

Abstract
An exploration of an abiotic approach to spirocalcaridines A and B is described centered on electrophile-induced dearomatizing spirocyclization of aryl enyne derivatives. Elaboration of the α–iodoenone via an Ullmann-like, copper-catalyzed amidation provided a formamide which upon treatment with methylamine undergoes a dienol-arene rearrangement, providing the corresponding kealiinine-like framework. This observation suggests a possible biosynthetic links between the spirocalcaridines and the naphthimidazole group of Leucetta alkaloids.
Keywords: 2-Aminoimidazole, Spirocyclization, Dearomatization, Ullman reaction, Rearrangement
Introduction
The Leucetta family of marine sponges produces a wide variety of secondary metabolites, including a growing number of 2-aminoimidazole alkaloids which have recently been reviewed.[1] Many of these imidazole-containing metabolites are biologically active, exhibiting amongst others anti-cancer and antibiotic activity, but their biological roles in Nature have yet to be rigorously defined.[1b] Structurally, these 2-aminoimidazoles fit into five broad subclasses depending on their substitution patterns and oxidation levels. The simplest systems are characterized by the presence of one benzyl moiety on the 2-aminoimidazole framework, e.g., preclathridine A (1); oftentimes there is a closely related congener in which the 2-amino moiety is functionalized with methyl parabanic acid, e.g., clathridine A (2).[2] Two groups contain two benzylic moieties; the naamine/naamidine group is substituted with benzyl groups at C4 and C5, e.g. naamidine H (3),[3] whereas the isonaamine/isonaamidine group is functionalized at N1 and C4, e.g., isonaamidine E (4).[4] The fourth group of Leucetta alkaloids contains a naphthimidazole framework such as that found in the kealiinines, e.g., 5[5] and kealiiquinones, e.g. 6[6] and 7.[7] The final and most recently isolated group of alkaloids belonging to this family of sponges is the most highly oxygenated comprising calcaridine A (8),[8] spirocalcaridines A (9) and B (10),[8b] spiroleucettadine (11)[9] and spironaamidine (12).[10]
To date, there have been no experimental studies directed towards unravelling the biosynthetic pathways which lead to the formation of these compounds. The lack of experimental support notwithstanding, obvious relationships can be mapped out among the various family members (Figure 2). The parent systems have been hypothesized to be derived from either octapamine (13), tyramine (14) or p-hydroxyphenyl pyruvate (16) leading to 17 and the 4,5- and 1,4-disubstituted systems result from the elaboration of this species.[1a] The remaining family members can then be derived from naamine A (19) or closely related analogs. For example, calcaridine A (8), which contains a 4,4-disubstituted 5-imidazolone fragment, derives from the oxidative rearrangement of naamine derivative.[8a, 8c, 11] The spirocalcaridines (9) and (10) can also be traced back to the same general precursor through a dearomatizing alkylation and oxidation of the resulting intermediate. Spiroleucettadine (11) and the related spironaamidine (12) can be envisioned to derive from the corresponding naamine derivative by oxidation of the imidazole 4,5-bond followed by an oxidative dearomatization. The final group of Leucetta-derived alkaloids that can be derived from naamine-type precursors are the kealiinines[5c] and kealiiquinones.[6–7] The former can be derived from an intramolecular Friedel-Crafts like process followed by oxidation, delivering the kealiinines. Additional oxidation would then permit entry to the kealiiquinones. Our group has taken advantage of these putative relationships to accomplish total syntheses of calcaridine A,[8a, 8c] kealiinines A-C,[5a, 5b] kealiiquinone and 2-deoxy-2-aminokealiiquinone.[12]
Figure 2.

Putative biosynthesis and biosynthetic relationships among the Leucetta alkaloids.
Results and Discussion
In our studies towards both the Leucetta[13] and the oroidin alkaloids[14] we have adopted a general strategy of elaborating pre-existing imidazoles rather than the de novo construction of the heterocycle and using broadly bioinspired approaches to construct the key structural elements contained within the alkaloid framework. Our attempts to execute such a strategy towards the spirocalcaridines have been largely unsuccessful to date and thus we have pursued an alternative abiotic approach.[15] The general plan depicted in retrosynthetic form centered on the construction of the key spiro fused framework 23 through an electrophile-induced dearomatizing spirocyclization and then annulation of the imidazole ring (23→22, Figure 3). Adjustment of the oxidation state of the imidazole, where necessary would be accomplished based on chemistry developed in our lab using N-sulfonyloxaziridines (21→9 or 10, Figure 3).[11] This strategy was predicated on the availability of 23 through dearomatizing cyclization of aryl ynones, chemistry developed independently by Larock[16] and Li[17] and subsequently developed further by others.
Figure 3.
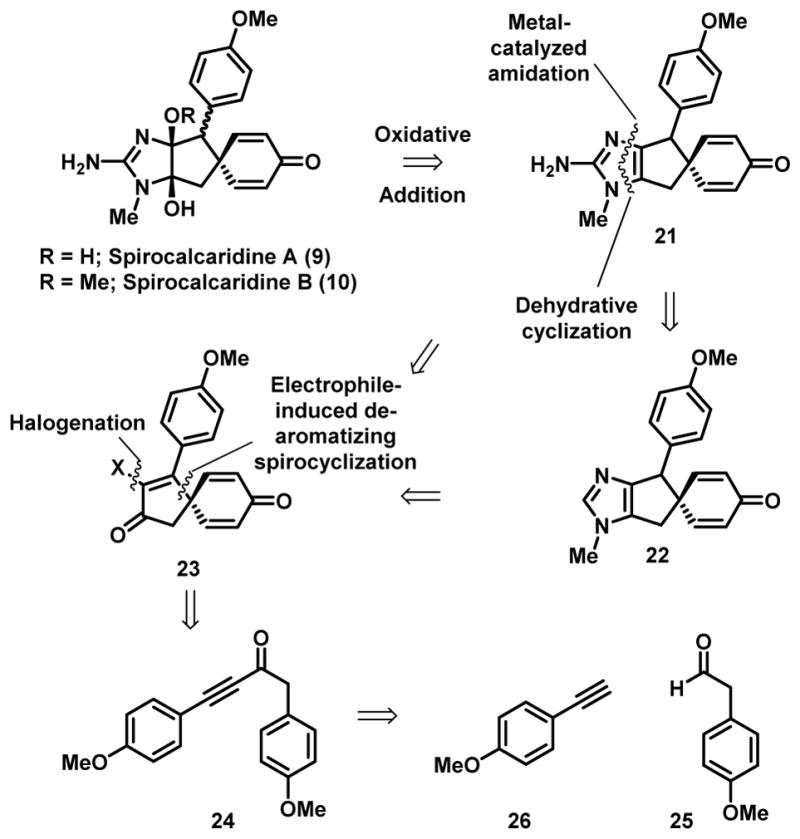
A retrosynthetic analysis of spirocalcaridines A and B
In an initial foray towards these natural product targets, we identified the α–diketone 32 as a potentially useful intermediate for further elaboration (Scheme 1). Taylor and coworkers have used a similar strategy in their recently published total synthesis of spirobacillene A.[18] In order to access this material, the spirofused derivative 28 was required which in turn necessitated ynone 24 as a precursor (Scheme 1). This was accomplished from deprotonation of p-ethynylanisole (26)[19] with n-BuLi and trapping of the resulting lithium acetylide with the Weinreb amide 27 (Scheme 1).[20] Gratifyingly, ynone 24 delivered the spirofused triene dione 28 in 94% yield upon treatment with TFA.[18] With the spiro derivative in hand, our plan involved the selective epoxidation of the cyclopentenone followed isomerization to give the α–diketone.[18] However, attempts to effect selective reduction of cyclopentenone were compromised by competitive reduction of the dienone carbonyl resulting in a complex mixture of alcohols and diols.[18] Presumably, the electron rich aryl ring retards the reduction rate of the cyclopentenyl carbonyl and thus competitive reduction of both enones occur. As a result, we directed our attention towards the preparation of derivatives already functionalized at the α–position.
