

Abstract
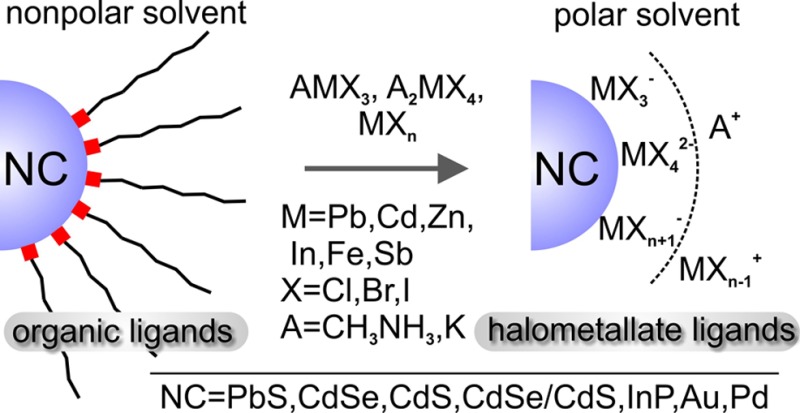
Lead halide perovskites (CH3NH3PbX3, where X = I, Br) and other metal halide complexes (MXn, where M = Pb, Cd, In, Zn, Fe, Bi, Sb) have been studied as inorganic capping ligands for colloidal nanocrystals. We present the methodology for the surface functionalization via ligand-exchange reactions and the effect on the optical properties of IV–VI, II–VI, and III–V semiconductor nanocrystals. In particular, we show that the Lewis acid–base properties of the solvents, in addition to the solvent dielectric constant, must be properly adjusted for successful ligand exchange and colloidal stability. High luminescence quantum efficiencies of 20–30% for near-infrared emitting CH3NH3PbI3-functionalized PbS nanocrystals and 50–65% for red-emitting CH3NH3CdBr3- and (NH4)2ZnCl4-capped CdSe/CdS nanocrystals point to highly efficient electronic passivation of the nanocrystal surface.
Surface chemistry largely dictates the physical and chemical properties of individual nanocrystals (NCs) as well as electronic communication between the NCs in their densely packed solids.1 Since the early 1990s, organic capping ligands have enabled tremendous progress in the colloidal synthesis of monodisperse NCs of a wide variety of metals, semiconductors, and magnetic materials.1a,2 However, long-chain insulating organic ligands must be removed for the integration of NCs into solids with tunable electronic properties, as required for NC-based solar cells,3 light-emitting diodes,4 photodetectors,5 thermoelectrics,6 transistors,5c,7 and integrated electronic circuits.8 Solution-phase exchange of organic ligands with smaller inorganic species has been recently demonstrated as a powerful methodology, as it preserves the integrity, size-tunable optical properties, and solution-processability of NCs, while greatly improving charge transport in NC solids. Common inorganic anions that have been successfully applied as inorganic capping ligands are metal chalcogenide complexes (MCCs, also known as chalcogenidometallates),6b,9 oxo- and polyoxometallates,10 and metal-free ions (S2–, HS–, Se2–, OH–, SCN–, etc.).8,11 The strong adsorption of an anion combined with dissociation of cations in highly polar organic solvents leads to efficient colloidal stabilization. Here, we present metal halide complexes (halometallates) as capping ligands for a variety of colloidal inorganic NCs (Scheme 1). The factors governing successful ligand exchange and colloidal stability are discussed, and the results are compared to the previous work of some of us related to chalcogenidometalate capping.6b,9 Importantly, we demonstrate the ability of halometallate ligands to efficiently passivate the surface of semiconductor NCs (PbS and CdSe/CdS), resulting in highly efficient excitonic photoluminescence (PL).
Scheme 1. Schematics of the Ligand-Exchange Methodology for Obtaining Halometallate-Capped Colloidal Nanocrystals.
On their own halometallates represent a very important class of inorganic compounds. For instance, methylammonium lead iodides and bromides (MAPbI3, MAPbBr3, where MA = CH3NH3+), known as hybrid metal halide perovskites due to their perovskite crystal structure, have attracted enormous interest in recent years as low-cost, solution-deposited photovoltaic materials with record power conversion efficiencies of 10–15%.12 Also CsSnI3 has been recently used as a solid-state electrolyte in 12%-efficient dye sensitized solar cells.13 Hence the combination of the two attractive classes of semiconductors, NCs and metal halide perovskites, may open new and exciting opportunities. In particular, we find that MAPbI3 is the first example of inorganic capping ligands that is able to retain the highly efficient near-infrared PL of PbS NCs (quantum yield of 20–30%).
