

Abstract
Major pulmonary disorders may occur after brain injuries as ventilator-associated pneumonia, acute respiratory distress syndrome or neurogenic pulmonary edema. They are key points for the management of brain-injured patients because respiratory failure and mechanical ventilation seem to be a risk factor for increased mortality, poor neurological outcome and longer intensive care unit or hospital length of stay. Brain and lung strongly interact via complex pathways from the brain to the lung but also from the lung to the brain. Several hypotheses have been proposed with a particular interest for the recently described “double hit” model. Ventilator setting in brain-injured patients with lung injuries has been poorly studied and intensivists are often fearful to use some parts of protective ventilation in patients with brain injury. This review aims to describe the epidemiology and pathophysiology of lung injuries in brain-injured patients, but also the impact of different modalities of mechanical ventilation on the brain in the context of acute brain injury.
Keywords: Brain-lung crosstalk, Brain injury, Lung injury, Protective ventilation, Double hit model
Core tip: Brain lung crosstalk is a complex interaction from the brain to the lung but also from the lung to the brain. Intensivists are often fearful to use some parts of protective ventilation in patients with brain injuries but if correctly applied, mechanical ventilation could have beneficial effect on brain oxygenation, even if positive end-expiratory pressure and recruitment maneuvers are used. This review aims to describe the epidemiology and pathophysiology of lung injuries in brain-injured patients, but also the impact of different modalities of mechanical ventilation on the brain in the context of acute brain injury.
INTRODUCTION
Brain lung crosstalk is a complex interaction from the brain to the lung but also from the lung to the brain. The occurrence of severe pulmonary injuries after experiencing a brain injury, such as severe traumatic brain injury (TBI), subarachnoid hemorrhage (SAH) or stroke, has been described[1-5]. These pulmonary injuries include ventilator-associated pneumonia (VAP), acute respiratory distress syndrome (ARDS) and neurogenic pulmonary edema (NPE). They are key points for the management of brain-injured patients because respiratory failure and mechanical ventilation seem to be a risk factor for increased mortality, poor neurological outcome and longer intensive care unit (ICU) or hospital length of stay (LOS)[4-9]. The pathophysiology of brain-lung interaction is complex and several hypotheses have been proposed with a particular interest for the recently described “double hit” model[1].
This review aims to describe the epidemiology and pathophysiology of lung injuries in brain-injured patients, but also the impact of different modalities of mechanical ventilation on the brain in the context of acute brain injury.
LUNG INJURIES AFTER BRAIN INJURIES
Major pulmonary disorders may occur after brain injuries as VAP, ARDS or NPE. In this review, the direct consequences of chest trauma, such as rib fractures, lung contusions or hemo/pneumothorax will not be discussed in the present review. Zygun et al[6], in an observational cohort study, reported non-neurologic organ dysfunctions in 209 patients with severe TBI. Eighty-nine percent of patients had at least one non-neurologic dysfunction (organ system component score ≥ 1), and 81% of patients developed respiratory dysfunction [arterial partial pressure of oxygen/inspired fraction of oxygen ratio (PaO2/FiO2) = 226-300]. Thirty-five percent of patients developed at least one organ failure (organ system component score ≥ 3), and the most common non-neurologic organ system failure was severe respiratory failure (PaO2/FiO2 ≤ 150), occurring in 23% of patients. Other multicenter studies have also reported high incidence of extracerebral organ dysfunctions after TBI[10] or SAH[11]. These extracerebral organ failures, especially respiratory failure and ICU-acquired sepsis, seem to be more frequent in patients with brain injuries than in patients with non-neurologic conditions[12].
Lung injuries are frequent and can lead to significant consequences for patients with brain injuries by directly altering outcomes. Respiratory failure and mechanical ventilation appear to be risk factors for increased mortality and poor neurological outcomes in patients with brain injuries[6-9] and are associated with longer ICU and hospital LOS[4,5]. Pelosi et al[13], in a recent prospective observational and multicenter study, described outcomes among mechanically ventilated patients with various types of brain injuries (362 patients with ischemic or hemorrhagic stroke and 190 patients with brain trauma) and compared them to non-neurologic patients. Respiratory failure was the most frequent extracerebral organ dysfunction in neurologic patients. Patients with neurologic disease who were mechanically ventilated had longer ICU and ventilator-days, more tracheostomy requirement, more VAP and higher mortality rates than non-neurologic patients.