Scheme 1.

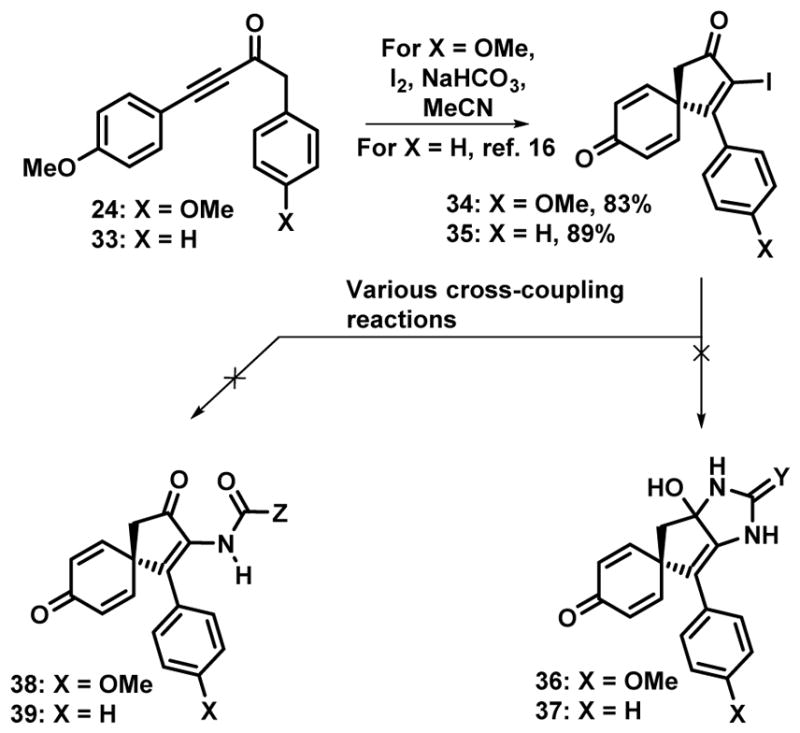
Larock has described previously the preparation of the α–iodocyclopentenone 35 from the dearomatizing spirocyclization of 33 with NIS,[16] similarly we found that the anisyl substituted derivative led to the related spirocycle 34 under similar conditions (Scheme 2). Brief attempts were made to react these intermediates with either guanidine or urea as means to install the imidazole component, but these experiments were unsuccessful. Similarly, attempts to introduce a nitrogen substituent in place of the iodine through a variety of palladium[21] or copper-catalyzed cross-coupling reactions were unsuccessful (see Scheme 3 for successful implementation of this strategy).[22] While the failure of the direct installation of the imidazole moiety was not unexpected,[23] given the general robustness of cross-coupling chemistry, we suspected the presence of the cyclohexadienone was causing problems. As a result, we decided to investigate the corresponding dienol derivative which would not be susceptible to Michael additions.
Scheme 2.
Scheme 3.
Dearomatizing spirocyclization
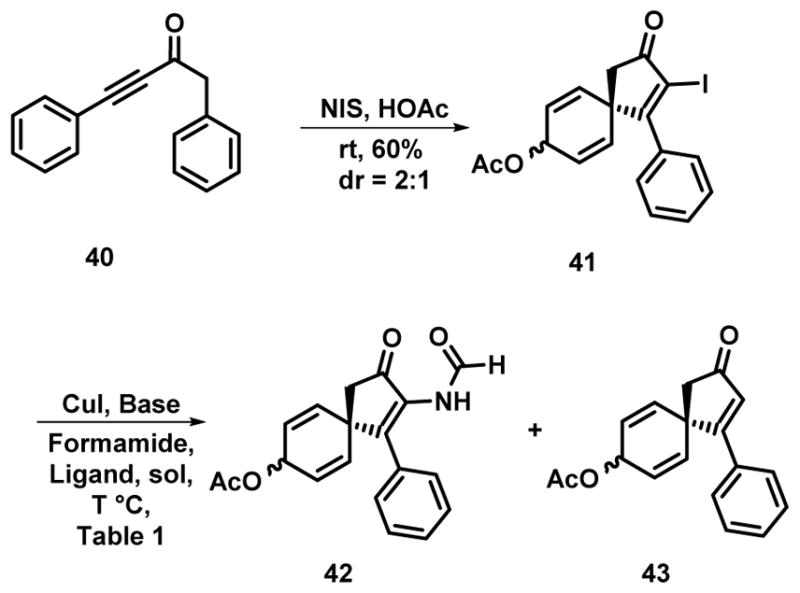
Initial studies were performed with the diphenyl substituted derivative 40 as this could be prepared via known procedures from phenyl acetylene. Treatment of the alkynone 40 with NIS in acetic acid provided the spirocyclic enone 41 as a 2:1 mixture of diastereomers (Scheme 3). The stereochemistry of the major diastereomer was not assigned and this mixture was used directly in the next step. Given that the acetoxy bearing center would be eventually be converted into a carbonyl in the synthetic scheme, this stereoisomeric mixture was of no consequence. It was our intention to use the iodo moiety as a means to introduce a substituted nitrogen via a cross-coupling strategy. While several options are available in a general sense, the use of palladium catalysis was likely to be compromised by the presence of the allylic acetate and thus our efforts were directed towards the use of copper-mediated methods.[22] The Buchwald lab has demonstrated that cross-coupling between vinyl iodides and acetamides occurs on treatment with 5 mol% CuI, N,N′-dimethylethylene diamine (DMEDA), Cs2CO3 in THF at temperatures between 50–70 °C.[22a] Subjection of iodide 41 to these reaction conditions but with the use of formamide resulted in the formation of the coupling product 42 in 50% yield along with 25% of 43 derived from the net reductive deiodination of 41 (Entry 1, Table 1 and Scheme 3).[24] A screen of various solvents (entries 2–5, Table 1) resulted in no improvement in the cross-coupling yields, although it is of note that in acetonitrile there is an increase in the amount of the reduction product 43. Attempts to improve the yield of the cross-coupling product 42 by stoichiometric quantities of copper and DMEDA were not successful (entry 6, Table 1); reduction of the reaction temperature under catalyst loadings still resulted in appreciable reductive deiodination. Changing the base to K2CO3 or the copper salt to the bromide has little to no effect on the yield. We also briefly investigated using other coupling partners under the best conditions, but found that neither urea nor N-methyl urea delivered the cross-coupled product, only reductive deiodination being observed.[25]
Table 1.