In a typical ligand-exchange procedure [for details, see the Supporting Information(SI)], a 0.05 M solution of a metal halide complex or neutral metal halide salt in N-methylformamide (MFA, 1 mL) was stirred with a hexane solution of NCs (2–5 mg in 1 mL) for several hours until the NCs were completely transferred to the polar phase (Figure 1a). NCs were precipitated from MFA by adding a nonsolvent such as acetone, centrifuged, and redissolved in propylene carbonate (PC) or MFA. No air- or moisture-free techniques were needed, except for FeCl2, SnX2, and BiX3 (X = Br, I). Completeness of the exchange of the initial organic ligands with halometallates was confirmed by Fourier-transform infrared (FTIR) spectroscopy (Figure 1b), as seen from the disappearance of the characteristic C–H and O–H stretching modes (2800–3500 cm–1), C–H bending vibrations, and carboxylic C–O and vinyl C=C stretching modes (600–1500 cm–1). Transmission electron microscopy (TEM) images for PbS NCs capped with MAPbI3 (Figure 1d) and PbI2 (Figure 1e), as well as for other NC-ligand combinations (Figures S1 and S2 in SI), confirmed the integrity of the NCs and retention of their narrow size distribution. The true colloidal nature of halometallate-capped NCs, apart from the photographs (Figure 1a), was also confirmed by single-particle population in the measurements of dynamic light scattering (DLS, Figure S3). Colloidal solutions of PbS NCs stabilized with MAPbI3 were stable for months without noticeable aggregation or precipitation. Pb and Cd chalcogenide NCs showed high affinity to nearly all studied ligands, whereas much fewer ligands formed stable colloidal solutions with metallic NCs (Table S1). Importantly, only in the case of CdSe NCs metal-free halide ions I–, Br– (as salts with K+ and MA+ cations) can partially displace oleate ligands, yet without the formation of colloidally stable solutions. Elemental analysis of purified halometallate-capped NCs confirmed the expected overall compositions (7–32 at% of ligand atoms for 3–5 nm NCs, Table S2). Halometallate-capped NCs are negatively charged, as seen from electrophoretic measurements providing ξ-potentials of at least −40 mV for MAPbI3-stabilized PbS NCs (Figures 1c and S4; see Table S3 for other NC-ligand combinations). Highly negative ξ-potentials, caused by the surface-bound anions such as [PbI3]− or [PbBr3]−, were measured for both preformed complexes (e.g., KPbI3) and for neutral halide salts MXn (e.g., PbI2). The latter can be attributed to the well-known self-ionization in polar solvents:14
| 1 |
Figure 1.
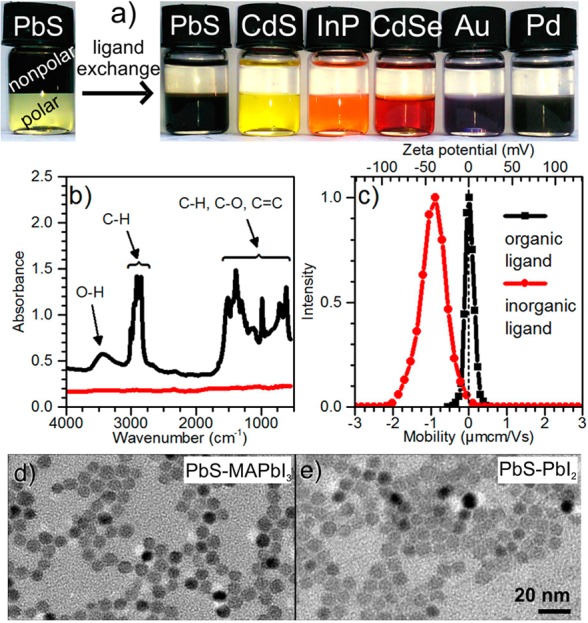
(a, from left to right) Illustration of the phase-transfer of PbS NCs from nonpolar solvent (hexane) to polar solvent (MFA) caused by the exchange of oleate capping with MAPbI3; other examples of halometallate-capped NCs (CdS-SbCl3, InP-InCl3, CdSe-FeCl2, Au-InCl3, Pd-InCl3 in PC as a solvent). (b) FTIR spectra before and after exchange of oleate-capping on the surface of PbS NCs with KPbI3. (c) Electrophoretic mobility for oleate-capped PbS NCs in toluene (black) and MAPbI3-stabilized PbS NCs in PC. (d, e) TEM images of PbS-MAPbI3 and PbS-PbI2 NCs.
