

Abstract
MicroRNAs play important regulatory roles in eukaryotic lineages. In this paper, we employed deep sequencing technology to sequence and identify microRNAs in M. incognita genome, which is one of the important plant parasitic nematodes. We identified 102 M. incognita microRNA genes, which can be grouped into 71 nonredundant miRNAs based on mature sequences. Among the 71 miRANs, 27 are known miRNAs and 44 are novel miRNAs. We identified seven miRNA clusters in M. incognita genome. Four of the seven clusters, miR-100/let-7, miR-71-1/miR-2a-1, miR-71-2/miR-2a-2 and miR-279/miR-2b are conserved in other species. We validated the expressions of 5 M. incognita microRNAs, including 3 known microRNAs (miR-71, miR-100b and let-7) and 2 novel microRNAs (NOVEL-1 and NOVEL-2), using RT-PCR. We can detect all 5 microRNAs. The expression levels of four microRNAs obtained using RT-PCR were consistent with those obtained by high-throughput sequencing except for those of let-7. We also examined how M. incognita miRNAs are conserved in four other nematodes species: C. elegans, A. suum, B. malayi and P. pacificus. We found that four microRNAs, miR-100, miR-92, miR-279 and miR-137, exist only in genomes of parasitic nematodes, but do not exist in the genomes of the free living nematode C. elegans. Our research created a unique resource for the research of plant parasitic nematodes. The candidate microRNAs could help elucidate the genomic structure, gene regulation, evolutionary processes, and developmental features of plant parasitic nematodes and nematode-plant interaction.
Introduction
Meloidogyne incognita is a world-wide serious plant pathogen that can infect almost all cultivated plants and cause billions of dollars in losses annually [1]. Currently, the draft genomic sequences of M. incognita [2] are available, which can help elucidate the biology of RKN and their interaction with hosts.
MicroRNAs are small (~22 nt) RNAs that target the mRNAs and regulate their degradation and transcription [3, 4]. Increasing evidence has demonstrated that microRNAs play a key function in many biological processes such as tissue identity, response to environmental stress, and developmental timing [5]. MicroRNAs were first identified in C. elegans [6] and are highly evolutionarily conserved in other species. MicroRNAs are found in various eukaryotes, including plants [7, 8], animals [5, 9] and viruses [10]. It is an important step to identify microRNAs in organisms for elucidating their genome biology and evolution [11]. Although there are hundreds of microRNAs identified in different nematodes, such as C. elegans [12] and C. briggsae [13], to our knowledge, there is no report of microRNAs of plant parasitic nematodes yet. The availability of draft genomic sequences of M. incognita makes it possible to identify its microRNAs on a genome-wide level.
This research used two major approaches to identify microRNAs: (1) the direct cloning approach by cloning and sequencing the microRNAs enriched libraries [14, 15] and (2) computational prediction [16–19]. Although increasing sequences available in the public databases, including expressed sequence tags (ESTs), genome survey sequences (GSS), and high throughput genomic sequences (HTGS), made it possible to identify microRNAs by computational prediction, there are two drawbacks for computational prediction of microRNAs. First, the available nucleotide sequences in the database are limited. Computational prediction methods based on a homology search cannot predict new microRNAs if they do not exist in the database. Second, it is hard to validate the predictions using experiments because of the high false positive rate in computational prediction results. Recently, deep sequencing technology has been extensively used in microRNA genes discoveries in many species [20–27]. In this study, we employed the deep sequencing technology to sequence and identify microRNAs in the M. incognita genome.
Methods
Preparation of specimens
Nematode inoculums were obtained from a population of Meloidogyne incognita (Kofoid and White) isolated from pepper root and were reproduced in greenhouse with pepper cultivar Qiemen (Capsicum annuum L. cv), a RKN-susceptible cultivar. Inoculums consisted of freshly hatched juveniles from egg masses. RKN J2 were concentrated and filtered off foreign matter through a 1 mm pore size nylon sieve. Seedlings of pepper cultivars were grown in a greenhouse (25°C–28°C). The samples were collected quickly. Then, the samples were snap-frozen in liquid nitrogen and stored at -80°C.
Small RNA libraries construction and DNA sequencing
We extracted total RNA from each RKN samples (about 1 × 106 individuals) using TRIzol reagent following the manufacturer’s recommended protocol (Invitrogen, USA). The RNA quality was examined using Bioanalyzer (Agilent2100) with RIN>8.0. We collected and purified RNAs between 10–30 nt using 15% polyacrylamide gel electrophoresis (PAGE) for the sample. After PAGE purification, we added a pair of adaptors to the ends of the small RNAs according to the Illumina TruSeq Small RNA Library Prep protocol. In briefly, a 5’ adaptor (Illumina, San Diego, CA, USA) was ligated to the 5’ ends of the small RNAs and the ligation products were purified on Novex 15% PAGE. Then, a 3’ adaptor (Illumina) was ligated to the first ligation product and further purified on Novex 10% PAGE. The small RNAs were converted to cDNA by RT-PCR and then 6% TBE-Urea gel (Invitrogen) was used to purify the amplification products. Finally, the DNA fragments were used for the high-throughput sequencing. The sequencing process was done in BGI (Beijing Genome Institute at Shenzhen) using the Illumina Genome Analyzer according to the manufacturer’s instructions.
Preprocessing of microRNAs Sequencing Data
The raw data was processed by a bioinformatics’ pipeline and include the following steps: (1) Remove low quality reads. Reads with quality score lower than 20 were removed. (2) Trim 3' prime adaptor sequences. (3) Remove adaptor-only contaminants. (4) Collect short RNAs ranging from 10 to 30 nt. Too short (<10 nt) and too long (>30 nt) reads were removed. (5) Remove sequences with polyA tails. Raw data are available at NCBI-GEO with accession number: GSE24833.
Analysis of M. incognita microRNAs
We grouped the identical clean reads into unique sequence tags (unitags). The abundance of each unitag was indicated by the number of reads belonging to it. We used bowtie [28] to map the unitags to the draft genome of M. incognita, which was downloaded from wormbase (WS205). We only used perfectly matched reads to identify microRNA genes. We employed the miRDeep2 [29] to map sequencing tags. We only kept the candidate precursors with hairpin-like structures, which were perfectly mapped by sequencing tags. We then used the default parameters of miRDeep2 to predict the precursors and the mature sequences of microRNA genes. Finally, the candidate precursor and mature microRNAs were checked manually for secondary structure and sequenced profiles.
