

Abstract
Multiple sclerosis (MS) is an autoimmune disease that represents a primary cause of neurological disability in the young adult population. Converging evidence support the importance of genetic determinants for MS etiology. However, with the exception of the major histocompatibility complex, their nature has been elusive for more than 20 years. In the last decade, the advent of large genome-wide association studies has significantly improved our understanding of the disease, leading to the golden era of MS genetic research. To date more than 110 genetic variants have been firmly associated to an increased risk of developing MS. A large part of these variants tag genes involved in the regulation of immune response and several of them are shared with other autoimmune diseases, suggesting a common etiological root for this class of disorders. Despite the impressive body of data obtained in the last years, we are still far from fully decoding MS genetic complexity. For example, we ignore how these genetic factors interact with each other and with the environment. Thus, the biggest challenge for the next era of MS research will consist in identifying and characterizing the molecular mechanisms and the cellular pathways in which these risk variants play a role.
Keywords: multiple sclerosis, GWAS, risk variant, complex disease
1. Introduction
Multiple sclerosis (MS) is an autoimmune disorder affecting the central nervous system (CNS). MS CNS pathology is characterized by well-demarked inflammation, breakdown of myelin sheaths (demyelination), microglia activation, proliferation of astrocytes with ensuing gliosis, and variable grades of axonal degeneration. Demyelinated lesions are disseminated within the CNS, involving both the white and gray matter, causing axonal and neuronal loss and consequently, a plethora of clinical deficits such as weakness in one or more limbs, sensory disturbances, optic neuritis, ataxia, bladder dysfunction, fatigue and cognitive deficits [1]. MS typically starts as an episodic, relapsing-remitting disease (RR-MS) with complete or partial recovery in between relapses. Over time, it evolves in many of the afflicted individuals into a secondary progressive phase (SP-MS) characterized by irreversible deterioration of both motor and cognitive functions. However, up to 15% of MS patients show a progressive course without relapses and remissions from the onset of clinical signs, which is defined as primary progressive MS (PP-MS) [2]. Ten FDA-approved treatments for MS are now available. However, none convincingly alters the long-term prognosis of the disease. Furthermore, these therapies have very diverse safety and toxicity profiles and no comparative data exist to guide how to select amongst the available options. No therapy exists for the progressive form of MS, the subtype most responsible for progression of severe disability.
The age of symptoms onset is typically between 20 and 40 years (slightly later in men than in women), but the disease can present at any time across the lifetime of the individual. Women are affected most frequently than men [3]. Worldwide, over 2.5 million people suffer from MS, making it the most common cause of acquired neurologic disability among young adults. The prevalence of MS varies with geography and ethnicity. Indeed, with some notable exceptions, MS is more frequent in high latitude regions and northern European populations [4, 5]. Remarkably, disease incidence has been increasing in the last century as seen for other autoimmune diseases [3]. The etiology of MS is still largely unknown but multiple lines of evidence suggest that the interaction between genetic and environmental factors underlies the risk of developing MS. In recent years, fueled by significant advances in high-throughput genotyping technologies, a considerable effort has been dedicated to the discovery of the genetic determinants of MS susceptibility. We will summarize the major findings of this endeavor and discuss immediate challenges in the field of MS genetics research.
2. MS is a genetic disease
Early evidence that MS holds a genetic component comes from the observation that the disease aggregates within families; first-degree relatives of MS patients are at greater risk for developing the disease compared to the general population [6]. The age-adjusted life-time risk of MS positively correlates with the degree of shared genetic identity, ranging from 0.2% in the general population to 2–4% in siblings and up to 30% in monozygotic twins of MS patients [7, 8]. Consistently, adoptees and spouses show risks comparable to the general population, corroborating the idea that shared genetic factors are the main cause of familial disease aggregation [9, 10]. However, the fact that even genetically identical individuals (monozygotic twins) are not always concordant for MS, strongly suggests that other risk factors exist. These include environmental factors such as smoking, Epstein-Barr virus (EBV) infection and sun exposure, and epigenetic determinants such as DNA methylation patterns, histone modifications and non-coding RNAs [11, 12].
Studies of MS prevalence in ethnic groups residing at the same latitudes support a genetic etiology in MS as well. For instance, the prevalence among non-white population groups in the United States appears to be lower compared to northern European descendants [13, 14]. Early estimates suggested that the disease is significantly less prevalent in African Americans than in European Americans (relative risk of 0.64) [15]. In contrast, contemporary incidence studies are challenging the long-held belief that African Americans are at a reduced risk for developing MS [16, 17]. Native Americans exhibit significantly lower incidence rates of MS in both the United States and Canada [18, 19]. In Europe, emblematic is the case of the genetically divergent Sardinian population that shows higher MS prevalence compared to other southern European groups [20]. The enrichment in specific genetic traits conferring higher risk for MS could explain the discrepancies in MS prevalence among ethnic groups living in the same geographical regions. Interestingly, compared to whites, African Americans are more likely to have a more severe disease course, which at least in part appears to be genetically determined [14, 21, 22].
The working model for MS heritability that best fits the available genetic and epidemiology data is the so-called common disease-common variant (CD-CV) hypothesis (Fig. 1). According to this model, MS risk is determined by cumulative effects of a large number of allelic variants. Each variant is relatively common within the population (minor allele frequency, MAF>1%) and contributes small portions of the risk [23]. Moreover, epistatic interactions among some of these risk variants are likely to take place as suggested by the non-linear correlation between familial risk and degree of relatedness [24]. Genetic heterogeneity (different variants might cause identical or similar forms of the disease in different subjects) represents an additional layer of complexity. This multifactorial, non-Mendelian pattern of heritability is not exclusive of MS but it is shared with other common disorders such as type II diabetes and obesity [25].
Figure 1. Working model for multiple sclerosis risk inheritance.
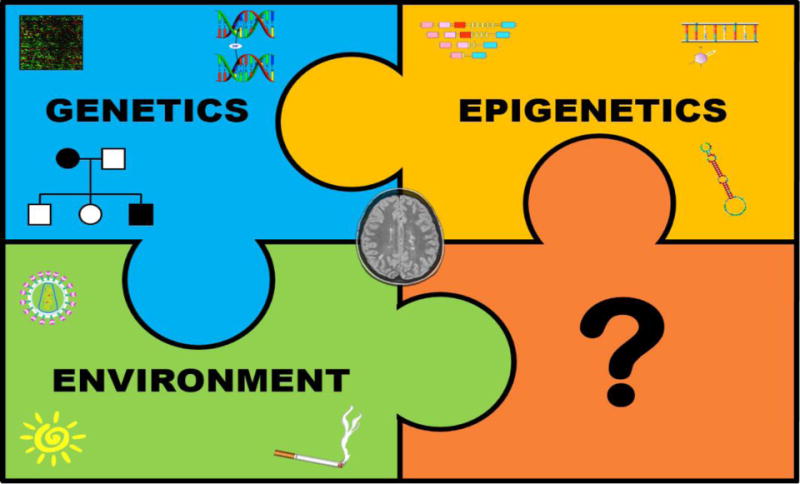
Multiple sclerosis is a complex genetic disorder in which multiple common allelic variants interact with non-genetic risk factors to determine disease susceptibility. Such non-genetic determinants can be either epigenetic or environmental. Several environmental risk factors for MS have been convincingly linked to MS risk; these include vitamin D deficiency, exposure to the Epstein Barr virus (EBV) after early childhood and manifestations of infectious mononucleosis, cigarette smoking and obesity.