We can emphasize the following factors governing efficient ligand exchange and colloidal stabilization. (i) The affinity of the incoming anionic ligands to the NC surface. Chalcogenidometalate ions (Sn2S64–, AsS43–, In2Se42–) possess both X- and L-type ligand functions, while halometallates can be viewed as almost purely L-type ligands. In other words, a halide ion cannot establish two σ-bonds (one with the central atom of the anion and the other with the metal on the NC surface), but a chalcogenide-based ligand can. Although the atomistic details of NC-ligand binding still have to be revealed for all known inorganic ligands, this simple consideration explains a much slower ligand-exchange reaction for halometallates compared to that for chalcogenidometalates (hours vs minutes for CdSe-oleate NCs treated with MACdBr3 and K3AsS4, as an example). (ii) The solvent dielectric constant, which allows electrolytic dissociation of cations, leading to the electrostatic mechanism of colloidal stabilization. Correspondingly, MFA and very similar N-methylacetamide are often the best solvents for performing ligand-exchange reactions due to very high static dielectric constants of 182 and 191, respectively. (iii) The solvation of anions. Once adsorbed, anions should stay on the surface. The Gibbs energy of anion adsorption on the solid/liquid interface changes with the solvent acidity due to the solvation of anions.15 A convenient parameter ET(30) and its normalized version ETN for the evaluation of the solvent Lewis acidity were proposed by Dimroth and Reichardt.16ETN indicates the ability of a given solvent to solvate anions and lies between 0 (tetramethylsilane) and 1 (water). In our case, PbS-MAPbI3 NCs formed concentrated (25 mg/mL) solutions in MFA (a good Lewis acid with ETN = 0.722),17 but immediately precipitated upon dilution. At the same time, PbS-MAPbI3 solutions in PC (weak Lewis acid, ETN = 0.472)17 were stable in a wide range of concentrations (0.1–50 mg/mL). This can be explained by the much stronger desorption of anions in MFA. (iv) The solvation of the cations. This factor is especially important for neutral metal halide salts because efficient solvation of complex cations such as [PbI]+ stabilizes the [PbI3]− anion as well (reaction 1). The ability of a given solvent to solvate cations strongly correlates with the solvent donor number (DN),17 which reflects the Lewis basicity of the solvent. PC (DN = 15.1) can solvate CH3NH3+ or K+. On the other hand, much less stable [PbI]+ cations formed by reaction 1 cannot be efficiently solvated by PC and thus PbS NCs stabilized with PbI2 were not soluble in neat PC. At the same time, addition of a cosolvent (∼1 wt %) with a high DN (dimethylformamide, DMF, DN = 26.6 or hexamethylphosphoric triamide, HMPA, DN = 38.8) immediately yields very stable colloids in PC.
For PbS NCs, there is growing recognition that halide ions reduce the density of surface trapping states and therefore enhance the performance of solar cells based on PbS NCs, reaching power conversion efficiencies of 6% and 7%.18 The authors of ref (18) used solid-state ligand exchange or partial halide passivation via solution-phase treatment of oleate-capped PbS NCs, maintaining most of the oleate capping for efficient colloidal stabilization in nonpolar solvents. Our present study shows the possibility of obtaining fully inorganic, halide-covered PbS NCs in the form of stable colloidal solutions. The integrity of PbS NC cores after the ligand exchange with MAPbI3 is evidenced by absorption spectra (Figure 2), which contain sharp excitonic features, slightly red-shifted with respect to the oleate-capped NCs. The extent of electronic passivation was monitored with steady-state and time-resolved PL measurements. Colloidal solutions of MAPbI3-PbS NCs exhibit PL quantum yields (QY) of 20–30%, comparable with the QYs of NCs capped with oleic acid before the ligand exchange and much higher than the QY of PbS NCs capped with [AsS4]3– (QY ≤ 1%). A similar perovskite compound, MAPbBr3, also preserved the efficient and stable PL properties of PbS NCs. Enhanced surface passivation, suggested by the high PL QY, is also evidenced by the retention of a long-lived component in the corresponding PL decays of PbS NCs (Figure 2B). The long component, usually on the order of sub-μs, is characteristic of the intrinsic recombination rate of the exciton, screened by the high dielectric constant of PbS. Time-resolved PL traces for NCs capped with the three different ligand materials are adequately fitted with double exponential fits (see Figure S5 and Table S4 in SI for 3.8 and 4.5 nm PbS NCs). Oleate-capped NCs are characterized by two time constants in the range of 300 ns and 80 ns. The faster component can be attributed to recombination on the surface states. MAPbI3-capped NCs show average PL lifetimes of similar magnitude to the oleate-capped ones, in agreement with the comparable QY of the two NC materials. The decays consist of a fast component of 10–30 ns during which 20–30% of the photogenerated carriers recombine, presumably via trapping, and a longer decay in the range 600–700 ns that can be attributed to excitonic recombination. Contrary to the above two cases, the fast PL decays of K3AsS4-capped NCs (two time constants in the range of 10 ns and 20–40 ns, respectively) strongly suggest fast carrier trapping as the dominant recombination channel, consistent with the low QYs.
