

Abstract
The U.S. Food and Drug Administration approved enzalutamide for the treatment of patients with chemotherapy-naïve metastatic castration-resistant prostate cancer (mCRPC). At the prespecified interim analysis, a statistically significant improvement in overall survival was demonstrated for patients in the enzalutamide arm compared with patients in the placebo arm. The overall benefit-risk profile supports the expanded indication for enzalutamide. On September 10, 2014, the U.S. Food and Drug Administration approved enzalutamide for the treatment of patients with chemotherapy-naïve metastatic castration-resistant prostate cancer (mCRPC). Enzalutamide was initially approved in 2012 for use in patients with mCRPC who had previously received docetaxel. The current approval was based on the results of a randomized, placebo-controlled, double-blind trial conducted in 1,717 asymptomatic or minimally symptomatic patients with chemotherapy-naïve mCRPC. Patients were assigned to receive either enzalutamide 160 mg or placebo orally once daily. The coprimary endpoints were overall survival (OS) and radiographic progression-free survival (rPFS), which was assessed by independent central radiology review. At the prespecified interim analysis, a statistically significant improvement in OS was demonstrated for patients in the enzalutamide arm compared with patients in the placebo arm (hazard ratio [HR], 0.71; 95% confidence interval [CI], 0.60–0.84). The median OS was 32.4 and 30.2 months in the enzalutamide and placebo arms, respectively. A statistically significant prolongation of rPFS was observed in patients in the enzalutamide arm (HR, 0.17; 95% CI, 0.14–0.21). In addition, the time to initiation of cytotoxic chemotherapy was prolonged in the enzalutamide arm (HR, 0.35; 95% CI, 0.30–0.40), with median times of 28.0 and 10.8 months in the enzalutamide and placebo arms, respectively. The safety profile was similar to that previously reported for enzalutamide. Adverse reactions of interest included seizure, hypertension, and falls. Enzalutamide should be discontinued if a seizure occurs during treatment. The overall benefit-risk profile supports the expanded indication for enzalutamide.
Implications for Practice:
This new approval expands the enzalutamide indication, allowing health care providers and patients to use enzalutamide for the treatment of metastatic castration-resistant prostate cancer either before or after cytotoxic chemotherapy.
Keywords: Metastatic castration-resistant prostate cancer, Enzalutamide, Androgen receptor inhibitor, Chemotherapy-naïve
Introduction
Prostate cancer is the most common malignancy in men, with an estimated 29,480 deaths in the United States in 2014 [1]. No treatment prolonged overall survival (OS) in patients with metastatic castration-resistant prostate cancer (mCRPC) until docetaxel combined with prednisone received Food and Drug Administration (FDA) approval in 2004 [2]. That approval was based on a clinically meaningful improvement in OS in patients treated with docetaxel (administered every 3 weeks) plus prednisone when compared with patients treated with mitoxantrone plus prednisone (hazard ratio [HR], 0.76; 95% confidence interval [CI], 0.62–0.94; p < .009) [2, 3]. The median OS was 18.9 and 16.5 months in the docetaxel and mitoxantrone arms, respectively. Of note, 45% of the patients had active pain (with a score of 2 or more on the Present Pain Intensity scale or an analgesic score of at least 10), and 22% of patients had visceral metastases at study entry [3]. Since its approval, docetaxel has generally been used to treat patients with symptomatic disease and/or visceral metastases. For asymptomatic patients with mCRPC, concerns have been raised regarding when to initiate docetaxel-based cytotoxic chemotherapy [4].
Two treatments have received FDA approval for use in asymptomatic or minimally symptomatic patients with mCRPC who have not received chemotherapy for metastatic disease. One is sipuleucel-T, an autologous cellular product consisting of peripheral blood mononuclear cells (PBMCs) obtained from patients by leukapheresis and cultured at 37°C with a recombinant fusion protein (prostatic acid phosphatase fused with granulocyte-macrophage colony-stimulating factor) for 36–44 hours. Its approval was primarily based on the results of a randomized, double-blind phase III trial of sipuleucel-T in asymptomatic or minimally symptomatic patients with mCRPC [5, 6]. Patients with visceral metastases were excluded. The control arm consisted of one third of leukapheresis-isolated autologous PBMCs cultured at 2°C–8°C without the recombinant fusion protein for 36–44 hours. Patients underwent 3 leukapheresis procedures (at approximately weeks 0, 2, and 4), followed 3 days later by an infusion of either sipuleucel-T or the study control. The final analysis of the primary endpoint showed that patients treated with sipuleucel-T had a statistically significant prolongation of OS compared with patients treated with the control (HR, 0.78; 95% CI, 0.61–0.98; p = .032). The median OS was 25.8 and 21.7 months in the sipuleucel-T and control arms, respectively.