2.1 MS genetic research before the advent of genome-wide association studies (GWAS)
The landscape of early discovery efforts for MS susceptibility factors was dominated by case-control studies of allelic variants in candidate genes. Candidate genes were defined as genes that are reasonable possibilities to play a role in a disease; for immune-mediated diseases, candidate genes might encode cytokines, immune-receptors, and proteins involved in pathogen clearance. With the notable exception of the human leukocyte antigen (HLA) gene cluster within the major histocompatibility complex (MHC) region, the direct testing by association of possibly relevant candidate genes selected based upon concepts of pathogenesis has been in general, unproductive.
2.2 Human leukocyte antigen (HLA) genes in MS
Following the discovery of the MHC in mice in 1936 [26], the human equivalent was subsequently mapped to the short arm of chromosome 6 (6p21.3) and extensively studied for both gene and allelic content variation. The first full sequence of this region was completed and reported in 1999 by the MHC-Sequencing Consortium [27]. Gene density was greater than expected; of the 224 identified loci, approximately 150 were predicted to be expressed and about 40% to have immunological functions. Among these, the highly polymorphic classical HLA class I (HLA-A, -B, -C) and class II (HLA-DPB1, -DQB1, -DRB1) gene clusters are now well characterized in terms of structure, diversity, and function. An extraordinary amount of data confirms their central role in the allogeneic response to tissue and hematological transplantation and risk for autoimmunity [28, 29].
The main genome-wide MS susceptibility signal maps to the HLA-DRB1 gene in the class II region of the MHC, and explains up to 10.5% of the genetic variance underlying risk. The HLA association with MS, which was first described several decades ago, is consistent with the idea that MS is -at its core- an antigen-specific autoimmune disease. HLA-DRB1*15:01 has the strongest effect with an average odds ratio (OR; a measure of association) of 3.08. HLA-DRB1*15:01 has been confirmed to have the strongest association with MS risk also in African Americans and northern Han Chinese population as well [30, 31]. HLA allelic heterogeneity is well documented in MS. A recent study pinpointed HLA-DRB1*04:05 as the primary risk variant in Japanese population while HLA-DRB1*15:01 is the top risk allele among Japanese individuals without HLA-DRB1*04:05 [32]. Moreover, additional alleles within the MHC locus have been found associated to MS in different populations. For instance, HLA-DRB1*03:01, HLA-DQB1*02:01 and HLA-DRB1*13:03 were confirmed to be secondary risk alleles for northern European populations [33] while in addition to HLA-DRB1*15:01, HLA-DRB1*15:03 and HLA-DRB1*04:05 confer susceptibility to MS in African Americans [30, 34]. Interestingly, some MHC alleles exhibit protective effects against MS risk. Class I alleles HLA-A*02 and HLA-B*44 are protective in northern Europeans with the former showing protective effects also in African Americans [33–35]. Class II HLA-DRB1*04:01 is also protective in both populations whereas HLA-DRB1*11:01 seems to exert protection in African Americans [34, 36]. HLA-DRB1*09:01 was instead reported as protective in both Japanese and Chinese populations [31, 37].
2.3 The GWAS era
The early association studies of candidate genes were followed by the application of the linkage strategy using multiple-case families. This classic analytical method relies on using genomic markers such as microsatellites or single nucleotide polymorphisms (SNPs) to track the co-segregation of a particular chromosomal segment with the trait of interest in families with more than one member affected in different generations. Such approach had been extremely successful in the mapping of monogenic diseases genes with high penetrance and Mendelian patterns of heritability. However, its application to a complex genetic disease like MS was confounded by the low penetrance of each risk variant and the rarity of families with affected relatives across multiple generations [38].
In the 1990s, a number of linkage studies have been conducted with different sample sizes and levels of resolution. In all of them, the only genomic region that reached the required statistical significance was the MHC class II locus. This finding was later replicated in a definitive linkage study employing 730 multi-case families that was conducted by the International Multiple Sclerosis Consortium (IMSGC) [39]. Although several genetic loci showed a suggestive linkage with the disease, the only locus achieving reliable statistical significance was again the MHC class II locus, specifically the HLA-DRB1*15:01 allele. However, the high number of genes and degree of polymorphism, and the locus-wide extensive linkage disequilibrium (a statistical association of variants due to physical proximity in the genome) hindered at the time any further characterization of MHC contribution to MS. Moreover, the fact that no other locus reached formal significance suggested that the effect size of other risk variants was substantially lower than the MHC contribution. Thus, it became clear that a much bigger sample size was needed in order to gain enough statistical power to detect other risk loci. Given the intrinsic difficulty of recruiting several thousands of families, a different analytical approach was proposed to overcome this problem, leading to the era of genome-wide association studies.
GWAS is a hypothesis-free approach that examines single nucleotide variation across the genome to identify DNA variants with allelic frequencies unevenly distributed between a group of unrelated individuals carrying a quantifiable trait and unrelated controls [40]. Compared to linkage analysis, GWAS can reach higher resolution and statistical power, given the possibility to collect large cohorts of unrelated subjects. Assuming no significant biases in the selection of cases and controls, these advantages make GWASs particularly suitable for the identification of small effect-size risk loci in multifactorial disorders [41].
Most importantly, GWAS studies were able to identify for the first time MS risk variants outside the MHC region (Table 1). In the first GWAS from the IMSGC employing 12,360 individuals of European ancestry, two variants in non-MHC loci –interleukin-2 receptor (IL2Rα) and interleukin-7 receptor (IL7Rα)– reached pre-established statistically significant levels of association [42]. This milestone achievement supported the CD-CV hypothesis as well as the importance of sample size. Indeed, the two novel variants along with the MHC locus were insufficient to explain most of MS heritability, suggesting the study was still underpowered. For this reason, a number of GWASs and meta-analyses were subsequently performed. Among them, the most informative effort was the second GWAS from IMSGC in collaboration with the Wellcome Trust Case Control Consortium 2 (WTCCC2) in which a total of 38,662 subjects of European descent were screened [33]. In this study 23 previously reported risk variants were replicated and additional 29 novel variants were identified as genome-wide significant. Remarkably, the majority of these SNPs was located nearby genes encoding for immune-related proteins and more than one third of them was previously associated to other autoimmune diseases [33]. Altogether, the data support an autoimmune etiology for MS and underline the possibility that common pathways may be dysregulated in different autoimmune diseases. The ideal follow-up to the 2011 GWAS was conducted two years later. By employing a custom genotyping array called ImmunoChip, 184 non-MHC loci with genome-wide significant association to at least one autoimmune disease were deeply interrogated in 80,095 subjects with European ancestry [43]. In this landmark study, 48 new MS susceptibility loci outside the MHC region were discovered. In addition, data from the UCSF experience indicates that quantitative traits derived from magnetic resonance imaging (MRI) of the brain, such as glutamate concentration, topologic localization of lesions, and degradation of cortical thickness are promising endophenotypic traits to be used in GWAS studies of MS [44–46].
