

A phase I/II trial was conducted of bevacizumab and imatinib in patients with metastatic melanoma. The results showed that bevacizumab and imatinib can be safely combined at the maximum doses used for each agent; however, significant clinical activity with this regimen was not observed in melanoma patients.
Keywords: Melanoma, Bevacizumab, Imatinib, Vascular endothelial growth factor, Platelet-derived growth factor
Abstract
Background.
Vascular endothelial growth factor and platelet-derived growth factor signaling in the tumor microenvironment appear to cooperate in promoting tumor angiogenesis.
Patients and Methods.
We conducted a phase I trial combining bevacizumab (i.v. every 2 weeks) and imatinib (oral daily). Once a recommended phase II dose combination was established, a phase II trial was initiated in patients with metastatic melanoma. A Simon 2-stage design was used with 23 patients required in the first stage and 41 patients in total should the criteria to proceed be met. We required that 50% of the patients be progression-free at 16 weeks. Dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) and power Doppler ultrasonography were performed in patients with metastatic tumors amenable to imaging with these methods at baseline and after 4 weeks.
Results.
A total of 17 patients were accrued to 4 dose and combination levels. Bevacizumab 10 mg/kg every 2 weeks could be safely combined with imatinib 800 mg daily. Common toxicities included fatigue, nausea, vomiting, edema, proteinuria, and anemia, but were not commonly severe. A total of 23 patients with metastatic melanoma (48% with American Joint Commission on Cancer stage M1c; median age, 63 years) were enrolled in the first stage of phase II. The 16-week progression-free survival rate was 35%, leading to termination of phase II after the first stage. In the small subset of patients who remained on study with lesions evaluable by DCE-MRI, significant decreases in tumor vascular permeability were noted, despite early disease progression using the Response Evaluation Criteria In Solid Tumors.
Conclusion.
Bevacizumab and imatinib can be safely combined at the maximum doses used for each agent. We did not observe significant clinical activity with this regimen in melanoma patients.
Implications for Practice:
Vascular endothelial growth factor (VEGF)-targeted antiangiogenic therapy has proven clinical efficacy as a standalone therapy in renal cell carcinoma and glioblastoma multiforme. Also, enhancement of conventional cytotoxic chemotherapy efficacy has been observed in colorectal, non-small-cell lung, breast, and ovarian cancers. Optimal strategies to cotarget angiogenic cytokines combined with VEGF have not been defined. It was found that bevacizumab could be safely combined with imatinib, which was used as a platelet-derived growth factor receptor inhibitor in our study. High-dose imatinib-related edema was not observed when paired with bevacizumab. This regimen might be suitable for further investigation in other cancers but apparently not in melanoma.
Introduction
Angiogenesis is ubiquitous in cancer pathogenesis, at the site of both primary tumor formation and metastases. However, angiogenesis involves numerous cell types and is initiated by numerous cytokines produced by tumor cells. Hypoxia inducible factor (HIF) activity is at the root of transcriptional regulation of the best-described proangiogenic cytokines, including vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) [1]. However, numerous additional secreted factors, such as angiopoetins, ephrins, transforming growth factor-β, hepatocyte growth factor, and fibroblast growth factor, are similarly under HIF control and linked to angiogenesis [2–6]. The relative importance of each proangiogenesis cytokine in each cancer type has not been resolved in model systems or in the clinic.
VEGF has been described as the most potent endothelial cell mitogen and essential in the initiating steps of angiogenesis [7]. PDGF is essential to the recruitment of pericytes, which are derived from mesenchymal stromal cells and are essential to the maturation and stabilization of these immature blood vessels [8]. Microvessels that are endowed with pericytes are no longer dependent on VEGF for their survival [9]. Under hypoxic conditions, pericytes are dependent on PDGF for survival, and treatment of tumors with PDGF inhibitors inhibits blood vessel formation and tumor growth in human tumor xenografts [10]. Melanoma expresses PDGF, suggesting that it represents a relevant point of intervention to inhibit angiogenesis in this disease [11–14].
Bevacizumab is a human, monoclonal antibody that is highly selective VEGF-A, the isoform that binds VEGF receptor (VEGFR)1 and VEGFR2 [15]. Doses up to 5 mg/kg per week, generally given every 2 or 3 weeks have proved to be efficacious in colorectal, non-small-cell lung, breast, and renal cell carcinoma and glioblastoma multiforme [16–20].
Imatinib is a tyrosine kinase inhibitor with potency against abl, c-kit, and PDGF receptor-β (PDGFRβ) [21]. Mouse xenograft models have established that imatinib can inhibit tumor progression in tumors that are not driven by abl or c-kit signaling [22]. The safety and efficacy of doses ranging from 400 mg to 800 mg daily have been well established [23, 24]. However, the efficacy of imatinib against chronic myelogenous leukemia and gastrointestinal stromal tumor has been attributed to its abl and c-kit potency. The PDGFRβ activity of imatinib has been most clearly demonstrated in dermatofibroma protuberans, hypereosinophilic syndrome, and chronic myelogenous leukemia with translocations involving PDGFRβ [25–27].
In metastatic melanoma, VEGF-targeted therapies have had modest clinical activity, raising the possibility that additional angiogenesis mediators would need to be targeted to develop effective antiangiogenic therapy for this disease [28]. Imatinib has been evaluated as a single-agent in two phase II trials in melanoma, neither of which was associated with significant clinical activity [29, 30]. However, evidence in animal models suggests that the combination of VEGF and PDGF inhibition has significantly more antiangiogenic activity than blockade of either alone [31, 32].
VEGF-targeted therapeutics are the most advanced for cancer therapy and include a monoclonal antibody targeting VEGF and three VEGFR tyrosine kinase inhibitors, all Food and Drug Administration-approved drugs [33–35]. Each of the approved VEGFR inhibitors also inhibit PFGFRβ, but to a varying degree. Numerous additional VEGFR inhibitors, which also antagonize PDGFRβ, are in clinical development, but the ratio of VEGFR/PDGFRβ potency is fixed. For that reason, we sought to evaluate the combination of bevacizumab and imatinib, a small molecular inhibitor of PDGFRβ with no activity against VEGFR. In doing so, we would have the ability to vary the dose of each agent to define the maximum tolerated doses for the regimen that could optimize the inhibition of both VEGF and PFGFRβ.
