

Abstract
A palladium(II) catalyst system has been identified for aerobic dehydrogenation of substituted cyclohexenes to the corresponding arene derivatives. Use of sodium anthraquinone-2-sulfonate (AMS) as a co-catalyst enhances the product yields. A wide range of functional groups are tolerated in the reactions, and the scope and limitations of the method are described. The catalytic dehydrogenation of cyclohexenes is showcased in an efficient route to a phthalimide-based TRPA1 activity modulator.
Aromatic rings are ubiquitous in industrial chemicals, ranging from commodities to pharmaceuticals. New reactions for the preparation of substituted aromatic molecules, particularly those that access selectivity patterns different from existing methods, could have a major impact on organic chemical synthesis.1 The synthesis of substituted aromatic molecules is often achieved via sequential introduction of substituents around the periphery of the aromatic ring. Common synthetic methods include classical nucleophilic and electrophilic substitution reactions, catalytic cross-coupling reactions, as well as modern C–H functionalization methods.2 Recently, we have pursued a complementary strategy, involving oxidative dehydrogenation of (partially) saturated carbocycles to afford substituted aromatic compounds.3 This concept was recently illustrated in Pd-catalyzed methods for dehydrogenation of cyclohexanones to phenols (Scheme 1, A).3a,b,4 Analogous approaches have been used to access other aromatic compounds, such as aryl ethers 5 and aniline derivatives6,7 (Scheme 1, B). Many of these methods utilize cyclohexanone and cyclohexenone derivatives as starting materials (cf. Scheme 1). Substituted cyclohexenes represent another appealing class of precursors to arenes. Cyclohexenes are readily available through a variety of methods, such as Diels-Alder cycloadditions, that install diverse substituent patterns around six-membered carbon rings.
Scheme 1.
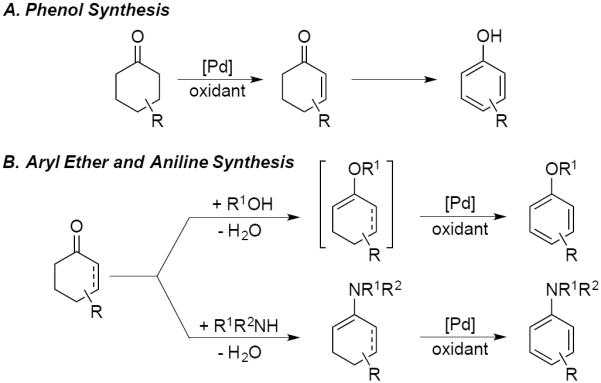
Oxidative Dehydrogenation of Cyclic Ketones to Access Aromatic Compounds
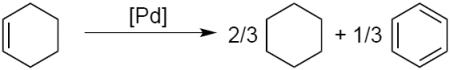
The dehydrogenation of cyclohexenes could proceed by PdII-mediated activation of an allylic C-H bond, followed by β-hydrogen elimination from the resulting PdII-allyl intermediate (Scheme 2, bottom pathway).8 The latter step differs from the more established reactivity of π-allyl-PdII species with nucleophiles (Scheme 2, top pathway).9 Previous studies of allylic C–H oxidation of cyclohexene have observed formation of benzene as a side-product.10 Development of the latter oxidative dehydrogenation chemistry as a synthetically useful method has received very little attention, however, with precedents typically limited to cyclohexene or similarly simple precursors.11 One exception is a recent study by Kandukuri and Oestrich, who observed aromatization of a cyclohexene substituent in the study of intramolecular oxidative C–C coupling reactions with indole substrates.12 Earlier studies were often complicated by competing disproportionation of the cyclohexene into cyclohexane and benzene (eq 1; i.e., with cyclohexene serving as the hydrogen acceptor).11a,b,f The present study describes an effective PdII-catalyzed method for aerobic dehydrogenation of diverse cyclohexenes to substituted aromatics, without competing disproportionation.13 The results illustrate important new dehydrogenative reactions that use O2 as the terminal oxidant, which provide the basis for replacement of undesirable, yet widely used, stoichiometric oxidants such as DDQ14 and Mn oxides.15
 |
(1) |
Scheme 2.