VAP
Pneumonia and VAP are frequently encountered in neurologic patients due to decrease in the level of consciousness and massive aspiration or even microaspirations[14]. Risk factors for developing VAP in brain-injured patients have been identified: polytransfusion, age, obesity, diabetes, immunocompromized status, chronic pulmonary disease and use of barbiturates[15]. Moreover, mechanical ventilation, sedation and myorelaxant use, previous antibiotic therapy and the absence of proclive position during mechanical ventilation increase the risk of developing VAP[16]. Additionally, brain injury-induced immunosuppression promotes the development of infectious diseases[17-20].
The incidence of VAP in patients with severe TBI is 21% to 60%[15,21,22]. Methicillin-susceptible Staphylococcus aureus is the most common pathogen reported in VAP in patients with severe TBI. Early enteral feeding and oral care has been shown to decrease the incidence of VAP in the neuro-ICU[22,23]. Pelosi et al[13] reported a higher rate of VAP in patients with TBI compared to patients with ischemic or hemorrhagic stroke and non-neurologic patients.
Cinotti et al[24] reported a retrospective analysis of 193 patients with SAH who were mechanically ventilated. VAP occurred in 48.7% of the patients, and the main responsible pathogen was also Methicillin-susceptible Staphylococcus aureus. This study did not find an increase in the mortality for these patients, but a longer duration of mechanical ventilation and ICU LOS[24]. Frontera et al[25] analyzed data of 573 patients with SAH (with or without mechanical ventilation) and quantified the prevalence of nosocomial infectious complications. The most common complication was pneumonia with a prevalence of 20%. Pneumonia was an independent factor for mortality or severe disability at 3 mo[25].
Kasuya et al[26] observed a 28% rate of VAP in 111 stroke patients on mechanical ventilation. VAP prolonged the duration mechanical ventilation and ICU LOS. Chronic lung disease, National Institute of Health Stroke Score at admission and hemorrhagic transformation were independent risk factors for VAP. The most common responsible bacteria were Methicillin-resistant Staphylococcus aureus and Methicillin-susceptible Staphylococcus aureus[26]. In patients with severe ischemic stroke, VAP increased mortality by 3-fold[27].
ARDS
ARDS occur with a high incidence rate in patients with brain injuries. The definition of ARDS used in most of the studies is the American-European consensus conference criteria[28]. A recent study reported an incidence of 35% of ARDS in a cohort of 192 patients with neurologic disorders (hemorrhagic stroke, SAH, subdural hematoma, TBI and ischemic stroke)[29]. Other studies have shown an ARDS incidence of 19% to 35% in patients with a glasgow coma scale (GCS) score < 9[12,29,30].
Patients with isolated TBI present 20%-25% of ARDS[31,32], and patients with SAH present 20%-38% of ARDS[3,7,33]. A recent retrospective study conducted from 1994 to 2008 in the United States of America reported an incidence of ARDS in admissions of patients with acute ischemic stroke of 4%[4]. Aspiration-related ARDS was diagnosed in 3.6% patients in another recent retrospective cohort study on 1495 patients with acute stroke[34].
In all cases, ARDS impacts the morbidity and mortality of patients with brain injuries[4,7,30,35,36]. Occurrence of ARDS after TBI leads to a 3-fold increase in hospital mortality[32]. ARDS is an independent risk factor for increased mortality and poor neurologic outcomes and is associated with longer ICU and hospital LOS[4,30]. Risk factors have been identified for the development of ARDS. First, the severity of the initial brain injury revealed by low Glasgow coma score (GCS 3-4) and initial cerebral computed tomography (CT) scan abnormalities (midline shift and global CT findings)[31,35,36]. Secondly, induced hypertension, administration of vasoactive drugs and a history of drug abuse have been reported as independent factors for ARDS in severe TBI[35]. Finally, general risk factors have been identified such as young age, male gender, ethnicity, history of chronic arterial hypertension, diabetes, chronic obstructive pulmonary disease, development of sepsis, cardiovascular, renal and hematological dysfunctions[4,32,37]. Recently, Mascia et al[30] described the ventilatory management of 82 patients with severe TBI in a prospective multicenter observational study. Twenty-two percent of the patients developed ARDS, and these patients initially had higher tidal volumes (Vt) than patients without ARDS. The proportion of ARDS increased with Vt settings in a dose-response relationship. In the days preceding ARDS, 72% of patients with ARDS had a mean Vt ≥ 10 mL/kg predicted body weight (PBW)[30]. The ventilator management of patients with severe TBI seems to be a key point in ARDS development and fits into the “double hit” model which will be detailed later in this review.