The other treatment is abiraterone acetate, an oral CYP17 inhibitor approved in combination with prednisone for the treatment of patients with mCRPC either before or after cytotoxic therapy. The product received FDA approval initially in 2011 for use in patients with mCRPC who have received previous docetaxel [7]. Compared with placebo plus prednisone, abiraterone acetate plus prednisone was associated with a 3.8-month prolongation of median OS (HR, 0.65; 95% CI, 0.54–0.77; p < .0001). In 2012, the indication was modified after FDA review of the results from a randomized, double-blind, phase III trial in asymptomatic or mildly symptomatic patients with chemotherapy-naïve mCRPC and no visceral metastases [8, 9]. The coprimary endpoints were OS and radiographic progression-free survival (rPFS). Patients received abiraterone acetate plus prednisone or placebo plus prednisone. The results showed a statistically significant improvement in rPFS favoring the abiraterone acetate arm with a hazard ratio of 0.43 (95% CI, 0.35–0.52; p < .0001). In addition, OS was prolonged, favoring the abiraterone arm (HR, 0.79; 95% CI, 0.66–0.96) in the third prespecified interim analysis. However, this interim analysis of OS did not cross the O'Brien-Fleming boundary for statistical significance. Prespecified secondary endpoints such as the time to initiation of cytotoxic chemotherapy showed statistically significant improvements in favor of abiraterone acetate [8, 9]. The totality of the findings, along with the established OS benefit in patients previously treated with docetaxel, justified the approval of abiraterone acetate for this indication. The results of the prespecified final analysis for OS were recently reported and showed a statistically significant improvement in OS for patients treated with abiraterone acetate compared with patients treated with placebo (HR, 0.81; 95% CI, 0.70–0.93; p = .003). The median OS was 34.7 and 30.3 months in the abiraterone acetate and placebo arms, respectively [10].
Enzalutamide (Xtandi, Astellas Pharma US Inc., http://www.astellas.us) is an androgen receptor inhibitor that received initial FDA approval on August 31, 2012, for the treatment of patients with mCRPC who have previously received docetaxel [11]. This was based on a 4.8-month improvement in median OS with enzalutamide in a randomized, placebo-controlled trial (AFFIRM) (HR, 0.63; 95% CI, 0.53–0.75; p < .0001) [11, 12]. At the time of approval, the applicant was conducting another randomized, placebo-controlled, phase III trial (MDV3100-03) in asymptomatic or mildly symptomatic patients with chemotherapy-naïve mCRPC [13]. Its key objective was to assess the efficacy and safety of enzalutamide in patients who had not received cytotoxic chemotherapy. This was the same disease setting that was studied in the sipuleucel-T and abiraterone acetate trials discussed in previous paragraphs. The enzalutamide trial was reported to have positive results and was first presented in February 2014 [14].
The applicant submitted a supplemental New Drug Application (sNDA) on March 18, 2104, based on the results of the MDV3100-03. This sNDA received a Priority Review designation and, on September 10, 2014, the product’s indication was modified to include patients with mCRPC irrespective of the use of previous docetaxel or other cytotoxic chemotherapy. The present report summarizes the key clinical and statistical review findings that support this new indication for enzalutamide and expand the FDA experience, with the use of rPFS as a coprimary endpoint.
Trial Design
MDV3100-03 was a randomized, double-blind, placebo-controlled, phase III trial of enzalutamide in patients with mCRPC who had not previously received chemotherapy for prostate cancer [14, 15]. To be eligible for the trial, patients were required to also have evidence of prostate-specific antigen (PSA) and/or radiographic progression [16], an Eastern Cooperative Oncology Group (ECOG) performance status of 0–1 and be asymptomatic or mildly symptomatic, determined by a Brief Pain Inventory-Short Form score of <4 at study entry. Patients with visceral metastases were eligible. The exclusion criteria included a history of seizure or any condition that might predispose to seizure, previous use of androgen synthesis inhibitors (e.g., abiraterone acetate, TAK-700), and the use of opiate analgesics for tumor pain within 4 weeks of study entry. The patients were permitted to use medications that can lower the seizure threshold. The trial was designed to assess an improvement in both OS and rPFS.
Patients were randomly assigned (1:1) to receive enzalutamide 160 mg or placebo orally once daily. Study treatment continued until disease progression (i.e., evidence of radiographic progression, a skeletal-related event [SRE], or clinical progression), along with the initiation of a cytotoxic chemotherapy or an investigational agent, unacceptable toxicity, or consent withdrawal. Radiographic scans were performed at baseline, every 8 weeks for the first 24 weeks after randomization, and every 12 weeks thereafter. After study treatment discontinuation, patients were monitored long term for survival, subsequent treatments for prostate cancer, skeletal-related events, and radiographic progression (if radiographic progression was not confirmed before treatment discontinuation).
OS was defined as the interval from randomization to death from any cause. For patients who were alive at the analysis data cutoff date, the survival time was censored at the last date the patient was known to be alive or the data cutoff date, whichever occurred first. Radiographic progression-free survival was defined as the time from randomization to the first objective evidence of radiographic progression assessed by independent central radiology (ICR) review or death from any cause within 168 days after study treatment discontinuation, whichever occurred first. The ICR review of the electronically transmitted imaging data from the study sites was blinded to treatment assignment and nonimaging clinical information. The blinded results of the ICR review were transmitted to the study sites. Radiographic progression was defined by bone scan identification of 2 or more new bone lesions with confirmation as per the Prostate Cancer Clinical Trials Working Group 2 (PCWG2) criteria [16]. For 2 or more new lesions on a bone scan observed at week 9, radiographic progression required 2 additional new lesions on a confirmatory scan performed at least 6 weeks later. Radiographic progression of soft tissue lesions was determined according to the Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1. For the primary rPFS analysis, events determined by the ICR review were used. For patients who did not have evidence of radiographic progression, their rPFS time was censored to the date of the last scan showing no disease progression. Patients who had a SRE (including radiation to metastases), discontinued study treatment, or initiated a new treatment before radiographic progression were censored to the date of the last radiographic assessment without evidence of progression before the incidence of the SRE, treatment discontinuation, or initiation of a new treatment.