Then, we aligned mature microRNAs of M. incognita to known microRNAs downloaded from the miRBase database (version 21) [30] (http://www.mirbase.org/) with BlastN [31]. Those microRNAs with 80% identities and shared the same seed sequences (2–7 nt) and were supposed to be orthologous and named after the known microRNAs. If the mature sequences of two microRNA genes were identical, we treated them as the same microRNA genes with two copies in the genome, such as miR-100a-1 and miR-100a-2 in M. incognita with mature sequences both as uacccguagauccgaacuaguc. If the mature sequences of two microRNA genes were different from less than three bases, we labeled them as derivations of the same microRNA gene, such as miR-100a-1 and miR-100b, with mature sequences such as uacccguagauccgaacuaguc and aacccguagauccgaacuagucu, respectively.
We grouped the microRNA genes into a cluster if their distances in the genome were less than 2000 bp.
Abundance estimate of each class of small RNAs
Clean reads were aligned to each class of small RNAs sequences, including miRNAs, rRNA, tRNA, snRNA and mRNA, using bowtie [28] with default parameters except the perfect match (-v 0). Then total reads of each class were counted to estimate the abundance of expression.
Validation of the expressions of five M. incognita microRNAs using RT-PCR
Nematodes of fresh hatched J2 were firstly exposed to freeze thaw cycles fusing liquid nitrogen and a 30°C water bath three to four times. Then the total RNAs were extracted using the TRIzol reagent (Invitrogen). The cDNA fragment was synthesized from total RNA using Superscript III reverse transcriptase (Invitrogen). The microRNA primers designed according to the premature microRNA sequences (S2 Table).
Real-time qPCR was performed on CFX96 Real-time PCR Detection System (Bio-Rad, USA) with 1.1 software, as follows: 95°C for 30 s, followed by 40 cycles of 95°C for 10 s, 58°C for 30 s, using qPCR SYBR Premix Ex Taq II for fluorophore SYBR green with fluorescein (Takara, Japan). Standard curves were established with five serial dilutions of first-strand cDNAs, ranging from 1 to 1/10 000. As reference, the M. incognita 18S ribosomal cDNA (GeneBank accession number U81578) was amplified using the primers 18S-F-852 and 18S-R-966 [32]. Relative quantity of gene expression was calculated and normalized to 18S ribosomal. The real-time qPCR were carried out with 5 technical repeats. We used the values of 2-ΔCt as the gene expression abundance level [33].
Identification and phylogenetic analysis of Argonaute protein family in M. incognita
The Argonaute protein family, which is defined by the presence of PAZ (Piwi-Argonaute-Zwille) and PIWI domains, was first identified in plants [34]. The Argonaute protein family could be phylogenetic divided into the Ago subfamily and the Piwi subfamily [35]. In general, the expression of Piwi proteins is restricted to the germ line, where they bind Piwi-interacting proteins (piRNAs). We identified the Argonaute family proteins based on the Pfam domains. Firstly, we searched the PAZ (PF02170) and Piwi (PF02171) domains in the M. incognita proteins using hmmsearch [36] against Pfam database with the e-value less than 0.01 [37]. The proteins with both PAZ and Piwi domains were identified as Argonaute family proteins. In total, we identified 15 Argonaute family proteins in the M. incognita draft genome. We also used the same method to identify Argonaute proteins from the genome of C. elegans, B. malayi, B. xylophilus and M. hapla, which were downloaded from WormBase (version WS243). We aligned Argonaute family proteins from five genomes using MAFFT [38]. Then we trimmed the alignment using trimAL [39] with parameter–automated1. Finally, we constructed a phylogenetic tree using PhyML [40] with default parameters.
Results
Overview of the small RNA sequencing results
We obtained 18,509,803 raw reads from the small RNA library of J2 juveniles of M. incognita. After removing low-quality bases, contaminants and masking adaptor sequences, we obtained 16,020,648 clean reads. The clean reads were mainly distributed between 15 and 23 nt (15,005,173, 93.7%) and had a peak length of 23 nt (Fig 1A). We were able to group the clean reads into 761,538 unique tags based on their sequence similarity. The most abundance sequence tag had 1,925,637 reads. 90.6% of the clean reads (14,515,814) can be mapped onto the draft genome sequences of M. incognita using bowtie [28] with no mismatches (-v 0).
Fig 1. General description of the small RNA sequences of M. incognita.
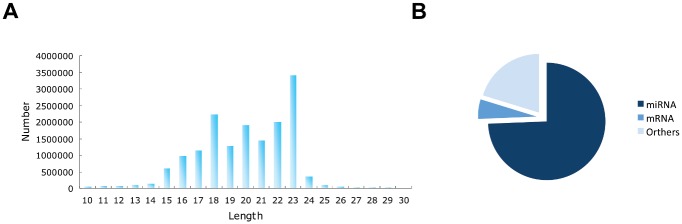
(A) Size distribution of the raw reads of M. incognita small RNAs. (B) Classification of the small RNA reads.
To annotate the small RNAs, we aligned the clean reads against the microRNAs, tRNA, rRNA, snRNA and mRNA sequences of M. incognita and then counted the reads of each class. There were 74.35%, 5.38% and 1.10% of clean reads mapped to predicted microRNA, protein encoding and tRNA genes, respectively. There were 19.14% of clean reads mapped to other classes, including rRNA, snRNA, and siRNA (Fig 1B). There were 9.4% of clean reads that were unable to be mapped to the M. incognita genome with no mismatches. The top 10 most abundance sequence tags were all microRNAs. The lengths of the microRNA reads were mainly distributed between 18 and 23 nt, which include 86.34% of the total microRNA reads (Fig 2).
Fig 2. Size distribution of reads from microRNA, protein encoding genes and others.