Optimization of cross-coupling reaction of 41
| Entry | Solvent | Ligand | Base | Catalyst | Amide | Temp/°C | Time/h | %-42 | %-43 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | THF | 10% DMEDA | 1.5 equiv. Cs2CO3 | 5% CuI | Formamide | 70 | 22 | 50 | 25 |
| 2 | 1,4-dioxane | 10% DMEDA | 2.0 equiv. Cs2CO3 | 5% CuI | Formamide | 110 | 28 | NRa | - |
| 3 | Toluene | 10% DMEDA | 1.5 equiv. Cs2CO3 | 5% CuI | Formamide | 80 | 24 | NRa | - |
| 4 | DMF | 10% DMEDA | 1.5 equiv. Cs2CO3 | 5% CuI | Formamide | 70 | 30 | 10 | - |
| 5 | CH3CN | 10% DMEDA | 1.5 equiv. Cs2CO3 | 5% CuI | Formamide | 70 | 30 | 45 | 40 |
| 6 | THF | 2 equiv. DMEDA | 2 equiv. Cs2CO3 | 1 equiv. CuI | Formamide | 80 | 48 | 52 | 30 |
| 7 | THF | 2 equiv. DMEDA | 2 equiv. Cs2CO3 | 1 equiv. CuI | Formamide | 40 | 10 | 21 | 20 |
| 8 | THF | 10% TMEDA | 2 equiv. K2CO3 | 5% CuI | Formamide | 25 | 12 | - | - |
| 9 | THF | 10% DMEDA | 2 equiv. K2CO3 | 5% CuI | Formamide | 70 | 15 | 40 | 30 |
| 10 | THF | 10% DMEDA | 2 equiv. Cs2CO3 | 5% CuBr | Formamide | 70 | 15 | 50 | 30 |
| 11 | THF | 10% DMEDA | 2 equiv. Cs2CO3 | 5% CuI | Formamide | 70 | 15 | 30 | - |
| 12 | THF | 10% TMEDA | 2 equiv. Cs2CO3 | 5% CuCN | Formamide | 70 | 15 | - | - |
| 13 | THF | 10% DMEDA | 2 equiv. Cs2CO3 | 5% CuI | Urea | 70 | 15 | - | 30 |
| 14 | THF | 10% DMEDA | 2 equiv. Cs2CO3 | 5% CuI | N-Methylurea | 70 | 15 | - | 30 |
NR = no reaction, starting material recovered unreacted
With formamide 42 in hand, we attempted to install the remaining nitrogen atom in the basic alkaloid framework ideally providing 44 (Scheme 4). Initial experiments were conducted with methylamine as this would provide an avenue for the direct introduction of the methyl group in a site specific manner to provide 44. Subsequent regioselective and chemoselective hydration of the carbon-carbon double bond in the cyclopentene would then provide the entire ABring system of spirocalcaridine. However, when 42 was reacted with methylamine.HCl in the presence of K2CO3 hydrolysis of both the acetate and formamide occurred, providing the aminoalcohol 45 in low, but unoptimized, yield. The efficiency notwithstanding, we attempted to convert 45 into the corresponding 2-aminoimidazole 47 by treatment with cyanamide, but this resulted in rearrangement of the spiro fused system into the aminonaphthol derivative 46 rather than the desired 2-aminoimidazole (Scheme 4). A related rearrangement occurred upon treatment of 42 with methylamine.HCl in the presence of Et3N and EtOH at reflux providing aminonaphthol derivative 48. Notably in this case the product does contain a methyl imidazole moiety, suggesting that it may be possible to annulate 42 under the appropriate circumstances (see below).
Scheme 4.

Attempted annulation of imidazole moiety
Given that both spirocalcaridines A (9) and B (10) possess a methoxy group on the aryl ring we prepared the corresponding substrate 53 via the iodine-induced ipso cyclization of the known alkynone 52,[26] delivering 53 in good yield as an almost 1:1 mixture of diastereomers (Scheme 5). It is of note that the reaction temperature has to be controlled very carefully during the addition of NIS as otherwise formation of the iodo- and diiodonaphthol 54 and 55 are observed. An X-ray structure of 55 unequivocally confirms the location of the iodides in the diiodinated naphthol (Scheme 5).[27] The α–iodoenone 53 was subjected to the cross-coupling chemistry with CuI and formamide to provide the corresponding amide 57 in 70% yield along with 20% of the reductively deiodinated product 56. With the formamide 57 in hand we explored a slightly different approach in which the oxidation state of the system was adjusted by reducing the C4-C8 double bond (spirocalcaridine numbering). A wide variety of conditions were evaluated, but none of them delivered the reduction product. An alternative strategy involved the formation and reduction of an imine (or iminium species), an ensuing cyclization would result in the formation of the key framework elements found in spirocalcaridines A and B. However, we found that although an N-methylimidazole was formed, the spiro ring system had again undergone rearrangement to form a naphthimidazole framework 58 (Scheme 5).
Scheme 5.

Attempted iodine-induced ipso addition reaction of alkyne 51.
The propensity of these spiro systems to undergo rearrangement under acidic conditions or presence of Lewis acid is not surprising per se given their close relationship to substrates that participate in the cyclohexadienol-benzene rearrangement. Mechanistically, we assume that the methylamine reacts to form the imine 60 which then undergoes rearrangement to form the naphthyl framework 62 after activation by protonation via 61 (Scheme 6). Deprotonation provides 63, which undergoes tautomerization and dehydrative cyclization to form the imidazole ring and 40.
Scheme 6.

Putative mechanism for the conversion of spiro fused system to the naphthimidazole framework.
This propensity to undergo rearrangement raises the possibility that the spirocalcaridines (and perhaps as yet unidentified congeners) may in fact serve as the biosynthetic precursors to the naphthimidazole group of Leucetta alkaloids. As a result the biosynthetic relationships among the more highly functionalized congeners may require some reanalysis. Naamine A (65, X = Y = H) or naamine G (65, X = Y = MeO) derivatives after N-methylation may serve as precursors to the spirofused derivative 66 via an oxidative-dearomatizing spirocyclization. Subsequent rearrangement via hydrolytic ring opening, ring-closure of the 2-aminoimidazole to the 2-imidazolone then delivers the two spiroleucettadine congeners. The 14-functionalized naamine derivative (65, Z = OH) may serve as a biosynthetic precursor to calcaridine A (8) through O-methylation and oxidative rearrangement. It is of note that we have accomplished the latter in the lab through the use of an N-sulfonyloxaziridine.[11] The same precursor (65, Z = OH) upon activation of the hydroxyl group may engage in a dearomatizing spirocyclization, which up dihydroxylation would lead to the formation of the spirocalcaridine framework. It has been speculated that derivatives related to 65 (Z = OH) can serve as precursors to the naphthimidazole framework via electrophilic addition (Friedel-Crafts-like) and subsequent oxidation. However, other pathways are feasible, including via ipso addition, followed by rearrangement via an intermediate related to 67 and then oxidation.[5a] Our own studies with this system points to another possibility in which spirocalcaridine (or closely related derivative) may undergo dehydration to 68; protonation of the carbonyl oxygen primes the system for rearrangement via a dienone-phenol rearrangement (69→70, Scheme 7). Rearomatization to 71 and a second dehydration delivers the naphthimidazole derivatives, the kealiinines. Alternatively, oxidation of 67 provides the same intermediate 68 for the dienone-phenol rearrangement to deliver 70. Further oxidation of the naphthimidazole provides the naphthoquinone derivatives kealiiquinone and 2-deoxy-2-aminokealiiquinone. We would note that our lab has recently accomplished the total synthesis of both kealiiquinone and 2-aminokealiiquinone through oxidative functionalization of kealiinine derivatives, thus there is a direct synthetic connection between three types of Leucetta alkaloids.[12b] When these results and ideas are taken in concert with previous results from our lab, the direct connection between almost all the Leucetta alkaloids can be mapped out, specifically the relationship between the naamidine framework and the other members can be envisioned. In fact, only the feasibility of the conversion of the naamine to spirocalcaridine framework remains to be demonstrated in the lab. This is a challenge that we are presently pursuing. It is important to note that the scheme outlined in Scheme 7 is purely hypothetical as far as the biosynthesis is concerned and there is no evidence for this sequence in biological systems. However, while our experiments do not address the biosynthesis directly, we have a growing body of evidence to suggest that the postulated transformations are feasible. Based on these hypotheses, it raises the possibility of additional family members related to 66, 67, 68 where (X = Y = OMe) and non-symmetrical variants where either X = H and Y = OMe or X = OMe and Y = H.
