et al. [76] demonstrated that c-FLIP overexpression is also significantly related to the presence of HR-HPV infection during the progression of cervical squamous cell cancer and that c-FLIP is an early marker of cervical carcinogenesis. Moreover, c-FLIP overexpression accelerated progression to androgen independence by inhibiting apoptosis in LNCaP prostate tumors implanted in nude mice [77]. Similarly, c-FLIP is also an independent prognostic indicator in colorectal carcinomas [56], and its expression in Burkitt’s lymphomas is also associated with a poor clinical outcome and could be a reliable prognostic factor in this type of cancer [78]. Furthermore, recent studies show that c-FLIP expression may contribute to the carcinogenesis and aggressiveness of endometrial carcinomas and may serve as a useful prognostic factor for this tumor [79, 80]. Overexpression of c-FLIPL also increases the hypoxia-inducible factor-1α (HIF1α) [81]. Overexpression of HIF1α can result in genes responsible for global changes in cell proliferation, metastasis, and invasion.
TRANSCRIPTION AND TRANSLATION REGULATION OF c-FLIP
Several transcription factors are known to downregulate c-FLIPL and c-FLIPS expression. E2F1, a transcription factor that plays a crucial role during S phase progression and apoptosis, triggers apoptosis in various lung adenocarcinoma cell lines by specific downregulation of c-FLIPS leading to caspase-8 activation at the DISC. c-FLIPS downregulation by E2F1 occurs at the transcriptional level though its mechanism remains to be found. The specific overexpression of c-FLIPS is also seen in human lung adenocarcinomas with low levels of E2F1 [82].
Androgen receptor (AR) is a ligand-activated transcription factor that mediates androgen action and is essential for the growth, function, and cell differentiation of the prostate gland. Recently, Gao et al. [77] demonstrated that the prostate apoptosis response factor-4 (Par-4) functions as an androgen receptor coactivator. This factor directly interacts with the DNA-binding domain of the androgen receptor, increases interaction of this receptor with DNA, and elevates androgen receptor-dependent transcription. Interestingly, Par-4 is recruited onto the c-FLIP promoter in the presence of androgens. The binding to Par-4 to the promoter enhanced c-FLIP expression and this elevated expression was reversed by the expression of a dominant-negative Par-4. Enhanced c-FLIP expression was associated with prostate cancer progression to the androgen-resistant stage [77].
Another regulator of c-FLIP expression is p53 tumor suppressor protein. p53 may exert transcriptional upregulating effects on the c-FLIP gene and also has potent effects on promoting the degradation of c-FLIP protein [83-86]. Moreover, the gene encoding c-FLIP is a direct target of c-myc-mediated transcriptional repression. Chromatin immunoprecipitation and luciferase reporter analyses have shown that c-myc binds and represses the c-FLIP promoter. Overexpression of c-myc or activation of myc-estrogen receptor (ER) decreased c-FLIP levels in both cell culture and mouse models of c-myc-induced tumorigenesis, while knocking down c-myc using siRNA increased c-FLIP expression [87].
Li et al. [88] reported that c-FLIPL is transcriptionally regulated by the activator protein-1 (AP-1) family member protein c-Fos and that MG-132, an inhibitor of the proteasome, sensitizes TRAIL-resistant prostate cancer cells by inducing c-Fos and repressing c-FLIPL. Moreover, c-Fos, which is activated by MG-132, negatively regulates c-FLIPL by direct binding to the putative promoter region of the c-FLIPL gene. In addition to activating c-Fos, MG-132 activates c-Jun, another AP-1 family member. c-Fos heterodimerizes with c-Jun to form AP-1 and repress transcription of c-FLIPL.
Recent results demonstrated that c-FLIP is also regulated at the translational level. Panner et al. [89, 90] showed that TRAIL resistance in glioblastoma multiforme cells (GBM) is the result of overexpression of c-FLIPS, and that c-FLIPS expression is in turn translationally increased by activation of the Akt-mammalian target of rapamycin (mTOR)-p70 S6 kinase 1 (S6K1) pathway. Conversely, inhibition of mTOR or its target S6K1 suppressed polyribosomal accumulation of c-FLIPS mRNA, c-FLIPS protein expression, and promoted TRAIL sensitivity in GBM cells. An mTOR-independent pathway can also act through a Ral effector protein, RalBP1, to suppress cdc42-mediated activation of S6 kinase and the translation of the protein c-FLIPS [90, 91]. However, not much is known about Ral except that it is an effector of Ras oncogenesis. The Ral proteins (RalA and RalB) are small GTPases and their activity is controlled by the RalGEF family of proteins: RalGDS, Rgl, and Rlf [92]. Activated Ral molecules interact with Ral effectors, including RalBP1 (a cdc42 GTPase-activating protein) [93]. Recently, Panner et al. have shown that (1) the RalA pathway is linked to both the translational control and the tumor-suppressive pathways, (2) RalA activation is linked to TRAIL sensitivity via the suppression of c-FLIPS levels, and (3) RalA activation is linked to c-FLIPS and the extrinsic cell death pathway via RalBP1 and cdc42 [90]. These interesting results define an mTOR-independent link between RalA and translation. These authors also demonstrated that RalBP1 serves in a membrane-bound, GTPase-activating protein (GAP)-dependent manner to link RalA activation to cdc42 suppression, thereby leading to a decrease in c-FLIPS expression and enhancing TRAIL-induced apoptosis.
c-FLIP DEGRADATION
c-FLIP is predominantly degraded by the ubiquitin-proteasome degradation system [94]. Both c-FLIP isoforms can be degraded by the proteasome, but c-FLIPS appears to be particularly sensitive to ubiquitylation and proteasomal degradation, partly due to two crucial lysine residues in the C-terminal 20 amino acids that are unique to c-FLIPS [48]. The sensitivity of c-FLIPS to ubiquitin-mediated degradation adds a novel concept to DISC regulation and its control of apoptosis [48]. Downregulation of c-FLIPL and c-FLIPS due to degradation is observed in cells treated with various apoptosis-inducing agents. The cyclin-dependent kinase inhibitor flavopiridol degrades c-FLIP and increases apoptosis, which in turn can be reversed by an inhibitor of the ubiquitin-proteasome system [95]. Combined treatment with the anticancer agent 3,3′-diindolylmethane and TRAIL led to significant degradation of c-FLIP, predominantly via the ubiquitin-proteasome degradation system [94]. Moreover, Peroxisome Proliferator-Activated Receptor γ (PPARγ) agonists sensitize cancer cells to TRAIL by ubiquitination and proteasome-dependent c-FLIP degradation [96, 97].
Proteasome inhibitors are a new class of drugs that decrease proliferation and induce apoptosis in a variety of hematologic and solid malignancies [98]. Interestingly, several proteasome inhibitors lead to the downregulation of c-FLIPL and c-FLIPS [99, 100].
c-FLIP VARIANTS AS TARGETS FOR CANCER THERAPY
Strategies to lower the threshold for triggering cancer cell apoptosis may lead to new and more effective therapeutic regimens for cancer. TRAIL holds enormous promise as a cancer therapeutic due to its highly selective apoptosis-inducing action on neoplastic versus normal cells [2]. However, to exploit this opportunity, the problem of TRAIL resistance in cancer must first be overcome. While c-FLIPL may function as a fine-tuned regulator that manifests dual pro- and anti-apoptotic actions during development and in normal adult tissues, accumulating evidence indicates an anti-apoptotic role for c-FLIP in diverse types of human cancer [11, 13, 16, 18, 20, 101-104]. Notably, recent reports demonstrated that small interfering RNAs (siRNAs) that specifically knocked down the expression of c-FLIPL or c-FLIPS in cancer cell lines of various types augmented TRAIL- and chemotherapy-induced apoptosis [11, 17, 25, 105]. Therefore, the outlook for the therapeutic index of c-FLIP-targeted drugs appears excellent, not only because of the therapeutic efficacy observed in models of c-FLIP targeting in cancer therapy with TRAIL, but also because the current understanding of c-FLIP action in normal tissues supports the notion that c-FLIP-targeted cancer therapy will be well tolerated.