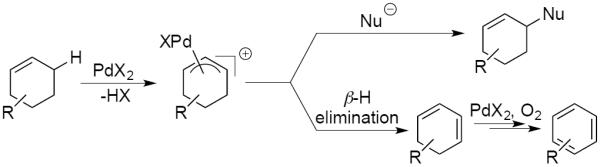
Pd-Catalyzed Reactions of Cyclohexene: Allylic C–H Oxidation or Oxidative Dehydrogenation
A cyclohexene bearing a remote carboxylic acid (1a, Table 1) was used in our initial evaluation of oxidative dehydrogenation conditions with PdCl2, Pd(OAc)2, and Pd(TFA)2 (TFA = trifluoroacetate) at 5 mol % loading (see Table S1 in the Supporting Information for full screening data). Pd(TFA)2 led to complete conversion of the substrate and a slightly higher yield than Pd(OAc)2, and it was evaluated under additional reaction conditions. Significantly improved yields were observed with diglyme (62%) and chlorobenzene (71%) as solvents. Use of CuII and AgI cocatalysts led to a significant reduction in yield, and the use of benzoquinone also had an inhibitory effect. A notable improvement was observed, however, with cocatalytic quantities of anthraquinone (entry 10). The best result was obtained with sodium anthraquinone-2-sulfonate (AMS), a quinone cocatalyst previously used by Sheldon to avoid disproportionation in the dehydrogenation of the parent cyclohexene.11b These conditions were successfully implemented on larger scale (10 mmol, entry 12). Use of PhCF3 rather than chlorobenzene as the solvent led to similarly good results (entry 13), and a high product yield was possible even with a 1 mol % Pd catalyst loading (entry 14).
Table 1.
Optimization of Reaction Conditionsa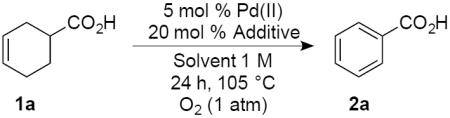
| Entry | Pd(II) | Solvent | Additive | Yield (Conv.)b |
|---|---|---|---|---|
| 1 | PdCl2 | Mesitylene | - | 0 (21) |
| 2 | Pd(OAc)2 | Mesitylene | - | 30 (82) |
| 3 | Pd(TFA)2 | Mesitylene | - | 32 (98) |
| 4 | Pd(TFA)2 | Diglyme | - | 62 (100) |
| 5 | Pd(TFA)2 | DMSO | - | 4 (29) |
| 6 | Pd(TFA)2 | PhCl | - | 71 (93) |
| 7 | Pd(TFA)2 | PhCl | Cu(TFA)2 | 5 (100) |
| 8 | Pd(TFA)2 | PhCl | AgTFA | 3 (27) |
| 9 | Pd(TFA)2 | PhCl | benzoquinone | 36 (64) |
| 10 | Pd(TFA)2 | PhCl | anthraquinone | 92 (100) |
| 11 | Pd(TFA)2 | PhCl | AMS c | 99 (100) |
| 12d | Pd(TFA)2 | PhCl | AMSc | 89 (100) |
| 13 | Pd(TFA)2 | PhCF3 | AMSc | 89 (100) |
| 14e | Pd(TFA)2 | PhCl | AMSc | 85 (100) |
Conditions: 1a (0.25 mmol), 1 atm O2, orbital mixing, 105 °C.
% Determined by 1H NMR using a standard.
AMS = sodium anthraquinone-2-sulfonate.
Reaction performed on 10 mmol scale; isolated yield.
Reaction performed with 1 mol% Pd(TFA)2 and 4 mol% AMS.
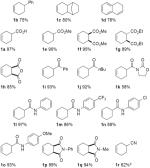
With these conditions in hand, we evaluated a number of readily available cyclohexenes containing diverse functional groups, mainly in the 4- and 5-position of the cyclohexene ring (Table 2).16 Aliphatic and alkyl substituents are well tolerated (1b–d), and dehydrogenation of substrates containing ester, diester, cyclic anhydride, ketone and imide functionalities also proceed well under the standard reaction conditions (1e–k). The relative stereochemistry of the ester functionalities in 1f and 1g did not have an effect on the reaction yield. N-Aryl amides with -CF3, -Cl and -OMe groups on the aryl moiety, as well as a nitrile, were well tolerated (1l–k, 1r). N-Substituted imides were also successfully dehydrogenated to the corresponding phthalimides (1p, q). In all cases, the competing cyclohexane disproportionation product was not observed. Overall, the reactions are consistent with the allylic C–H activation pathway shown in Scheme 2; however, initiation of the reaction by activation of an acidic C–H bond adjacent to a carbonyl group cannot be excluded with a number of the substrates.