Table 1.
Genome wide association studies and meta-analyses in multiple sclerosis.
| Study | Number of Cases | Number of Controls | Tested polymorphisms | Significant non-MHC loci (novel) |
|---|---|---|---|---|
| IMSGC (2007) [42] | 931 | 1862 | 334923 | 2 |
| WTCCC1 (2007) [84] | 975 | 1466 | 12374 | 0 |
| Comabella (2008) [85] | 242 | 242 | 528867 | 0 |
| Aulchenko (2008) [86] | 45 | 195 | 250000 | 1* |
| Baranzini (2009) [87] | 978 | 883 | 551642 | 0 |
| De Jager¶ (2009) [88] | 860 | 1720 | 709690 | 3* |
| ANZgene (2009) [89] | 1618 | 3413 | 302098 | 2 |
| Sanna (2010) [90] | 882 | 872 | 555335 | 1* |
| Nischwitz (2010) [91] | 590 | 825 | 300000 | 3 |
| Jakkula (2010) [92] | 68 | 136 | 297343 | 1* |
| IMSGC and WTCCC2 (2011) [33] | 9772 | 17376 | 475806 | 29* |
| Patsopoulos¶ (2011) [93] | 1453 | 2176 | 906600 | 3* |
| Matesanz¶ (2012) [94] | 296 | 801 | 130903 | 0 |
| Martinelli-Boneschi (2012) [95] | 197 | 234 | 277866 | 0 |
Case and control numbers refer to discovery datasets.
Studies that applied the conventional genome-wide significant threshold (P< 5×10−8); for the others, an arbitrary threshold (P≤10−7) was used to identify the significant loci.
Studies based on meta-analysis.
To date, 110 non-MHC risk variants in 103 loci have been sequentially identified (Fig. 2). Surprisingly, they still explain only 27% of the predicted MS heritability [43]. What is left –the so-called “missing heritability”– is most likely due to still unknown common variants whose effect size is so small that falls below the threshold of the current detecting power. Additionally, a consistent part of it might come from interactions between known risk variants [47]. Alternatively, it has been proposed that rare variants (MAF<0.5%) exerting much larger effects than common ones might possibly account for most of MS missing heritability. However, experimental evidence has so far failed to confirm the existence of such rare high-risk variants [48]. The recent advent of more accurate and affordable whole-genome sequencing technologies will allow testing this hypothesis more systematically.
Figure 2. 2014 genetic atlas of multiple sclerosis.
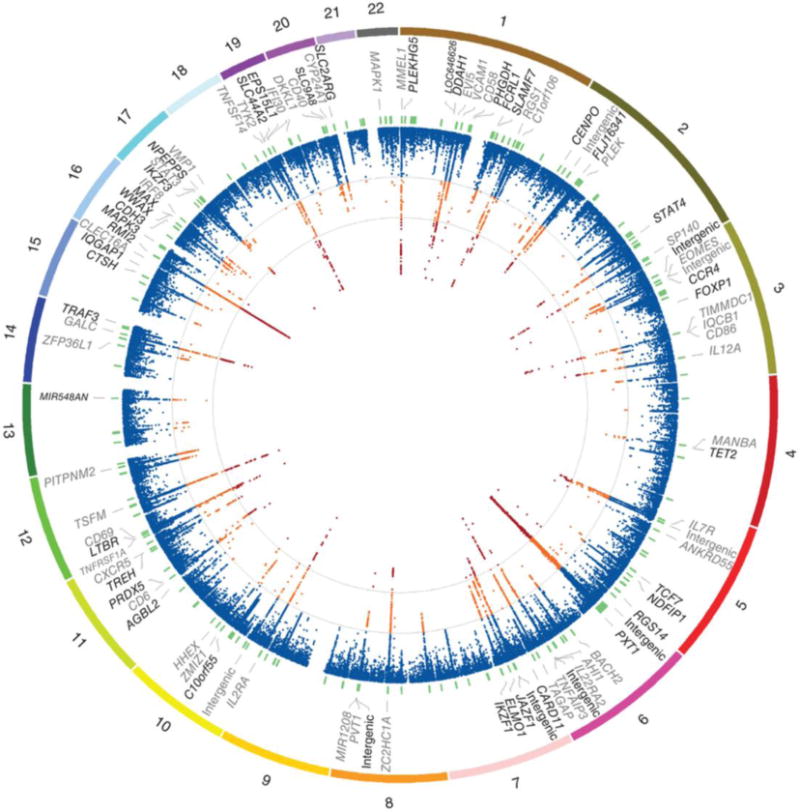
The outer most track of the circus plot indicates the numbered autosomal chromosomes. The second track shows the gene closest to the most associated SNP. Previously identified associations are indicated in grey. The third track indicates the physical position of the 184 fine-mapping intervals (green). The inner most track indicates −log(p) for each SNP (scaled from 0–12 which truncates the signal in several regions). Additionally, contour lines are given at the a priori discovery (−log(p) = 4) and genome-wide significance (−log(p) = 7.3) thresholds. Orange indicates −log(p) ≥ 4 and < 7.3, while red indicates −log(p) ≥ 7.3. Reproduced from [43].
2.4 The post-genomic era: from variants to functions
Although the GWAS approach has been essential to decipher the genetic basis of MS, we are still far from fully understanding the geno-pathological processes underlying the disease. Indeed, it should be pointed out that the GWAS risk variants may just be proxy SNPs for the causal variants due to the presence of extended blocks of linkage disequilibrium across the genome. Moreover, GWAS outcomes suggest that the vast majority of risk variants targets regulatory DNA elements which may be located at great distance from the genes they control, further muddling the functional analysis of genomic data [49]. Therefore, additional effort is required to correctly interpret GWAS results.
So far, only a limited number of bona fide causative variants and related pathogenic mechanisms have been identified with a high degree of confidence (Table 2). For instance, the MS-associated SNP rs6897932, located within the alternatively spliced exon 6 of IL7Rα, was found to alter the ratio between the soluble and membrane-bound isoforms of the protein by disrupting an exonic splicing enhancer [50]. Similarly, the risk variant rs1800693 in the tumor necrosis factor 1A (TNFRSF1A) gene was shown to drive the expression of a novel soluble form of the receptor that can inhibit TNF signaling, mimicking the effects of TNF-blocking drugs that are known to exacerbate MS pathology [51]. In the case of the exonic risk variant rs34536433, the relative amino acidic substitution in the tyrosine kinase 2 (TYK2) protein results in enhanced activation of its kinase function. High TYK2 activity promotes lymphoblasts to differentiate into T-helper 1 (Th1) cells, which are critical in mediating the autoimmune response in MS [52]. More recently, the risk variant rs12487066 has been mechanistically associated to decreased levels of Casitas B-lineage lymphoma proto-oncogene b (CBLB) in CD4+ T cells due to increased binding of the transcription factor C/EBPβ [53].