There does not appear to be a “handle” to inhibit c-FLIP function with small molecule ligands, since the cytoprotective DISC binding is mediated by highly conserved DEDs which function by homotypic binding. However, small molecules that induce c-FLIP downregulation and sensitize neoplastic cells to apoptosis induction by TRAIL have been identified in global, unbiased chemical screens. Our results revealed that forced expression of c-FLIPL or c-FLIPS caused resistance to apoptosis in the CCRF-HSB-2 leukemia cell line [68] and the MCF-7 breast cancer cell line (Wu CH, Day TW, Najafi F, and Safa AR, unpublished results). Moreover, c-FLIPL, but not c-FLIPS, inhibited apoptosis induced by the fluoropyrimidine 5-fluorouracil (5-FU), the topoisomerase-1 inhibitor irinotecan (CPT-11), and the DNA damaging agent oxaliplatin (OXA) in HCT116 colorectal cancer cells, suggesting that c-FLIPL is the more important splice form in mediating chemoresistance [20]. Furthermore, c-FLIP inhibits TRAIL-independent, DR5- and caspase 8-dependent apoptosis in response to chemotherapy in colorectal cancer cells. We (Wu CH, Day TW, and Safa AR, unpublished results) and others [106] have found that MCF-7 cells overexpressing c-FLIP are resistant to Taxol. The c-FLIPL variant protects against apoptosis induced by a diverse group of chemotherapeutic drugs with different mechanisms of action [102, 103, 107]. c-FLIP also inhibits spontaneous death ligand-independent cell death [25, 102], further supporting the notion that targeting c-FLIP may have therapeutic potential for cancer treatment. Cisplatin, camptothecin, and etoposide were all shown to downregulate c-FLIPS expression in human malignant glioma cells [103, 108]. Moreover, doxorubicin decreased levels of c-FLIP, particularly c-FLIPS in prostate cancer cell lines, and sensitized these cells to TRAIL [109]. Overexpression of the c-FLIPL variant has been found in ovarian cancer cells resistance to cisplatin [110]. Interestingly, the damaged DNA-binding protein 2 (DDB2), a component involved in the genomic repair of UV damage, was recently shown to be involved in the cross-resistance of a cisplatin-selected HeLa cell line (HR3) to Fas-inducing antibody and tumor necrosis factor (TNF)-α-mediated apoptosis [110]. The HR3 cell line exhibits enhanced expression of DDB2 and cross-resistance to UV-induced activation of caspases and apoptosis. The overexpression of DDB2 was shown to increase both endogenous and exogenous c-FLIP mRNA levels, and inhibition of c-FLIP by antisense oligonucleotides suppressed DDB2 protection, revealing that DDB2 regulates TNF signaling-mediated apoptosis via c-FLIP. Therefore, therapeutics capable of modulating the death-inducing ligands and/or chemotherapeutic agents by downregulating c-FLIP variants should hold enormous promise in future clinical trials.
We recently published the novel findings that Taxol downregulates c-FLIP (both c-FLIPS and c-FLIPL) in cancer cells [68], and that doses of the drug that induced concentration-dependent apoptosis were sufficient to abrogate the expression of both c-FLIP variants (Fig. 5). The significance of these findings to paclitaxel-induced neoplastic cell apoptosis was demonstrated by our results showing that transfection with an antisense c-FLIP plasmid abrogated c-FLIPS and c-FLIPL expression and sensitized cancer cells to Taxol [68]. These results demonstrate that c-FLIP isoforms are major contributing factors to cancer cell resistance to paclitaxel-induced apoptosis.
Fig. (5). Caspase-independent downregulation of c-FLIPL and c-FLIPS levels by Taxol.

(a) CCRF-HSB-2 cells were treated with 0.5, 1, 5, 10, or 50 nM Taxol for 24 h. Cell lysates were subjected to immunoblot analysis using an anti-c-FLIP antibody. (b) MCF-7 cells were treated with 10, 100, or 500 nM Taxol for 48 h. Cell lysates were subjected to immunoblot analysis using an anti-c-FLIP antibody. (c) CCRF-HSB-2 cells were treated with 100 μM of z-VAD-fmk for 3 h followed by 10 nM Taxol for an additional 24 h. Immunoblot analysis was conducted on cell lysates treated with or without z-VAD-fmk using anti-c-FLIP antibodies. Equivalent loading was confirmed by reprobing the same blot with anti-β-actin.
AGENTS KNOWN TO DOWNREGULATE c-FLIP
Many therapeutics capable of downregulating c-FLIP levels enhance the expression of the TRAIL receptor DR5, and in some cases enhance levels of both the DR4 and DR5 TRAIL receptors. One such agent is lexatumumab, a humanized agonistic monoclonal antibody specific for the DR5 receptor, which is currently in Phase I/II clinical trials in patients with advanced malignancies. Recently, lexatumumab was shown to decrease the expression of c-FLIPL and increase the level of DR5 in primary and metastatic renal cell carcinoma [111]. Similarly, the induction of apoptosis by the proteasome inhibitors MG-132 and PS-341 (bortezomib, VelcadeR) in primary chronic lymphocytic leukemia (CLL) cells and the Burkitt lymphoma cell line BJAB was associated with upregulation of TRAIL and its death receptors, DR4 and DR5, and decreased c-FLIP protein expression [99]. As mentioned earlier, c-FLIP is degraded via a ubiquitin-proteosome system. Therefore, PS-341 should increase c-FLIP and prevent apoptosis. Interestingly, Zhao et al. have shown that PS-341 decreases c-FLIP at the gene level [112]. In contrast, Liu et al. reported that PS-341 upregulates DR5 as well as c-FLIP and survivin in human non-small cell lung carcinomas (NSCLC) cells [113]. Thus, the effect of PS-341 on the regulation of c-FLIP expression may be cancer cell-type specific. A proposed central mechanism of action of PS-341 is the inhibition of NF-κB activation through inhibition of IκB degradation [114]. NF-κB activation results in upregulation of c-FLIP [115]. Thus, downregulation of the c-FLIP gene by PS-341 may be due to inhibition of NF-κB activation by this agent. PPARγ ligands exert PPARγ-independent effects on inducing DR5 expression and downregulating c-FLIP levels, leading to enhanced TRAIL-induced apoptosis [97]. Synthetic triterpenoids sensitize TRAIL-resistant T47D and MDA-MB-468 breast cancer cells to TRAIL-mediated apoptosis by downregulating c-FLIPL, and upregulate cell surface TRAIL receptors DR4 and DR5 in breast cancer cell [116]. Similarly, the p53 protein increased the sensitivity of testicular germ cells to mono-(2-ethylhexyl)phthalate-induced apoptosis by elevating the membrane levels of DR5 and Fas and decreasing the intracellular level of c-FLIP [86]. Progesterone induces apoptosis in TRAIL-resistant ovarian cancer cells by circumventing c-FLIPL overexpression [117].
Another class of agents known to downregulate c-FLIP expression are the histone deacetylases inhibitors (HDACi). These compounds are considered to be among the most promising agents for the developing novel cancer therapeutics [118, 119]. In diverse types of cancer cells, HDACi induce growth arrest, differentiation, and apoptosis. The results of clinical trials with several of these agents indicate that they are well tolerated at doses that have antitumor activity [119, 120]. Importantly, normal cells are almost always more resistant to HDACi than tumor cells [119, 120]. The mechanism of their antitumor activity is likely related at least in part to hyperacetylation of the promoter regions of the TRAIL and DR5 genes [121-123]. Co-treatment with the histone deacetylase inhibitor LAQ824 enhanced the mRNA and protein expression of the death receptors DR5 and/or DR4, but reduced the c-FLIP mRNA and protein levels and increased TRAIL-triggered apoptosis [124].