Scheme 7.

Putative biosynthetic relationship between the naamine, calcaridines and the naphthimidazole group of Leucetta alkaloids.
Conclusions
In summary, we have described some exploratory experiments towards the total synthesis of spirocalcaridine A and B and while these have not been successful in this context due to dienone-phenol rearrangements, these results provide circumstantial evidence for alternative biosynthetic relationships between the spiro fused systems and the naphthimidazole systems. This type of rearrangement has been posited to account for the biosynthesis of several aporphinoid alkaloids.[28] Recent results from our lab have demonstrated that oxidation of the kealiinine framework gives rise to the quinone moiety within the kealiiquinone group and thus in the lab we have demonstrated experimentally a link between four subfamilies of these aminoimidazole natural products. While this does not prove the biogenesis of these Leucetta alkaloids, it provides evidence that these pathways are at least feasible.
Experimental Section
All chemicals were obtained from commercial vendors and were used as received unless stated otherwise. All reactions were conducted under an atmosphere of dry nitrogen in oven-dried glassware. Solvents were dried using a Pure-Solv 400 solvent purification system (Innovative Technology, Inc.), except for DMF, which was dried over CaH2 and then distilled under vacuum. The 1H NMR spectra were acquired at 500 MHz in CDCl3, unless indicated otherwise, using residual CHCl3 as reference. 13C NMR spectra were obtained at 125 MHz in CDCl3, unless otherwise indicated, using the central absorbance of CDCl3 as internal standard. In cases where diastereomers were isolated as inseparable mixtures, 1H NMR signals for related absorptions are integrated together and the minor isomer signal is underlined. For 13C NMR data, the minor isomer is reported in parentheses. High resolution mass spectra were obtained at the University of Florida by electrospray ionization (HRMS-ESI) or at the Shimadzu Center for Advanced Analytical Chemistry, University of Texas at Arlington.
1,4-bis(4-methoxyphenyl)but-3-yn-2-one (24)
To a solution of alkyne 26 (3.00 g, 22.7 mmol) in anhydrous THF (20 mL) at −78 °C was added n-butyllithium (2.5 M solution in hexane, 10.9 mL, 27.2 mmol). The yellow solution was stirred at −78 °C for 30 min, before adding into a pre-cooled (−78 °C) solution of Weinreb amide 27 (5.22 g, 25.0 mmol) in anhydrous THF (20 mL) via cannula. The mixture was stirred at −78 °C for 1 h, then the cooling bath was removed and stirring continued for another 15 min. The mixture was then recooled to −78 °C and quenched with sat. aq. NH4Cl (40 mL). Upon warming to room temperature the mixture was diluted with water (100 mL) and extracted with ethyl acetate (3 × 100 mL), dried over Na2SO4 and concentrated under vacuum. The obtained crude material was subjected to purification by flash column chromatography (ethyl acetate:hexane = 1:4) to afford the desired compound 24 as yellow solid (4.83 g, 76%). m.p. = 78–80 °C; 1H NMR (500 MHz, CDCl3): 7.41 (d, J = 8.6 Hz, 2H), 7.22 (d, J = 8.6 Hz, 2H), 6.90 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 3.84 (s, 2H), 3.82 (s, 3H), 3.80 (s, 3H); 13C NMR (125 MHz, CDCl3): δ = 185.8, 161.8, 159.0, 135.3, 130.0, 125.6, 114.4, 111.7, 94.1, 87.8, 55.5, 55.4, 51.3; FT-IR (neat, cm−1): 2964, 2936, 2899, 2839, 2188, 1667, 1599, 1508, 1462, 1439, 1396, 1297, 1247, 1173, 1068, 1020, 818, 795, 688, 567, 534; HR-MS (m/z): calc for [M-H]+ C18H15O3 279.1027 found 279.1020.
4-(4-methoxyphenyl)spiro[4, 5]deca-3,6,9-triene-2,8-dione (28)
To a solution of compound 24 (1.00 g, 3.57 mmol) in anhydrous CH2Cl2 (25 mL) at 0 °C was added TFA (5.5 mL, 71.35 mmol) and the resulting solution was stirred at 0 °C for 3 h. During this period of time, the reaction color changed to red wine from light yellow. The volatiles were removed under vacuum, ethyl acetate (150 mL) was added and the organic layer was washed with sat aq. NaHCO3 (100 mL), dried over Na2SO4 and concentrated under vacuum. Obtained crude material was purification by flash column chromatography (ethyl acetate: hexane = 3:7) to afford the compound 28 as a black solid (0.90 g, 94%). m.p. = 70–72 °C; 1H NMR (500 MHz, CDCl3): 7.46 (d, J = 8.6 Hz, 2H,), 6.93 (d, J = 10.3 Hz, 2H,), 6.83 (d, J = 8.6 Hz, 2H,), 6.60 (s, 1H), 6.40 (d, J = 10.3 Hz, 2H), 2.73 (s, 2H); 13C NMR (125 MHz, CDCl3): δ = 203.2, 184.9, 173.2, 162.5, 152.2, 129.8, 129.7, 127.6, 125.5, 114.5, 55.6, 51.1, 46.9; FT-IR (neat, cm−1): 3086, 3043, 2936, 2841, 2245, 1737, 1697, 1661, 1583, 1509, 1397, 1308, 1245, 1185, 1041, 1020, 864, 836, 731, 592, 509; HR-MS (m/z): calc for [M+Na]+ C17H14O3Na 289.0835 found 289.0824.