Table 2.
Candidate causal variants and putative mechanisms.
| Gene | Variant | Putative Mechanism | Reference |
|---|---|---|---|
| IL7R | rs6897932 (exon 6) | Low level skipping of exon 6, changes in the soluble/membrane bound ratio with higher sIL7R | [50] |
| IL2RA | rs2104286 (intronic) | Changes in the soluble/membrane bound ratio with higher sIL2RA | [96] |
| TNFRSF1A | rs1800693 (intronic) | Skipping of exon 6, changes in the soluble/membrane bound ratio with higher sTNFR1 | [51] |
| CD58 | rs2300747 (intronic) | Higher membrane expression of CD58 and correction of CD4+ regulatory cell function | [97] |
| IRF8 | 17445836 (intronic) | Widespread effect on the type I interferon transcriptional responses | [88] |
| TYK2 | rs34536443 (exon 21) | Decrease TYK2 kinase activity and cytokine shifting towards Th2 | [52] |
| CD6 | rs1782933 (intronic) | Decreased expression of full length CD6 in CD4+ cells leading to altered proliferation | [98] |
| EVI5 | rs11810217 (intronic) | Regulation of adjacent gene GFl1 | [99] |
| CYP27B1 | rs12368653 (intergenic) | Under-expression in tolerogenic dendritic cells (DC2) | [100] |
| PRKCA | GGTG ins/del (intergenic) | Inclusion/exclusion of exon 3. Risk associated with lower PRKCA expression | [101] |
| CBLB | rs12487066 (intergenic) | Diminished CBL-B expression and altered Type I IFN function in CD4+ cells | [53] |
The post-genomic era of MS is still in its infancy. To discover true causal variants, GWAS data need to be refined with fine-mapping approaches employing comprehensive batteries of markers saturating the confirmed regions of association. Studies in different population could be also useful to narrow down the initial GWAS regions due to different patterns of linkage disequilibrium between ethnic groups. For instance, African Americans are known to possess shorter haplotype blocks due to their longer ancestral population history [54]. This peculiarity has been successfully employed to replicate and refine asthma-and body mass index (BMI)-associated loci previously identified in European ancestry populations [55, 56] and it is also proven to be informative in MS [34].
3. The lesson from animal models
In nature MS affects only humans, thus no spontaneous animal model exists to study this disorder. However, a pathological phenotype that recapitulates many features of MS –named experimental autoimmune encephalomyelitis (EAE)– can be artificially induced in rodents by immunization with myelin proteins as well as by adoptive transfer of CD4+ activated T cells that are specific for myelin components [57]. Although not perfect, EAE has been providing a robust platform in which both genetic and environmental risk factors can be studied. Moreover, the possibility to manipulate the mouse genome by knocking-out specific genes or inserting DNA sequences from MS patients has made the EAE paradigm even more versatile in modelling disease risk [58].
Similar to MS, EAE shows a genetically complex phenotype in which multiple loci influence susceptibility. Among them, the MHC locus shows the biggest contribution to full manifestations of the disease [59]. By employing congenic mouse lines with different EAE susceptibilities, a recent study has identified for the first time several non-MHC loci relevant to EAE that overlap with known MS risk loci. These common loci belong to evolutionary conserved pathways that control TH-cell differentiation, underling the critical role of CD4+ T cells in both MS and EAE pathogenic processes [60]. Moreover, this finding corroborates the relevance of EAE as model for human MS. To this regard, it should be mentioned that an association between IL7R and disease was highlighted in the first mouse EAE genome scan several years before human studies revealed its role in MS [61].
The possibility to express in mice single human MHC alleles has allowed a more precise dissection of their contribution to MS risk, overcoming the confounding effects of linkage disequilibrium. For example, mice bearing the HLA-DRß1*1501 and human T-cell receptor (TCR) specific for the myelin basic protein (MBP) peptide 85–99, spontaneously develop EAE [62]. This result suggested that HLA-DRB1 *15:01 alone accounts for most of the risk within the extended haplotype DRB1 *15:01-DQA1 *01:02-DQB1 *06:02. On the contrary, a recent study employing the myelin proteolipid protein (PLP) peptides 139–151 and 175–194 has argued that DQB1*06:02 is the main allele associated to MS risk [63]. Furthermore, humanized mice co-expressing HLA-DRB1*15:01 and HLA-DRB5*01:01 experimentally demonstated that epistatic interactions can occur between alleles, whereas the latter allele was shown to reduce the severity of the disease by inducing cell death of encephalitogenic T cells [62]. Transgenic models have been also important to confirm the involvement of some MHC class I alleles in MS pathogenesis. For example, humanized mice carrying the HLA-A*03:01 allele and a TCR molecule from a MS patient recognizing the PLP peptide 36–44 were found to develop after immunization a mild form of MS-like disease, characterized by motor deficit and optic neuritis [64].
Finally, EAE studies are also useful to functionally validate candidate MS-associated genes. For instance, mice lacking the Stat3 gene in CD4+ T cells were shown to be completely resistant to EAE induction, highlighting a role for this candidate GWAS gene in generating pathogenic TH17 T cell responses [65]. Similar results were obtained with knock-out mice for other candidate genes such as p55, basic leucine zipper transcription factor ATF-like (Batf) and Il7rα [66–68].
4. Genetic determinants of disease progression
Clinical heterogeneity is a well-recognized property of MS, which can range from a relatively mild illness to a rapidly advancing, devastating condition. Currently, our understanding of the molecular events leading to neurodegeneration and disability in progressive MS is tenuous, and unbiased genetic approaches have enormous potential to elucidate disease modifier molecules and pathways. Familial clustering of phenotypic features of MS, including age of onset and disease course, has long been known, and supports a role for genetic factors affecting the disease phenotype [69–72]; similar associations are also present in animal models [73–76]. The identification of any genetic modifiers of disease expression is likely to have profound translational implications for the understanding and management of MS, and in particular for progressive MS, the most disabling and currently untreatable form of the disease.
In a series totaling 532 study participants, the putative protective HLA B*44 allele (but not HLA-A*02) shows a mild association with a better radiological outcome both in terms of brain parenchymal fraction and T2 hyperintense lesion volume (P=0.03 for each outcome) [77]. In another study employing 250 MS patients, a dose–dependent correlation between carriage of HLA-DRB1*15:01 and the presence of diffuse cord lesions was found [78]. Moreover, the minor allele HLA-DRB1 *07:01 was significantly associated with numbers of wedge-shaped lesions and lesions in the cervical cord, while HLA-DRB1 *11:04 and DRB1 *01:03 were associated with higher and lower numbers of thoracic cord lesions, respectively [78]. HLA-DRB1*15:01 also correlates with disease severity, as measured by spectroscopic metabolic differences [79]. Additionally, the carriage of HLA-DRB1*15:01 is associated with a lower age of onset as confirmed by studies in different cohorts [80, 81].