Table 2.
Dehydrogenation of Various Cyclohexenes to the Corresponding Arene Derivativesa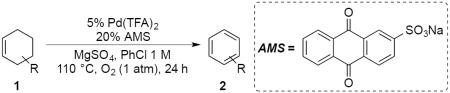
|
Conditions: Substrate (1.0 mmol), magnetic stirring, 110 °C. Yield represents amount of isolated product.
Low yield due to volatility of the product.
During the course of this study, several less-successful substrates were identified (Chart 1). For the diacid 1s, only 28% yield of o-phthalic acid was obtained, together with significant amounts of the decarboxylation product, benzoic acid. The N-Aryl amide bearing a bromide substituent 1t and the cyclohexenyl imide with a free N-H group 1u proceed in very low yield, accompanied by unidentified side-products.17 Finally, dehydrogenation of 4-vinyl cyclohexene 1v (a dimer of 1,3-butadiene) leads to a mixture of styrene and ethylbenzene. The latter reactivity has been observed previously with other Pd-based catalyst systems and presumably arises from isomerization of the exocyclic double bond into the ring prior to dehydrogenation.11c,g Additional substrates bearing N-heterocycles, such as pyridine (1w) and benzimidazole (1x), as well as a thiophene containing substrate (1y) were also tested under the reaction conditions, but provided low yields of product.
Chart 1.
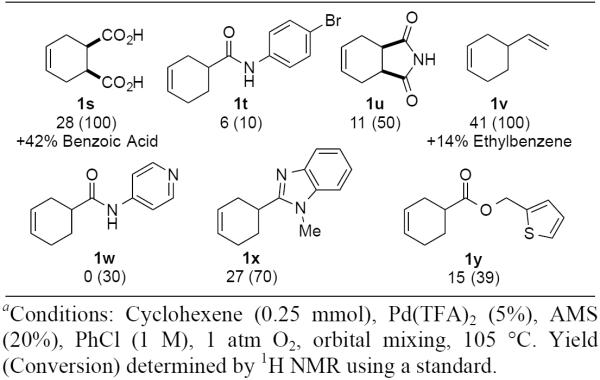
Less Successful Dehydrogenation Substratesa
The N-phenylsuccinimide substrates 3a and 3b derived from Diels-Alder cycloadditions of N-phenylmaleimide (Scheme 3) also led to relatively poor product yields with the parent catalyst system (≤ 45%). The lower reactivity of these substrates, with substituents adjacent to the alkene, seemed likely to be associated with steric effects. We reasoned that this complication could potentially be overcome with a more electrophilic catalyst that has higher affinity for the alkene. Addition of pTsOH to Pd(OAc)2 forms Pd(OTs)2 in situ,18 and the Pd(OAc)2/pTsOH combination proved to be effective for dehydrogenation of the cyclohexene precursors to phthalimides 4a and 4b (79% and 84% yields, respectively).
Scheme 3.
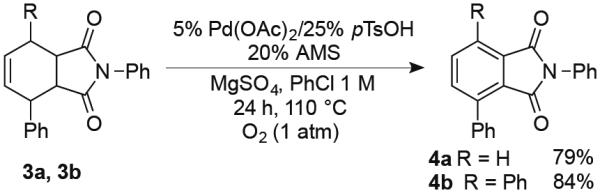
Synthesis of Tri- and Tetrasubstituted Phthalimide Derivatives
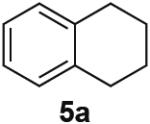
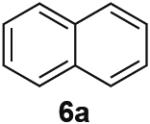
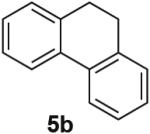
PdII-mediated benzylic C–H bond activation has less precedent and is more challenging than allylic C–H activation.19 A preliminary effort to achieve dehydrogenation of 1,2,3,4-tetrahydronaphthalene, 9,10-dihydrophenanthrene and 2,3-dihydrobenzofuran as prototypical substrates reveals that the initial Pd(TFA)2 catalyst system was ineffective. The Pd(OAc)2/pTsOH catalyst system showed promising results (Table 3). Although product yields from these reactions are modest, they represent a good starting point for future catalyst development efforts and show marked improvement over a previously attempted PdII-catalyzed aerobic dehydrogenation of tetrahydronaphthalene, which exhibited only stoichiometric reactivity with respect to PdII (i.e., 2 equiv).11f
Table 3.