Figure 2.
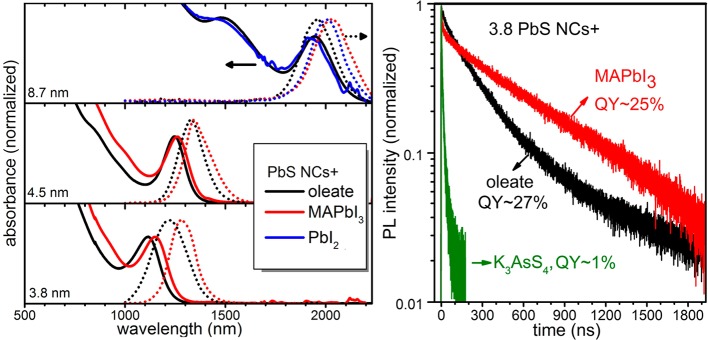
(a) Absorption and steady-state PL spectra of PbS NCs before and after ligand exchange. (b) Time-resolved PL spectra of the ∼3.8 nm PbS NC solutions capped with oleic acid (black), MAPbI3 (red), and K3AsS4 (blue) ligands.
For the visible spectral region, we prepared two sizes of CdSe/CdS NCs (emission peaks at 608 and 640 nm, CdS shell thickness of 2.8–3 nm), according to the recent method of Bawendi et al.19 These NCs feature a high PL QY of ∼72–75% in combination with a very narrow ensemble PL line width of ≤30 nm (≤90 meV). MACdBr3 and (NH4)2ZnCl4 ligands retain bright PL with a QY ≈ 50–65% (higher for (NH4)2ZnCl4, Figures 3A and S6). In contrast, the capping with chalcogenidometallate ligands such as K4Sn2S6 and K3AsS4 results in significantly quenched PL QYs of 10–15%.
Figure 3.
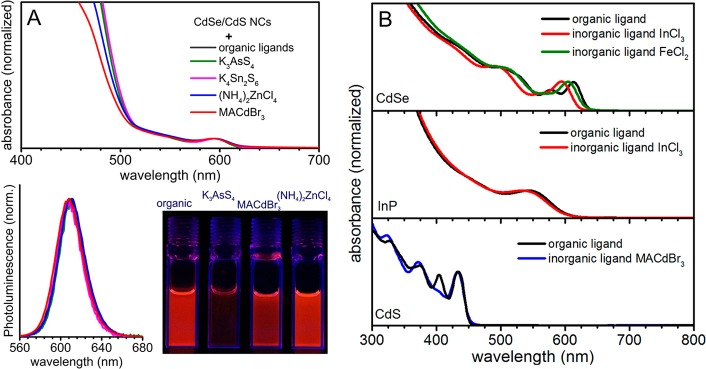
(A) Absorption and emission spectra of CdSe/CdS NCs with 3.4 nm CdSe core and 2.6 nm thick CdS shell (∼8 ML), capped with initial organic ligands and ligand-exchanged with halometallates and chalcogenidometallates. The photograph taken under UV light (254 nm) illustrates the differences in quantum efficiencies between various surface chemistries (solutions have identical optical density at the 1st abs. peak). (B) Absorption spectra for 5.6 nm CdSe, 2.4 nm InP, and 4.7 nm CdS NCs capped with initial organic and various halometallate ligands.