4-(4-Methoxyphenyl)-3-iodospiro[4, 5]deca-3,6,9-triene-2,8-dione (34)
To a solution of compound 24 (1.00 g, 3.57 mmol) in anhydrous acetonitrile (35 mL) at room temperature was added NaHCO3 (0.60 g, 7.13 mmol) followed by iodine (1.81 g, 7.13 mmol). The resulting brown mixture was stirred at room temperature for 30 min. The reaction was quenched with sat. aq. Na2S2O3 (100 mL), extracted with ethyl acetate (3 × 100 mL), dried over Na2SO4 and concentrated under vacuum. The crude material thus obtained was purified by flash column chromatography (ethyl acetate: hexane = 2.5:7.5) to afford the compound 34 as a black solid (1.16 g, 83%); mp. 133–135 °C; 1H NMR (500 MHz, CDCl3): δ = 7.38 (d, J = 10.0 Hz, 2H), 6.87 (d, J = 10.0 Hz, 2H), 6.80 (d, J = 10.0 Hz, 2H), 6.37 (d, J = 10.0 Hz, 2H), 3.82 (s, 3H), 2.93 (s, 2H); 13C NMR (75 MHz, CDCl3): δ = 199.3, 184.5, 175.1, 161.5, 149.8, 130.5, 129.2, 126.3, 114.0, 104.3, 55.5, 54.3, 43.7; FT-IR (neat, cm−1): 2960, 2919, 2838, 1718, 1693, 1660, 1622, 1601, 1562, 1503, 1415, 1293, 1247, 1178, 1024, 923, 857, 835, 766, 553, 524; HR-MS (m/z): calc for [M+Na]+ C17H13O3INa 414.9802, found 414.9804.
2-Iodo-3-oxo-1-phenylspiro[4.5]deca-1,6,9-trien-8-yl-acetate (41)
Alkynone 40 (1.35 g, 6.1 mmol) was dissolved in acetic acid (5 mL) followed by the careful addition of N-iodosuccinimide (NIS) (1.65 g, 7.3 mmol) to maintain ambient temperature. After stirring the resulting mixture for 30 min, ethyl acetate was added to it followed by aqueous saturated NaHCO3 to neutralize the reaction mixture. The EtOAc solution was separated and the aqueous layer was extracted with EtOAc two more times. The combined organic layers were dried (Na2SO4) and concentrated to provide a crude product which was separated chromatographically with 10% ethyl acetate in hexanes as the eluent to provide a ca. 2:1 mixture of 41 (1.46 g, 60%) as a pale brown oil: 1H NMR: δ = 7.42-7.35 (m, 3H), 7.24-7.22 (m, 2H), 5.95-5.88 (m, 2H), 5.87-5.81 (m, 2H), 5.55, 5.25 (m, 1H), 2.82, 2.76 (s, 2H), 2.04, 1.94 (s, 3H); 13C NMR: δ = 201.1 (201.0), 180.1 (178.9), 170.5 (170.5), 135.2 (135.0), 132.5 (133.8), 130.0 (129.9), 128.3 (128.0), 127.3 (127.9), 126.3 (125.4), 105.4 (104.8), 63.6 (63.4), 51.8 (51.4), 46.7 (47.6), 21.1 (21.0); IR (neat, cm−1): = 3033, 2924, 1721, 1369, 1236, 1016, 928, 739, 698; HRMS-ESI (m/z): Calc. for C18H15INaO3 [M+Na]+ 428.9958, found: 428.9926.
2-Formylamino-3-oxo-1-phenylspiro[4.5]deca-1,6,9-trien-8-yl-acetate (42)
A 10 mL resealable, thick-walled tube was charged with CuI (20 mg, 0.1 mmol), iodo-ketone 41 (841 mg, 2.1 mmol) and Cs2CO3 (1.01 g, 3.1 mmol). The tube was evacuated and back filled with nitrogen and N,N′-dimethylethylenediamine (0.02 mL, 0.2 mmol), formamide (0.10 mL, 2.5 mmol) and THF (5 mL) were added under nitrogen. The pressure tube was sealed with a Teflon cap, immersed in a preheated oil bath at 70 °C and the reaction mixture was stirred 48 h following reaction progress by TLC. After the resulting pale blue suspension was allowed to reach to rt, EtOAc was added. The reaction mixture was filtered through a pad of silica eluting with EtOAc. The filtrate was concentrated and the residue was purified by flash chromatography on silica gel with 10% ethyl acetate in hexanes to provide 42 as brown oil (334 mg, 50%): 1H NMR: δ = 8.88, 8.83 (d, J = 11.5 Hz, 1 H), 7.39 (s, 3H), 7.34 (m, 1H), 7.18 (m, 2H), 5.95-5.90 (m, 2H), 5.86-5.80 (m, 2H), 5.59, 5.32 (m, 1H), 2.73, 2.67 (s, 2H), 2.05, 1.95 (s, 3H); 13C NMR: δ = 199.9 (199.8), 170.5, 161.8 (162.0), 154.1 (153.5), 133.3 (134.5),133.9 (133.6), 132.3 (132.5), 129.6 (129.7), 129.1 (128.8), 127.6 (128.1), 126.1 (125.3), 63.6 (63.4), 47.4 (48.2), 46.2 (45.9), 21.2 (21.0); IR (neat, cm−1): = 3195, 3023, 2917, 2850, 1667, 1494, 1441, 1221, 1097, 748, 698; HRMS (m/z): Calcd. For C19H16NO4 [M-H] = 322.1085, found 322.1069.
3-Oxo-1-phenyl-spiro[4.5]deca-1,6,9-trien-8-yl-acetate (43)
From the above reaction a second product 43 (87 mg, 15%) was isolated as a dark brown oil: 1H NMR: δ = 7.75-7.72 (m, 1 H), 7.54-7.51 (m, 1H), 7.43-7.32 (m, 3H), 6.58, 7.51 (s, 1H), 6.01-5.93. (m, 4 H), 5.78-5.77 (m, 1 H), 2.67, 2.60 (s, 2H), 2.13, 2.11 (s, 3H); 13C NMR: δ =205.8 (205.7), 176.9 (175.8), 170.8 (170.6), 135.4 (135.2), 133.9 (133.4), 131.0 (130.8), 129.9 (129.3), 128.7 (128.5), 128.5 (127.8), 124.4 (124.3), 64.2 (64.0), 51.3 (50.7), 48.5 (47.9), 21.3 (21.3); IR (neat, cm−1)= 3022, 1726, 1691, 1590, 1568, 1368, 1229, 1012, 966, 863, 754, 647; HRMS Calcd for [M+Na] C18H16O3Na 303.0992, found 303.0966.