Elucidating the mechanisms regulating c-FLIP expression in neoplastic disease may lead to c-FLIP-downregulating targeted therapeutics that are efficacious and well tolerated in combination with TRAIL, conventional chemotherapeutic agents, or other therapies that activate the caspase-8/10-dependent apoptosis pathway. This approach may be especially effective in human malignancies that overexpress c-FLIP, i.e., glioblastoma multiforme, prostate, colorectal, endometrial, ovarian, mesothelial, and lung cancer [11, 17, 20, 41, 79, 89, 101, 125].
In an exciting twist in identifying regulators of TRAIL-triggered apoptosis, Jin et al. [126] used the intracellular death domain (DD) of DR5 as a target to screen a phage-displayed combinatorial peptide library. The DD of DR5 identified a peptide from the library that showed sequence similarity to a stretch of amino acids in the C-terminus of c-FLIPL. In vitro binding assays revealed that this peptide selectively interacted with the DD of DR5. Similarly, these authors demonstrated that full-length c-FLIPL and the C-terminal p12 domain of c-FLIP interacted with DR5 both in vitro and in mammalian cells, and that this interaction was independent of TRAIL. Moreover, when cancer cells were treated with TRAIL, c-FLIPL was released from DR5 and FADD was recruited to the active DR5 signaling complex. Of particular significance, a cellular membrane-permeable version of this peptide induces apoptosis in mammalian cells. Therefore, these results provide a basis for rationally designing peptidomimetic or small molecule targeted therapeutics which, by interacting with the C-terminal domain of c-FLIPL, release DR5 and subsequently form the active DR5 signaling complex for triggering apoptosis by TRAIL and/or chemotherapeutic agents in cancer cells resistant to these agents.
Selective c-FLIPL and c-FLIPS knockdown strategies to sensitize cancer cells to TRAIL may lead to combination therapies consisting of TRAIL plus innovative c-FLIP variant-selective gene therapy strategies. c-FLIPL and c-FLIPS knockdown could provide significant clinical advances in the use of TRAIL in treating lung cancer. Similarly, many agents mentioned above abrogate expression of the c-FLIP variants, suggesting that these experimental therapeutics have potential value to reverse cancer cell resistance to TRAIL, paclitaxel and cisplatin.
Oncolytic viruses are known to infect, replicate in, and eventually eliminate tumor cells without harming normal cells [127]. Reoviruses are oncolytic viruses that multiply preferentially in tumor cells that harbor activated ras family or ras-signaling pathways, while sparing normal cells [127]. Recently, Clarke et al. [128] demonstrated that reovirus infection results in the downregulation of c-FLIP and increased TRAIL-induced apoptosis in ovarian cancer cells, and overexpression of c-FLIP blocked reovirus-induced sensitization of these cells to TRAIL-induced apoptosis. Using oncolytic viruses aimed at downregulating c-FLIP may be potentially useful in combination with TRAIL and/or chemotherapeutic agents to eradicate cancer cells.
c-FLIPS and/or c-FLIPL serve as rational target(s) in various cancers for novel small molecule targeted therapeutics and/or gene therapy strategies., e.g., small molecule therapeutics that selectively downregulate c-FLIPS or c-FLIPL and gene therapy strategies that knock down a specific c-FLIP variant. Developing these innovative therapeutic strategies in conjunction with TRAIL and Taxol will potentially overcome the barrier of dose-limiting toxicity in cancer chemotherapy. Furthermore, rather than empirically combining cytotoxic drugs to attempt to enhance their therapeutic response, modulating specific targets in cancer cells based on rational preclinical design is the most logical approach to eradicate cancer. The validity of this approach in treating metastatic solid tumors is exemplified by the efficacy of targeted inhibition of the protein kinase c-Kit by imatinib mesylate (GleevecR) in the first-line treatment of metastatic gastrointestinal stromal tumor (GIST) [129]. TRAIL or chemotherapy resistance in diverse cancer cell types can be reversed by parallel treatment with other substances, such as agents known to downregulate c-FLIP variants.
CONCLUSION
Apoptosis is a mechanism of programmed cell death involving cellular signaling pathways that induce cells to self-destruct in response to specific cellular cues or environmental hazards. The mechanism of action of chemotherapeutic agents often involves the induction of cancer cell apoptosis, and resistance to apoptosis in these cells is a major contributing factor in drug resistance. Therefore, using targeted therapeutics to restore apoptosis signaling in cancer cells has great potential to improve the outcome of cancer chemotherapy. Several studies have demonstrated elevated c-FLIP levels in various tumor types suggesting that c-FLIP may be a relevant clinical target. The current state of the art reviewed in this article suggests that c-FLIP may represent an important clinical marker of drug resistance, and that targeting c-FLIP in combination with TRAIL or standard chemotherapies has therapeutic potential for treating cancer. The foregoing discussion justifies optimism that future cancer therapy will be improved by innovations that replace current standard chemotherapy with targeted therapy or combined chemotherapy with drug resistance-reversing targeted therapy, e.g., combination regimens of chemotherapy and small molecule drugs that downregulate c-FLIP.
ACKNOWLEDGEMENTS
We would like to thank Dr. Mary D. Kraeszig for her editorial assistance. The authors have been supported by research grants from the National Cancer Institute (CA 080734, CA 90878, and CA 101743), Department of Defense (DOD) (OC 06095), the Lung Cancer Working Group (LCWG), and the Indiana University Cancer Center Translational Research Acceleration Collaboration (ITRAC) initiative to Ahmad R. Safa.