Attempts have been also carried out to link the genetic load of risk variants in MS patients with disease clinical parameters. Higher MS genetic burden (MSGB) scores were demonstrated to correlate with the presence of oligoclonal bands in the cerebrospinal fluid and with an early age at symptom onset [82]. On the contrary, MSGB assessment has shown limited discriminatory ability in predicting susceptibility.
5. Future directions and conclusive remarks
Since their introduction in the last decade, GWAS studies have radically changed the field of MS genetics. More than 100 genetic variants are currently associated to MS susceptibility and in the next few years others will be added to the list. Despite the significant progress, the knowledge of MS genetics remains incomplete. The major challenge in bridging the gap from genetic association to functional understanding and eventual translational application consists in fitting the increasing number of available data into a coherent biological frame. Given the complex nature of the disease, only a multidisciplinary approach will be able to gain insight into MS pathological mechanisms. To serve this scope, it will be necessary to develop novel bioinformatic tools and statistical methods with the capability to integrate large-scale datasets from omics-based experiments, exposures, and clinical outcomes.
A rational plan to decipher MS complexity would include as first step the systematic fine-mapping of all MS risk loci identified in GWAS studies by means of SNP-saturating custom arrays or direct sequencing. The results should be then replicated and further refined across different ancestral groups. Subsequently, the most promising candidate causative variants need to be validated by robust and meaningful functional assays. Furthermore, epistatic interactions between variants and the interplay of the genome with the environment should be systematically explored as well. For this purpose, genotype by genotype (G × G) and genotype by environment (G × E) interaction profiles represent the most promising approach. Finally, the hypothesis that high-impact rare variants represent a huge part of the MS missing heritability could be carefully addressed by employing well-designed whole-genome sequencing screenings in collections of multi-case families.
In parallel, better in vivo models need to be developed as well. Given the confines of modelling MS complexity in vitro, so far EAE represents the only available system to study the effects of risk variants in a complex biological environment. In this light, the creation of transgenic mice bearing human genes has helped to mimic MS more closely. However, strict limitations exist regarding the size of the insert that can be integrated into the mouse genome with traditional methods. Thus, the in vivo study of longdistance interactions within large portions of the human genome can be problematic. For this reason, the next generation of MS mouse models should take advantage of alternative technologies such as bacterial artificial chromosome (BAC) recombineering, that allow the integration of larger human sequences (100–300 kb) into the recipient genome [83]. These models will be particularly helpful in elucidating the role of risk variants mapping to intergenic regions, usually associated to regulatory elements.
In conclusion, a holistic approach across different disciplines represents the best way to tackle MS. Such coordinated effort will translate into shorter times to move from the bench to the bedside and into more effective therapies for MS patients.
Research highlights.
Multiple sclerosis (MS) is a complex autoimmune disorder with a genetic component
The major histocompatibility complex (MHC) is the most prominent genetic determinant for MS
Genome-wide association studies have substantially improved our knowledge of MS genetics
To date, 110 risk variants have been identified outside the MHC locus
Abbreviations
- MS
multiple sclerosis
- GWAS
genome wide association study
- RR-MS
relapsing-remitting MS
- SP-MS
secondary progressive MS
- PP-MS
primary progressive MS
- CD-CV
common disease-common variant
- MAF
minor allele frequency
- MHC
major histocompatibility complex
- HLA
human leukocyte antigen
- OR
odds ratio
- SNPs
single nucleotide polymorphisms
- IMSGC
International Multiple Sclerosis Consortium
- IL2Rα
interleukin-2 receptor
- IL7Rα
interleukin-7 receptor
- WTCCC2
Wellcome Trust Case Control Consortium 2
- TNFRSF1A
tumor necrosis factor 1A
- TYK2
tyrosine kinase 2
- Th1
T-helper 1
- CBLB
Casitas B-lineage lymphoma proto-oncogene b
- BMI
body mass index
- EAE
experimental autoimmune encephalomyelitis
- TCR
T-cell receptor
- MBP
myelin basic protein
- PLP
proteolipid protein
- Batf
basic leucine zipper transcription factor ATF-like
- MSGB
MS genetic burden
- G × G
genotype by genotype
- G × E
genotype by environment
- BAC
bacterial artificial chromosome
- EBV
Epstein-Barr virus
- MRI
magnetic resonance imaging
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. Conflict of interest: None.
References
- 1.Hauser SL, Goodin DS. Harrison’s Principle of Internal Medicine. 18. McGraw-Hill; 2012. Multiple sclerosis and other demyelinating diseases. [Google Scholar]
- 2.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 3.Koch-Henriksen N, Sorensen PS. The changing demographic pattern of multiple sclerosis epidemiology. Lancet Neurol. 2010;9:520–532. doi: 10.1016/S1474-4422(10)70064-8. [DOI] [PubMed] [Google Scholar]
- 4.Simpson S, Jr, Blizzard L, Otahal P, Van der Mei I, Taylor B. Latitude is significantly associated with the prevalence of multiple sclerosis: a meta-analysis. J Neurol Neurosurg Psychiatry. 2011;82:1132–1141. doi: 10.1136/jnnp.2011.240432. [DOI] [PubMed] [Google Scholar]
- 5.Pugliatti M, Sotgiu S, Rosati G. The worldwide prevalence of multiple sclerosis. Clin Neurol Neurosurg. 2002;104:182–191. doi: 10.1016/s0303-8467(02)00036-7. [DOI] [PubMed] [Google Scholar]
- 6.Sadovnick AD, Baird PA. The familial nature of multiple sclerosis: age-corrected empiric recurrence risks for children and siblings of patients. Neurol. 1988;38:990–991. doi: 10.1212/wnl.38.6.990. [DOI] [PubMed] [Google Scholar]
- 7.Robertson NP, Fraser M, Deans J, Clayton D, Walker N, Compston DA. Age-adjusted recurrence risks for relatives of patients with multiple sclerosis. Brain. 1996;119:449–455. doi: 10.1093/brain/119.2.449. [DOI] [PubMed] [Google Scholar]
- 8.Willer CJ, Dyment DA, Risch NJ, Sadovnick AD, Ebers GC, Canadian Collaborative Study Group Twin concordance and sibling recurrence rates in multiple sclerosis. Proc Natl Acad Sci U S A. 2003;100:12877–12882. doi: 10.1073/pnas.1932604100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ebers GC, Sadovnick AD, Risch NJ. A genetic basis for familial aggregation in multiple sclerosis. Canadian Collaborative Study Group. Nature. 1995;377:150–151. doi: 10.1038/377150a0. [DOI] [PubMed] [Google Scholar]