3-Amino-8-hydroxy-4-phenyl-spiro[4.5]deca-3,6,9-trien-2-one (45)
Methylamine hydrochloride (200 mg, 3.0 mmol) and K2CO3 (410 mg, 3.0 mmol) were added to a solution of 42 (192 mg, 0.6 mmol) in ethanol (10 mL) and the mixture was heated at reflux overnight. Water was added to the reaction mixture after evaporating the solvent and the aqueous solution was extracted with EtOAc (2×10 mL). The combined organic solutions were dried (Na2SO4) and concentrated to provide a crude product, which was purified by chromatography on silica gel using 2:3 mixture of EtOAc: hexanes to isolate 45 (30 mg, 20%) as a light brown solid: 1H NMR: δ = 7.40-7.38 (m, 2H), 7.35-7.32 (m, 2H), 7.28-7.27 (m, 1H), 5.95 (dd, J = 3.2, 10.1 Hz, 2H), 5.72 (dd, J = 1.8, 10.1 Hz, 2H), 4.37 (s, 1H), 8.84 (s, 2H), 2.58(s, 2H); 13C NMR δ = 201.6, 140.7, 140.1, 135.0, 134.4, 128.7, 128.6,128.2, 127.5, 61.9, 48.0, 44.9;
3-Amino-4-phenyl-2-naphthol (46)
Aminol 45 (23 mg, 0.1 mmol) was added to a solution of cyanamide (90 mg, 2.1 mmol) in water. This mixture was acidified to pH 4.5 by careful addition of 10% HCl and the resulting mixture was heated at 90 °C for 3 h. After cooling, the resulting mixture was made basic to pH 10 with 20% NaOH and the aqueous layer was extracted with EtOAc (2 × 10 mL). The combined organic layers were dried (Na2SO4), concentrated to provide 46 (60%) as a brown solid: m.p. = 162-164 °C; 1H NMR (Acetone-d6): δ = 7.58-7.53 (m, 3H), 7.46-7.43 (m, 1H), 7.34-7.32 (m, 2H), 7.17 (s, 1H), 7.10-7.07 (m, 2H), 7.06-7.03 (m, 1H),4.25 (brd, 2H), 2.85 (brd, 2H); 13C NMR (Methanol-d3): δ = 145.8, 137.5, 133.7, 130.6, 128.9, 128.8, 128.7, 127.2, 125.7, 123.5, 122.7, 122.1, 120.1, 107.6. HRMS (m/z): Calcd. for [M-H] C16H12NO 234.0924, found 234.0924.
1-Methyl-4-phenyl-1H-naphtho[2,3-d]imidazole (48)
Methylamine hydrochloride (158 mg, 2.4 mmol) and Et3N (0.13 mL, 0.9 mmol) were added to a solution of 42 (153 mg, 0.5 mmol) in ethanol (5 mL) that was heated at reflux temperature overnight. Water was added to the reaction mixture after evaporating the solvent and the aqueous layer was extracted with EtOAc (2x 10 mL). The combined organic layers were dried (Na2SO4) and concentrated to provide a crude product, which was purified over silica gel using a 3:2 mixture of EtOAc: hexanes to produce 48 (43 mg, 35%) as a green solid: m.p. = 185–188 °C; 1H NMR: δ = 8.70 (d, J = 10.1 Hz, 1H), 8.27 (d, J = 9.2 Hz, 1H), 8.01 (s, 1H), 7.97 (d, J = 7.8 Hz 2H), 7.54 (t, J = 7.8 Hz, 2H), 7.47 (t, J = 10.1 Hz, 1H), 7.34 (t, J = 7.8 Hz, 1H), 7.09 (t, J = 9.2 Hz, 1H), 7.01 (t, J = 10.1 Hz, 1H), 4.21 (s, 3H); 13C NMR: δ = 155.7, 147.1, 135.7, 135.4, 135.2, 133.9, 129.8, 128.8, 128.0, 126.8, 126.5, 122.7, 121.9, 121.4, 117.1, 33.8; IR (neat, cm−1): = 3039, 2918, 2844, 1607, 1592, 1572, 1538, 1495, 1456, 1372, 1261, 1171, 1044, 896, 740; HRMS-ESI (m/z): Calcd. for C18H14N2 [M+H]+: 259.1230, found 259.1227; Calcd. for C36H29N4 [2M+H]+ 517.2387, found 517.2390.
2-iodo-1-(4-methoxy-phenyl)-3-oxo-spiro[4.5]deca-1,6,9-trien-8-yl-acetate (53)
Alkynone 52 (530 mg, 2.1 mmol) was dissolved in acetic acid (5 mL) followed by careful addition of NIS (577 mg, 5.5 mmol) to maintain ambient temperature. After stirring the resulting mixture for 15 min, ethyl acetate (75 mL) was added to it followed by addition of sufficient satd. aq. NaHCO3 to neutralize the reaction mixture. The organic solution was separated and the aqueous layer was extracted with ethyl acetate (2 × 75 mL). The combined organic layers were dried (Na2SO4) and concentrated to provide a crude product, which was separated chromatographically with 20% ethyl acetate in hexanes as the eluent to provide a 5:4 mixture of 53 (747 mg, 81%) as a reddish brown oil: 1H NMR: δ = 7.57, 7.32 (d, J = 8.7 Hz, 2H)), 6.88, 6.87 (d, J = 8.7 Hz, 2H), 5.95-5.92, 5.94-5.91 (m, 2H), 5.86-5.82, 5.85-5.80 (m, 2H), 5.59, 5.39 (m, 1H), 3.82, 3.82 (s, 3H), 2.78, 2.71 (s, 2H), 2.05, 2.01 (s, 3H); 13C NMR: δ = 201.1 (200.9), 179.1 (177.6), 170.5 (170.5), 161.1 (160.8), 133.3 (134.3), 129.2 (130.2), 127.3 (127.0), 125.8 (125.0), 113.7 (113.4), 104.2 (103.0), 63.8 (63.5), 55.3, 51.7 (51.3), 47.0 (47.9), 21.2 (21.1); IR (neat, cm−1)= 2930, 2837, 1728, 1704, 1603, 1504, 1330, 1226, 1175, 1020, 900, 835, 760; HRMS-ESI (m/z): Calcd. for C19H18IO4 [M+H]+ 437.0244, found 437.0243.
3-Iodo-4-(4-methoxyphenyl)naphthalen-2-ol (54) and 1,3-Diiodo-4-(4-methoxyphenyl)naphthalen-2-ol (55)
Alkynone 52 (530 mg, 2.1 mmol) was dissolved in acetic acid (5 mL) and NIS (577 mg, 5.5 mmol) was added at once to the reaction at rt (the reaction mixture became warm). After stirring the resulting mixture for 15 min, usual workup provided the crude material, which was purified over silica gel (1:9 EtOAc: hexanes) resulting in the isolation of 54 (245 mg, 31%) and 55 (179 mg, 17%).
Characterization data for compound 54, reddish brown solid: m.p. = 139–141 °C; 1H NMR: δ = 7.72 (d, J = 7.8 Hz, 1H), 7.43 (t, J = 7.8 Hz, 2H), 7.34 (d, J = 8.7 Hz, 1H), 7.19 (d, J = 7.8 Hz, 1H), 7.18 (d, J = 8.7 Hz, 2H), 7.06 (d, J = 8.7 Hz, 2H), 5.66 (s, 1H), 3.92 (s,3H); 13C NMR (DEPT 135): δ = 159.4 (C), 151.1 (C), 146.2 (C), 138.9 (C), 134.7 (C), 131.0 (CH), 129.1 (C), 127.5 (CH), 127.2 (CH), 126.7 (CH), 124.4 (CH), 114.0 (CH), 108.9 (CH), 97.7 (C), 55.4 (CH3); IR (neat, cm−1): = 3478 (br), 2954, 2833, 1604, 1583, 1510, 1328, 1242, 1213, 1170, 1026, 872, 843, 795, 773, 753; HRMS-ESI (m/z): Calcd. for C17H14IO2 [M+H]+ 377.0033, found 377.0046; Calcd. for C17H13INaO2 [M+Na]+ 398.9853, found 398.9867.