Dehydrogenation at Benzylic Positionsa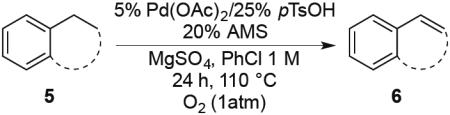
| Substrate | Product | Yield (Conv.) |
|---|---|---|
|
|
31 (34) |
|
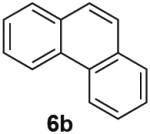
|
53 (61) |
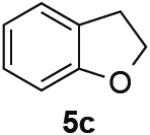
|
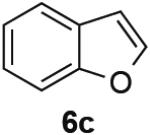
|
62 (100) |
Conditions: Substrate (0.25 mmol), orbital stirring, 110 °C. Yield (Conversion) determined by 1H NMR using a standard.
The dehydrogenation reactions described above present unique opportunities to access substituted aromatics via more convergent synthetic routes. Phthalimides are featured in a number of important biologically active compounds, such as modulators of TRPA1 (Transient Receptor Potential subfamily A1) activity, which are peripheral damage receptors involved in prolonged pain responses. 20 Scheme 4A highlights a patented cross-coupling route to the substituted phthalimide fragment in one these TRPA1 modulators (4c).21 The nitrogroup of 3-nitrophthalimide is converted to the 3-iodophthalimide cross-coupling partner via high-pressure hydrogenation of the nitro group, diazotization of the resulting aniline, and nucleophilic aromatic substitution by iodide. The desired carboxymethyl substituent is then introduced via Pdcatalyzed cross-coupling of the aryl iodide with allyl-BPin using 10 mol % Pd(PPh3)4, followed by oxidative cleavage of the double bond. In contrast, the dehydrogenation route in Scheme 4B accesses the desired substitution pattern in a single step via Diels-Alder cycloaddition of N-methylmaleimide and a methyl sorbate-derived diene.22 The starting materials used in this route are very inexpensive and access the substituted phthalimide precursor 3d in excellent yield. The Pd(TFA)2/AMS catalyst system then mediates dehydrogenation of 3d to the desired phthalimide product 4d in 71% yield. This route illustrates how aerobic dehydrogenation of cyclohexenes, like the previous dehydrogenation of cyclohexanones and related derivatives,3–7 offers complementary appeal or significant advantages relative to cross-coupling reactions in the preparation of substituted aromatics. The dehydrogenation methods exploit classical organic transformations, such as Diels-Alder cycloadditions (to access cyclohexenes) and Robinson annulations (to access cyclohexenones3a), as versatile and efficient routes to core structures that are excellent precursors to selectively substituted aromatic compounds.
Scheme 4.

Synthesis of a Precursor to a TRPA1 Modulator via Oxidative Dehydrogenation
In conclusion, we have identified new Pd catalyst systems for the oxidative dehydrogenation of cyclohexenes. The method enables efficient synthesis of substituted arene derivatives and shows good functional group tolerance. Use of this method in the preparation of a substituted phthalimide showcases the strategic opportunity to use this transformation in the synthesis of biologically active compounds.
Supplementary Material
ACKNOWLEDGMENT
We thank Yusuke Izawa for conducting preliminary research in the area. We thank Jack W. Kruper and Anna Davis for useful discussions. This work was supported by the NIH (R01-GM100143) and The Dow Chemical Company. NMR spectroscopy facilities were partially supported by the NSF (CHE-0342998, CHE-1048642) and NIH (S10 RR08389). Mass spectrometry instrumentation was partially supported by NIH (S10 RR024601).