The generality of the halometallate-capping method can be further illustrated for CdSe, CdS, and InP NCs stabilized with various metal halides (Figure 3B), including magnetic transition metal ions (FeCl2). Contrary to chalcogenidometallates, halometallates are able in some cases to extract cations from the NCs due to the solubility of the corresponding metal halides. This was clearly revealed for Cd-chalcogenides: treatment with “isocationic” ligands such as MACdBr3 leads to the ligand exchange only, whereas treatment with InCl3 can slowly extract one monolayer of Cd ions forming soluble Cd halide species and Se–In bonds. This process is evidenced by a blue shift in absorption spectra (Figure 3B) and by a reversal of CdSe stoichiometry from Cd-rich (Cd/Se ≈ 1.2 for organic-capped NCs, ICP-MS analysis) to Se-rich (Cd/Se ≈ 0.8). Removal of the surface Cd atoms in the form of soluble salts has been also reported for organic-ligand exchanges.20 Furthermore, complete cation exchange can also take place: treatment with MAPbI3 fully converts CdSe NCs into PbSe NCs. Ligand exchange with K4Sn2S6 and K3AsS4 retains the Cd-rich stoichiometry of CdSe NCs.
In conclusion, a general methodology for the surface functionalization of nanocrystals with metal halides was demonstrated, allowing efficient electronic passivation for highly luminescent PbS and CdSe/CdS NCs. Halide complexes are available for many metals for which soluble chalcogenide complexes are unknown or proved to be very unstable. Consequently, there is the possibility of “isocationic” ligation of II–VI, IV–VI, and III–V semiconductor NCs (e.g., PbS-PbI2, CdS-MACdBr3, InP-InCl3), thus excluding the use of a foreign metal in the ligand shell (e.g., CdSe-K4Sn2S6). Future work should focus on the electronic characterization and optoelectronic applications of NC solids composed of densely packed halometallate-cappped NCs.
Acknowledgments
We thank the European Union for financial support via ERC Starting Grant 2012 (Project NANOSOLID, GA No.306733) and a Marie Curie Fellowship (IIF-GA-2012-330524). We thank F. Krumeich and K. Kravchyk for EDX analysis of all samples.
Supporting Information Available
Experimental details and additional figures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- a Talapin D. V.; Lee J.-S.; Kovalenko M. V.; Shevchenko E. V. Chem. Rev. 2009, 110, 389–458. [DOI] [PubMed] [Google Scholar]; b Guyot-Sionnest P. J. Phys. Chem. Lett. 2012, 3, 1169–1175. [DOI] [PubMed] [Google Scholar]
- (2); a Murray C. B.; Norris D. J.; Bawendi M. G. J. Am. Chem. Soc. 1993, 115, 8706–8715. [Google Scholar]; b Yin Y.; Alivisatos A. P. Nature 2005, 437, 664–670. [DOI] [PubMed] [Google Scholar]
- Kramer I. J.; Sargent E. H. Chem. Rev. 2013, 114, 863–882. [DOI] [PubMed] [Google Scholar]
- Anikeeva P. O.; Halpert J. E.; Bawendi M. G.; Bulović V. Nano Lett. 2007, 7, 2196–2200. [DOI] [PubMed] [Google Scholar]
- a Keuleyan S.; Lhuillier E.; Brajuskovic V.; Guyot-Sionnest P. Nat. Photonics 2011, 5, 489–493. [Google Scholar]; b Clifford J. P.; Konstantatos G.; Johnston K. W.; Hoogland S.; Levina L.; Sargent E. H. Nat. Nanotechnol. 2009, 4, 40–44. [DOI] [PubMed] [Google Scholar]; c Jong-Soo L.; Kovalenko M. V.; Jing H.; Dae Sung C.; Talapin D. V. Nat. Nanotechnol. 2011, 6, 348–352. [DOI] [PubMed] [Google Scholar]