Characterization data for compound 55 reddish brown solid: m.p. = 104–106 °C; 1H NMR: δ = 8.06 (d, J = 8.7 Hz, 1H), 7.53 (tt, J = 1.4, 7.8 Hz, 1H), 7.36 (d, J = 8.2 Hz, 1H), 7.22 (tt, J = 1.4, 7.8 Hz, 1H), 7.14 (d, J = 8.7 Hz, 2H), 7.08 (d, J = 8.7 Hz, 2H), 6.36 (s, 1H), 3.92 (s,3H); 13C NMR (DEPT 135): δ = 159.5 (C), 151.1 (C), 147.5 (C), 135.5 (C), 134.9 (C), 131.1 (CH), 131.0 (CH), 129.6 (C), 128.9 (CH), 128.5 (CH), 125.1 (CH), 114.1 (CH), 93.5 (C), 83.2 (C), 55.6 (CH3); IR (neat, cm−1): = 3391 (br), 3062, 2953, 2840, 1605, 1511, 1486, 1371, 1234, 1171, 1027, 749; HRMS-ESI (m/z): Calcd. for C17H13I2O2 [M+H]+ 502.9005, found 341.1512; Calcd. for C17H12I2NaO2 [M+Na]+ 524.8824, found 363.1328. observed ion consistent with MW of 340 Da. This structure was confirmed by X-ray crystallography.
2-Formylamino-1-(4-methoxy-phenyl)-3-oxo-spiro[4.5]deca-1,6,9-trien-8-yl-acetate (57)
Following the procedure for 42; CuI (2 mg, 0.01 mmol), iodo-ketone 53 (102 mg, 0.23 mmol) and Cs2CO3 (152 mg, 0.50 mmol), N, N′-dimethylethylenediamine (0.01 mL, 0.02 mmol), formamide (0.02 mL, 0.50 mmol) and THF (2 mL) were heated at reflux temperature for 12 h. The crude product was purified with 1:1 mixture of EtOAc:hexanes to isolate a 1:1 mixture of 57 (58 mg, 70%) as light brown oil; 1H NMR: δ = 8.87, 8.83 (two overlapping doublets, 1H), 7.38, 7.38 (d, J = 8.5 Hz, 1H), 7.19, 7.19 (d, J = 8.5 Hz, 2H), 6.89, 6.89 (d, J = 8.5 Hz, 2H), 5.98-5.90 (m, 2H), 5.90-5.78 (m, 2H), 5.64, 5.44 (s, 1H), 3.81, 3.81 (s, 3H), 2.69, 2.62 (s, 2H), 2.06, 2.01 (s, 3H); 13C NMR: δ = 199.8 (199.7), 170.6 (170.5), 162.0 (162.3), 160.6 (160.8), 154.4 (153.9), 133.9 (134.8), 133.3 (132.7), 129.1 (129.8), 125.8 (125.7), 124.4 (124.5), 114.6 (114.3), 63.8 (63.6), 55.4 (55.4), 47.6 (48.5), 46.1 (45.7), 21.2 (21.1); IR (neat, cm−1): = 3317, 3011, 2965, 2828, 1699, 1662, 1604, 1507, 1384, 1283, 1245, 1175, 1030, 804, 734; HRMS-ESI (m/z): Calcd. for C20H20NO5 [M+H]+ 354.1341, found 354.1311; Calcd. for C20H19NNaO5 [M+Na]+ 376.1161, found 376.1129.
1-(4-Methoxy-phenyl)-3-oxo-spiro[4.5]deca-1,6,9-trien-8-yl-acetate (56)
From the above reaction a small amount of the reductive dehalogenation product 56 (15 mg, 20%) was isolated as a 1:1 mixture of a dark brown solid: m.p. = 92–95 °C; 1H NMR: δ = 7.72, 7.42 (d, J = 8.8 Hz, 2H), 6.75 (d, J = 8.3 Hz, 4 H), 6.37, 6.34 (s, 1H), 5.89-5.81 (m, 4H), 5.74, 5.64 (m, 1H), 3.70, 3.68 (s, 3H), 2.49,2.42 (s, 2H), 2.04,2.00(s, 3H); 13C NMR: δ = 205.6 (205.5), 176.0 (175.2), 170.9 (170.6), 161.9 (161.8), 136.1 (135.5), 130.5 (129.8), 127.7 (127.1), 126.2 (125.9), 124.1 (123.9), 114.2 (114.0), 64.3 (64.1), 55.5, 51.3 (50.8), 48.2 (47.7), 21.3; IR (neat, cm−1): = 2841, 1728, 1680, 1600, 1585, 1508, 1238, 1176, 1022, 869, 806, 757; HRMS (m/z): Calcd. for C19H18O4Na 333.1097, found 333.1072.
1-Methyl-4-(4-methoxyphenyl)-1H-naphth[2,3-d]imidazole (58)
A solution of methylamine in methanol (8.03 M, 0.05 mL, 0.4 mmol) was added to amide 58 (120 mg, 0.4 mmol) pre-absorbed on silica gel (~1 g). After stirring at rt. overnight, Zn(BH4)2 in THF (4.0 M, 0.10 mL, 0.4 mmol) was added to above reaction, and stirred for 2 h. After removing the solvent, crude product was purified over silica gel (1:1 EtOAc: hexanes) to isolate 59 (30 mg, 30%) as a light brown solid: m.p. = 158–160 °C; 1H NMR: δ = 8.07 (d, J = 8.7 Hz, 1H), 8.00 (d, J = 8.7 Hz, 2H), 7.79 (s, 1H), 7.57 (d, J = 8.7 Hz, 2H), 7.45 (tt, J = 1.4, 7.3 Hz, 1H), 7.35 (tt, J = 1.4, 7.3 Hz, 1H), 7.12 (d, J = 8.7 Hz, 2H), 3.92 (s, 3H), 3.91 (s, 1H); 13C NMR δ = 159.1, 147.5, 142.5, 134.9, 132.3, 131.1, 129.6, 128.9, 128.3, 127.8, 126.5 124.4, 123.5, 114.0, 104.6, 55.4, 31.2; IR (neat, cm−1): = 3054, 2920, 2844, 1667, 1513, 1241, 1174, 1024, 828, 746; HRMS (m/z): Calcd. for C19H16ONa [M+Na]+ 311.1155, found 311.1137.
X-ray crystallographic data
A suitable crystal covered with a layer of hydrocarbon/paratone-N oil was selected and mounted on a Cryo-loop, and immediately placed in the low temperature nitrogen stream. The X-ray intensity data for 55 were measured at 100(2) K on a SMART APEX CCD area detector system equipped with a Oxford Cryosystems 700 series cooler, a graphite monochromator, and a Mo-Kα fine-focus sealed tube (λ = 0.71073 Å). Intensity data were processed using the Saint Plus program. All the calculations for the structure determination were carried out using the SHELXTL package (version 6.14). Initial atomic positions were located by direct methods using XS, and the structure of the compound was refined by the least-squares method using XL. Absorption correction was applied by using SADABS. Hydrogen atoms were placed at calculated positions and refined riding on the corresponding carbons. All non-hydrogen atoms were refined anisotropically.
Supplementary Material
Figure 1.

Representative Leucetta alkaloids
Figure 4.

X-ray crystal structure of the diiodonaphthol derivative 55.