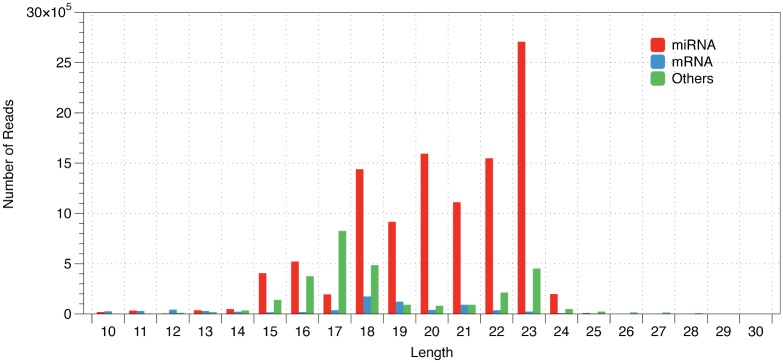
Identification and validation of microRNA genes of M. incognita
We identified 102 candidate microRNA genes of M. incognita (S1 Table) using miRDeep2 with a score of more than 0. We then predicted the mature microRNA from precursor microRNAs. The mature microRNAs were from 18 to 24 nt in length. We noted a significant bias of A and U at the first position of mature microRNAs (Fig 3B). 94 out of 102 mature microRNAs started with A or U (41 microRNAs started with A, 53 microRNAs started with U). The other parts of precursor microRNAs besides mature microRNAs usually undergo the degradation process. As a result, the mature microRNA had a significantly deeper coverage than those of the other parts of microRNA genes, such as the star microRNA (miRNA*), the other strand of mature microRNA on the hairpin structure of precursor microRNA, and loop sequences (the sequences between mature miRNA and miRNA*). Most of the putative pre-miRNAs had a very high read depth in mature arm and a much low depth in the other arm with a typical hairpin structure (Fig 4).
Fig 3. Distribution of bases at each position of mature microRNAs.

The first base of mature microRNA tend to be A and U.
Fig 4. min-miR-1 hairpin structure and sequencing profile.

(A) Hairpin structure predicted with RNAfold. (B) Proportion of small RNA tags mapped to the pre-miRNA of min-miR-1.
Based on mature microRNA sequence, we were able to group 102 microRNAs into 71 unique microRNA genes. Among them, 25 microRNA genes had multiple copies in the draft genome of M. incognita. From those 71 unique miRNAs, we identified 27 known miRNA families, which are known in the miRBase database (Table 1).
Table 1. Known miRNA families identified in M. incognita.
| Name | Mature | Length | GC% | Reads number |
|---|---|---|---|---|
| miR-71 | ugaaagacauggguaguugaga | 22 | 40.9% | 3489388 |
| miR-100b | aacccguagauccgaacuagucu | 23 | 47.8% | 2870101 |
| miR-124 | uaaggcacgcggugaaug | 18 | 55.6% | 1639977 |
| miR-1 | uggaauguaaagaaguau | 18 | 27.8% | 880854 |
| miR-72 | aggcaagauguuggcauugcuga | 23 | 47.8% | 495031 |
| miR-92 | uauugcacucguuucggccu | 20 | 50.0% | 313210 |
| miR-252 | cuaaguaguagugccgcauuuaa | 23 | 39.1% | 65196 |
| miR-2a | uaucacagccugcuuuagcgua | 22 | 45.5% | 57375 |
| miR-87 | gugagcaaaguuucaggugugc | 22 | 50.0% | 42872 |
| miR-100a | uacccguagauccgaacuaguc | 22 | 50.0% | 28784 |
| miR-2b | uaucacaguucgauauggcc | 20 | 45.0% | 25154 |
| miR-50 | ugauaugucuuguauucuug | 20 | 30.0% | 20539 |
| miR-184 | uggacggaagucugauaaggag | 22 | 50.0% | 17866 |
| miR-81 | ugagaucauaccagaucac | 19 | 42.1% | 15488 |
| miR-86 | uaagugaauaucuugccacaagcu | 24 | 37.5% | 9234 |
| miR-279 | ugacuagauccacacucaucu | 21 | 42.9% | 9908 |
| miR-137 | agguauucuccguggugaugaca | 23 | 47.8% | 6368 |
| miR-59 | acgaaucguuugcacaucgguguu | 24 | 45.8% | 6103 |
| miR-79 | auaaagcuagauuaccagag | 20 | 35.0% | 5635 |
| miR-67 | ucacaacccccuagaguucgcua | 23 | 52.2% | 5292 |
| miR-239 | uuuguacuagccaaaaugucugca | 24 | 37.5% | 4576 |
| miR-36 | ucaccgggaauuuauucaug | 20 | 40.0% | 698 |
| let-7 | ugagguaguagguuguauaguu | 22 | 36.4% | 223 |
| miR-242 | uugcguaggcaucuugucag | 20 | 50.0% | 121 |
| miR-240 | cacuggccuuucaaaccu | 18 | 50.0% | 60 |
| miR-76 | uucguuguuucugaaaccugaa | 22 | 36.4% | 12 |
| miR-790 | acgguuugacaaaguuau | 18 | 33.3% | 9 |
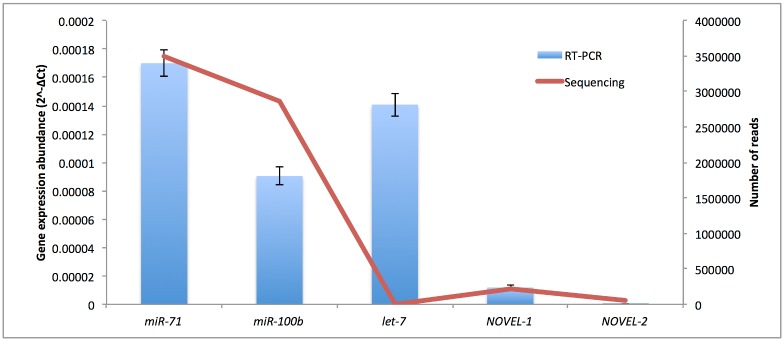
We selected 5 microRNAs for validation using RT-PCR, including 3 known microRNAs and 2 novel microRNAs of M. incognita. All of the 5 microRNAs could be detected using real time RT-PCR with the mature microRNAs as a primer. The microRNA expression was determined using real time RT-PCR by 2-ΔCt measurements. The expression levels of four microRNAs, miR-71, miR-100b, NOVEL-1 and NOVEL-2, were consistent with those obtained by high-throughput sequencing. The expression levels of miR-71 and miR-100b were much higher than those of NOVEL-1 and NOVEL-2 in both results of sequencing and qRT-PCR. However, the expression abundance of let-7 detected by real time RT-PCR is much higher than that by high-throughput sequencing (Fig 5).