- 10.Ebers GC, Yee IM, Sadovnick AD, Duquette P. Conjugal multiple sclerosis: population-based prevalence and recurrence risks in offspring. Canadian Collaborative Study Group. Ann Neurol. 2000;48:927–931. [PubMed] [Google Scholar]
- 11.Simon KC, Munger KL, Ascherio A. XVI European Charcot Foundation lecture: nutrition and environment: can MS be prevented? J Neurol Sci. 2011;311:1–8. doi: 10.1016/j.jns.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huynh JL, Casaccia P. Epigenetic mechanisms in multiple sclerosis: implications for pathogenesis and treatment. Lancet Neurol. 2013;12:195–206. doi: 10.1016/S1474-4422(12)70309-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Detels R, Visscher BR, Malmgren RM, Coulson AH, Lucia MV, Dudley JP. Evidence for lower susceptibility to multiple sclerosis in Japanese-Americans. Am J Epidemiol. 1977;105:303–310. doi: 10.1093/oxfordjournals.aje.a112387. [DOI] [PubMed] [Google Scholar]
- 14.Cree BA, Khan O, Bourdette D, et al. Clinical characteristics of African Americans vs Caucasian Americans with multiple sclerosis. Neurol. 2004;63:2039–2045. doi: 10.1212/01.wnl.0000145762.60562.5d. [DOI] [PubMed] [Google Scholar]
- 15.Wallin MT, Page WF, Kurtzke JF. Multiple sclerosis in US veterans of the Vietnam era and later military service: race, sex, and geography. Ann Neurol. 2004;55:65–71. doi: 10.1002/ana.10788. [DOI] [PubMed] [Google Scholar]
- 16.Wallin MT, Culpepper WJ, Coffman P, et al. The Gulf War era multiple sclerosis cohort: age and incidence rates by race, sex and service. Brain. 2012;135:1778–1785. doi: 10.1093/brain/aws099. [DOI] [PubMed] [Google Scholar]
- 17.Langer-Gould A, Brara SM, Beaber BE, Zhang JL. Incidence of multiple sclerosis in multiple racial and ethnic groups. Neurol. 2013;80:1734–1739. doi: 10.1212/WNL.0b013e3182918cc2. [DOI] [PubMed] [Google Scholar]
- 18.Hader WJ. Prevalence of multiple sclerosis in Saskatoon. Can Med Assoc J. 1982;127:295–297. [PMC free article] [PubMed] [Google Scholar]
- 19.Svenson LW, Woodhead SE, Platt GH. Regional variations in the prevalence rates of multiple sclerosis in the province of Alberta, Canada. Neuroepidemiol. 1994;13:8–13. doi: 10.1159/000110352. [DOI] [PubMed] [Google Scholar]
- 20.Cocco E, Sardu C, Massa R, et al. Epidemiology of multiple sclerosis in south-western Sardinia. Mult Scler. 2011;17:1282–1289. doi: 10.1177/1352458511408754. [DOI] [PubMed] [Google Scholar]
- 21.Cree BA, Reich DE, Khan O, et al. Modification of Multiple Sclerosis Phenotypes by African Ancestry at HLA. Arch Neurol. 2009;66:226–233. doi: 10.1001/archneurol.2008.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boster AL, Endress CF, Hreha SA, Caon C, Perumal JS, Khan OA. Pediatric-onset multiple sclerosis in African-American black and European-origin white patients. Pediatr Neurol. 2009;40:31–33. doi: 10.1016/j.pediatrneurol.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 23.Chakravarti A. Population genetics–making sense out of sequence. Nat Genet. 1999;21:56–60. doi: 10.1038/4482. [DOI] [PubMed] [Google Scholar]
- 24.O’Gorman C, Lin R, Stankovich J, Broadley SA. Modelling genetic susceptibility to multiple sclerosis with family data. Neuroepidemiol. 2013;40:1–12. doi: 10.1159/000341902. [DOI] [PubMed] [Google Scholar]
- 25.Barrett JC, Clayton DG, Concannon P, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gorer PA. The detection of a hereditary antigenic difference in the blood of mice by means of human group A serum. J Genet. 1936;32:17–31. [Google Scholar]
- 27.The MHC sequencing consortium. Complete sequence and gene map of a human major histocompatibility complex. Nature. 1999;401:921–923. doi: 10.1038/44853. [DOI] [PubMed] [Google Scholar]
- 28.Petersdorf EW. The major histocompatibility complex: a model for understanding graft-versus-host disease. Blood. 2013;122:1863–1872. doi: 10.1182/blood-2013-05-355982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsai S, Santamaria P. MHC Class II Polymorphisms, autoreactive T-Cells, and autoimmunity. Front Immunol. 2013;4:321. doi: 10.3389/fimmu.2013.00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oksenberg JR, Barcellos LF, Cree BA, et al. Mapping multiple sclerosis susceptibility to the HLA-DR locus in African Americans. Am J Hum Genet. 2004;74:160–167. doi: 10.1086/380997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qiu W, James I, Carroll WM, Mastaglia FL, Kermode AG. HLA-DR allele polymorphism and multiple sclerosis in Chinese populations: a meta-analysis. Mult Scler. 2011;17:382–388. doi: 10.1177/1352458510391345. [DOI] [PubMed] [Google Scholar]
- 32.McElroy JP, Isobe N, Gourraud PA, et al. SNP-based analysis of the HLA locus in Japanese multiple sclerosis patients. Genes Immun. 2011;12:523–530. doi: 10.1038/gene.2011.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Isobe N, Gourraud PA, Harbo HF, et al. Genetic risk variants in African Americans with multiple sclerosis. Neurology. 2013;81:219–227. doi: 10.1212/WNL.0b013e31829bfe2f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.International MHC Autoimmunity Genetics Network. Mapping of multiple susceptibility variants within the MHC region for 7 immune-mediated diseases. Proc Natl Acad Sci U S A. 2009;106:18680–18685. doi: 10.1073/pnas.0909307106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patsopoulos NA, Barcellos LF, Hintzen RQ, et al. Fine-mapping the genetic association of the major histocompatibility complex in multiple sclerosis: HLA and non-HLA effects. PLoS Genet. 2013;9:e1003926. doi: 10.1371/journal.pgen.1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoshimura S, Isobe N, Yonekawa T, et al. Genetic and infectious profiles of Japanese multiple sclerosis patients. PLoS One. 2012;7:e48592. doi: 10.1371/journal.pone.0048592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Willer CJ, Dyment DA, Cherny S, et al. A genome-wide scan in forty large pedigrees with multiple sclerosis. J Hum Genet. 2007;52:955–962. doi: 10.1007/s10038-007-0194-6. [DOI] [PubMed] [Google Scholar]