REFERENCES
- [1].Clarke R, Leonessa F, Trock B. Multidrug resistance/P-glycoprotein and breast cancer: review and meta-analysis. Semin. Oncol. 2005;32:S9–S15. doi: 10.1053/j.seminoncol.2005.09.009. [DOI] [PubMed] [Google Scholar]
- [2].Fojo AT, Menefee M. Microtubule targeting agents: basic mechanisms of multidrug resistance (MDR) Semin. Oncol. 2005;32:S3–S8. doi: 10.1053/j.seminoncol.2005.09.010. [DOI] [PubMed] [Google Scholar]
- [3].Roberti A, La Sala D, Cinti C. Multiple genetic and epigenetic interacting mechanisms contribute to clonally selection of drug-resistant tumors: current views and new therapeutic prospective. J. Cell. Physiol. 2006;207:571–581. doi: 10.1002/jcp.20515. [DOI] [PubMed] [Google Scholar]
- [4].Glavinas H, Krajcsi P, Cserepes J, Sarkadi B. The role of ABC transporters in drug resistance, metabolism and toxicity. Curr. Drug. Deliv. 2004;1:27–42. doi: 10.2174/1567201043480036. [DOI] [PubMed] [Google Scholar]
- [5].Safa AR. Identification and characterization of the binding sites of P-glycoprotein for multidrug resistance-related drugs and modulators. Curr. Med. Chem. Anticancer Agents. 2004;4:1–17. doi: 10.2174/1568011043482142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mashima T, Tsuruo T. Defects of the apoptotic pathway as therapeutic target against cancer. Drug Resist. Updat. 2005;8:339–343. doi: 10.1016/j.drup.2005.11.001. [DOI] [PubMed] [Google Scholar]
- [7].Vidal L, Attard G, Kaye S, De Bono J. Reversing resistance to targeted therapy. J. Chemother. 2004;16:7–12. doi: 10.1179/joc.2004.16.Supplement-1.7. [DOI] [PubMed] [Google Scholar]
- [8].Duiker EW, Mom CH, de Jong S, Willemse PH, Gietema JA, van der Zee AG, de Vries EG. The clinical trail of TRAIL. Eur. J. Cancer. 2006;42:2233–2240. doi: 10.1016/j.ejca.2006.03.018. [DOI] [PubMed] [Google Scholar]
- [9].Chaudhari BR, Murphy RF, Agrawal DK. Following the TRAIL to apoptosis. Immunol. Res. 2006;35:249–262. doi: 10.1385/IR:35:3:249. [DOI] [PubMed] [Google Scholar]
- [10].Bouralexis S, Findlay DM, Evdokiou A. Death to the bad guys: targeting cancer via Apo2L/TRAIL. Apoptosis. 2005;10:35–51. doi: 10.1007/s10495-005-6060-0. [DOI] [PubMed] [Google Scholar]
- [11].Rippo MR, Moretti S, Vescovi S, Tomasetti M, Orecchia S, Amici G, Catalano A, Procopio A. FLIP overexpression inhibits death receptor-induced apoptosis in malignant mesothelial cells. Oncogene. 2004;23:7753–7760. doi: 10.1038/sj.onc.1208051. [DOI] [PubMed] [Google Scholar]
- [12].Morales JC, Ruiz-Magana MJ, Ruiz-Ruiz C. Regulation of the resistance to TRAIL-induced apoptosis in human primary T lymphocytes: role of NF-kappaB inhibition. Mol. Immunol. 2007;44:2587–2597. doi: 10.1016/j.molimm.2006.12.015. [DOI] [PubMed] [Google Scholar]
- [13].Mathas S, Lietz A, Anagnostopoulos I, Hummel F, Wiesner B, Janz M, Jundt F, Hirsch B, Johrens-Leder K, Vornlocher HP, Bommert K, Stein H, Dorken B. c-FLIP mediates resistance of Hodgkin/Reed-Sternberg cells to death receptor-induced apoptosis. J. Exp. Med. 2004;199:1041–1052. doi: 10.1084/jem.20031080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Saulle E, Petronelli A, Pasquini L, Petrucci E, Mariani G, Biffoni M, Ferretti G, Scambia G, Benedetti-Panici P, Cognetti F, Humphreys R, Peschle C, Testa U. Proteasome inhibitors sensitize ovarian cancer cells to TRAIL induced apoptosis. Apoptosis. 2007;12:635–655. doi: 10.1007/s10495-006-0025-9. [DOI] [PubMed] [Google Scholar]
- [15].Shankar S, Srivastava RK. Enhancement of therapeutic potential of TRAIL by cancer chemotherapy and irradiation: mechanisms and clinical implications. Drug Resist. Updat. 2004;7:139–156. doi: 10.1016/j.drup.2004.03.002. [DOI] [PubMed] [Google Scholar]
- [16].Zhang X, Jin TG, Yang H, DeWolf WC, Khosravi-Far R, Olumi AF. Persistent c-FLIP(L) expression is necessary and sufficient to maintain resistance to tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in prostate cancer. Cancer Res. 2004;64:7086–7091. doi: 10.1158/0008-5472.CAN-04-1498. [DOI] [PubMed] [Google Scholar]
- [17].Galligan L, Longley DB, McEwan M, Wilson TR, McLaughlin K, Johnston PG. Chemotherapy and TRAIL-mediated colon cancer cell death: the roles of p53, TRAIL receptors, and c-FLIP. Mol. Cancer Ther. 2005;4:2026–2036. doi: 10.1158/1535-7163.MCT-05-0262. [DOI] [PubMed] [Google Scholar]
- [18].Van Geelen CM, de Vries EG, de Jong S. Lessons from TRAIL-resistance mechanisms in colorectal cancer cells: paving the road to patient-tailored therapy. Drug Resist. Updat. 2004;7:345–358. doi: 10.1016/j.drup.2004.11.002. [DOI] [PubMed] [Google Scholar]
- [19].Xiao C, Yang BF, Song JH, Schulman H, Li L, Hao C. Inhibition of CaMKII-mediated c-FLIP expression sensitizes malignant melanoma cells to TRAIL-induced apoptosis. Exp. Cell. Res. 2005;304:244–255. doi: 10.1016/j.yexcr.2004.11.002. [DOI] [PubMed] [Google Scholar]
- [20].Longley DB, Wilson TR, McEwan M, Allen WL, McDermott U, Galligan L, Johnston PG. c-FLIP inhibits chemotherapy-induced colorectal cancer cell death. Oncogene. 2005;25:838–848. doi: 10.1038/sj.onc.1209122. [DOI] [PubMed] [Google Scholar]
- [21].Malhi H, Gores GJ. TRAIL resistance results in cancer progression: a TRAIL to perdition? Oncogene. 2006;25:7333–7335. doi: 10.1038/sj.onc.1209765. [DOI] [PubMed] [Google Scholar]
- [22].Mezzanzanica D, Balladore E, Turatti F, Luison E, Alberti P, Bagnoli M, Figini M, Mazzoni A, Raspagliesi F, Oggionni M, Pilotti S, Canevari S. CD95-mediated apoptosis is impaired at receptor level by cellular FLICE-inhibitory protein (long form) in wild-type p53 human ovarian carcinoma. Clin. Cancer Res. 2004;10:5202–5214. doi: 10.1158/1078-0432.CCR-03-0537. [DOI] [PubMed] [Google Scholar]
- [23].Steele LP, Georgopoulos NT, Southgate J, Selby PJ, Trejdosiewicz LK. Differential susceptibility to TRAIL of normal versus malignant human urothelial cells. Cell Death Differ. 2006;13:1564–1576. doi: 10.1038/sj.cdd.4401846. [DOI] [PubMed] [Google Scholar]
- [24].Shin EC, Seong YR, Kim CH, Kim H, Ahn YS, Kim K, Kim SJ, Hong SS, Park JH. Human hepatocellular carcinoma cells resist to TRAIL-induced apoptosis, and the resistance is abolished by cisplatin. Exp. Mol. Med. 2002;34:114–122. doi: 10.1038/emm.2002.17. [DOI] [PubMed] [Google Scholar]
- [25].Sharp DA, Lawrence DA, Ashkenazi A. Selective knockdown of the long variant of cellular FLICE inhibitory protein augments death receptor-mediated caspase-8 activation and apoptosis. J. Biol. Chem. 2005;280:19401–19409. doi: 10.1074/jbc.M413962200. [DOI] [PubMed] [Google Scholar]