The ARDS distribution over the time is bimodal, with an early peak on day 2-3 after the onset of mechanical ventilation and a later peak on day 7-8[10], often related to pneumonia[15].
NPE
NPE has been described for more than 100 years[38]. It has been defined as a clinical entity with an acute onset of protein-rich lung edema after significant central nervous system injuries such as TBI, SAH, stroke, spinal cord injury, status epilepticus, meningitis or subdural hemorrhage and the exclusion of other plausible causes[39-42].
In a review on NPE cases reported from 1990 to 2003, the most frequent neurologic injury was SAH (42.9%) and symptom onset was < 4 h after brain injury in 71.4% of patients. The mortality rate of NPE was high, nearing 10%, but patients who survived usually recover very quickly (< 72 h for 52.4%)[41]. Rogers et al[40] reported a large autopsy database of patients with head injuries who died at the scene or within 96 h of injury. The diagnosis of NPE included the presence of edema, congestion and hemorrhage associated with an increase in lung weight. The incidence of NPE in isolated TBI patients who died at the scene was 32%. It reached 50% for patients who died within 96 h. An inverse correlation between cerebral perfusion pressure and the PaO2/FiO2 ratio was observed, even if the chest X-ray was considered normal[40]. The incidence of NPE in aneurysmal SAH varies from 2% to 25%[11,43]. The incidence seems to be higher in fatal SAH[44]. Risk factors identified are old age, delay to surgery, vertebral artery surgery and the severity of clinical and CT-scan scores (Hun-Hess and Fisher grades)[11,45]. The occurrence of NPE after SAH is associated with poor outcomes and higher mortality[46,47].
NPE can be considered as a form of ARDS with the consensus definition. So, some authors proposed the following diagnostic criteria: (1) bilateral infiltrates; (2) PaO2/FiO2 ratio < 200; (3) no evidence of left atrial hypertension; (4) presence of severe central nervous system injury that has caused increased intracranial pressure (ICP); and (5) absence of other common causes of ARDS (e.g., aspiration, massive blood transfusion or sepsis)[48].
PATHOPHYSIOLOGY OF BRAIN-LUNG CROSSTALK
Brain to lung pathway
The pathophysiology of lung injuries after an acute brain injury is still in debate, and several theories have been proposed; recently, the “double hit” model has been described[1].
The sympathetic response to increased ICP has an important role. Some authors explained some parts of NPE with neuro-cardiac and neuro-hemodynamic paradigms[48]. It has been well demonstrated that direct myocardial injury with Takotsubo’s cardiomyopathy, can participate to NPE[49-51]. Massive sympathetic discharge following brain injuries seems to induce direct myocyte injuries with wall motion abnormalities that follow a pattern of sympathetic nerve innervation[52]. The neuro-hemodynamic theory is defined by indirect ventricular compliance impairment resulting from rapid increases in systemic and pulmonary pressures. Indeed, translocation of blood flow from the highly resistant systemic circulation to the low resistance pulmonary circulation causes a hydrostatic form of pulmonary edema[53]. Animal models have shown an increase in left atrial, systemic and pulmonary pressures associated with NPE[54-56]. Although hydrostatic pressure and cardiac impairment most likely play a role in the pathogenesis of NPE, these theories do not explain the presence of red blood cells and protein in the alveolar fluid[57].