We conducted a phase I trial combining bevacizumab with imatinib in patients with advanced solid tumors. Once we determined the recommended phase II dose, we enrolled patients with metastatic melanoma in a phase II trial with this combination regimen. In order to investigate the effect of this therapy on tumor vascular permeability and overall tumor vascularity, where possible, we evaluated patients with dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) and dynamic vascular ultrasonography. Each method has been used in the context of clinical trials with antiangiogenic therapy, and decreases in these parameters have been associated with improved clinical outcomes [36–40].
Patients and Methods
Patients
Patients were eligible for enrollment in the phase I portion of the trial if they had histologically or cytologically confirmed advanced solid tumors that were metastatic or unresectable and for which curative or effective palliative measures were exhausted or did not exist. In the phase II portion of the study, eligible patients were required to have a diagnosis of metastatic melanoma and measurable disease. Previous systemic chemotherapy or immunotherapy was permitted in phase I. For phase II, the patients were allowed no more than one previous regimen containing cytotoxic chemotherapy for advanced disease. Other inclusion criteria were age ≥21 years, Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, life expectancy of at least 3 months, adequate bone marrow function (white blood cell count >3,000 cells per mm3, absolute neutrophil count (ANC) >1,500 cells per mm3, platelet count >100,000 cells per mm3), adequate renal function (serum creatinine <2× the upper limit of normal [ULN] or calculated serum creatinine clearance >40 mL/min), and adequate hepatic function (total bilirubin <1.5× ULN, alanine aminotransferase and aspartate aminotransferase <2.5× ULN or <5× ULN in the presence of liver metastasis, and international normalized ratio <1.5 and activated partial thromboplastin time within normal limits). Patients were excluded if they had received chemotherapy, radiation therapy, or immunotherapy within 4 weeks, nitrosoureas or mitomycin C within 6 weeks, or a monoclonal antibody within 8 weeks of study entry. Any toxicity from previous therapies must have resolved. Previous baseline brain involvement was not permitted. Anticoagulant medications were prohibited. Additional exclusion criteria were a history of bleeding diathesis or coagulopathy, uncontrolled hypertension, a serious nonhealing wound, ulcer, or fracture, a major surgical procedure, and open biopsy or traumatic injury within 4 weeks of study entry. Baseline proteinuria, acute infection, or other uncontrolled medical comorbidities were also prohibited. The institutional review board at the University of Pennsylvania approved the study protocol. All patients signed an informed consent form before enrollment.
The pretreatment evaluations included medical history, physical examination, ECOG performance status, a computed tomography (CT) scan of the chest, abdomen, and pelvis, an MRI scan of the brain, complete blood count with differential, serum chemistries, prothrombin time, partial thromboplastin time, and lactate dehydrogenase.
Treatment and Dose Modifications
Imatinib was administered orally once daily (400 mg or 600 mg) or twice daily in divided doses (800 mg) continuously. Bevacizumab was administered intravenously every 2 weeks. The first infusion was over 90 minutes, with the second over 60 minutes and third and subsequent infusions over 30 minutes in the absence of infusion reactions.
If the patient experienced a grade 3 or 4 nonhematologic toxicity, the study drugs were withheld until the toxicity had resolved to grade ≤1, and the doses were reduced by 1 dose level. For grade 3 or 4 hematologic toxicity, the drugs were withheld until the counts had recovered to grade ≤1. If recovery occurred within 2 weeks, the study drug would be resumed at the same dose. Gastrointestinal perforation, fistula formation, retrograde posterior leukoencephalopathy, grade 4 hypertension, and grade 3 or 4 hemorrhagic or thromboembolic events were grounds for discontinuation of bevacizumab. Proteinuria was monitored every 2 weeks, and bevacizumab was withheld for >2 g proteinuria per 24 hours.
Response and Toxicity Assessments
Response was assessed every 8 weeks with CT and/or MRI using the Response Evaluation Criteria In Solid Tumors. MRI scans of the brain were obtained during the follow-up period only if clinically indicated. Toxicities were graded using the National Cancer Institute Common Toxicity Criteria, version 3.0.
Pharmacodynamics
In selected melanoma patients enrolled in the phase II portion of the study, DCE-MRI and contrast-enhanced ultrasonography (CE-US) were performed before therapy and after 4 weeks of treatment. A target tumor (>3 cm) outside of the thorax was required for inclusion in this substudy. A single target lesion was identified from the baseline staging scans.
DCE-MRI
DCE-MRI was performed on a 1.5 T scanner (Sonata; Siemens, Munich, Germany, http://www.medical.siemens.com), with phased arrow torso coils. The target lesion was identified from routine T1- and T2-weighted imaging. Dynamic enhanced imaging was performed with a three-dimensional T1-weighted (repetition time, 4 milliseconds; excitation time, 1 milliseconds; flip angle, 30°) spoiled gradient echo slab (5 mm × 8–10 slices) to include both tumor and major regional artery. In 1 patient, a novel radial projection slab of 5 mm × 32 slices was used with identical acquisition parameters [41]. Imaging was performed continuously during i.v. administration of 0.1 mmol/kg gadolinium-diamide (Omniscan; GE Healthcare, Little Chalfont, U.K., http://www.gehealthcare.com) at 1 mL/sec. The total DCE-MRI scanning time was 8 minutes. Temporal resolution of imaging was approximately 3 seconds per repetition.
Intensity curves for the arterial region of interest were obtained for the input function. The tumor boundary was segmented manually on a single central slice, and pixel-wise tumor intensity-versus-time curves were generated, along with a single arterial intensity-versus-time curve. Because prior phantom studies using the identical sequence have demonstrated linearity between relaxivity and MR signal intensity in the physiologic range of gadolinium concentrations (data not shown), the gadolinium concentration for the tumor and artery was estimated based on the MRI signal intensity differences between pre- and postgadolinium images [42]. The time-intensity curves were fit to the two compartment models [43] as follows:
Least-square minimization was performed to obtain the kinetic parameters Ktrans and kep, representing the unidirectional influx and efflux rates of gadolinium between plasma and the tumor interstitium, respectively. Median pixel values for tumor Ktrans were identified.