- 39.Sawcer S, Ban M, Maranian M, et al. A high-density screen for linkage in multiple sclerosis. Am J Hum Genet. 2005;77:454–467. doi: 10.1086/444547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hardy J, Singleton A. Genomewide association studies and human disease. N Engl J Med. 2009;360:1759–1768. doi: 10.1056/NEJMra0808700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 42.International Multiple Sclerosis Genetics Consortium. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357:851–862. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 43.International Multiple Sclerosis Genetics Consortium. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. 2013;45:1353–1360. doi: 10.1038/ng.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baranzini SE, Srinivasan R, Khankhanian P, et al. Genetic variation influences glutamate concentrations in brains of patients with multiple sclerosis. Brain. 2010;133:2603–2611. doi: 10.1093/brain/awq192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gourraud PA, Sdika M, Khankhanian P, et al. A genome-wide association study of brain lesion distribution in multiple sclerosis. Brain. 2013;136:1012–1024. doi: 10.1093/brain/aws363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Azevedo CJ, Kornak J, Chu P, et al. In vivo evidence of glutamate toxicity in multiple sclerosis. Ann Neurol. 2014;76:269–278. doi: 10.1002/ana.24202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci U S A. 2012;109:1193–1198. doi: 10.1073/pnas.1119675109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hunt KA, Mistry V, Bockett NA, et al. Negligible impact of rare autoimmune-locus coding-region variants on missing heritability. Nature. 2013;498:232–235. doi: 10.1038/nature12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maurano MT, Humbert R, Rynes E, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gregory SG, Schmidt S, Seth P, et al. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat Genet. 2007;39:1083–1091. doi: 10.1038/ng2103. [DOI] [PubMed] [Google Scholar]
- 51.Gregory AP, Dendrou CA, Attfield KE, et al. TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature. 2012;488:508–511. doi: 10.1038/nature11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Couturier N, Bucciarelli F, Nurtdinov RN, et al. Tyrosine kinase 2 variant influences T lymphocyte polarization and multiple sclerosis susceptibility. Brain. 2011;134:693–703. doi: 10.1093/brain/awr010. [DOI] [PubMed] [Google Scholar]
- 53.Sturner KH, Borgmeyer U, Schulze C, Pless O, Martin R. A multiple sclerosis-associated variant of CBLB links genetic risk with type I IFN function. J immunol. 2014;193:4439–4447. doi: 10.4049/jimmunol.1303077. [DOI] [PubMed] [Google Scholar]
- 54.Tishkoff SA, Kidd KK. Implications of biogeography of human populations for ‘race’ and medicine. Nat Genet. 2004;36:S21–27. doi: 10.1038/ng1438. [DOI] [PubMed] [Google Scholar]
- 55.Kantor DB, Palmer CD, Young TR, et al. Replication and fine mapping of asthma-associated loci in individuals of African ancestry. Hum Genet. 2013;132:1039–1047. doi: 10.1007/s00439-013-1310-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gong J, Schumacher F, Lim U, et al. Fine Mapping and Identification of BMI Loci in African Americans. Am J Hum Genet. 2013;93:661–671. doi: 10.1016/j.ajhg.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller SD, Karpus WJ, Davidson TS. Experimental autoimmune encephalomyelitis in the mouse. Curr Protoc Immunol. 2010:11. doi: 10.1002/0471142735.im1501s88. Chapter 15: Unit 15. [DOI] [PubMed] [Google Scholar]
- 58.Attfield KE, Dendrou CA, Fugger L. Bridging the gap from genetic association to functional understanding: the next generation of mouse models of multiple sclerosis. Immunol Rev. 2012;248:10–22. doi: 10.1111/j.1600-065X.2012.01132.x. [DOI] [PubMed] [Google Scholar]
- 59.Weissert R, Wallstrom E, Storch MK, et al. MHC haplotype-dependent regulation of MOG-induced EAE in rats. J Clin Invest. 1998;102:1265–1273. doi: 10.1172/JCI3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Blankenhorn EP, Butterfield R, Case LK, et al. Genetics of experimental allergic encephalomyelitis supports the role of T helper cells in multiple sclerosis pathogenesis. Ann Neurol. 2011;70:887–896. doi: 10.1002/ana.22642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sundvall M, Jirholt J, Yang HT, et al. Identification of murine loci associated with susceptibility to chronic experimental autoimmune encephalomyelitis. Nat Genet. 1995;10:313–317. doi: 10.1038/ng0795-313. [DOI] [PubMed] [Google Scholar]
- 62.Gregersen JW, Kranc KR, Ke X, et al. Functional epistasis on a common MHC haplotype associated with multiple sclerosis. Nature. 2006;443:574–577. doi: 10.1038/nature05133. [DOI] [PubMed] [Google Scholar]
- 63.Kaushansky N, Altmann DM, David CS, Lassmann H, Ben-Nun A. DQB1*0602 rather than DRB1*1501 confers susceptibility to multiple sclerosis-like disease induced by proteolipid protein (PLP) J Neuroinflammation. 2012;9:29. doi: 10.1186/1742-2094-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Friese MA, Jakobsen KB, Friis L, et al. Opposing effects of HLA class I molecules in tuning autoreactive CD8+ T cells in multiple sclerosis. Nat Med. 2008;14:1227–1235. doi: 10.1038/nm.1881. [DOI] [PubMed] [Google Scholar]
- 65.Harris TJ, Grosso JF, Yen HR, et al. Cutting edge: An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J immunol. 2007;179:4313–4317. doi: 10.4049/jimmunol.179.7.4313. [DOI] [PubMed] [Google Scholar]
- 66.Suvannavejh GC, Lee HO, Padilla J, Dal Canto MC, Barrett TA, Miller SD. Divergent roles for p55 and p75 tumor necrosis factor receptors in the pathogenesis of MOG(35–55)-induced experimental autoimmune encephalomyelitis. Cell Immunol. 2000;205:24–33. doi: 10.1006/cimm.2000.1706. [DOI] [PubMed] [Google Scholar]
- 67.Schraml BU, Hildner K, Ise W, et al. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature. 2009;460:405–409. doi: 10.1038/nature08114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ashbaugh JJ, Brambilla R, Karmally SA, Cabello C, Malek TR, Bethea JR. IL7Ralpha contributes to experimental autoimmune encephalomyelitis through altered T cell responses and nonhematopoietic cell lineages. J immunol. 2013;190:4525–4534. doi: 10.4049/jimmunol.1203214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brassat D, Azais-Vuillemin C, Yaouanq J, et al. Familial factors influence disability in MS multiplex families. French Multiple Sclerosis Genetics Group. Neurol. 1999;52:1632–1636. doi: 10.1212/wnl.52.8.1632. [DOI] [PubMed] [Google Scholar]
- 70.Barcellos LF, Oksenberg JR, Green AJ, et al. Genetic basis for clinical expression in multiple sclerosis. Brain. 2002;125:150–158. doi: 10.1093/brain/awf009. [DOI] [PubMed] [Google Scholar]
- 71.Hensiek AE, Seaman SR, Barcellos LF, et al. Familial effects on the clinical course of multiple sclerosis. Neurology. 2007;68:376–383. doi: 10.1212/01.wnl.0000252822.53506.46. [DOI] [PubMed] [Google Scholar]
- 72.Wellek A, Korsukewitz C, Bach JP, et al. Sibling disability risk at onset and during disease progression in familial multiple sclerosis. Mult Scler. 2011;17:1060–1066. doi: 10.1177/1352458511405088. [DOI] [PubMed] [Google Scholar]
- 73.Butterfield RJ, Blankenhorn EP, Roper RJ, Zachary JF, Doerge RW, Teuscher C. Identification of genetic loci controlling the characteristics and severity of brain and spinal cord lesions in experimental allergic encephalomyelitis. Am J Pathol. 2000;157:637–645. doi: 10.1016/S0002-9440(10)64574-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marta M, Stridh P, Becanovic K, et al. Multiple loci comprising immune-related genes regulate experimental neuroinflammation. Genes Immun. 2010;11:21–36. doi: 10.1038/gene.2009.62. [DOI] [PubMed] [Google Scholar]