The trial was powered to evaluate an improvement in both OS and rPFS. The overall 2-sided α-level was 0.05, with 0.049 allocated to OS and 0.001 to rPFS. During the trial, the statistical plan was amended to prespecify an interim analysis for OS with approximately 516 deaths (67% of the 765 events required for the final analysis) and to clarify that the rPFS analysis was to be conducted at the interim OS analysis, with a minimum of the first 410 ICR-determined events. These planned analyses were to be conducted in the intent-to-treat population and were agreed on with the FDA in March 2013. A prespecified analysis plan for all the secondary endpoints was also agreed on. The key secondary endpoints included the time to initiation of cytotoxic chemotherapy, time to first skeletal-related event, PSA response, and RECIST responses.
Results
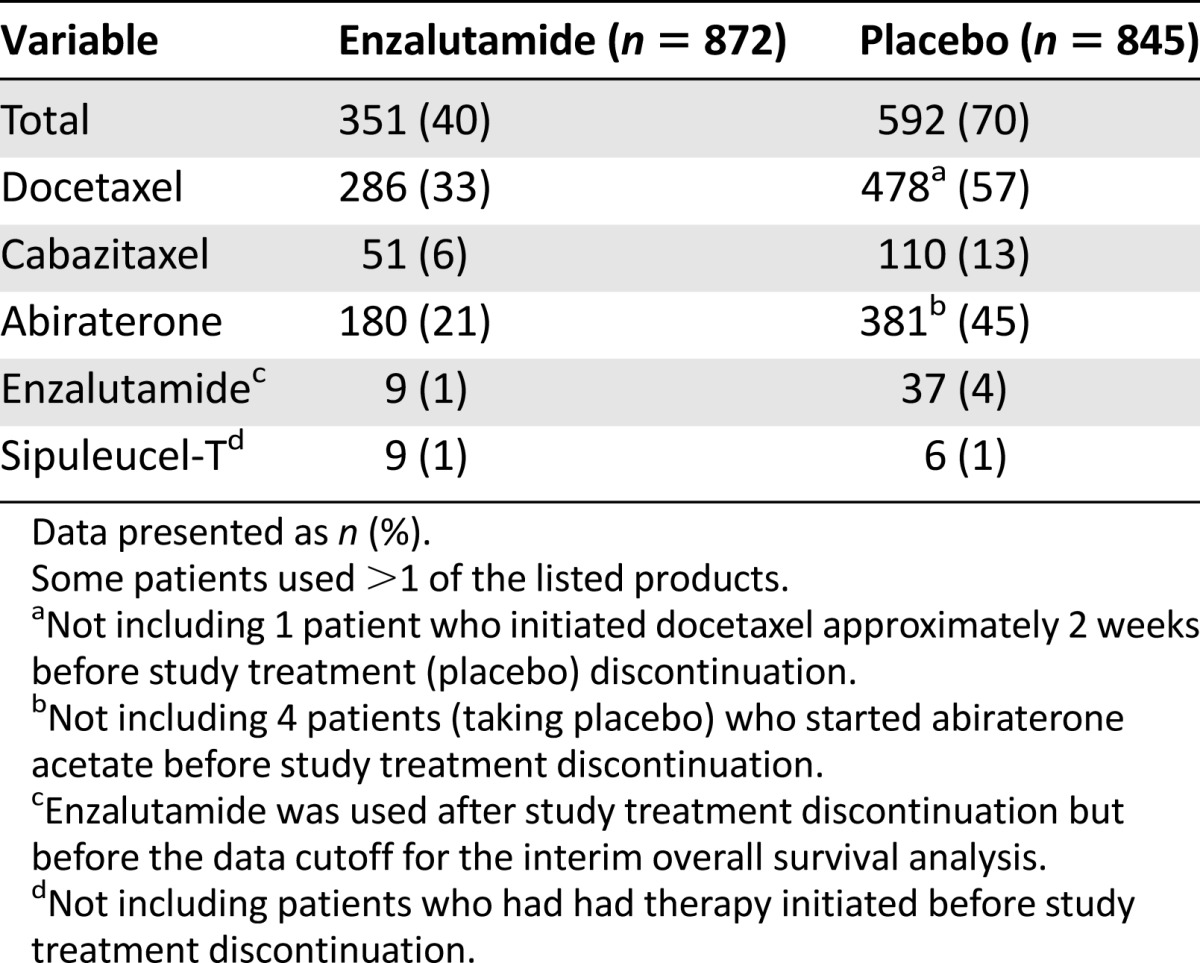
The trial enrolled 1,717 patients from 22 countries, with 872 patients assigned to receive enzalutamide and 845 patients to receive placebo. Fourteen percent of the patients were from the United States. The baseline patient characteristics were balanced between the two arms. The key demographic and disease characteristics for the two arms are summarized in Table 1. The baseline pain assessment was 0–1 (considered asymptomatic) in 67% of patients and 2–3 (considered mildly symptomatic) in 32%. The ECOG performance status score was 0 for 68% of the patients and 1 for 32%. Fifty percent of the patients had an initial Gleason score of 8–10. At study entry, 54% of patients had radiographic evidence of disease progression and 43% had PSA-only progression. Visceral metastases were present in 12% of the patients. One patient in each arm did not receive the study treatment. Approximately 12% of the patients in each arm had at least one protocol deviation and/or violation. The use of subsequent (after discontinuation of the study drug) prostate cancer treatments (limited to treatments that prolong OS) is tabulated in Table 2. More patients in the placebo arm (30% higher) received such treatments.