Contrast-enhanced ultrasonography was performed on a Philips HDI 5000 SonoCT (Philips Healthcare, Bothell, WA, http://www.healthcare.philips.com) unit. The target lesion was scanned in grayscale, followed by color and power Doppler imaging for vascular quantification. A microbubble contrast agent (Optison; GE Healthcare; or Definity; Lantheus Medical Imaging, North Billerica, MA, http://www.lantheus.com) was administered per the manufacturer’s recommendations. Power Doppler imaging was performed at 0.5 Hz to minimize microbubble destruction. The tumor regions were segmented manually, and the color-weighted volume fraction representing tumor vascularity was calculated using previously described methods [44].
Study Design
In the phase I portion of the trial, a standard 3+3 dose escalation design was used. The doses of imatinib ranged from 200 to 800 mg daily, and the doses of bevacizumab ranged from 5 to 10 mg/kg i.v. weekly. The last patient receiving a given dose level was observed for 4 weeks on treatment before enrolling patients at the subsequent dose level. Dose-limiting toxicities (DLTs) were defined as those at least probably related to the study drug(s) and were as follows: grade 4 neutropenia (ANC <500 cells per mm3 lasting longer than 7 days); grade 3 or 4 neutropenia (ANC <1,000 cells per mm3) with fever greater than 101°F; grade 3 thrombocytopenia (platelet count <50,000 cells per mm3); and nonhematologic toxicity grade ≥3, except for nausea, vomiting, and hypertension that was not optimally medically managed. The maximum tolerated dose (MTD) was defined as the dose level below the dose that induced DLTs in at least one third of the patients. If a MTD was not determined, the highest dose level (800 mg imatinib and 10 mg/kg bevacizumab) would be defined as the recommended phase II dose.
The primary endpoint of the phase I portion was to determine the MTD of the combination of imatinib and bevacizumab. In the phase II portion, the primary endpoint was progression-free survival (PFS). We considered a 16-week PFS rate of more than 50% to be evidence of efficacy in this patient population and worthy of further study. A Simon 2-stage design was used, requiring 11 of the first 23 patients to be progression-free at 8 weeks to proceed to full accrual of 39 patients [45].
Statistical Analysis
The statistical analyses for the baseline demographics, response rates, adverse events, and DCE-MRI and vascular ultrasound findings were descriptive, and Kaplan-Meier analysis was used for survival data. The association between continuous outcomes and categorical outcomes was assessed using Pearson’s or Spearman’s correlation coefficients. Kaplan-Meier estimates of progression-free survival and 95% confidence intervals (CIs) were calculated using GraphPad Prism software (GraphPad Software, Inc., San Diego, CA, http://www.graphpad.com).
Results
Phase I
From October 2003 to January 2008, 40 patients were enrolled in the present study. The characteristics of the 17 patients enrolled in the phase I portion of this trial are listed in Table 1. In the absence of DLTs to define an MTD, the recommended phase II dose was imatinib 400 mg b.i.d. and bevacizumab 10 mg/kg every 14 days. Eight patients were enrolled at this dose to have confidence in its tolerability before enrolling the phase II melanoma cohort. The common toxicities observed during the phase I portion are listed in Table 2; the most frequent was grade 1 or 2 fatigue, nausea, vomiting, edema, proteinuria, and anemia. No patients in the phase I portion had an objective response, although 10 of 14 patients at dose levels 2, 3, and 4 had stable disease (Table 3).
Table 1.
Phase I patient characteristics
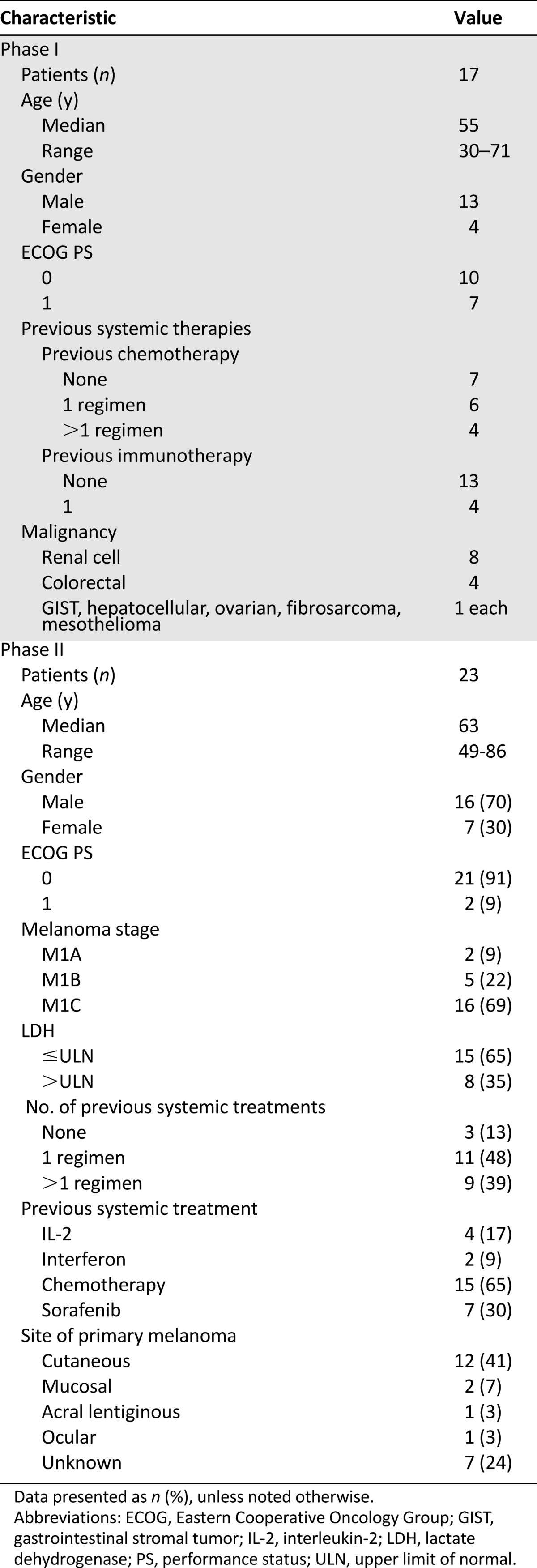
Table 2.