- 75.Thessen Hedreul M, Moller S, Stridh P, et al. Combining genetic mapping with genome-wide expression in experimental autoimmune encephalomyelitis highlights a gene network enriched for T cell functions and candidate genes regulating autoimmunity. Hum Mol Genet. 2013;22:4952–4966. doi: 10.1093/hmg/ddt343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Laaksonen H, Guerreiro-Cacais AO, Adzemovic MZ, et al. The multiple sclerosis risk gene IL22RA2 contributes to a more severe murine autoimmune neuroinflammation. Genes Immun. 2014;15:457–465. doi: 10.1038/gene.2014.36. [DOI] [PubMed] [Google Scholar]
- 77.Healy BC, Liguori M, Tran D, et al. HLA B*44: protective effects in MS susceptibility and MRI outcome measures. Neurol. 2010;75:634–640. doi: 10.1212/WNL.0b013e3181ed9c9c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Qiu W, Raven S, James I, et al. Spinal cord involvement in multiple sclerosis: a correlative MRI and high-resolution HLA-DRB1 genotyping study. J Neurol Sci. 2011;300:114–119. doi: 10.1016/j.jns.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 79.Okuda DT, Srinivasan R, Oksenberg JR, et al. Genotype-Phenotype correlations in multiple sclerosis: HLA genes influence disease severity inferred by 1HMR spectroscopy and MRI measures. Brain. 2009;132:250–259. doi: 10.1093/brain/awn301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Masterman T, Ligers A, Olsson T, Andersson M, Olerup O, Hillert J. HLA-DR15 is associated with lower age at onset in multiple sclerosis. Ann Neurol. 2000;48:211–219. [PubMed] [Google Scholar]
- 81.Smestad C, Brynedal B, Jonasdottir G, et al. The impact of HLA-A and -DRB1 on age at onset, disease course and severity in Scandinavian multiple sclerosis patients. Eur J Neurol. 2007;14:835–840. doi: 10.1111/j.1468-1331.2007.01825.x. [DOI] [PubMed] [Google Scholar]
- 82.Harbo HF, Isobe N, Berg-Hansen P, et al. Oligoclonal bands and age at onset correlate with genetic risk score in multiple sclerosis. Mult Scler. 2014;20:660–668. doi: 10.1177/1352458513506503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sharan SK, Thomason LC, Kuznetsov SG, Court DL. Recombineering: a homologous recombination-based method of genetic engineering. Nat Protoc. 2009;4:206–223. doi: 10.1038/nprot.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wellcome Trust Case Control Consortium, Australo-Anglo-American Spondylitis Consortium. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007;39:1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Comabella M, Craig DW, Camina-Tato M, et al. Identification of a novel risk locus for multiple sclerosis at 13q31.3 by a pooled genome-wide scan of 500,000 single nucleotide polymorphisms. PLoS One. 2008;3:e3490. doi: 10.1371/journal.pone.0003490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Aulchenko YS, Hoppenbrouwers IA, Ramagopalan SV, et al. Genetic variation in the KIF1B locus influences susceptibility to multiple sclerosis. Nat Genet. 2008;40:1402–1403. doi: 10.1038/ng.251. [DOI] [PubMed] [Google Scholar]
- 87.Baranzini SE, Wang J, Gibson RA, et al. Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum Mol Genet. 2009;18:767–778. doi: 10.1093/hmg/ddn388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.De Jager PL, Jia X, Wang J, et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet. 2009;41:776–782. doi: 10.1038/ng.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Australia, New Zealand Multiple Sclerosis Genetics Consortium. Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat Genet. 2009;41:824–828. doi: 10.1038/ng.396. [DOI] [PubMed] [Google Scholar]
- 90.Sanna S, Pitzalis M, Zoledziewska M, et al. Variants within the immunoregulatory CBLB gene are associated with multiple sclerosis. Nat Genet. 2010;42:495–497. doi: 10.1038/ng.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nischwitz S, Cepok S, Kroner A, et al. Evidence for VAV2 and ZNF433 as susceptibility genes for multiple sclerosis. J Neuroimmunol. 2010;227:162–166. doi: 10.1016/j.jneuroim.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 92.Jakkula E, Leppa V, Sulonen AM, et al. Genome-wide association study in a high-risk isolate for multiple sclerosis reveals associated variants in STAT3 gene. Am J Hum Genet. 2010;86:285–291. doi: 10.1016/j.ajhg.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Patsopoulos NA, Bayer Pharma MSGWG, Steering Committees of Studies Evaluating Interferon-b et al. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann Neurol. 2011;70:897–912. doi: 10.1002/ana.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Matesanz F, Gonzalez-Perez A, Lucas M, et al. Genome-wide association study of multiple sclerosis confirms a novel locus at 5p13.1. PLoS One. 2012;7:e36140. doi: 10.1371/journal.pone.0036140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Martinelli-Boneschi F, Esposito F, Brambilla P, et al. A genome-wide association study in progressive multiple sclerosis. Mult Scler. 2012;18:1384–1394. doi: 10.1177/1352458512439118. [DOI] [PubMed] [Google Scholar]
- 96.Maier LM, Lowe CE, Cooper J, et al. IL2RA genetic heterogeneity in multiple sclerosis and type 1 diabetes susceptibility and soluble interleukin-2 receptor production. PLoS Genet. 2009;5:e1000322. doi: 10.1371/journal.pgen.1000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.De Jager PL, Baecher-Allan C, Maier LM, et al. The role of the CD58 locus in multiple sclerosis. Proc Natl Acad Sci U S A. 2009;106:5264–5269. doi: 10.1073/pnas.0813310106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kofler DM, Severson CA, Mousissian N, De Jager PL, Hafler DA. The CD6 multiple sclerosis susceptibility allele is associated with alterations in CD4+ T cell proliferation. J immunol. 2011;187:3286–3291. doi: 10.4049/jimmunol.1100626. [DOI] [PubMed] [Google Scholar]
- 99.Martin D, Pantoja C, Fernandez Minan A, et al. Genome-wide CTCF distribution in vertebrates defines equivalent sites that aid the identification of disease-associated genes. Nat Struct Mol Biol. 2011;18:708–714. doi: 10.1038/nsmb.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shahijanian F, Parnell GP, McKay FC, et al. The CYP27B1 variant associated with an increased risk of autoimmune disease is underexpressed in tolerizing dendritic cells. Hum Mol Genet. 2014;23:1425–1434. doi: 10.1093/hmg/ddt529. [DOI] [PubMed] [Google Scholar]
- 101.Paraboschi EM, Rimoldi V, Solda G, et al. Functional variations modulating PRKCA expression and alternative splicing predispose to multiple sclerosis. Hum Mol Genet. 2014 doi: 10.1093/hmg/ddu392. [DOI] [PubMed] [Google Scholar]