- [26].Lane D, Robert V, Grondin R, Rancourt C, Piche A. Malignant ascites protect against TRAIL-induced apoptosis by activating the PI3K/Akt pathway in human ovarian carcinoma cells. Int. J. Cancer. 2007;29:29. doi: 10.1002/ijc.22840. [DOI] [PubMed] [Google Scholar]
- [27].Kataoka T. The caspase-8 modulator c-FLIP. Crit. Rev. Immunol. 2005;25:31–58. doi: 10.1615/critrevimmunol.v25.i1.30. [DOI] [PubMed] [Google Scholar]
- [28].Baier A, Meineckel I, Gay S, Pap T. Apoptosis in rheumatoid arthritis. Curr. Opin. Rheumatol. 2003;15:274–279. doi: 10.1097/00002281-200305000-00015. [DOI] [PubMed] [Google Scholar]
- [29].Thome M, Tschopp J. Regulation of lymphocyte proliferation and death by FLIP. Nat. Rev. Immunol. 2001;1:50–58. doi: 10.1038/35095508. [DOI] [PubMed] [Google Scholar]
- [30].Cereghetti GM, Scorrano L. The many shapes of mitochondrial death. Oncogene. 2006;25:4717–4724. doi: 10.1038/sj.onc.1209605. [DOI] [PubMed] [Google Scholar]
- [31].Meng XW, Lee SH, Kaufmann SH. Apoptosis in the treatment of cancer: a promise kept? Curr. Opin. Cell. Biol. 2006;18:668–676. doi: 10.1016/j.ceb.2006.10.008. [DOI] [PubMed] [Google Scholar]
- [32].Gogvadze V, Orrenius S. Mitochondrial regulation of apoptotic cell death. Chem. Biol. Interact. 2006;163:4–14. doi: 10.1016/j.cbi.2006.04.010. [DOI] [PubMed] [Google Scholar]
- [33].Ferri KF, Jacotot E, Blanco J, Este JA, Zamzami N, Susin SA, Xie Z, Brothers G, Reed JC, Penninger JM, Kroemer G. Apoptosis control in syncytia induced by the HIV type 1-envelope glycoprotein complex: role of mitochondria and caspases. J. Exp. Med. 2000;192:1081–1092. doi: 10.1084/jem.192.8.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- [35].Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- [36].Wolf BB, Green DR. Suicidal tendencies: apoptotic cell death by caspase family proteinases. J. Biol. Chem. 1999;274:20049–20052. doi: 10.1074/jbc.274.29.20049. [DOI] [PubMed] [Google Scholar]
- [37].Strasser A, O’Connor L, Dixit VM. Apoptosis signaling. Annu. Rev. Biochem. 2000;69:217–245. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- [38].Slee EA, Keogh SA, Martin SJ. Cleavage of BID during cytotoxic drug and UV radiation-induced apoptosis occurs downstream of the point of Bcl-2 action and is catalysed by caspase-3: a potential feedback loop for amplification of apoptosis-associated mitochondrial cytochrome c release. Cell Death Differ. 2000;7:556–565. doi: 10.1038/sj.cdd.4400689. [DOI] [PubMed] [Google Scholar]
- [39].Krueger A, Baumann S, Krammer PH, Kirchhoff S. FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol. Cell. Biol. 2001;21:8247–8254. doi: 10.1128/MCB.21.24.8247-8254.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ashkenazi A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat. Rev. Cancer. 2002;2:420–430. doi: 10.1038/nrc821. [DOI] [PubMed] [Google Scholar]
- [41].Shimada K, Nakamura M, Matsuyoshi S, Ishida E, Konishi N. Specific positive and negative effects of FLIP on cell survival in human prostate cancer. Carcinogenesis. 2006;27:1349–1357. doi: 10.1093/carcin/bgi380. [DOI] [PubMed] [Google Scholar]
- [42].Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386:517–521. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- [43].Hu S, Vincenz C, Buller M, Dixit VM. A novel family of viral death effector domain-containing molecules that inhibit both CD-95- and tumor necrosis factor receptor-1-induced apoptosis. J. Biol. Chem. 1997;272:9621–9624. doi: 10.1074/jbc.272.15.9621. [DOI] [PubMed] [Google Scholar]
- [44].Bertin J, Armstrong RC, Ottilie S, Martin DA, Wang Y, Banks S, Wang GH, Senkevich TG, Alnemri ES, Moss B, Lenardo MJ, Tomaselli KJ, Cohen JI. Death effector domain-containing herpesvirus and poxvirus proteins inhibit both Fas- and TNFR1-induced apoptosis. Proc. Natl. Acad. Sci. USA. 1997;94:1172–1176. doi: 10.1073/pnas.94.4.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- [46].Golks A, Brenner D, Fritsch C, Krammer PH, Lavrik IN. c-FLIPR, a new regulator of death receptor-induced apoptosis. J. Biol. Chem. 2005;280:14507–14513. doi: 10.1074/jbc.M414425200. [DOI] [PubMed] [Google Scholar]
- [47].Tschopp J, Irmler M, Thome M. Inhibition of Fas death signals by FLIPs. Curr. Opin. Immunol. 1998;10:552–558. doi: 10.1016/s0952-7915(98)80223-9. [DOI] [PubMed] [Google Scholar]
- [48].Poukkula M, Kaunisto A, Hietakangas V, Denessiouk K, Katajamaki T, Johnson MS, Sistonen L, Eriksson JE. Rapid turnover of c-FLIPS is determined by its unique C-terminal tail. J. Biol. Chem. 2005;10:10. doi: 10.1074/jbc.M504019200. [DOI] [PubMed] [Google Scholar]
- [49].Cohen GM. Caspases: the executioners of apoptosis. Biochem. J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Krueger A, Schmitz I, Baumann S, Krammer PH, Kirchhoff S. Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J. Biol. Chem. 2001;276:20633–20640. doi: 10.1074/jbc.M101780200. [DOI] [PubMed] [Google Scholar]
- [51].Kataoka T, Tschopp J. N-terminal fragment of c-FLIP(L) processed by caspase 8 specifically interacts with TRAF2 and induces activation of the NF-kappaB signaling pathway. Mol. Cell. Biol. 2004;24:2627–2636. doi: 10.1128/MCB.24.7.2627-2636.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Srinivasula SM, Ahmad M, Ottilie S, Bullrich F, Banks S, Wang Y, Fernandes-Alnemri T, Croce CM, Litwack G, Tomaselli KJ, Armstrong RC, Alnemri ES. FLAME-1, a novel FADD-like anti-apoptotic molecule that regulates Fas/TNFR1-induced apoptosis. J. Biol. Chem. 1997;272:18542–18545. doi: 10.1074/jbc.272.30.18542. [DOI] [PubMed] [Google Scholar]
- [53].Wang X, Wang Y, Zhang J, Kim HP, Ryter SW, Choi AM. FLIP protects against hypoxia/reoxygenation-induced endothelial cell apoptosis by inhibiting Bax activation. Mol. Cell. Biol. 2005;25:4742–4751. doi: 10.1128/MCB.25.11.4742-4751.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [54].Thorburn A. Death receptor-induced cell killing. Cell Signal. 2004;16:139–144. doi: 10.1016/j.cellsig.2003.08.007. [DOI] [PubMed] [Google Scholar]
- [55].Yeh WC, Itie A, Elia AJ, Ng M, Shu HB, Wakeham A, Mirtsos C, Suzuki N, Bonnard M, Goeddel DV, Mak TW. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity. 2000;12:633–642. doi: 10.1016/s1074-7613(00)80214-9. [DOI] [PubMed] [Google Scholar]
- [56].Burns TF, El-Deiry WS. Identification of inhibitors of TRAIL-induced death (ITIDs) in the TRAIL-sensitive colon carcinoma cell line SW480 using a genetic approach. J. Biol. Chem. 2001;276:37879–37886. doi: 10.1074/jbc.M103516200. [DOI] [PubMed] [Google Scholar]