Phase I toxicities (at least possibly related)
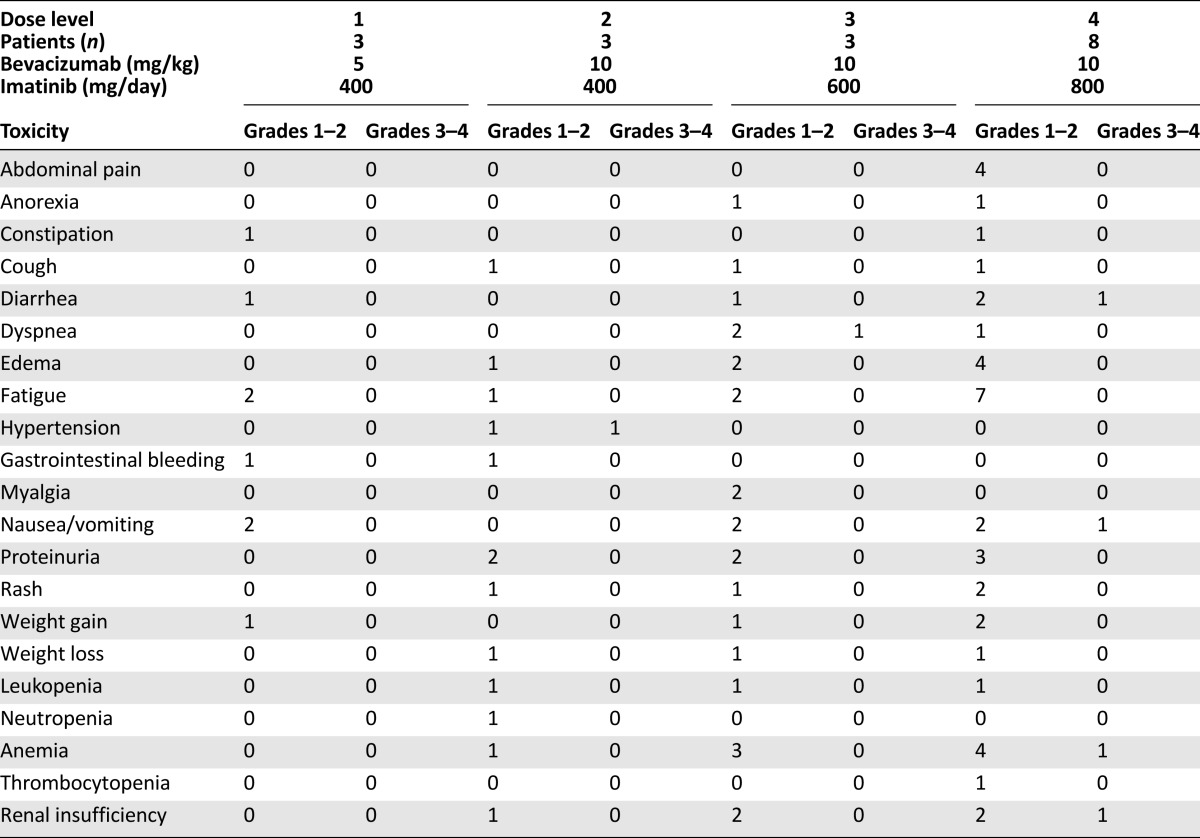
Table 3.
Phase I response data
Phase II
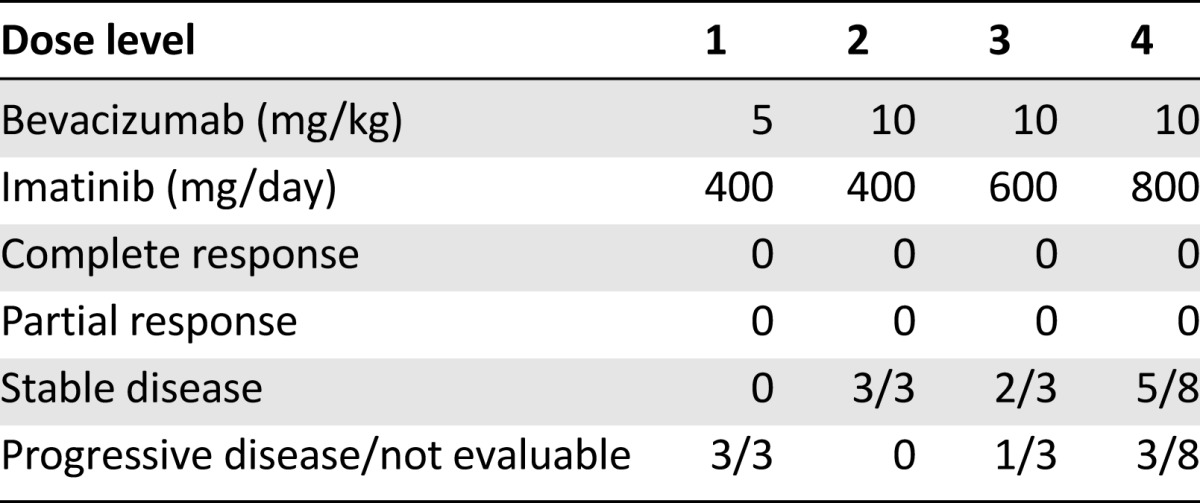
Twenty-three patients were treated in the phase II portion of the study (Table 1). The toxicity observed in this cohort is listed in Table 4. The common toxicities were similar in frequency and severity to those observed in phase I. Severe nausea and vomiting requiring antiemetics was observed in 14% of the patients. Mild to moderate edema was frequently observed but was manageable with diuretics. One patient developed grade 3 pneumonitis, which was attributed to imatinib, resulting in removal from the study. Two patients died during the study; both deaths were attributed to disease progression. Sixteen patients (57%) required a dose reduction of imatinib for intolerable grade 2 toxicity; six patients (21%) required dose interruption of bevacizumab for proteinuria. Thus, although the patients could tolerate this regimen sufficiently well in the first cycle to permit dose level 4 to be defined as the recommended phase II dose, for chronic administration, imatinib 600 mg/day and bevacizumab 10 mg/kg was the regimen most patients could tolerate.