- a Ibanez M.; Cadavid D.; Zamani R.; Garcia-Castello N.; Izquierdo-Roca V.; Li W. H.; Fairbrother A.; Prades J. D.; Shavel A.; Arbiol J.; Perez-Rodriguez A.; Morante J. R.; Cabot A. Chem. Mater. 2012, 24, 562–570. [Google Scholar]; b Kovalenko M. V.; Spokoyny B.; Lee J. S.; Scheele M.; Weber A.; Perera S.; Landry D.; Talapin D. V. J. Am. Chem. Soc. 2010, 132, 6686–6695. [DOI] [PubMed] [Google Scholar]
- Choi J. H.; Fafarman A. T.; Oh S. J.; Ko D. K.; Kim D. K.; Diroll B. T.; Muramoto S.; Gillen J. G.; Murray C. B.; Kagan C. R. Nano Lett. 2012, 12, 2631–2638. [DOI] [PubMed] [Google Scholar]
- a Kim D. K.; Lai Y. M.; Diroll B. T.; Murray C. B.; Kagan C. R. Nat. Commun. 2012, 3. [DOI] [PubMed] [Google Scholar]; b Koh W. K.; Saudari S. R.; Fafarman A. T.; Kagan C. R.; Murray C. B. Nano Lett. 2011, 11, 4764–4767. [DOI] [PubMed] [Google Scholar]
- a Kovalenko M. V.; Scheele M.; Talapin D. V. Science 2009, 324, 1417–1420. [DOI] [PubMed] [Google Scholar]; b Kovalenko M. V.; Bodnarchuk M. I.; Zaumseil J.; Lee J.-S.; Talapin D. V. J. Am. Chem. Soc. 2010, 132, 10085–10092. [DOI] [PubMed] [Google Scholar]; c Lee J.-S.; Kovalenko M. V.; Huang J.; Chung D. S.; Talapin D. V. Nat. Nanotechnol. 2011, 6, 348–352. [DOI] [PubMed] [Google Scholar]
- a Llordes A.; Hammack A. T.; Buonsanti R.; Tangirala R.; Aloni S.; Helms B. A.; Milliron D. J. J. Mater. Chem. 2011, 21, 11631–11638. [Google Scholar]; b Llordes A.; Garcia G.; Gazquez J.; Milliron D. J. Nature 2013, 500, 323–326. [DOI] [PubMed] [Google Scholar]
- a Nag A.; Kovalenko M. V.; Lee J.-S.; Liu W.; Spokoyny B.; Talapin D. V. J. Am. Chem. Soc. 2011, 133, 10612–10620. [DOI] [PubMed] [Google Scholar]; b Fafarman A. T.; Koh W. K.; Diroll B. T.; Kim D. K.; Ko D. K.; Oh S. J.; Ye X. C.; Doan-Nguyen V.; Crump M. R.; Reifsnyder D. C.; Murray C. B.; Kagan C. R. J. Am. Chem. Soc. 2011, 133, 15753–15761. [DOI] [PubMed] [Google Scholar]
- a Lee M. M.; Teuscher J.; Miyasaka T.; Murakami T. N.; Sna ith H. J. Science 2012, 338, 643–647. [DOI] [PubMed] [Google Scholar]; b Burschka J.; Pellet N.; Moon S.-J.; Humphry-Baker R.; Gao P.; Nazeeruddin M. K.; Gratzel M. Nature 2013, 499, 316–319. [DOI] [PubMed] [Google Scholar]
- Chung I.; Lee B.; He J.; Chang R. P. H.; Kanatzidis M. G. Nature 2012, 485, 486–489. [DOI] [PubMed] [Google Scholar]
- Addison C. C.Inorganic Chemistry of the Main-Group Elements; Royal Society of Chemistry: 1973. [Google Scholar]
- Fawcett W. R.; Motheo A. J. Electrochim. Acta 1991, 36, 1971–1977. [Google Scholar]
- Reichardt C. Chem. Rev. 1994, 94, 2319–2358. [Google Scholar]
- Reichardt C.Solvents and Solvent Effects in Organic Chemistry, 3rd ed.; Wiley-VCH Verlag: Weinheim, 2003. [Google Scholar]
- a Tang J.; Kemp K. W.; Hoogland S.; Jeong K. S.; Liu H.; Levina L.; Furukawa M.; Wang X.; Debnath R.; Cha D.; Chou K. W.; Fischer A.; Amassian A.; Asbury J. B.; Sargent E. H. Nat. Mater. 2011, 10, 765–771. [DOI] [PubMed] [Google Scholar]; b Ip A. H.; Thon S. M.; Hoogland S.; Voznyy O.; Zhitomirsky D.; Debnath R.; Levina L.; Rollny L. R.; Carey G. H.; Fischer A.; Kemp K. W.; Kramer I. J.; Ning Z.; Labelle A. J.; Chou K. W.; Amassian A.; Sargent E. H. Nat. Nanotechnol. 2012, 7, 577–582. [DOI] [PubMed] [Google Scholar]
- Chen O.; Zhao J.; Chauhan V. P.; Cui J.; Wong C.; Harris D. K.; Wei H.; Han H.-S.; Fukumura D.; Jain R. K.; Bawendi M. G. Nat. Mater. 2013, 12, 445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson N. C.; Hendricks M. P.; Choi J. J.; Owen J. S. J. Am. Chem. Soc. 2013, 135, 18536–18548. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.