Table 1.
Key baseline characteristics in the MDV3100-03 trial
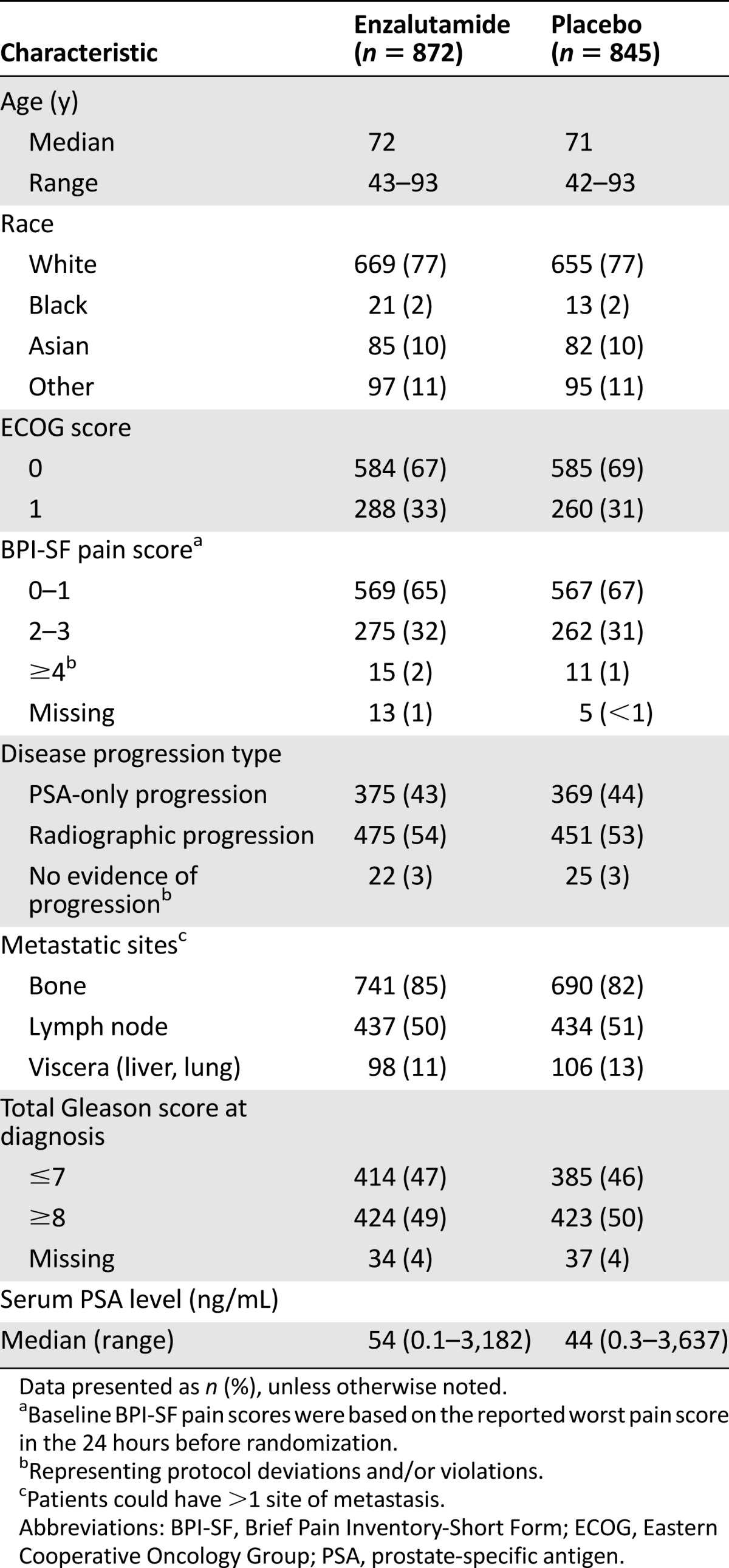
Table 2.
Products used after discontinuation of study treatment
Efficacy
A prespecified interim analysis for OS (with 540 deaths) showed a statistically significant improvement in patients in the enzalutamide arm compared with patients in the placebo arm (HR, 0.71; 95% CI, 0.60–0.84; p < .0001; Table 3). The median OS was 32.4 months (95% CI, 30.1–not reached [NR]) in the enzalutamide arm and 30.2 months (95% CI, 28.0–NR) in the placebo arm. The survival benefit demonstrated for enzalutamide was sustained in a number of sensitivity and subgroup analyses conducted by the investigators [14, 16] and confirmed by FDA review.
Table 3.