Table 4.
Phase II toxicities
A partial response was observed in 1 patient (4%; 95% CI, 0%–12%) and stable disease in 7 patients (30%; 95% CI, 11%–49%; Table 5). Five patients remained on study for more than 4 months. The median progression-free survival was 7.7 weeks, and the 6-month progression-free survival rate was 4% (Fig. 1). With only 8 patients progression-free at 16 weeks, the criteria for continuing to the second stage of accrual were not met.
Table 5.
Phase II response data
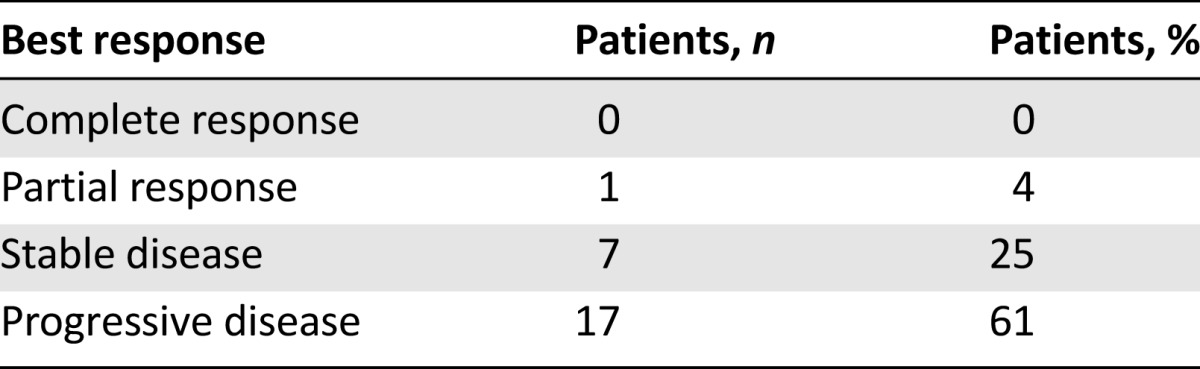
Figure 1.

Progression-free survival for phase II cohort.
Imaging-Based Vascular Response Assessment
A minority of patients met the criteria for inclusion in the DCE-MRI/CE-US portion of the study owing to the lack of eligible tumors for assessment. Because of disease progression in several patients eligible for imaging, only baseline DCE-MRI or CE-US studies were available for most patients imaged. Five patients underwent CE-US before and after therapy, and four underwent DCE-MRI evaluations at both time points. Three patients underwent both assessments. The results of these functional imaging studies are shown in Table 6, with a representative MRI scan shown for 1 patient with stable disease (Fig. 2). Of the 5 patients imaged with CE-US before and after therapy, 2 had significant vascular responses (absolute declines in color-weighted fractional area [CWFA] of 77% and 56%), 1 had a minor vascular response (14% decline in CWFA), and 2 had no response (1% and 5% increase in CWFA). Of the 4 patients with paired DCE-MRI studies, the target tumor for 1 patient demonstrated a major reduction in Ktrans (72% decline). For 3 other patients with paired imaging studies, minor decreases in Ktrans were observed when comparing the on-treatment and pretreatment imaging findings (Ktrans declines of 23%, 9%, and 4%). However, 1 of these patients demonstrated remarkable intertumor variability (Fig. 2). Despite the evidence of an effect on tumor vascularity in this subset of patients, disease control was not observed.
Table 6.
Results of functional imaging studies
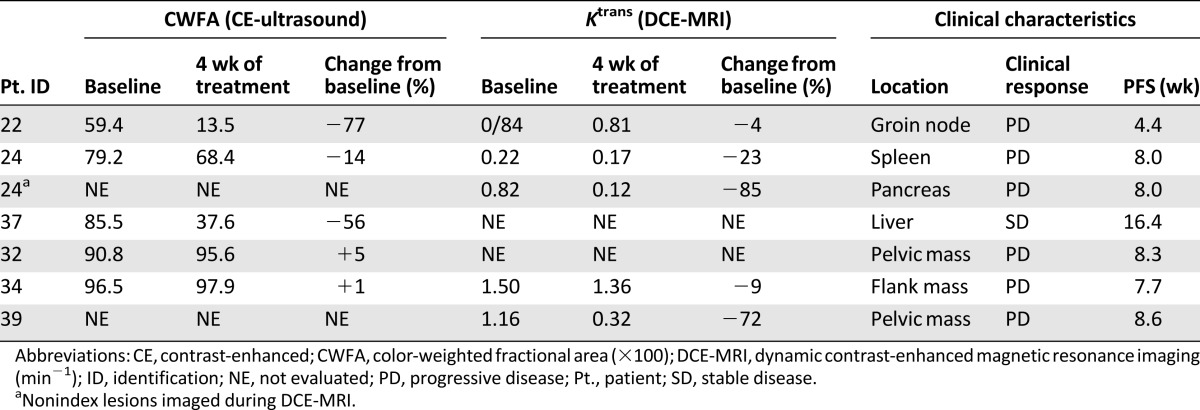
Figure 2.

Pharmacodynamic assessment of change in tumor vasculature. Baseline (A) and week 4 (B) postgadolinium T1-weighted imaging of the abdomen in a patient with several larger metastatic tumor deposits, including splenic (single arrow) and pancreatic (double arrow) lesions. The week 4 image demonstrates marked vascular inhibition of the pancreatic lesion, but not the splenic lesion. Ktrans changes in these 2 lesions are listed in Table 6.
Discussion
The most rational strategy for assembling combination regimens targeting multiple angiogenesis mediators is not known. Preclinical studies have suggested that VEGF signaling is the single most important angiogenesis target; however, angiogenesis can still proceed even in the face of optimal VEGF inhibition [46–48]. Based on evidence that PDGF signaling plays an important secondary role in tumor angiogenesis, we hypothesized that maximizing inhibition of each pathway might provide more effective short-term effects on tumor blood vessels and long-lasting effects, if PDGF signaling is an important mechanism of resistance to VEGF targeted therapy.