Fig 5. The expression abundance of microRNAs detected by real time RT-PCR (bars) and by high-throughput sequencing (lines).
Identification of microRNA clusters in M. incognita
MicroRNAs are often clustered in the genome [30]. We identified seven microRNA clusters in M. incognita genome (Table 2), and four of them were also found in other species.
Table 2. List of the miRNA clusters of M. incognita.
| Contig | Position | Strand | Cluster | Reported in other nematode |
|---|---|---|---|---|
| MiV1ctg20 | 43319–43619 | + | miR-71-1 | H. contortus [48] |
| miR-2a-1 | ||||
| MiV1ctg221 | 69290–69588 | - | miR-71-2 | |
| miR-2a-2 | ||||
| MiV1ctg1924 | 3647–4158 | + | let-7 | B. malayi [41] |
| miR-100 | ||||
| MiV1ctg644 | 18822–19030 | - | miR-279 | B. pahangi [48] |
| miR-2b | ||||
| MiV1ctg27 | 99326–99923 | - | NOVEL-1-1 | |
| NOVEL-39 | ||||
| MiV1ctg2865 | 1792–2014 | + | miR-240 | |
| NOVEL-30 | ||||
| MiV1ctg1143 | 4187–4580 | + | NOVEL-12 | |
| NOVEL-11 | ||||
| NOVEL-14 |
MiR-100 orthologues are often found in clusters with let-7 and the clusters range in size from ~300 to 4000 bp [41]. The microRNA cluster let-7 and miR-100 has been found in Brugia malayi [41], Drosophila [42] and humans [43]. In the M. incognita genome, the miR-100 is clustered within ~350 bp of let-7. Many organisms express multiple miR-100 paralogues. There are four paralogues (miR-100a through 100d) in B. malayi. We have identified 3 miR-100 paralogues in M. incognita. The sequences alignment of the miR-100 orthologues from human, fly, B. malayi, A. suum, B. xylophilus, and M. incognita showed that the seed sequence, ACCCGUA, conserved in these species (Fig 6A). It is interesting that miR-100 has been lost in free living nematodes such as C. elegans [44] and P. pacificus (Fig 6B).
Fig 6. Conservation of miR-100, in sequence and genomic organization.
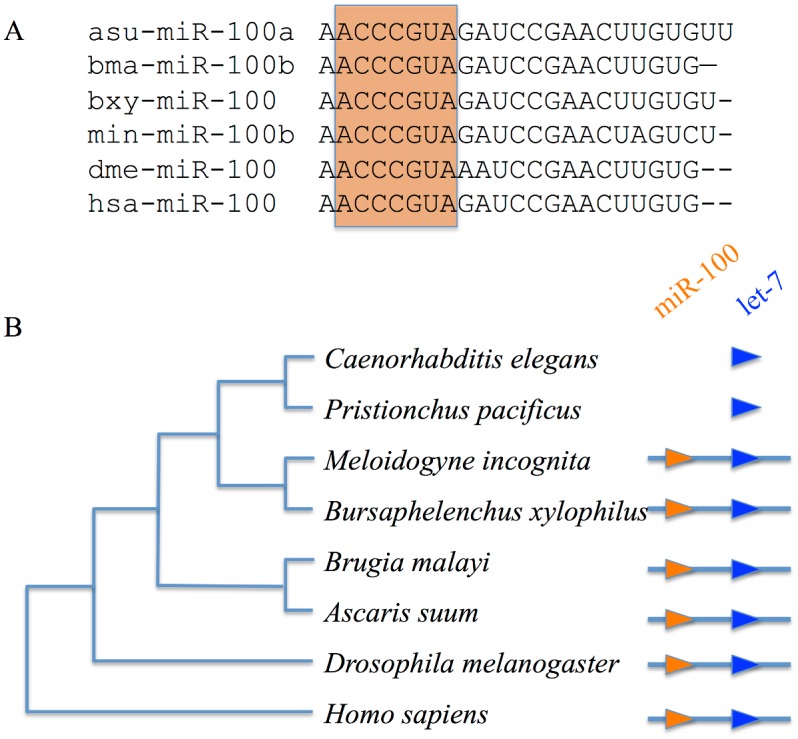
(A) Sequence alignment of miR-100 microRNAs among different species (asu-: A. suum, bma-: B. malayi, bxy-: B. xylophilus, min-: M. incognita, dme-: D. melanogaster, has-: H. sapiens). The seed sequences indicated with red square area. (B) miR-100 and let-7 were clustered together in diverse animals but the miR-100 had lost in the common ancestor of C. elegans and P. pacificus.
The miR-71/miR-2 cluster is found in two locations in M. incognita. Previous functional analysis showed that Drosophila miR-2 is associated with the suppression of embryonic apoptosis [45]. The miR-71 of C. elegans is related to lifespan, stress response [46]. The miR-71 of C. elegans also was reported to function in neurons to promote germline-mediated longevity and facilitates the localization and activity of DAF-16 in the intestine [47]. The miR-71/miR-2 cluster was also found in H. contortus [48], which also suggest the functional linkage of these two miRNAs.
The miR-279 and miR-2b were also in a close cluster in M. incognita. The miR-279 was reported to regulate the JAK/STAT pathway to drive rest:activity rhythms in Drosophila [49].
The highly expressed microRNA genes in M. incognita
Few microRNA genes are highly expressed in our sequencing data. The numbers of reads for the top 10 abundance microRNAs are shown in Fig 7. The first two abundance microRNAs, miR-71 and miR-100b, have 6,359,489 reads, which are approximately 50% of the total clean reads. The very high expression level indicated that these miRNAs may be important to the life of M. incognita. Interestingly, there are two microRNAs, miR-100 and miR-92, that were highly expressed in M. incognita, but were lost in C. elegans.
Fig 7. The top 10 abundance miRNAs in M. incognita J2 library.