Key efficacy results of the MDV3100-03 trial
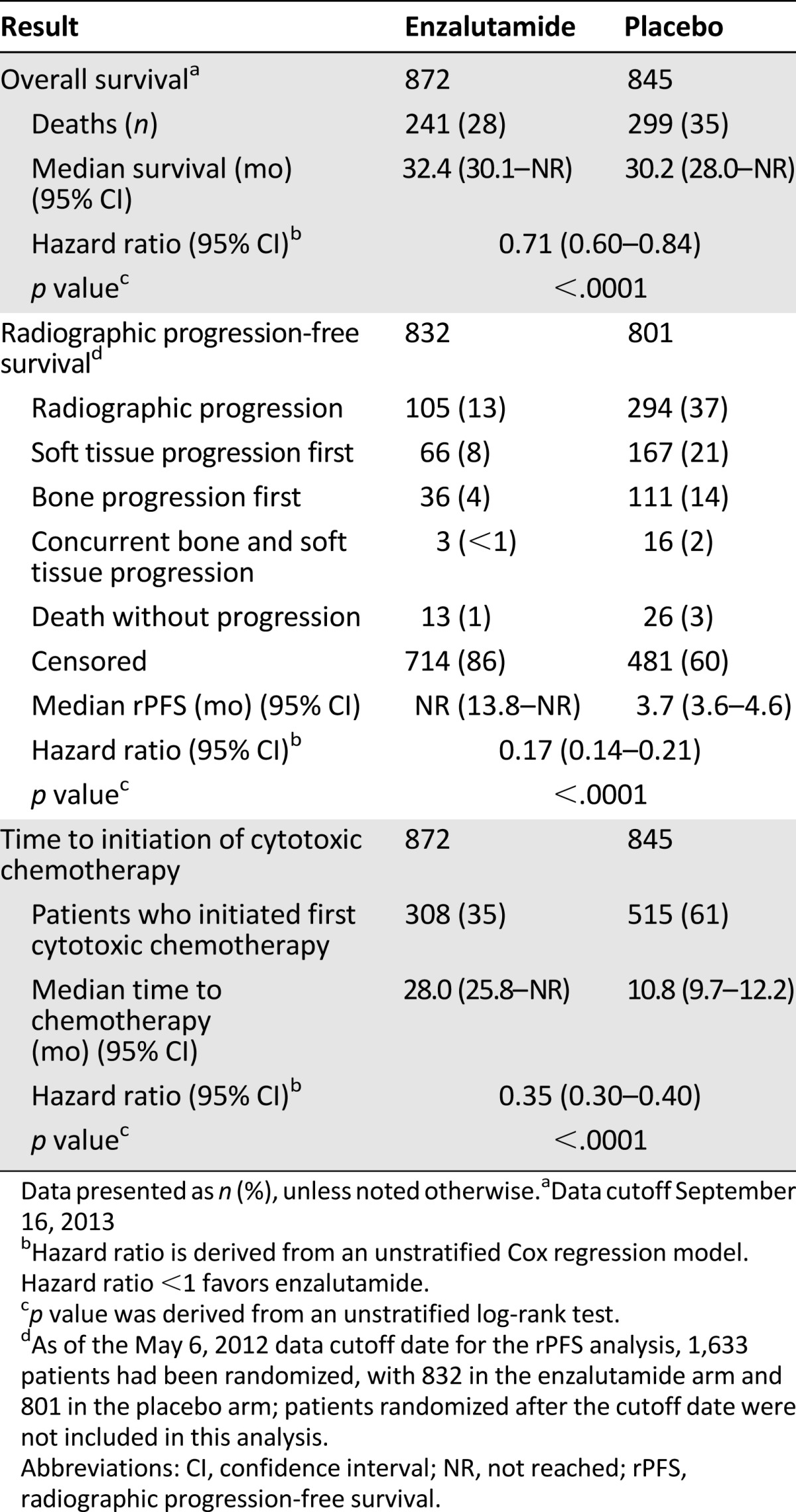
At the time of the interim OS analysis, the primary rPFS analysis was conducted with 438 rPFS events as determined by ICR. At that time, 1,633 patients had been randomized. Treatment with enzalutamide resulted in a statistically significant improvement in rPFS (HR, 0.17; 95% CI, 0.14–0.21; p < .0001; Table 3). The median rPFS was not reached (95% CI, 13.8–NR) in the enzalutamide arm and was 3.7 months (95% CI, 3.6–4.6) in the placebo arm. The results are consistent with the investigator-determined rPFS analysis, which showed an 11-month improvement in rPFS with enzalutamide compared with placebo (HR, 0.21; 95% CI, 0.17–0.26; p < .0001). In considering the reliability of this point estimate, it is important to note that only 14% of patients in the enzalutamide arm had developed disease progression at the time of the primary analysis of rPFS. In gathering experience with this endpoint, it is also important to note that most patients had first developed progression in areas of soft tissue rather than bone. This might have been because of the PCWG2 requirement for a confirmatory scan to determine bone progression.