We found that the highest doses of imatinib and bevacizumab, used as single agents or in combination with other therapies, were able to be combined safely. This was somewhat surprising, because imatinib at 800 mg/day has been reported to be associated with significant fluid retention and nausea [30, 49, 50]. We did observe nausea as a frequent toxicity, but it was not dose-limiting in the traditional sense. Also, although we did observe fluid retention in some patients, the frequency and severity appeared to be less than that reported for imatinib at 800 mg/day. Although dose reductions were required for some patients, the toxicity profile for this combination compared favorably to that of single-agent imatinib treatment at 800 mg daily. We speculate that bevacizumab ameliorates some of the edema associated with high-dose imatinib treatment by reducing the permeability of the normal vasculature.
Efficacy of this combination in patients with metastatic melanoma was not apparent. In some respects, this is not surprising given the lack of significant single-agent activity with either agent in this patient population. However, we hoped that more complete inhibition of angiogenesis than is achievable with VEGF-targeted therapy might result in slower disease progression. Single-agent activity of imatinib has been recently reported in a small subgroup (approximately 1%) of melanoma patients whose tumors harbored activating KIT mutations [51]. We did not enroll patients in the clinical subgroups where these mutations are found. The efficacy of bevacizumab has been explored in combination with carboplatin/paclitaxel in a randomized phase II trial of patients with metastatic melanoma [52]. Marginal and statistically nonsignificant improvements in progression-free survival were observed. In the adjuvant setting, single-agent bevacizumab showed no benefit with regard to overall survival among patients with resected stage IIb, IIc, and III disease [53]. These trials suggest that VEGF is not the same central mediator of tumor angiogenesis that it is in renal cell carcinoma and other solid tumors. In the years since this trial was first conceived, VEGFR/PDFGR inhibitors have proliferated and demonstrated notable efficacy in certain cancers as single agents, but not in melanoma.
We performed functional imaging evaluations with DCE-MRI and CE-US to correlate the clinical outcome with changes in tumor vasculature. We hypothesized that significant changes in tumor vascular permeability or overall vascularity, assessed by either method, would correlate with clinical efficacy. However, so few patients demonstrated evidence of disease control, such a relationship could not be established. Furthermore, a significant number of patients who had undergone baseline DCE-MRI or CE-US quickly deteriorated clinically, prompting early response assessment with conventional imaging and removal from the study because of progressive disease. Although clinical progression is commonly observed in metastatic melanoma patients in the absence of a treatment response, it is possible that this regimen accelerated disease progression in some instances. Importantly, instances were seen of significant changes in tumor vasculature, but a lack of disease control, suggesting a lack of utility for measures of tumor vasculature as meaningful surrogate biomarkers in melanoma. It is possible that therapies that affect tumor vascularity will have more efficacy in one tumor type than in another (e.g., melanoma vs. renal cell carcinoma) even when a comparable magnitude of effect is observed [37]. However, so far, too few of our patients were able to undergo imaging at both of the intended time points to further investigate that hypothesis. An alternative in future studies would be to perform DCE-MRI or CE-US after a much shorter interval of therapy. We did not do that in favor of a time interval we thought would be more informative regarding the maximum effect on the tumor vascular architecture.
Conclusion
Our results do not rule out the possibility that antiangiogenic therapy could be effectively combined with other modalities of treatment of metastatic melanoma, including novel oncogene-targeted therapy, chemotherapy, or immunotherapy. Our aim was to investigate the pharmacodynamics and clinical efficacy associated with dual-targeted antiangiogenic therapy. The safety of this regimen and evidence of vascular response, albeit in a subset of the patients treated, supports further investigation of this combination in other tumor types, particularly those for which bevacizumab has proved a useful adjunct to conventional cytotoxic chemotherapy.
Acknowledgment
This study was supported by Grant K23 CA104884-02 from the NIH (Bethesda, MD).
Author Contributions
Conception/Design: Keith T. Flaherty, Mark A. Rosen, Ravi K. Amaravadi, Helen Chen, Chandra Sehgal, Peter J. O’Dwyer
Provision of study material or patients: Keith T. Flaherty, Mark A. Rosen, Ravi K. Amaravadi, Lynn M. Schuchter, Maryann Gallagher, Chandra Sehgal, Peter J. O’Dwyer
Collection and/or assembly of data: Keith T. Flaherty, Betty K. Hamilton, Mark A. Rosen, Ravi K. Amaravadi, Maryann Gallagher, Chandra Sehgal, Peter J. O’Dwyer
Data analysis and interpretation: Keith T. Flaherty, Betty K. Hamilton, Mark A. Rosen, Ravi K. Amaravadi, Lynn M. Schuchter, Maryann Gallagher, Helen Chen, Chandra Sehgal, Peter J. O’Dwyer
Manuscript writing: Keith T. Flaherty, Betty K. Hamilton, Mark A. Rosen, Ravi K. Amaravadi, Lynn M. Schuchter, Chandra Sehgal, Peter J. O’Dwyer
Final approval of manuscript: Keith T. Flaherty, Betty K. Hamilton, Mark A. Rosen, Ravi K. Amaravadi, Lynn M. Schuchter, Maryann Gallagher, Helen Chen, Chandra Sehgal, Peter J. O’Dwyer
Disclosures
Keith T. Flaherty: Genentech, Novartis (C/A); Peter J. O’Dwyer: Tetralogic Pharmaceuticals, PrECOG, Sanofi-Aventis, Genentech (C/A), Genentech (H), Pfizer, Bristol-Meyers Squibb, Methylgene, Novartis, Genentech, Boston Biomedical, FibroGen, AstraZeneca, Incyte, GlaxoSmithKline (RF), Tetralogic Pharmaceuticals (OI). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Kaelin WG., Jr The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8:865–873. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]