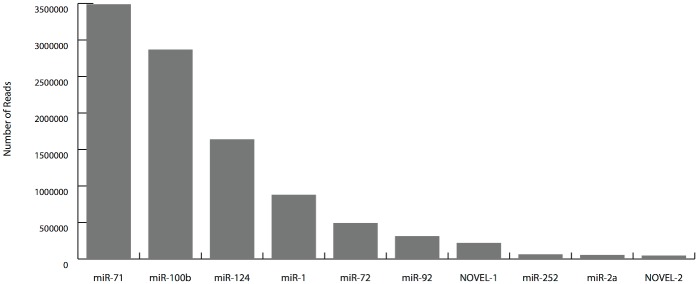
The most expressed microRNA, miR-71, has important roles in extending the life span in C. elegans after germline removal [47]. The miR-71 regulates the DAF-16/FOXO in neurons to enhance germline-mediated longevity [47]. It has also been reported that miR-71 can target the TIR-1/Sarm1 adaptor protein to inhibit calcium signaling pathway [50].
The second most expressed microRNA, miR-100, was found to be an oncogene in human, which is differently expressed in many cancer cells [51]. In nasopharyngeal cancer, miR-100 regulates the expression of Polo-like kinase 1 [52]. In adrenocortical cancer cells and in clear cell ovarian cancer, miR-100 targets mTOR [53]. In acute myeloblastic leukemia, miR-100 targets the RBSP3 to regulate cell differentiation and survival [51].
The third highly expressed microRNA, miR-124, may function in the neural cell. The human miR-124 is the most abundant microRNA expressed in neuronal cells although the differentiation was not affected by the changing of miR-124 expression in neural cells [54]. The mice miR-124 regulated the temporal progression of adult neurogenesis. Suppressing miR-124 function during regeneration caused hyperplasias and neurogenesis delay in mice [55].
The fourth abundant microRAN, miR-1, is a muscle-specific microRNA. The miR-1 controls both pre- and postsynaptic function in C. elegans neuromuscular junctions [56]. The miR-92 gene has been found in B. malayi and A. suum. It is also a key oncogenic gene in colon cancer in humans [57].
Lack of piRNA and piRNA pathway components in M. incognita
piRNAs are critical microRNAs for germ line cell development in many species. The generation of piRNAs employed a distinct mechanism that does not involve Dicer [58]. In C. elegans, piRNA orthologs are only 21 nt in length with a 5' terminal U, which are known as 21U-RNAs. The piRNAs are not conserved at the sequence level among other Caenorhabditis species and do not have significant complementarity to targets [20]. Piwi proteins and piRNAs have been found in worms, flies, sponges, and humans [59]. PiRNAs interact with Piwi proteins to form RNA-protein complexes. The Piwi Argonaute ortholog PRG-1 is required for interaction with piRNAs [60]. The piRNA-Piwi protein complex has been reported to play an important role in silencing the retrotransposons in germ line cells through regulating both epigenetic and post-transcriptional pathways, particularly those in spermatogenesis [61].
However, in our small RNA sequencing results of the J2 library for M. incognita, we did not found any small RNAs with characteristics of piRNAs. Moreover, the Piwi-clade Argonaute orthologs could not be found in M. incognita genome (Fig 8). Recently, the HEN1 ortholog henn-1 was identified and proved to be required in piRNAs pathway in C. elegans [62]. However, we failed to detect any ortholog of the HEN1 methyltransferase in M. incognita genome. Notably, the Piwi-clade Argonaute and HEN1 orthologs are also not found in the parasite nematode B. malayi [63] and A. suum [64]. It was hypothesized that the piRNA pathway may be lost in A. suum [64]. In a very recent study, it is also indicated that piRNAs exist only in nematode C. elegans and closely related nematodes, and absent in all other nematode lineages [65]. The lacking of piRNAs and piRNA pathway components imply that the piRNA pathway may also be lost in M. incognita.
Fig 8. M. incognita lacks Piwi-clade argonaute proteins, which is essential for Pi-RNA biosynthesis.

All of argonaute proteins, containing Piwi and PAZ domain, were identified from the genome of C. elegans, B. xylophilus, B. malayi, M. hapla and M. incognita and the multiple sequences alignment were carried out using mafft with default parameters. The phylogenetic tree was constructed using PhyML and the number on the branch indicated the bootstrap values.
Conservation of miRNAs of M. incognita in other nematodes
We examined how M. incognita miRNAs were conserved in other four nematodes species: C. elegans, A. suum, B. malayi and P. pacificus. Table 3 shows how M. incognita microRNAs exist at least in one other nematodes genome. There are 26 M. incognita microRNAs conserved in at least one other nematode genome. However, only seven M. incognita microRNAs are conserved in all five nematodes: let-7, miR124, miR-2, miR-71, miR-72, miR-79, miR-87. There are four microRNAs, miR-100, miR-92, miR-279 and miR-137, which exist only in genomes of parasitic nematodes A. suum, B. malayi, P. pacificus and M. incognita, but do not exist in the genomes of the free living nematode C. elegans. These four microRNAs may have an important function in the parasite process.
Table 3. miRNAs of M. incognita conserved in other nematodes.
| mir | min | cel | asu | bma | ppc |
|---|---|---|---|---|---|
| let-7 | * | * | * | * | * |
| miR-100 | * | * | * | ||
| miR-124 | * | * | * | * | * |
| miR-137 | * | * | * | ||
| miR-184 | * | * | |||
| miR-1 | * | * | * | * | |
| miR-239 | * | * | * | ||
| miR-240 | * | * | * | ||
| miR-242 | * | * | * | ||
| miR-252 | * | * | * | * | |
| miR-279 | * | * | * | * | |
| miR-2 | * | * | * | * | * |
| miR-36 | * | * | * | * | |
| miR-49 | * | * | * | ||
| miR-50 | * | * | * | * | |
| miR-59 | * | * | |||
| miR-67 | * | * | * | * | |
| miR-71 | * | * | * | * | * |
| miR-72 | * | * | * | * | * |
| miR-76 | * | * | * | ||
| miR-790 | * | * | |||
| miR-79 | * | * | * | * | * |
| miR-81 | * | * | * | * | |
| miR-86 | * | * | * | * | |
| miR-87 | * | * | * | * | * |
| miR-92 | * | * | * |
Note: Star (*) indicates that the specific miRNAs family has been found in this species. min: M. incognita, cel: C. elegans, asu: A. suum, bma: B. malayi, ppc: P. pacificus
Discussion and Conclusions
In this study, we generated about 18 million raw microRNA reads. After preprocessing, we eventually obtained a total of about 0.5 million non-redundant unitags with high quality reads. However, only 43.44% (232307 out of 534834) of these non-redundant small RNA unitags have a perfect match in the draft M. incognita genome. This could due to the following reasons: (1) Genetic polymorphisms: It is well known that the genetic variation of M. incognita is due to the heteroploid phenomenon; (2) Incompleteness of the genome: The public-released draft genome of M. incognita was supposed to be only part of the genome [2]. There are lots of gaps in the assembled genome; (3) Systemic errors: Sequencing errors can block perfect alignment.