The number of patients in whom the findings of the investigator and ICR review differed was smaller than that seen in many other FDA-reviewed trials but was not balanced between arms. In the present study, the ICR communicated blinded information on progression versus no progression to the site, and this could explain the low level of discordance compared with similar trials. The discordance rate of rPFS events and timing between the ICR and investigator assessments was 13% for the enzalutamide arm and 26% for the placebo arm. An examination of the data from the patients for whom a discordance was present found that progression was most often due to a difference in the interpretation of changes in soft tissue disease. The extent to which the PSA findings might have contributed to the interpretation of disease progression by the investigator within the soft tissue is unknown.
Analyses of key secondary endpoints supported the improvements in OS and rPFS with enzalutamide treatment. The most important secondary endpoint was the time to the initiation of cytotoxic chemotherapy after randomization. The time to the initiation of cytotoxic chemotherapy (Table 3) was significantly delayed with enzalutamide (HR, 0.35; 95% CI, 0.30–0.40; p < .0001), with a median of 28.0 months in the enzalutamide arm versus 10.8 months in the placebo arm. No significant difference was found between the treatment arms in the time to the initiation of cytotoxic chemotherapy from study treatment discontinuation, suggesting that the 17-month delay in the median time to the initiation of cytotoxic chemotherapy resulted from the prolonged use of enzalutamide before progression. In addition, antitumor activity was demonstrated for enzalutamide in the analysis of the soft tissue response by RECIST. Among the 45% of patients with measurable disease, 59% of the patients treated with enzalutamide and 5% of the patients who had received placebo had an investigator-determined soft tissue response. These results are consistent with those previously reported for enzalutamide [11, 12].
Toxicity
As of the January 15, 2014, data cutoff, patients in the enzalutamide arm had a mean treatment duration of 17.3 months compared with 7.2 months for patients in the placebo arm. Dose reduction and interruption were uncommon, occurring in 2% and 12% of patients in the enzalutamide arm, respectively.
The most common (≥5%) grade 1–4 adverse reactions included asthenia or fatigue, back pain, diarrhea, arthralgia, hot flush, peripheral edema, musculoskeletal pain, headache, upper respiratory infection, muscular weakness, dizziness, insomnia, lower respiratory infection, spinal cord compression and cauda equina syndrome, hematuria, paresthesia, anxiety, and hypertension. Grade 3–4 adverse reactions were reported in 46% of the patients treated with enzalutamide and 37% of the patients receiving placebo. Adverse reactions leading to study treatment discontinuation occurred at a similar rate (6%) between the arms.
Seizures occurred in 1 patient in each arm of the trial; however, both of these patients had a history of a seizure disorder. Seizures occurred in 0.9% of the patients in the enzalutamide arm of the AFFIRM (postdocetaxel) trial and in no patients in the placebo arm. In all the trials, patients experiencing a seizure were permanently discontinued from therapy. The present study allowed the use of concomitant medications known to lower the seizure threshold and included patients with a history of a cerebrovascular accident. The safety of enzalutamide in patients with a history of seizure or with a transient ischemic attack or loss of consciousness within 12 months of the initiation of enzalutamide is unknown and is being studied as a postmarketing requirement.
Hypertension (and related terms) was identified as an adverse drug reaction when enzalutamide was studied in the postdocetaxel setting. In the present study, the incidence of treatment-emergent hypertension was 13.9% in the enzalutamide arm and 4.7% in the placebo arm. Treatment-emergent hypertension with enzalutamide required dose modification or discontinuation in <1% of the patients. When adjusted for patient-years on study, hypertension remained increased in the enzalutamide arm. In addition to hypertension, the pooled safety analysis of the two placebo-controlled trials (before and after cytotoxic chemotherapy) showed that falls, including fall-related injuries, occurred in 9% of patients treated with enzalutamide compared with 4% of patients receiving placebo.
Discussion
The FDA’s review and analyses of the clinical data from the MDV3100-03 trial found that compared with placebo, enzalutamide significantly improved OS, rPFS, and time to initiation of cytotoxic chemotherapy in asymptomatic or minimally symptomatic patients with chemotherapy-naïve mCRPC. The OS benefit was supported by a large magnitude of improvement in rPFS, which translated into a significant delay in the use of cytotoxic chemotherapy. The improvement in both OS and rPFS in patients with chemotherapy-naïve mCRPC is consistent with that detected in patients who had received previous docetaxel in the AFFIRM trial, indicating that the treatment effect of enzalutamide is irrespective of previous docetaxel treatment [11, 12]. The safety profile is acceptable and is generally consistent between the two trials. Given the totality of evidence in the two trials, expansion of the enzalutamide indication was considered justified.
The improvement in both OS and rPFS in patients with chemotherapy-naïve mCRPC is consistent with that detected in patients who had received previous docetaxel in the AFFIRM trial, indicating that the treatment effect of enzalutamide is irrespective of previous docetaxel treatment
The estimate of median OS was not robust in the prespecified interim analysis of OS, because it contained 71% of the information required for the protocol-specified final analysis. To better determine the survival estimate for those patients, the final OS analysis will be provided as a postmarketing commitment.