- [57].Peter ME. The flip side of FLIP. Biochem. J. 2004;382:e1–e3. doi: 10.1042/BJ20041143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Micheau O, Thome M, Schneider P, Holler N, Tschopp J, Nicholson DW, Briand C, Grutter MG. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J. Biol. Chem. 2002;277:45162–45171. doi: 10.1074/jbc.M206882200. [DOI] [PubMed] [Google Scholar]
- [59].Chang DW, Xing Z, Pan Y, Algeciras-Schimnich A, Barnhart BC, Yaish-Ohad S, Peter ME, Yang X. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. Embo J. 2002;21:3704–3714. doi: 10.1093/emboj/cdf356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Boatright KM, Deis C, Denault JB, Sutherlin DP, Salvesen GS. Activation of caspases-8 and -10 by FLIP(L) Biochem. J. 2004;382:651–657. doi: 10.1042/BJ20040809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Cottet S, Dupraz P, Hamburger F, Dolci W, Jaquet M, Thorens B. cFLIP protein prevents tumor necrosis factor-alpha-mediated induction of caspase-8-dependent apoptosis in insulin-secreting betaTc-Tet cells. Diabetes. 2002;51:1805–1814. doi: 10.2337/diabetes.51.6.1805. [DOI] [PubMed] [Google Scholar]
- [62].Fang LW, Tai TS, Yu WN, Liao F, Lai MZ. Phosphatidylinositide 3-kinase priming couples c-FLIP to T cell activation. J. Biol. Chem. 2004;279:13–18. doi: 10.1074/jbc.M303860200. [DOI] [PubMed] [Google Scholar]
- [63].Chaudhary PM, Eby MT, Jasmin A, Kumar A, Liu L, Hood L. Activation of the NF-kappaB pathway by caspase 8 and its homologs. Onco-gene. 2000;19:4451–4460. doi: 10.1038/sj.onc.1203812. [DOI] [PubMed] [Google Scholar]
- [64].Kataoka T, Budd RC, Holler N, Thome M, Martinon F, Irmler M, Burns K, Hahne M, Kennedy N, Kovacsovics M, Tschopp J. The caspase-8 inhibitor FLIP promotes activation of NF-kappaB and Erk signaling pathways. Curr. Biol. 2000;10:640–648. doi: 10.1016/s0960-9822(00)00512-1. [DOI] [PubMed] [Google Scholar]
- [65].Dohrman A, Kataoka T, Cuenin S, Russell JQ, Tschopp J, Budd RC. Cellular FLIP (long form) regulates CD8+ T cell activation through caspase-8-dependent NF-kappa B activation. J. Immunol. 2005;174:5270–5278. doi: 10.4049/jimmunol.174.9.5270. [DOI] [PubMed] [Google Scholar]
- [66].Golks A, Brenner D, Krammer PH, Lavrik IN. The c-FLIP-NH2 terminus (p22-FLIP) induces NF-kappaB activation. J. Exp. Med. 2006;203:1295–1305. doi: 10.1084/jem.20051556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, Liu YC, Karin M. The E3 ubiquitin ligase Itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell. 2006;124:601–613. doi: 10.1016/j.cell.2006.01.021. [DOI] [PubMed] [Google Scholar]
- [68].Day TW, Najafi F, Wu CH, Safa AR. Cellular FLICE-like inhibitory protein (c-FLIP): A novel target for Taxol-induced apoptosis. Biochem. Pharmacol. 2006;71:1551–1561. doi: 10.1016/j.bcp.2006.02.015. [DOI] [PubMed] [Google Scholar]
- [69].Kim YY, Park BJ, Seo GJ, Lim JY, Lee SM, Kimm KC, Park C, Kim J, Park SI. Long form of cellular FLICE-inhibitory protein interacts with Daxx and prevents Fas-induced JNK activation. Biochem. Biophys. Res. Commun. 2003;312:426–433. doi: 10.1016/j.bbrc.2003.10.144. [DOI] [PubMed] [Google Scholar]
- [70].Nakajima A, Komazawa-Sakon S, Takekawa M, Sasazuki T, Yeh WC, Yagita H, Okumura K, Nakano H. An antiapoptotic protein, c-FLIPL, directly binds to MKK7 and inhibits the JNK pathway. Embo J. 2006;25:5549–5559. doi: 10.1038/sj.emboj.7601423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Yang BF, Xiao C, Roa WH, Krammer PH, Hao C. Calcium/calmodulin-dependent protein kinase II regulation of c-FLIP expression and phosphorylation in modulation of Fas-mediated signaling in malignant glioma cells. J. Biol. Chem. 2003;278:7043–7050. doi: 10.1074/jbc.M211278200. [DOI] [PubMed] [Google Scholar]
- [72].Higuchi H, Yoon JH, Grambihler A, Werneburg N, Bronk SF, Gores GJ. Bile acids stimulate c-FLIP phosphorylation enhancing TRAIL-mediated apoptosis. J. Biol. Chem. 2003;278:454–461. doi: 10.1074/jbc.M209387200. [DOI] [PubMed] [Google Scholar]
- [73].Misra RS, Russell JQ, Koenig A, Hinshaw-Makepeace JA, Wen R, Wang D, Huo H, Littman DR, Ferch U, Ruland J, Thome M, Budd RC. Caspase-8 and c-FLIPL Associate in Lipid Rafts with NF-{kappa}B Adaptors during T Cell Activation. J. Biol. Chem. 2007;282:19365–19374. doi: 10.1074/jbc.M610610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Budd RC, Yeh WC, Tschopp J. cFLIP regulation of lymphocyte activation and development. Nat. Rev. Immunol. 2006;6:196–204. doi: 10.1038/nri1787. [DOI] [PubMed] [Google Scholar]
- [75].Naito M, Katayama R, Ishioka T, Suga A, Takubo K, Nanjo M, Hashimoto C, Taira M, Takada S, Takada R, Kitagawa M, Matsuzawa S, Reed JC, Tsuruo T. Cellular FLIP inhibits beta-catenin ubiquitylation and enhances Wnt signaling. Mol. Cell. Biol. 2004;24:8418–8427. doi: 10.1128/MCB.24.19.8418-8427.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wang W, Wang S, Song X, Sima N, Xu X, Luo A, Chen G, Deng D, Xu Q, Meng L, Lu Y, Ma D. The relationship between c-FLIP expression and human papillomavirus E2 gene disruption in cervical carcino-genesis. Gynecol. Oncol. 2007;105:571–577. doi: 10.1016/j.ygyno.2007.01.051. [DOI] [PubMed] [Google Scholar]
- [77].Gao S, Wang H, Lee P, Melamed J, Li CX, Zhang F, Wu H, Zhou L, Wang Z. Androgen receptor and prostate apoptosis response factor-4 target the c-FLIP gene to determine survival and apoptosis in the prostate gland. J. Mol. Endocrinol. 2006;36:463–483. doi: 10.1677/jme.1.01991. [DOI] [PubMed] [Google Scholar]
- [78].Valnet-Rabier MB, Challier B, Thiebault S, Angonin R, Margueritte G, Mougin C, Kantelip B, Deconinck E, Cahn JY, Fest T. c-FLIP protein expression in Burkitt’s lymphomas is associated with a poor clinical outcome. Br. J. Haematol. 2005;128:767–773. doi: 10.1111/j.1365-2141.2005.05378.x. [DOI] [PubMed] [Google Scholar]
- [79].Dolcet X, Llobet D, Pallares J, Rue M, Comella JX, Matias-Guiu X. FLIP is frequently expressed in endometrial carcinoma and has a role in resistance to TRAIL-induced apoptosis. Lab. Invest. 2005;85:885–894. doi: 10.1038/labinvest.3700286. [DOI] [PubMed] [Google Scholar]
- [80].Chen HX, Liu YJ, Zhou XD, Luo RY. Expression of cellular FLICE/caspase-8 inhibitory protein is associated with malignant potential in endometrial carcinoma. Int. J. Gynecol. Cancer. 2005;15:663–670. doi: 10.1111/j.1525-1438.2005.00122.x. [DOI] [PubMed] [Google Scholar]
- [81].Ishioka T, Katayama R, Kikuchi R, Nishimoto M, Takada S, Takada R, Matsuzawa S, Reed JC, Tsuruo T, Naito M. Impairment of the ubiquitin-proteasome system by cellular FLIP. Genes Cells. 2007;12:735–744. doi: 10.1111/j.1365-2443.2007.01087.x. [DOI] [PubMed] [Google Scholar]