In summary, this first report of microRNAs of plant parasitic nematodes and the genome-wide identification of M. incognita microRNAs have created a unique resource for the research of plant parasitic nematode. The candidate microRNAs will help researchers better understand and refine their approach to studies on genomic structure, gene regulation, evolutionary processes, and developmental features of plant parasitic nematodes and nematode-plant interaction. However, further biological experiments are needed to verify the functionalities of M. incognita microRNAs and how they regulate their target genes in the developing process.
Supporting Information
(XLSX)
(DOCX)
Acknowledgments
This work was partially supported by National Natural Science Foundation of China (NSFC) (30900925) and National Basic Research and Development Program of China (2009CB119000). The authors would like to thanks Jun Wang at the Beijing Genome Institute (BGI) of Shenzhen for cDNA library construction and DNA sequencing. Thanks are also given to Dr. Xinqiu Tan for efforts in manuscript improvement.
Data Availability
Raw data are available at NCBI-GEO with accession number: GSE24833.
Funding Statement
This work was partially supported by National Natural Science Foundation of China (NSFC) (30900925) and National Basic Research and Development Program of China (2009CB119000).
References
- 1. Trudgill DL, Blok VC. Apomictic, polyphagous root-knot nematodes: exceptionally successful and damaging biotrophic root pathogens. Annu Rev Phytopathol. 2001;39:53–77. Epub 2001/11/10. doi: 39/1/53 [pii] 10.1146/annurev.phyto.39.1.53 . [DOI] [PubMed] [Google Scholar]
- 2. Abad P, Gouzy J, Aury JM, Castagnone-Sereno P, Danchin EG, Deleury E, et al. Genome sequence of the metazoan plant-parasitic nematode Meloidogyne incognita. Nat Biotechnol. 2008;26(8):909–15. Epub 2008/07/29. doi: nbt.1482 [pii] 10.1038/nbt.1482 . [DOI] [PubMed] [Google Scholar]
- 3. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97. Epub 2004/01/28. doi: S0092867404000455 [pii]. . [DOI] [PubMed] [Google Scholar]
- 4. Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205. Epub 2007/05/18. 10.1146/annurev.cellbio.23.090506.123406 . [DOI] [PubMed] [Google Scholar]
- 5. Zhang B, Wang Q, Pan X. MicroRNAs and their regulatory roles in animals and plants. J Cell Physiol. 2007;210(2):279–89. Epub 2006/11/11. 10.1002/jcp.20869 . [DOI] [PubMed] [Google Scholar]
- 6. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–54. Epub 1993/12/03. doi: 0092-8674(93)90529-Y [pii]. . [DOI] [PubMed] [Google Scholar]
- 7. Jones-Rhoades MW, Bartel DP, Bartel B. MicroRNAS and their regulatory roles in plants. Annu Rev Plant Biol. 2006;57:19–53. Epub 2006/05/04. 10.1146/annurev.arplant.57.032905.105218 . [DOI] [PubMed] [Google Scholar]
- 8. Zhang B, Pan X, Cobb GP, Anderson TA. Plant microRNA: a small regulatory molecule with big impact. Dev Biol. 2006;289(1):3–16. Epub 2005/12/06. doi: S0012-1606(05)00764-5 [pii] 10.1016/j.ydbio.2005.10.036 . [DOI] [PubMed] [Google Scholar]
- 9. Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–5. Epub 2004/09/17. 10.1038/nature02871 nature02871 [pii]. . [DOI] [PubMed] [Google Scholar]
- 10. Cullen BR. Viruses and microRNAs. Nat Genet. 2006;38 Suppl:S25–30. Epub 2006/06/01. doi: ng1793 [pii] 10.1038/ng1793 . [DOI] [PubMed] [Google Scholar]
- 11. Carrington JC, Ambros V. Role of microRNAs in plant and animal development. Science. 2003;301(5631):336–8. Epub 2003/07/19. 10.1126/science.1085242 301/5631/336 [pii]. . [DOI] [PubMed] [Google Scholar]
- 12. Warf MB, Johnson WE, Bass BL. Improved annotation of C. elegans microRNAs by deep sequencing reveals structures associated with processing by Drosha and Dicer. Rna. 2011;17(4):563–77. 10.1261/rna.2432311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shi Z, Montgomery TA, Qi Y, Ruvkun G. High-throughput sequencing reveals extraordinary fluidity of miRNA, piRNA, and siRNA pathways in nematodes. Genome research. 2013;23(3):497–508. 10.1101/gr.149112.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boissonneault V, St-Gelais N, Plante I, Provost P. A polymerase chain reaction-based cloning strategy applicable to functional microRNA studies. Anal Biochem. 2008;381(1):166–8. Epub 2008/07/09. doi: S0003-2697(08)00414-4 [pii] 10.1016/j.ab.2008.06.026 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sunkar R, Zhu JK. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell. 2004;16(8):2001–19. Epub 2004/07/20. 10.1105/tpc.104.022830 tpc.104.022830 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang X, Zhang J, Li F, Gu J, He T, Zhang X, et al. MicroRNA identification based on sequence and structure alignment. Bioinformatics. 2005;21(18):3610–4. Epub 2005/07/05. doi: bti562 [pii] 10.1093/bioinformatics/bti562 . [DOI] [PubMed] [Google Scholar]
- 17. Li Y, Li W, Jin YX. Computational identification of novel family members of microRNA genes in Arabidopsis thaliana and Oryza sativa. Acta Biochim Biophys Sin (Shanghai). 2005;37(2):75–87. Epub 2005/02/03. . [PubMed] [Google Scholar]
- 18. Lai EC, Tomancak P, Williams RW, Rubin GM. Computational identification of Drosophila microRNA genes. Genome Biol. 2003;4(7):R42 Epub 2003/07/08. 10.1186/gb-2003-4-7-r42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yousef M, Showe L, Showe M. A study of microRNAs in silico and in vivo: bioinformatics approaches to microRNA discovery and target identification. FEBS J. 2009;276(8):2150–6. Epub 2009/03/03. doi: EJB6933 [pii] 10.1111/j.1742-4658.2009.06933.x . [DOI] [PubMed] [Google Scholar]