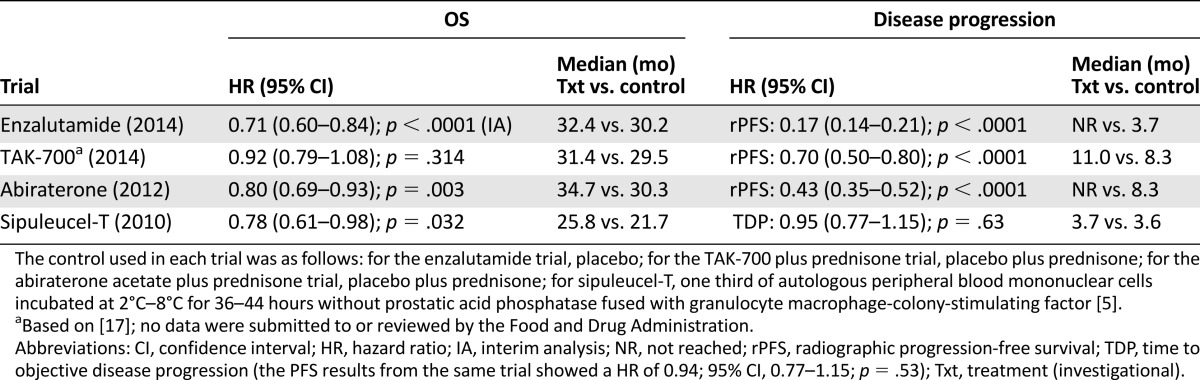
The relationship between OS and rPFS improvements has not been well characterized in patients with chemotherapy-naïve mCRPC. The current approval is the second application that used rPFS and OS as coprimary endpoints. The first application to use these endpoints in this disease setting was abiraterone acetate. For both applications, the approvals were not based on the improvement in rPFS alone, but rather on the totality of the data, including the data from trials of abiraterone and enzalutamide in the postdocetaxel setting. Available evidence (Table 4) suggests that for products targeting the androgen-androgen receptor pathway, a large magnitude of improvement in rPFS is associated with an improvement in OS. This is reflected in the enzalutamide and abiraterone trials, but not in the trial of TAK-700 plus prednisone versus placebo plus prednisone, in which a moderate, but statistically significant, prolongation in rPFS (difference in median of approximately 3 months at the final analysis) was reported, without a statistically significant improvement in OS [17]. TAK-700 is a CYP17 inhibitor that did not significantly improve OS compared with placebo in patients with mCRPC either before or after docetaxel-based therapy [17, 18]. In contrast to the three hormonal agents, the observed OS prolongation in the sipuleucel-T trial was not associated with a prolongation in the time to disease progression or progression-free survival [5, 6]. The possible mechanisms of this disassociation have been discussed in a number of scientific reports [19–21].
Table 4.
Key results from trials conducted in asymptomatic or minimally symptomatic patients with chemotherapy-naïve mCRPC
With the current expanded indication, practitioners might question whether it is best to administer enzalutamide before or after chemotherapy. In addition, it is not known whether patients can benefit from retreatment with enzalutamide after chemotherapy, in particular, patients who have responded well to enzalutamide before chemotherapy, and this might be worthy of investigation. It is important to note that approximately 20%–40% of patients with chemotherapy-naïve mCRPC receiving enzalutamide had either no response or the response was short-lived. The optimal treatment for these patients remains unknown and additional research to determine the mechanism(s) of resistance is needed. Recent evidence suggests that patients with circulating tumors cells that tested positive for an androgen-receptor isoform encoded by splice variant 7 (AR-V7) might be resistant to enzalutamide and abiraterone acetate [22]. If prospectively validated, this marker could help identify patients who might not respond to enzalutamide and could facilitate the design of clinical trials tailored to these patients. This could lead to a new framework for selection of the optimum treatment for mCRPC in the evolving, biomarker-driven oncology era.
It is important to note that approximately 20%–40% of patients with chemotherapy-naïve mCRPC receiving enzalutamide had either no response or the response was short-lived. The optimal treatment for these patients remains unknown and additional research to determine the mechanism(s) of resistance is needed.
Conclusion
The current approval expands the indication for enzalutamide, allowing health care providers and patients to use it for the treatment of patients with mCRPC either before or after cytotoxic chemotherapy. This is well supported by the totality of the data and the consistency of findings from two large randomized trials of enzalutamide in patients with mCRPC.
This article is available for continuing medical education credit at CME.TheOncologist.com.
Acknowledgment
We acknowledge Dr. Robert Justice’s constructive comments and edits.
Footnotes
For Further Reading: Axel S. Merseburger, Joaquim Bellmunt, Cheryl Jenkins et al. Perspectives on Treatment of Metastatic Castration-Resistant Prostate Cancer. The Oncologist 2013;18:558–567.
Implications for Practice: With so many novel agents available to treat patients with metastatic castration-resistant prostate cancer (mCRPC), a better understanding of factors to consider when assessing the clinical utility of treatment options for patients is needed. This review article discusses treatment strategies for mCRPC in the first- and second-line setting, and highlights the role of clinical markers, patient history, and assessing fitness for treatment when making treatment decisions. Prostate cancer is a heterogeneous disease, therefore, treatment must consider the characteristics of the disease as it manifests in an individual patient. In addition, assessments of patient response to treatment should reflect the mechanism of the drug. Further study is needed to identify predictive biomarkers to indicate patient response to novel agents.