- [82].Salon C, Eymin B, Micheau O, Chaperot L, Plumas J, Brambilla C, Brambilla E, Gazzeri S. E2F1 induces apoptosis and sensitizes human lung adenocarcinoma cells to death-receptor-mediated apoptosis through specific downregulation of c-FLIP(short) Cell Death Differ. 2006;13:260–272. doi: 10.1038/sj.cdd.4401739. [DOI] [PubMed] [Google Scholar]
- [83].Zhou XD, Yu JP, Chen HX, Yu HG, Luo HS. Expression of cellular FLICE-inhibitory protein and its association with p53 mutation in colon cancer. World J. Gastroenterol. 2005;11:2482–2485. doi: 10.3748/wjg.v11.i16.2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Bartke T, Siegmund D, Peters N, Reichwein M, Henkler F, Scheurich P, Wajant H. p53 upregulates c-FLIP, inhibits transcription of NF-kappaB-regulated genes and induces caspase-8-independent cell death in DLD-1 cells. Oncogene. 2001;20:571–580. doi: 10.1038/sj.onc.1204124. [DOI] [PubMed] [Google Scholar]
- [85].Chandrasekaran Y, McKee CM, Ye Y, Richburg JH. Influence of TRP53 status on FAS membrane localization, CFLAR (c-FLIP) ubiquitinylation, and sensitivity of GC-2spd (ts) cells to undergo FAS-mediated apoptosis. Biol. Reprod. 2006;74:560–568. doi: 10.1095/biolreprod.105.045146. [DOI] [PubMed] [Google Scholar]
- [86].Chandrasekaran Y, Richburg JH. The p53 protein influences the sensitivity of testicular germ cells to mono-(2-ethylhexyl) phthalate-induced apoptosis by increasing the membrane levels of Fas and DR5 and decreasing the intracellular amount of c-FLIP. Biol. Reprod. 2005;72:206–213. doi: 10.1095/biolreprod.104.030858. [DOI] [PubMed] [Google Scholar]
- [87].Ricci MS, Jin Z, Dews M, Yu D, Thomas-Tikhonenko A, Dicker DT, El-Deiry WS. Direct repression of FLIP expression by c-myc is a major determinant of TRAIL sensitivity. Mol. Cell. Biol. 2004;24:8541–8555. doi: 10.1128/MCB.24.19.8541-8555.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Li W, Zhang X, Olumi AF. MG-132 sensitizes TRAIL-resistant prostate cancer cells by activating c-Fos/c-Jun heterodimers and repressing c-FLIP(L) Cancer Res. 2007;67:2247–2255. doi: 10.1158/0008-5472.CAN-06-3793. [DOI] [PubMed] [Google Scholar]
- [89].Panner A, James CD, Berger MS, Pieper RO. mTOR controls FLIPS translation and TRAIL sensitivity in glioblastoma multiforme cells. Mol. Cell. Biol. 2005;25:8809–8823. doi: 10.1128/MCB.25.20.8809-8823.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Panner A, Nakamura JL, Parsa AT, Rodriguez-Viciana P, Berger MS, Stokoe D, Pieper RO. mTOR-independent translational control of the extrinsic cell death pathway by RalA. Mol. Cell. Biol. 2006;26:7345–7357. doi: 10.1128/MCB.00126-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Panner A, Parsa AT, Pieper RO. Translational regulation of TRAIL sensitivity. Cell Cycle. 2006;5:147–150. doi: 10.4161/cc.5.2.2359. [DOI] [PubMed] [Google Scholar]
- [92].Wolthuis RM, Bos JL. Ras caught in another affair: the exchange factors for Ral. Curr. Opin. Genet. Dev. 1999;9:112–117. doi: 10.1016/s0959-437x(99)80016-1. [DOI] [PubMed] [Google Scholar]
- [93].Jullien-Flores V, Dorseuil O, Romero F, Letourneur F, Saragosti S, Berger R, Tavitian A, Gacon G, Camonis JH. Bridging Ral GTPase to Rho pathways. RLIP76, a Ral effector with CDC42/Rac GTPase-activating protein activity. J. Biol. Chem. 1995;270:22473–22477. doi: 10.1074/jbc.270.38.22473. [DOI] [PubMed] [Google Scholar]
- [94].Zhang S, Shen HM, Ong CN. Down-regulation of c-FLIP contributes to the sensitization effect of 3,3′-diindolylmethane on TRAIL-induced apoptosis in cancer cells. Mol. Cancer Ther. 2005;4:1972–1981. doi: 10.1158/1535-7163.MCT-05-0249. [DOI] [PubMed] [Google Scholar]
- [95].Palacios C, Yerbes R, Lopez-Rivas A. Flavopiridol induces cellular FLICE-inhibitory protein degradation by the proteasome and promotes TRAIL-induced early signaling and apoptosis in breast tumor cells. Cancer Res. 2006;66:8858–8869. doi: 10.1158/0008-5472.CAN-06-0808. [DOI] [PubMed] [Google Scholar]
- [96].Kim Y, Suh N, Sporn M, Reed JC. An inducible pathway for degradation of FLIP protein sensitizes tumor cells to TRAIL-induced apoptosis. J. Biol. Chem. 2002;277:22320–22329. doi: 10.1074/jbc.M202458200. [DOI] [PubMed] [Google Scholar]
- [97].Zou W, Liu X, Yue P, Khuri FR, Sun SY. PPARgamma ligands enhance TRAIL-induced apoptosis through DR5 upregulation and c-FLIP downregulation in human lung cancer cells. Cancer Biol. Ther. 2007;6:99–106. doi: 10.4161/cbt.6.1.3555. [DOI] [PubMed] [Google Scholar]
- [98].Milano A, Iaffaioli RV, Caponigro F. The proteasome: a worthwhile target for the treatment of solid tumours? Eur. J. Cancer. 2007;43:1125–1133. doi: 10.1016/j.ejca.2007.01.038. [DOI] [PubMed] [Google Scholar]
- [99].Kabore AF, Sun J, Hu X, McCrea K, Johnston JB, Gibson SB. The TRAIL apoptotic pathway mediates proteasome inhibitor induced apoptosis in primary chronic lymphocytic leukemia cells. Apoptosis. 2006;11:1175–1193. doi: 10.1007/s10495-006-8048-9. [DOI] [PubMed] [Google Scholar]
- [100].Sayers TJ, Brooks AD, Koh CY, Ma W, Seki N, Raziuddin A, Blazar BR, Zhang X, Elliott PJ, Murphy WJ. The proteasome inhibitor PS-341 sensitizes neoplastic cells to TRAIL-mediated apoptosis by reducing levels of c-FLIP. Blood. 2003;102:303–310. doi: 10.1182/blood-2002-09-2975. [DOI] [PubMed] [Google Scholar]
- [101].Abedini MR, Qiu Q, Yan X, Tsang BK. Possible role of FLICE-like inhibitory protein (FLIP) in chemoresistant ovarian cancer cells in vitro. Oncogene. 2004;23:6997–7004. doi: 10.1038/sj.onc.1207925. [DOI] [PubMed] [Google Scholar]
- [102].Wilson TR, McLaughlin KM, McEwan M, Sakai H, Rogers KM, Redmond KM, Johnston PG, Longley DB. c-FLIP: a key regulator of colorectal cancer cell death. Cancer Res. 2007;67:5754–5762. doi: 10.1158/0008-5472.CAN-06-3585. [DOI] [PubMed] [Google Scholar]
- [103].Rogers KM, Thomas M, Galligan L, Wilson TR, Allen WL, Sakai H, Johnston PG, Longley DB. Cellular FLICE-inhibitory protein regulates chemotherapy-induced apoptosis in breast cancer cells. Mol. Cancer Ther. 2007;6:1544–1551. doi: 10.1158/1535-7163.MCT-06-0673. [DOI] [PubMed] [Google Scholar]
- [104].Dutton A, Young LS, Murray PG. The role of cellular FLICE inhibitory protein (c-FLIP) in the pathogenesis and treatment of cancer. Expert Opin. Ther. Targets. 2006;10:27–35. doi: 10.1517/14728222.10.1.27. [DOI] [PubMed] [Google Scholar]
- [105].White SJ, Lu P, Keller GM, Voelkel-Johnson C. Targeting the short form of cFLIP by RNA interference is sufficient to enhance TRAIL sensitivity in PC3 prostate carcinoma cells. Cancer Biol. Ther. 2006;5:1618–1623. doi: 10.4161/cbt.5.12.3352. [DOI] [PubMed] [Google Scholar]
- [106].Wang Z, Goulet R, 3rd, Stanton KJ, Sadaria M, Nakshatri H. Differential effect of anti-apoptotic genes Bcl-xL and c-FLIP on sensitivity of MCF-7 breast cancer cells to paclitaxel and docetaxel. Anticancer Res. 2005;25:2367–2379. [PubMed] [Google Scholar]