- 2.Skuli N, Liu L, Runge A, et al. Endothelial deletion of hypoxia-inducible factor-2alpha (HIF-2alpha) alters vascular function and tumor angiogenesis. Blood. 2009;114:469–477. doi: 10.1182/blood-2008-12-193581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamashita T, Ohneda K, Nagano M, et al. Hypoxia-inducible transcription factor-2alpha in endothelial cells regulates tumor neovascularization through activation of ephrin A1. J Biol Chem. 2008;283:18926–18936. doi: 10.1074/jbc.M709133200. [DOI] [PubMed] [Google Scholar]
- 4.Gunaratnam L, Morley M, Franovic A, et al. Hypoxia inducible factor activates the transforming growth factor-alpha/epidermal growth factor receptor growth stimulatory pathway in VHL(-/-) renal cell carcinoma cells. J Biol Chem. 2003;278:44966–44974. doi: 10.1074/jbc.M305502200. [DOI] [PubMed] [Google Scholar]
- 5.Chu SH, Feng DF, Ma YB, et al. Stabilization of hepatocyte growth factor mRNA by hypoxia-inducible factor 1. Mol Biol Rep. 2009;36:1967–1975. doi: 10.1007/s11033-008-9406-1. [DOI] [PubMed] [Google Scholar]
- 6.Calvani M, Rapisarda A, Uranchimeg B, et al. Hypoxic induction of an HIF-1alpha-dependent bFGF autocrine loop drives angiogenesis in human endothelial cells. Blood. 2006;107:2705–2712. doi: 10.1182/blood-2005-09-3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer. 2002;2:795–803. doi: 10.1038/nrc909. [DOI] [PubMed] [Google Scholar]
- 8.Lindahl P, Johansson BR, Levéen P, et al. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 9.Shih SC, Ju M, Liu N, et al. Transforming growth factor beta1 induction of vascular endothelial growth factor receptor 1: Mechanism of pericyte-induced vascular survival in vivo. Proc Natl Acad Sci USA. 2003;100:15859–15864. doi: 10.1073/pnas.2136855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blaskovich MA, Lin Q, Delarue FL, et al. Design of GFB-111, a platelet-derived growth factor binding molecule with antiangiogenic and anticancer activity against human tumors in mice. Nat Biotechnol. 2000;18:1065–1070. doi: 10.1038/80257. [DOI] [PubMed] [Google Scholar]
- 11.Barnhill RL, Xiao M, Graves D, et al. Expression of platelet-derived growth factor (PDGF)-A, PDGF-B and the PDGF-alpha receptor, but not the PDGF-beta receptor, in human malignant melanoma in vivo. Br J Dermatol. 1996;135:898–904. doi: 10.1046/j.1365-2133.1996.d01-1092.x. [DOI] [PubMed] [Google Scholar]
- 12.Rofstad EK, Halsør EF. Vascular endothelial growth factor, interleukin 8, platelet-derived endothelial cell growth factor, and basic fibroblast growth factor promote angiogenesis and metastasis in human melanoma xenografts. Cancer Res. 2000;60:4932–4938. [PubMed] [Google Scholar]
- 13.Forsberg K, Valyi-Nagy I, Heldin CH, et al. Platelet-derived growth factor (PDGF) in oncogenesis: Development of a vascular connective tissue stroma in xenotransplanted human melanoma producing PDGF-BB. Proc Natl Acad Sci USA. 1993;90:393–397. doi: 10.1073/pnas.90.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ugurel S, Rappl G, Tilgen W, et al. Increased serum concentration of angiogenic factors in malignant melanoma patients correlates with tumor progression and survival. J Clin Oncol. 2001;19:577–583. doi: 10.1200/JCO.2001.19.2.577. [DOI] [PubMed] [Google Scholar]
- 15.Ferrara N. Role of vascular endothelial growth factor in physiologic and pathologic angiogenesis: Therapeutic implications. Semin Oncol. 2002;29(suppl 16):10–14. doi: 10.1053/sonc.2002.37264. [DOI] [PubMed] [Google Scholar]
- 16.Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 17.Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 18.Miller K, Wang M, Gralow J, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666–2676. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- 19.Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 2003;349:427–434. doi: 10.1056/NEJMoa021491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vredenburgh JJ, Desjardins A, Herndon JE, II, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13:1253–1259. doi: 10.1158/1078-0432.CCR-06-2309. [DOI] [PubMed] [Google Scholar]
- 21.Buchdunger E, Zimmermann J, Mett H, et al. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996;56:100–104. [PubMed] [Google Scholar]
- 22.Buchdunger E, Cioffi CL, Law N, et al. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther. 2000;295:139–145. [PubMed] [Google Scholar]
- 23.van Oosterom AT, Judson I, Verweij J, et al. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: A phase I study. Lancet. 2001;358:1421–1423. doi: 10.1016/s0140-6736(01)06535-7. [DOI] [PubMed] [Google Scholar]
- 24.Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–1042. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 25.McArthur GA, Demetri GD, van Oosterom A, et al. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J Clin Oncol. 2005;23:866–873. doi: 10.1200/JCO.2005.07.088. [DOI] [PubMed] [Google Scholar]
- 26.Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201–1214. doi: 10.1056/NEJMoa025217. [DOI] [PubMed] [Google Scholar]
- 27.Apperley JF, Gardembas M, Melo JV, et al. Response to imatinib mesylate in patients with chronic myeloproliferative diseases with rearrangements of the platelet-derived growth factor receptor beta. N Engl J Med. 2002;347:481–487. doi: 10.1056/NEJMoa020150. [DOI] [PubMed] [Google Scholar]
- 28.Varker KA, Biber JE, Kefauver C, et al. A randomized phase 2 trial of bevacizumab with or without daily low-dose interferon alfa-2b in metastatic malignant melanoma. Ann Surg Oncol. 2007;14:2367–2376. doi: 10.1245/s10434-007-9389-5. [DOI] [PubMed] [Google Scholar]