- 20. Ruby JG, Jan C, Player C, Axtell MJ, Lee W, Nusbaum C, et al. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell. 2006;127(6):1193–207. Epub 2006/12/19. 10.1016/j.cell.2006.10.040 . [DOI] [PubMed] [Google Scholar]
- 21. Bar M, Wyman SK, Fritz BR, Qi J, Garg KS, Parkin RK, et al. MicroRNA discovery and profiling in human embryonic stem cells by deep sequencing of small RNA libraries. Stem Cells. 2008;26(10):2496–505. Epub 2008/06/28. doi: 2008–0356 [pii] 10.1634/stemcells.2008-0356 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burnside J, Ouyang M, Anderson A, Bernberg E, Lu C, Meyers BC, et al. Deep sequencing of chicken microRNAs. BMC Genomics. 2008;9:185. Epub 2008/04/24. doi: 1471-2164-9-185 [pii] 10.1186/1471-2164-9-185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Glazov EA, Cottee PA, Barris WC, Moore RJ, Dalrymple BP, Tizard ML. A microRNA catalog of the developing chicken embryo identified by a deep sequencing approach. Genome Res. 2008;18(6):957–64. Epub 2008/05/13. doi: gr.074740.107 [pii] 10.1101/gr.074740.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang J, Hao P, Chen H, Hu W, Yan Q, Liu F, et al. Genome-wide identification of Schistosoma japonicum microRNAs using a deep-sequencing approach. PLoS One. 2009;4(12):e8206 Epub 2009/12/10. 10.1371/journal.pone.0008206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wei B, Cai T, Zhang R, Li A, Huo N, Li S, et al. Novel microRNAs uncovered by deep sequencing of small RNA transcriptomes in bread wheat (Triticum aestivum L.) and Brachypodium distachyon (L.) Beauv. Funct Integr Genomics. 2009;9(4):499–511. Epub 2009/06/06. 10.1007/s10142-009-0128-9 . [DOI] [PubMed] [Google Scholar]
- 26. Zhang J, Xu Y, Huan Q, Chong K. Deep sequencing of Brachypodium small RNAs at the global genome level identifies microRNAs involved in cold stress response. BMC Genomics. 2009;10:449. Epub 2009/09/24. doi: 1471-2164-10-449 [pii] 10.1186/1471-2164-10-449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhao CZ, Xia H, Frazier TP, Yao YY, Bi YP, Li AQ, et al. Deep sequencing identifies novel and conserved microRNAs in peanuts (Arachis hypogaea L.). BMC Plant Biol. 2010;10:3. Epub 2010/01/06. doi: 1471-2229-10-3 [pii] 10.1186/1471-2229-10-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10(3):R25 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Friedlander MR, Mackowiak SD, Li N, Chen W, Rajewsky N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic acids research. 2012;40(1):37–52. Epub 2011/09/14. 10.1093/nar/gkr688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic acids research. 2008;36(Database issue):D154–8. Epub 2007/11/10. 10.1093/nar/gkm952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of molecular biology. 1990;215(3):403–10. 10.1016/S0022-2836(05)80360-2 . [DOI] [PubMed] [Google Scholar]
- 32. Dubreuil G, Magliano M, Deleury E, Abad P, Rosso MN. Transcriptome analysis of root-knot nematode functions induced in the early stages of parasitism. The New phytologist. 2007;176(2):426–36. 10.1111/j.1469-8137.2007.02181.x . [DOI] [PubMed] [Google Scholar]
- 33. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods. 2001;25(4):402–8. 10.1006/Meth.2001.1262 WOS:000173949500003. [DOI] [PubMed] [Google Scholar]
- 34. Bohmert K, Camus I, Bellini C, Bouchez D, Caboche M, Benning C. AGO1 defines a novel locus of Arabidopsis controlling leaf development. The EMBO journal. 1998;17(1):170–80. 10.1093/emboj/17.1.170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hock J, Meister G. The Argonaute protein family. Genome biology. 2008;9(2):210 10.1186/gb-2008-9-2-210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eddy SR. Accelerated Profile HMM Searches. PLoS computational biology. 2011;7(10):e1002195 10.1371/journal.pcbi.1002195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic acids research. 2014;42(Database issue):D222–30. 10.1093/nar/gkt1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic acids research. 2005;33(2):511–8. Epub 2005/01/22. 10.1093/nar/gki198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25(15):1972–3. Epub 2009/06/10. 10.1093/bioinformatics/btp348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guindon S, Delsuc F, Dufayard JF, Gascuel O. Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol. 2009;537:113–37. Epub 2009/04/21. 10.1007/978-1-59745-251-9_6 . [DOI] [PubMed] [Google Scholar]
- 41. Poole CB, Davis PJ, Jin J, McReynolds LA. Cloning and bioinformatic identification of small RNAs in the filarial nematode, Brugia malayi. Molecular and biochemical parasitology. 2010;169(2):87–94. Epub 2009/10/31. 10.1016/j.molbiopara.2009.10.004 . [DOI] [PubMed] [Google Scholar]
- 42. Sempere LF, Sokol NS, Dubrovsky EB, Berger EM, Ambros V. Temporal regulation of microRNA expression in Drosophila melanogaster mediated by hormonal signals and broad-Complex gene activity. Developmental biology. 2003;259(1):9–18. . [DOI] [PubMed] [Google Scholar]
- 43. Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(9):2999–3004. 10.1073/pnas.0307323101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roush S, Slack FJ. The let-7 family of microRNAs. Trends in cell biology. 2008;18(10):505–16. Epub 2008/09/09. 10.1016/j.tcb.2008.07.007 . [DOI] [PubMed] [Google Scholar]
- 45. Leaman D, Chen PY, Fak J, Yalcin A, Pearce M, Unnerstall U, et al. Antisense-mediated depletion reveals essential and specific functions of microRNAs in Drosophila development. Cell. 2005;121(7):1097–108. Epub 2005/07/02. 10.1016/j.cell.2005.04.016 . [DOI] [PubMed] [Google Scholar]