- [107].Shivapurkar N, Reddy J, Matta H, Sathyanarayana UG, Huang CX, Toyooka S, Minna JD, Chaudhary PM, Gazdar AF. Loss of expression of death-inducing signaling complex (DISC) components in lung cancer cell lines and the influence of MYC amplification. Oncogene. 2002;21:8510–8514. doi: 10.1038/sj.onc.1205941. [DOI] [PubMed] [Google Scholar]
- [108].Song JH, Song DK, Herlyn M, Petruk KC, Hao C. Cisplatin down-regulation of cellular Fas-associated death domain-like interleukin-1beta-converting enzyme-like inhibitory proteins to restore tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human melanoma cells. Clin. Cancer Res. 2003;9:4255–4266. [PubMed] [Google Scholar]
- [109].Kelly MM, Hoel BD, Voelkel-Johnson C. Doxorubicin pretreatment sensitizes prostate cancer cell lines to TRAIL induced apoptosis which correlates with the loss of c-FLIP expression. Cancer Biol. Ther. 2002;1:520–527. doi: 10.4161/cbt.1.5.169. [DOI] [PubMed] [Google Scholar]
- [110].Sun CL, Chao CC. Cross-resistance to death ligand-induced apoptosis in cisplatin-selected HeLa cells associated with overexpression of DDB2 and subsequent induction of cFLIP. Mol. Pharmacol. 2005;67:1307–1314. doi: 10.1124/mol.104.008797. [DOI] [PubMed] [Google Scholar]
- [111].Zhang L, Zhang X, Barrisford GW, Olumi AF. Lexatumumab (TRAIL-receptor 2 mAb) induces expression of DR5 and promotes apoptosis in primary and metastatic renal cell carcinoma in a mouse orthotopic model. Cancer Lett. 2007;251:146–157. doi: 10.1016/j.canlet.2006.11.013. [DOI] [PubMed] [Google Scholar]
- [112].Zhao X, Qiu W, Kung J, Zhao X, Peng X, Yegappan M, Yen-Lieberman B, Hsi ED. Bortezomib induces caspase-dependent apoptosis in Hodgkin lymphoma cell lines and is associated with reduced c-FLIP expression: A gene expression profiling study with implications for potential combination therapies. Leuk. Res. 2007;18:18. doi: 10.1016/j.leukres.2007.05.024. [DOI] [PubMed] [Google Scholar]
- [113].Liu X, Yue P, Chen S, Hu L, Lonial S, Khuri FR, Sun SY. The proteasome inhibitor PS-341 (bortezomib) up-regulates DR5 expression leading to induction of apoptosis and enhancement of TRAIL-induced apoptosis despite up-regulation of c-FLIP and survivin expression in human NSCLC cells. Cancer Res. 2007;67:4981–4988. doi: 10.1158/0008-5472.CAN-06-4274. [DOI] [PubMed] [Google Scholar]
- [114].Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, Munshi N, Dang L, Castro A, Palombella V, Adams J, Anderson KC. NF-kappa B as a therapeutic target in multiple myeloma. J. Biol. Chem. 2002;277:16639–16647. doi: 10.1074/jbc.M200360200. [Epub 12002 Feb 16628] [DOI] [PubMed] [Google Scholar]
- [115].Okamoto K, Fujisawa J, Reth M, Yonehara S. Human T-cell leukemia virus type-I oncoprotein Tax inhibits Fas-mediated apoptosis by inducing cellular FLIP through activation of NF-kappaB. Genes Cells. 2006;11:177–191. doi: 10.1111/j.1365-2443.2006.00927.x. [DOI] [PubMed] [Google Scholar]
- [116].Hyer ML, Croxton R, Krajewska M, Krajewski S, Kress CL, Lu M, Suh N, Sporn MB, Cryns VL, Zapata JM, Reed JC. Synthetic triterpenoids cooperate with tumor necrosis factor-related apoptosis-inducing ligand to induce apoptosis of breast cancer cells. Cancer Res. 2005;65:4799–4808. doi: 10.1158/0008-5472.CAN-04-3319. [DOI] [PubMed] [Google Scholar]
- [117].Syed V, Mukherjee K, Godoy-Tundidor S, Ho SM. Progesterone induces apoptosis in TRAIL-resistant ovarian cancer cells by circumventing c-FLIP(L) overexpression. J. Cell. Biochem. 2007;102:442–452. doi: 10.1002/jcb.21304. [DOI] [PubMed] [Google Scholar]
- [118].Komatsu N, Kawamata N, Takeuchi S, Yin D, Chien W, Miller CW, Koeffler HP. SAHA, a HDAC inhibitor, has profound anti-growth activity against non-small cell lung cancer cells. Oncol. Rep. 2006;15:187–191. [PubMed] [Google Scholar]
- [119].Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- [120].Liu T, Kuljaca S, Tee A, Marshall GM. Histone deacetylase inhibitors: multifunctional anticancer agents. Cancer Treat. Rev. 2006;32:157–165. doi: 10.1016/j.ctrv.2005.12.006. [DOI] [PubMed] [Google Scholar]
- [121].Nakata S, Yoshida T, Horinaka M, Shiraishi T, Wakada M, Sakai T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene. 2004;23:6261–6271. doi: 10.1038/sj.onc.1207830. [DOI] [PubMed] [Google Scholar]
- [122].Kim YH, Park JW, Lee JY, Kwon TK. Sodium butyrate sensitizes TRAIL-mediated apoptosis by induction of transcription from the DR5 gene promoter through Sp1 sites in colon cancer cells. Carcinogenesis. 2004;25:1813–1820. doi: 10.1093/carcin/bgh188. [DOI] [PubMed] [Google Scholar]
- [123].Nebbioso A, Clarke N, Voltz E, Germain E, Ambrosino C, Bontempo P, Alvarez R, Schiavone EM, Ferrara F, Bresciani F, Weisz A, de Lera AR, Gronemeyer H, Altucci L. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat. Med. 2005;11:77–84. doi: 10.1038/nm1161. [DOI] [PubMed] [Google Scholar]
- [124].Guo F, Sigua C, Tao J, Bali P, George P, Li Y, Wittmann S, Moscinski L, Atadja P, Bhalla K. Cotreatment with histone deacetylase inhibitor LAQ824 enhances Apo-2L/tumor necrosis factor-related apoptosis inducing ligand-induced death inducing signaling complex activity and apoptosis of human acute leukemia cells. Cancer Res. 2004;64:2580–2589. doi: 10.1158/0008-5472.can-03-2629. [DOI] [PubMed] [Google Scholar]
- [125].Liu X, Yue P, Schonthal AH, Khuri FR, Sun SY. Cellular FLICE-inhibitory protein down-regulation contributes to celecoxib-induced apoptosis in human lung cancer cells. Cancer Res. 2006;66:11115–11119. doi: 10.1158/0008-5472.CAN-06-2471. [DOI] [PubMed] [Google Scholar]
- [126].Jin TG, Kurakin A, Benhaga N, Abe K, Mohseni M, Sandra F, Song K, Kay BK, Khosravi-Far R. Fas-associated protein with death domain (FADD)-independent recruitment of c-FLIPL to death receptor 5. J. Biol. Chem. 2004;279:55594–55601. doi: 10.1074/jbc.M401056200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Figova K, Hrabeta J, Eckschlager T. Reovirus - possible therapy of cancer. Neoplasma. 2006;53:457–462. [PubMed] [Google Scholar]
- [128].Clarke P, Tyler KL. Down-regulation of cFLIP following reovirus infection sensitizes human ovarian cancer cells to TRAIL-induced apoptosis. Apoptosis. 2007;12:211–223. doi: 10.1007/s10495-006-0528-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Joensuu H, Fletcher C, Dimitrijevic S, Silberman S, Roberts P, Demetri G. Management of malignant gastrointestinal stromal tumours. Lancet Oncol. 2002;3:655–664. doi: 10.1016/s1470-2045(02)00899-9. [DOI] [PubMed] [Google Scholar]