- 29.Ugurel S, Hildenbrand R, Zimpfer A, et al. Lack of clinical efficacy of imatinib in metastatic melanoma. Br J Cancer. 2005;92:1398–1405. doi: 10.1038/sj.bjc.6602529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wyman K, Atkins MB, Prieto V, et al. Multicenter phase II trial of high-dose imatinib mesylate in metastatic melanoma: Significant toxicity with no clinical efficacy. Cancer. 2006;106:2005–2011. doi: 10.1002/cncr.21834. [DOI] [PubMed] [Google Scholar]
- 31.Bergers G, Song S, Meyer-Morse N, et al. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Erber R, Thurnher A, Katsen AD, et al. Combined inhibition of VEGF and PDGF signaling enforces tumor vessel regression by interfering with pericyte-mediated endothelial cell survival mechanisms. FASEB J. 2004;18:338–340. doi: 10.1096/fj.03-0271fje. [DOI] [PubMed] [Google Scholar]
- 33.Grothey A, Galanis E. Targeting angiogenesis: Progress with anti-VEGF treatment with large molecules. Nat Rev Clin Oncol. 2009;6:507–518. doi: 10.1038/nrclinonc.2009.110. [DOI] [PubMed] [Google Scholar]
- 34.Stein MN, Flaherty KT. CCR drug updates: Sorafenib and sunitinib in renal cell carcinoma. Clin Cancer Res. 2007;13:3765–3770. doi: 10.1158/1078-0432.CCR-06-2844. [DOI] [PubMed] [Google Scholar]
- 35.Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–1068. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- 36.Morgan B, Thomas AL, Drevs J, et al. Dynamic contrast-enhanced magnetic resonance imaging as a biomarker for the pharmacological response of PTK787/ZK 222584, an inhibitor of the vascular endothelial growth factor receptor tyrosine kinases, in patients with advanced colorectal cancer and liver metastases: Results from two phase I studies. J Clin Oncol. 2003;21:3955–3964. doi: 10.1200/JCO.2003.08.092. [DOI] [PubMed] [Google Scholar]
- 37.Flaherty KT, Rosen MA, Heitjan DF, et al. Pilot study of DCE-MRI to predict progression-free survival with sorafenib therapy in renal cell carcinoma. Cancer Biol Ther. 2008;7:496–501. doi: 10.4161/cbt.7.4.5624. [DOI] [PubMed] [Google Scholar]
- 38.Hahn OM, Yang C, Medved M, et al. Dynamic contrast-enhanced magnetic resonance imaging pharmacodynamic biomarker study of sorafenib in metastatic renal carcinoma. J Clin Oncol. 2008;26:4572–4578. doi: 10.1200/JCO.2007.15.5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lassau N, Koscielny S, Albiges L, et al. Metastatic renal cell carcinoma treated with sunitinib: Early evaluation of treatment response using dynamic contrast-enhanced ultrasonography. Clin Cancer Res. 2010;16:1216–1225. doi: 10.1158/1078-0432.CCR-09-2175. [DOI] [PubMed] [Google Scholar]
- 40.Lamuraglia M, Escudier B, Chami L, et al. To predict progression-free survival and overall survival in metastatic renal cancer treated with sorafenib: Pilot study using dynamic contrast-enhanced Doppler ultrasound. Eur J Cancer. 2006;42:2472–2479. doi: 10.1016/j.ejca.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 41.Lin W, Guo J, Rosen MA, et al. Respiratory motion-compensated radial dynamic contrast-enhanced (DCE)-MRI of chest and abdominal lesions. Magn Reson Med. 2008;60:1135–1146. doi: 10.1002/mrm.21740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vallée JP, Lazeyras F, Kasuboski L, et al. Quantification of myocardial perfusion with FAST sequence and Gd bolus in patients with normal cardiac function. J Magn Reson Imaging. 1999;9:197–203. doi: 10.1002/(sici)1522-2586(199902)9:2<197::aid-jmri7>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 43.Tofts PS, Brix G, Buckley DL, et al. Estimating kinetic parameters from dynamic contrast-enhanced T(1)-weighted MRI of a diffusable tracer: Standardized quantities and symbols. J Magn Reson Imaging. 1999;10:223–232. doi: 10.1002/(sici)1522-2586(199909)10:3<223::aid-jmri2>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 44.Wood AK, Bunte RM, Cohen JD, et al. The antivascular action of physiotherapy ultrasound on a murine tumor: Role of a microbubble contrast agent. Ultrasound Med Biol. 2007;33:1901–1910. doi: 10.1016/j.ultrasmedbio.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simon R. Optimal two-stage designs for phase II clinical trials. Control Clin Trials. 1989;10:1–10. doi: 10.1016/0197-2456(89)90015-9. [DOI] [PubMed] [Google Scholar]
- 46.Helfrich I, Scheffrahn I, Bartling S, et al. Resistance to antiangiogenic therapy is directed by vascular phenotype, vessel stabilization, and maturation in malignant melanoma. J Exp Med. 2010;207:491–503. doi: 10.1084/jem.20091846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ferrara N. Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev. 2010;21:21–26. doi: 10.1016/j.cytogfr.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 48.Crawford Y, Kasman I, Yu L, et al. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell. 2009;15:21–34. doi: 10.1016/j.ccr.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 49.Rutkowski P, Van Glabbeke M, Rankin CJ, et al. Imatinib mesylate in advanced dermatofibrosarcoma protuberans: Pooled analysis of two phase II clinical trials. J Clin Oncol. 2010;28:1772–1779. doi: 10.1200/JCO.2009.25.7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Groot JW, Zonnenberg BA, van Ufford-Mannesse PQ, et al. A phase II trial of imatinib therapy for metastatic medullary thyroid carcinoma. J Clin Endocrinol Metab. 2007;92:3466–3469. doi: 10.1210/jc.2007-0649. [DOI] [PubMed] [Google Scholar]
- 51.Carvajal RD, Chapman PB, Wolchok JD. A phase II study of imatinib mesylate (IM) for patients with advanced melanoma harboring somatic alterations of KIT. J Clin Oncol. 2009;27:9001. [Google Scholar]
- 52.Kim KB, Sosman JA, Fruehauf JP, et al. BEAM: A randomized phase II study evaluating the activity of bevacizumab in combination with carboplatin plus paclitaxel in patients with previously untreated advanced melanoma. J Clin Oncol. 2012;30:34–41. doi: 10.1200/JCO.2011.34.6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corrie PG, Marshall A, Dunn JA, et al. Adjuvant bevacizumab in patients with melanoma at high risk of recurrence (AVAST-M): Preplanned interim results from a multicentre, open-label, randomised controlled phase 3 study. Lancet Oncol. 2014;15:620–630. doi: 10.1016/S1470-2045(14)70110-X. [DOI] [PubMed] [Google Scholar]