Author Contributions
Conception/Design: Yang-Min Ning, Michael Brave, V. Ellen Maher, Lijun Zhang, Richard Pazdur
Collection and/or assembly of data: Yang-Min Ning, Michael Brave, V. Ellen Maher, Lijun Zhang, Shenghui Tang
Data analysis and interpretation: Yang-Min Ning, Michael Brave, V. Ellen Maher, Lijun Zhang, Shenghui Tang, Rajeshwari Sridhara, Geoffrey Kim, Amna Ibrahim, Richard Pazdur
Manuscript writing: Yang-Min Ning, Michael Brave, V. Ellen Maher, Lijun Zhang
Final approval of manuscript: Yang-Min Ning, Michael Brave, V. Ellen Maher, Lijun Zhang, Shenghui Tang, Rajeshwari Sridhara, Geoffrey Kim, Amna Ibrahim, Richard Pazdur
Disclosures
The authors indicated no financial relationships.
References
- 1.Siegel R, Ma J, Zou Z, et al. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Dagher R, Li N, Abraham S, et al. Approval summary: Docetaxel in combination with prednisone for the treatment of androgen-independent hormone-refractory prostate cancer. Clin Cancer Res. 2004;10:8147–8151. doi: 10.1158/1078-0432.CCR-04-1402. [DOI] [PubMed] [Google Scholar]
- 3.Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–1512. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 4.Hamberg P, Verhagen PC, de Wit R. When to start cytotoxic therapy in asymptomatic patients with hormone refractory prostate cancer? Eur J Cancer. 2008;44:1193–1197. doi: 10.1016/j.ejca.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 5.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 6.FDA Clinical and Statistical Reviews of Sipuleucel-T. Available at http://www.fda.gov/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/ucm213554.htm. Accessed March 6, 2015.
- 7.Medical FDA review of abiraterone acetate. Available at http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202379Orig1s000MedR.pdf. Accessed March 6, 2015.
- 8.Ryan CJ, Smith MR, de Bono JS, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–148. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kluetz PG, Ning YM, Maher VE, et al. Abiraterone acetate in combination with prednisone for the treatment of patients with metastatic castration-resistant prostate cancer: U.S. Food and Drug Administration drug approval summary. Clin Cancer Res. 2013;19:6650–6656. doi: 10.1158/1078-0432.CCR-13-2134. [DOI] [PubMed] [Google Scholar]
- 10.Ryan CJ, Smith MR, Fizazi K, et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): Final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015;16:152–160. doi: 10.1016/S1470-2045(14)71205-7. [DOI] [PubMed] [Google Scholar]
- 11.Ning YM, Pierce W, Maher VE, et al. Enzalutamide for treatment of patients with metastatic castration-resistant prostate cancer who have previously received docetaxel: U.S. Food and Drug Administration drug approval summary. Clin Cancer Res. 2013;19:6067–6073. doi: 10.1158/1078-0432.CCR-13-1763. [DOI] [PubMed] [Google Scholar]
- 12.Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 13.Medivation, Inc. A multinational phase 3, randomized, double-blind, placebo-controlled efficacy and safety study of oral MDV3100 in chemotherapy-naive patients with progressive metastatic prostate cancer who have failed androgen deprivation therapy. ClinicalTrials.gov Identifier: NCT01212991; first received on September 29, 2010.
- 14.Beer TM, Armstrong AJ, Sternberg CN, et al. Enzalutamide in men with chemotherapy-naive metastatic prostate cancer (mCRPC): Results of phase III PREVAIL study. J Clin Oncol. 2014;32(suppl 4; abstract LBA1) [Google Scholar]
- 15.Beer TM, Armstrong AJ, Rathkopf DE, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371:424–433. doi: 10.1056/NEJMoa1405095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scher HI, Halabi S, Tannock I, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: Recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008;26:1148–1159. doi: 10.1200/JCO.2007.12.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saad F, Fizazi K, Jinga V, et al. Orteronel plus prednisone in patients with chemotherapy-naive metastatic castration-resistant prostate cancer (ELM-PC 4): A double-blind, multicentre, phase 3, randomised, placebo-controlled trial. Lancet Oncol. 2015;16:338–348. doi: 10.1016/S1470-2045(15)70027-6. [DOI] [PubMed] [Google Scholar]
- 18.Fizazi K, Jones R, Oudard S, et al. Phase III, randomized, double-blind, multicenter trial comparing orteronel (TAK-700) plus prednisone with placebo plus prednisone in patients with metastatic castration-resistant prostate cancer that has progressed during or after docetaxel-based therapy: ELM-PC 5. J Clin Oncol. 2015;33:723–731. doi: 10.1200/JCO.2014.56.5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Longo DL. New therapies for castration-resistant prostate cancer. N Engl J Med. 2010;363:479–481. doi: 10.1056/NEJMe1006300. [DOI] [PubMed] [Google Scholar]
- 20.Huber ML, Haynes L, Parker C, et al. Interdisciplinary critique of sipuleucel-T as immunotherapy in castration-resistant prostate cancer. J Natl Cancer Inst. 2012;104:273–279. doi: 10.1093/jnci/djr514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madan RA, Gulley JL, Fojo T, et al. Therapeutic cancer vaccines in prostate cancer: The paradox of improved survival without changes in time to progression. The Oncologist. 2010;15:969–975. doi: 10.1634/theoncologist.2010-0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Antonarakis ES, Lu C, Wang H, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028–1038. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]