Footnotes
Supporting Information Additional catalyst screening data, experimental procedures, compound characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
REFERENCES
- (1).Astruc D, editor. Modern Arene Chemistry: Concepts, Synthesis and Applications. Wiley-VCH; Weinheim: 2002. [Google Scholar]
- (2).(a) Yu J-Q, Shi Z, editors. Topics in Current Chemistry: C-H Activation. Springer; New York: 2010. [PubMed] [Google Scholar]; (b) Ribas X, editor. C-H and C-X Bonds Functionalization: Transition Metal Mediation. RSC Publishing; London: 2013. [Google Scholar]
- (3).(a) Izawa Y, Pun D, Stahl SS. Science. 2011;333:209. doi: 10.1126/science.1204183. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Izawa Y, Zheng C, Stahl SS. Angew. Chem. Int. Ed. 2013;52:3672. doi: 10.1002/anie.201209457. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Pun D, Diao T, Stahl SS. J. Am. Chem. Soc. 2013;135:8213. doi: 10.1021/ja403165u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Imahori T, Tokuda T, Taguchi T, Takahata H. Org. Lett. 2012;14:1172. doi: 10.1021/ol300145g. [DOI] [PubMed] [Google Scholar]; (b) Kikushima K, Nishina Y. RSC Adv. 2013;3:20150. [Google Scholar]; For contributions by other groups, see:
- (5).(a) Simon M-O, Girard SA, Li C-J. Angew. Chem. Int. Ed. 2012;51:7537. doi: 10.1002/anie.201200698. [DOI] [PubMed] [Google Scholar]; (b) Sutter M, Sotto N, Raoul Y, Métay E, Lemaire M. Green Chem. 2013;15:347. [Google Scholar]
- (6).(a) Girard SA, Hu X, Knauber T, Zhou F, Simon M-O, Deng G-J, Li C-J. Org. Lett. 2012;14:5606. doi: 10.1021/ol3027279. [DOI] [PubMed] [Google Scholar]; (b) Hajra A, Wei Y, Yoshikai N. Org. Lett. 2012;14:5488. doi: 10.1021/ol302568b. [DOI] [PubMed] [Google Scholar]; (c) Xie Y, Liu S, Liu Y, Wen Y, Deng G-J. Org. Lett. 2012;14:1692. doi: 10.1021/ol3002442. [DOI] [PubMed] [Google Scholar]; (d) Hong WP, Iosub AV, Stahl SS. J. Am. Chem. Soc. 2013;135:13664. doi: 10.1021/ja4073172. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sutter M, Duclos M-C, Guicheret B, Raoul Y, Métay E, Lemaire M. ACS Sustainable Chem. Eng. 2013 [Google Scholar]; (f) Cao X, Bai Y, Xie Y, Deng G-J. J. Mol. Catal. A: Chem. 2014:383–384. 94. [Google Scholar]
- (7).(a) Kim D, Min M, Hong S. Chem. Commun. 2013;49:4021. doi: 10.1039/c3cc41296b. [DOI] [PubMed] [Google Scholar]; (b) Chen S, Liao Y, Zhao F, Qi H, Liu S, Deng G-J. Org. Lett. 2014;16:1618. doi: 10.1021/ol500231c. [DOI] [PubMed] [Google Scholar]; (c) Liao Y, Peng Y, Qi H, Deng G-J, Gong H, Li C-J. Chem. Commun. 2015;51:1031. doi: 10.1039/c4cc08370a. [DOI] [PubMed] [Google Scholar]; For oxidative dehydrogenation methods that use cyclohexanones and cyclohexenones as precursors to other aromatic compounds, such as aryl sulfides, coumarins, and aryl indoles, see:
- (8).Takacs JM, Lawson EC, Clement F. J. Am. Chem. Soc. 1997;119:5956. [Google Scholar]; This reaction could occur via a traditional β-hydride elimination pathway or via an anti-elimination process, which has been observed previously:
- (9).(a) McMurry JE, Kočovský P. Tet. Lett. 1984;25:4187. [Google Scholar]; (b) Hansson S, Heumann A, Rein T, Åkermark B. J. Org. Chem. 1990;55:975. [Google Scholar]; (c) Bäckvall J-E, Hopkins RB, Grennberg H, Mader M, Awasthi AK. J. Am. Chem. Soc. 1990;112:5160. [Google Scholar]; (d) Byström S, Larsson EM, Åkermark B. J. Org. Chem. 1990;55:5674. [Google Scholar]; (e) Larsson ME, Åkermark BA. Tet. Lett. 1993;34:2523. [Google Scholar]; (f) Åkermark B, Larsson EM, Oslob JD. J. Org. Chem. 1994;59:5729. [Google Scholar]; (g) Grennberg H, Bergstad K, Bäckvall J-E. J. Mol. Catal. A: Chem. 1996;113:355. [Google Scholar]; (h) Grennberg H, Bäckvall J-E. Chem. Eur. J. 1998;4:1083. [Google Scholar]; (i) Pilarski LT, Selander N, Böse D, Szabó KLNJ. Org. Lett. 2009;11:5518. doi: 10.1021/ol9023369. [DOI] [PubMed] [Google Scholar]; For allylic C-H activation of cyclohexene rings see:
- (10).Wolfe S, Campbell PGC. J. Am. Chem. Soc. 1971;93:1499. [Google Scholar]
- (11).(a) Trost BM, Metzner PJ. J. Am. Chem. Soc. 1980;102:3572. [Google Scholar]; (b) Sheldon RA, Sobczak JM. J. Mol. Catal. 1991;68:1. [Google Scholar]; (c) Neumann R, De bruyn M. Adv. Synth. Catal. 2007;349:1624. [Google Scholar]; (d) Williams TJ, Caffyn AJM, Hazari N, Oblad PF, Labinger JA, Bercaw JE. J. Am. Chem. Soc. 2008;130:2418. doi: 10.1021/ja076740q. [DOI] [PubMed] [Google Scholar]; (e) Bercaw JE, Hazary N, Labinger JA, Oblad PF. Angew. Chem. Int. Ed. 2008;47:9941. doi: 10.1002/anie.200804455. [DOI] [PubMed] [Google Scholar]; (f) Bercaw JE, Hazari N, Labinger JA. J. Org. Chem. 2008;73:8654. doi: 10.1021/jo8016296. [DOI] [PubMed] [Google Scholar]; (g) Jaekel C, Horillo-Martinez P;, Virolleaud M-A. ChemCatChem. 2010;2:175. [Google Scholar]
- (12).Kandukuri SR, Oestreich M. J. Org. Chem. 2012;77:8750. doi: 10.1021/jo301088f. [DOI] [PubMed] [Google Scholar]
- (13).Stang EM, White MC. J. Am. Chem. Soc. 2011;133:14892. doi: 10.1021/ja2059704. [DOI] [PMC free article] [PubMed] [Google Scholar]; A PdII catalyst system for dehydrogenation of terminal alkenes to 1,3-dienes, with a stoichiometric quinone oxidant, was reported recently:
- (14).(a) Fu PP, Harvey RG. Dehydrogenation of Polycyclic Hydroaromatic Compounds. Chem. Rev. 1978;78:317. [Google Scholar]; (b) Buckle DR. Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons, Inc; New York: 2010. [Google Scholar]
- (15).Cahiez G, Alami M, Taylor RJK, Reid M, Foot JS, Fader L. Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons, Ltd; 2001. [Google Scholar]
- (16).For the 1 mmol-scale reactions, use of MgSO4 was found to enhance the yields by 5–10%, presumably by serving as a drying agent. See supporting information for the experimental procedure.
- (17).1H NMR analysis of the crude reaction mixture with 1t reveals new alkene peaks, possibly arising from a Heck reaction product, but we were not successful in isolating the by-products. Peaks associated with the independently prepared hydrodebromination products of 1t/2t are not observed.
- (18).Houlden CE, Bailey CD, Ford JG, Gagné MR, Lloyd-Jones GC, Booker-Milburn KI. J. Am. Chem. Soc. 2008;130:10066. doi: 10.1021/ja803397y. [DOI] [PMC free article] [PubMed] [Google Scholar]; For an example where Pd(OAc)2 and pTsOH were shown to generate Pd(OTs)2 in situ see:
- (19).(a) Rong Y, Li R, Lu W. Organometallics. 2007;26:4376. [Google Scholar]; (b) Jiang H, Chen H, Wang A, Liu X. Chem. Commun. 2010;46:7259. doi: 10.1039/c0cc00841a. [DOI] [PubMed] [Google Scholar]; (c) Liu H, Shi G, Pan S, Jiang Y, Zhang Y. Org. Lett. 2013;15:4098. doi: 10.1021/ol401687f. [DOI] [PubMed] [Google Scholar]; (d) Curto JM, Kozlowski MC. J. Am. Chem. Soc. 2015;137:18. doi: 10.1021/ja5093166. [DOI] [PMC free article] [PubMed] [Google Scholar]; For recent PdII-catalyzed methods for benzylic functionalization, see:
- (20).Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, Cravatt BF, Patapoutian A. Nature. 2007;445:541. doi: 10.1038/nature05544. [DOI] [PubMed] [Google Scholar]
- (21).Muthuppal-Niappan M, Kumar S, Thomas A, Khairatkar-Joshi N, Mukhopadhyay I. World Patent WO 09/118596. 2009 Oct 1; [Google Scholar]
- (22).Kimura M, Ezoe A, Mori M, Tamaru Y. J. Am. Chem. Soc. 2005;127:201. doi: 10.1021/ja0469030. [DOI] [PubMed] [Google Scholar]; For preparation of the diene, see:
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.