- 46. de Lencastre A, Pincus Z, Zhou K, Kato M, Lee SS, Slack FJ. MicroRNAs both promote and antagonize longevity in C. elegans. Current biology: CB. 2010;20(24):2159–68. Epub 2010/12/07. 10.1016/j.cub.2010.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Boulias K, Horvitz HR. The C. elegans microRNA mir-71 acts in neurons to promote germline-mediated longevity through regulation of DAF-16/FOXO. Cell metabolism. 2012;15(4):439–50. 10.1016/j.cmet.2012.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Winter AD, Weir W, Hunt M, Berriman M, Gilleard JS, Devaney E, et al. Diversity in parasitic nematode genomes: the microRNAs of Brugia pahangi and Haemonchus contortus are largely novel. BMC genomics. 2012;13:4 Epub 2012/01/06. 10.1186/1471-2164-13-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Luo W, Sehgal A. Regulation of circadian behavioral output via a MicroRNA-JAK/STAT circuit. Cell. 2012;148(4):765–79. Epub 2012/02/07. 10.1016/j.cell.2011.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hsieh YW, Chang C, Chuang CF. The microRNA mir-71 inhibits calcium signaling by targeting the TIR-1/Sarm1 adaptor protein to control stochastic L/R neuronal asymmetry in C. elegans. PLoS genetics. 2012;8(8):e1002864 10.1371/journal.pgen.1002864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zheng YS, Zhang H, Zhang XJ, Feng DD, Luo XQ, Zeng CW, et al. MiR-100 regulates cell differentiation and survival by targeting RBSP3, a phosphatase-like tumor suppressor in acute myeloid leukemia. Oncogene. 2012;31(1):80–92. 10.1038/onc.2011.208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nagaraja AK, Creighton CJ, Yu Z, Zhu H, Gunaratne PH, Reid JG, et al. A link between mir-100 and FRAP1/mTOR in clear cell ovarian cancer. Mol Endocrinol. 2010;24(2):447–63. Epub 2010/01/19. 10.1210/me.2009-0295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. ZUO T, LIU H-s, HAN J-f, XU S-x, WANG X-j, YUAN S-h, et al. Effects of Degradation Bacteria on Reactive Oxygen Species and Protective Enzymes of Tobacco Leaves under Quinclorac Stress. Journal of Henan Agricultural Sciences. 2010;12:013. [Google Scholar]
- 54. Cao X, Pfaff SL, Gage FH. A functional study of miR-124 in the developing neural tube. Genes & development. 2007;21(5):531–6. Epub 2007/03/09. 10.1101/gad.1519207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cheng LC, Pastrana E, Tavazoie M, Doetsch F. miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nature neuroscience. 2009;12(4):399–408. Epub 2009/03/17. 10.1038/nn.2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Simon DJ, Madison JM, Conery AL, Thompson-Peer KL, Soskis M, Ruvkun GB, et al. The microRNA miR-1 regulates a MEF-2-dependent retrograde signal at neuromuscular junctions. Cell. 2008;133(5):903–15. Epub 2008/05/31. 10.1016/j.cell.2008.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tsuchida A, Ohno S, Wu W, Borjigin N, Fujita K, Aoki T, et al. miR-92 is a key oncogenic component of the miR-17-92 cluster in colon cancer. Cancer science. 2011;102(12):2264–71. Epub 2011/09/03. 10.1111/j.1349-7006.2011.02081.x . [DOI] [PubMed] [Google Scholar]
- 58. Thomson T, Lin H. The biogenesis and function of PIWI proteins and piRNAs: progress and prospect. Annual review of cell and developmental biology. 2009;25:355–76. Epub 2008/01/10. 10.1146/annurev.cellbio.24.110707.175327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Grimson A, Srivastava M, Fahey B, Woodcroft BJ, Chiang HR, King N, et al. Early origins and evolution of microRNAs and Piwi-interacting RNAs in animals. Nature. 2008;455(7217):1193–7. Epub 2008/10/03. 10.1038/nature07415 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Batista PJ, Ruby JG, Claycomb JM, Chiang R, Fahlgren N, Kasschau KD, et al. PRG-1 and 21U-RNAs interact to form the piRNA complex required for fertility in C. elegans. Molecular cell. 2008;31(1):67–78. Epub 2008/06/24. 10.1016/j.molcel.2008.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nagao A, Mituyama T, Huang H, Chen D, Siomi MC, Siomi H. Biogenesis pathways of piRNAs loaded onto AGO3 in the Drosophila testis. RNA. 2010;16(12):2503–15. Epub 2010/10/29. 10.1261/rna.2270710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Montgomery TA, Rim YS, Zhang C, Dowen RH, Phillips CM, Fischer SE, et al. PIWI Associated siRNAs and piRNAs Specifically Require the Caenorhabditis elegans HEN1 Ortholog henn-1. PLoS genetics. 2012;8(4):e1002616 Epub 2012/04/27. 10.1371/journal.pgen.1002616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ghedin E, Wang S, Spiro D, Caler E, Zhao Q, Crabtree J, et al. Draft genome of the filarial nematode parasite Brugia malayi. Science. 2007;317(5845):1756–60. Epub 2007/09/22. 10.1126/science.1145406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang J, Czech B, Crunk A, Wallace A, Mitreva M, Hannon GJ, et al. Deep small RNA sequencing from the nematode Ascaris reveals conservation, functional diversification, and novel developmental profiles. Genome research. 2011;21(9):1462–77. Epub 2011/06/21. 10.1101/gr.121426.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sarkies P, Selkirk ME, Jones JT, Blok V, Boothby T, Goldstein B, et al. Ancient and novel small RNA pathways compensate for the loss of piRNAs in multiple independent nematode lineages. PLoS biology. 2015;13(2):e1002061 10.1371/journal.pbio.1002061 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
(DOCX)
Data Availability Statement
Raw data are available at NCBI-GEO with accession number: GSE24833.