Acknowledgments
This work has been supported by the Robert A Welch Foundation (Y-1362) and in part by NIH (GM065503). The NSF (CHE-0234811 and CHE-0840509) is thanked for funding the purchase of the NMR spectrometers used in this research. The authors acknowledge the Texas Advanced Computing Center (TACC) at The University of Texas at Austin for providing resources that have contributed to the research results reported within this paper. URL: http://www.tacc.utexas.edu
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/ejoc.xxxxxxxxx.
Supporting Information Copies of 1H and 13C NMR spectra for all new compounds.
References
- 1.a Koswatta PB, Lovely CJ. Nat Prod Rep. 2011;28:511–528. doi: 10.1039/c0np00001a. [DOI] [PubMed] [Google Scholar]; b Sullivan JD, Giles RL, Looper RE. Curr Bioact Cpds. 2009;5:39–78. [Google Scholar]; c Roue M, Quevrain E, Domart-Coulon I, Bourguet-Kondracki ML. Nat Prod Rep. 2012;29:739–751. doi: 10.1039/c2np20040f. [DOI] [PubMed] [Google Scholar]
- 2.a Koswatta PB, Lovely CJ. Tetrahedron Lett. 2009;50:4998–5000. [Google Scholar]; b Zavesky BP, Babij NR, Wolfe JP. Org Lett. 2014;16:4952–4955. doi: 10.1021/ol502471x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koswatta PB, Lovely CJ. Chem Commun. 2010;46:2148–2150. doi: 10.1039/b926285g. [DOI] [PubMed] [Google Scholar]
- 4.Lima HM, Garcia-Barboza BJ, Khatibi NN, Lovely CJ. Tetrahedron Lett. 2011;52:5725–5727. doi: 10.1016/j.tetlet.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a Gibbons JB, Gligorich KM, Welm BE, Looper RE. Org Lett. 2012;14:4734–4737. doi: 10.1021/ol3019242. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Das J, Koswatta PB, Yousufuddin M, Jones JD, Lovely CJ. Org Lett. 2012;14:6210–6213. doi: 10.1021/ol302958e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hassan W, Edrada R, Ebel R, Wray V, Berg A, Van Soest R, Wiryowidagdo S, Proksch P. J Nat Prod. 2004;67:817–822. doi: 10.1021/np0305223. [DOI] [PubMed] [Google Scholar]
- 6.Akee RK, Carroll TR, Yoshida WY, Scheuer PJ, Stout TJ, Clardy J. J Org Chem. 1990;55:1944–1946. [Google Scholar]
- 7.Fu X, Barnes JR, Do T, Schmitz FJ. J Nat Prod. 1997;60:497–498. [Google Scholar]
- 8.a Koswatta PB, Sivappa R, Dias HVR, Lovely CJ. Org Lett. 2008;10:5055–5058. doi: 10.1021/ol802018r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Edrada RA, Stessman CC, Crews P. J Nat Prod. 2003;66:939–942. doi: 10.1021/np020503d. [DOI] [PubMed] [Google Scholar]; c Koswatta PB, Sivappa R, Dias HVR, Lovely CJ. Synthesis. 2009:2970–2982. [Google Scholar]
- 9.White KN, Amagata T, Oliver AG, Tenney K, Wenzel PJ, Crews P. J Org Chem. 2008;73:8799–8722. doi: 10.1021/jo800960w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagasawa Y, Kato H, Rotinsulu H, Mangindaan REP, Voogd NJd, Tsukamoto S. Tetrahedron Lett. 2011;52:5342–5344. [Google Scholar]
- 11.Sivappa R, Koswatta P, Lovely CJ. Tetrahedron Lett. 2007;48:5771–5774. doi: 10.1016/j.tetlet.2007.06.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a Lima HM, Sivappa R, Yousufuddin M, Lovely CJ. Org Lett. 2012;14:2274–2277. doi: 10.1021/ol300704w. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Das J, Bhan A, Mandal S, Lovely CJ. Bioorg Med Chem Lett. 2013;23:6183–6187. doi: 10.1016/j.bmcl.2013.08.093. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lima HM, Sivappa R, Yousufuddin M, Lovely CJ. J Org Chem. 2014;79:2481–2490. doi: 10.1021/jo4027337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lovely CJ. In: Strategies and Tactics in Organic Synthesis. Harmata MA, editor. Vol. 8. Academic Press; 2012. [Google Scholar]
- 14.Du H, He Y, Rasapalli S, Lovely CJ. Synlett. 2006:965–992. [Google Scholar]
- 15.Koswatta PB. PhD Dissertation. University of Texas at Arlingron; 2010. [Google Scholar]
- 16.Zhang X, Larock RC. J Am Chem Soc. 2005;127:12230–12231. doi: 10.1021/ja053079m. [DOI] [PubMed] [Google Scholar]
- 17.Tang B, Tang D, Tang S, Yu Q, Zhang Y, Liang Y, Zhong P, Li J. Org Lett. 2008;10:1063–1066. doi: 10.1021/ol703050z. [DOI] [PubMed] [Google Scholar]
- 18.Unsworth WP, Cuthbertson JD, Taylor RJK. Org Lett. 2013;15:3306–3309. doi: 10.1021/ol4013958. [DOI] [PubMed] [Google Scholar]
- 19.Although p-ethynylanisole is commercially available, it is rather expensive and so this was prepared via a Sonogashira reaction between TMS-acetylene and p-iodoanisole followed by base induced desilylation.
- 20.Gupta MK, Li Z, Snowden TS. Org Lett. 2014;16:1602–1605. doi: 10.1021/ol500200n. [DOI] [PubMed] [Google Scholar]
- 21.Kotecki BJ, Fernando DP, Haight AR, Lukin KA. Org Lett. 2009;11:449–450. doi: 10.1021/ol802931m. [DOI] [PubMed] [Google Scholar]
- 22.a Jiang L, Job GE, Klapars A, Buchwald SL. Org Lett. 2003;5:3667–3669. doi: 10.1021/ol035355c. [DOI] [PubMed] [Google Scholar]; b Zheng N, Buchwald SL. Org Lett. 2007;9:4749–4751. doi: 10.1021/ol7020737. [DOI] [PubMed] [Google Scholar]
- 23.A further issue that would require a solution would be the selective N-methylation of the 2-aminoimidazole which would likely be challenging.
- 24.Focken T, Charette AB. Org Lett. 2006;8:2985–2988. doi: 10.1021/ol0609006. [DOI] [PubMed] [Google Scholar]
- 25.a Nandakumar MV. Tetrahedron Lett. 2004;45:1989–1990. [Google Scholar]; b Hosseinzadeh R, Sarrafi Y, Mohadjerani M, Mohammadpourmir F. Tetrahedron Lett. 2008;9:840–843. [Google Scholar]
- 26.a Rosiak A, Frey W, Christoffers J. Eur J Org Chem. 2006:4044–4054. [Google Scholar]; b Zhang X, Sarkar S, Larock RC. J Org Chem. 2006;71:236–243. doi: 10.1021/jo051948k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.The CIF for compound 55 has been deposited with the Cambridge Crystallographic Data Centre; accession number CCDC 1040263.
- 28.Shamma M, Guinaudeau H. Tetrahedron. 1984;40:4795–4822. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.