The blast theory
Theodore and Robin first defined the “blast theory” of NPE as an impairment of vascular permeability[58]. The transient increase of intravascular pressure, caused by an acute increase in ICP, damages the capillary-alveolar membrane. So, pulmonary endothelium injuries cause a leak of protein-rich plasma[58]. This theory includes the coexistence of high hydrostatic pressure and pulmonary endothelium injury. Some degree of capillary hypertension seems necessary for the occurrence of this pulmonary edema, and a pressure-dependent increase in permeability may be a common point in NPE[59,60]. Animal models have allowed the exploration of this theory. Maron et al[59] reported in canine isolated perfused lung lobes, a minimum of 70 torr of venous pressure is necessary to have protein permeability and to note a linear correlation between the increase in venous pressure and the osmotic reflection coefficient for total proteins[59]. Bosso et al[60] explored the relationship between the degree of pulmonary hypertension and post-mortem extravascular lung water content (EVLW) in rabbits with intracranial hypertension. The pulmonary arterial pressure had to exceed 25 torr to observe an increase in extravascular lung water[60]. In contrast, Bowers et al[61] determined the effects of intracranial hypertension in a sheep model by measuring the flow rate and protein content of lung lymph. They noted a constant increase in lung vascular permeability but with inconstant increase in pulmonary vascular pressure[61]. Few reports are available in humans because hemodynamic monitoring at the time of the initial severe increase in ICP is rare. After this initial hemodynamic instability and massive sympathetic response, systemic and pulmonary pressures could return to normal values, whereas capillary-alveolar membrane damage persists[58,62]. Some authors observed no changes in systemic pressure, despite the occurrence of NPE underlying direct pulmonary endothelial damage following brain injury[63]. This concept has been called “pulmonary venule adrenergic hypersensitivity”.
Pulmonary venule adrenergic hypersensitivity
Some human cases with continuous hemodynamic monitoring reported NPE without hemodynamic instability[63,64]. So, the NPE may result, in part, from select pulmonary venoconstriction after massive sympathetic discharge following brain injury. Pulmonary vessels have α- and β-adrenergic receptors that may be activated leading to endothelial integrity changes[65]. Animal models demonstrate an increase in pulmonary vascular permeability and edema formation that could not be explained by hemodynamic changes alone[61,66]. In anesthetized dogs with raised ICP, McClellan et al[66] noted a 3-fold increase in pulmonary vascular permeability (exudative edema) with a moderate increase in pulmonary arterial pressures and cardiac output. However, when they reproduced these hemodynamic changes in dogs without intracranial hypertension, they did not report any changes in the protein leak index[66]. Peterson et al[67] administered α-adrenergic blockers to anesthetized sheep with progressive levels of intracranial hypertension. They reported the prevention of pulmonary edema formation with minor systemic arterial pressure effects supporting a direct adrenergic action on the pulmonary vascular bed[67].
Double hit model
Systemic inflammatory response appeared to play a major role in the development of pulmonary failure after acute brain injury. This pathophysiological process completes the blast injury theory[1,68]. Intracranial inflammatory response occurs after brain injury, and pro-inflammatory cytokines [interleukin 1 (IL-1), IL-6), tumor necrosis factor (TNF), IL-8] are produced locally in cerebral injured tissue[69]. Microglia and astrocytes are the principal source of inflammatory mediators. Then, alteration of the blood brain barrier (BBB) permeability allows their discharge into the systemic circulation with a transcranial gradient. This could be responsible for extracerebral dysfunctions[70-72]. This systemic production of inflammatory mediators constitutes an inflammatory environment: the “first hit”. Organ are therefore more susceptible to subsequent events, the “second hit”, such as mechanical ventilation, infections or surgical procedures, that are in normal condition harmless[1] (Figure 1). López-Aguilar et al[73] randomized rabbits to control or brain injured group with a 120 min mechanical ventilation with the same ventilator settings followed by aggressive mechanical ventilation. In the brain-injured group, lungs had more changes in the ultrafiltration coefficient, weight and alveolar hemorrhage[73]. Hyperactivated neutrophils and leukocyte-endothelial cell interactions could probably have contributed to this pathological process[74]. Acute inflammatory response in both brain and lung after brain injury has been shown in human and animal. Experimental intracerebral hemorrhage injury is accompanied by an increase in intracellular adhesion molecule-1 and tissue factor in both brain and lung. Progressive neutrophil recruitment and morphological pulmonary damage such as disruption of alveolar structures has been observed[75]. Kalsotra et al[76] showed a large migration of macrophages and neutrophils in the major airways and alveolar spaces after brain injury in rats, with an increase of leukotriene B4 production within the lung[76]. Brain-dead human donors have significantly higher IL-8 levels in the broncho-alveolar lavage compared to healthy subjects or ventilated non brain-dead patients. Moreover, neutrophil infiltration in the lungs well correlates with levels of IL-8[77]. In a rat weight-drop model of TBI, ultrastructural damage in type II pneumocytes with important intracellular vacuoles and increased lipid peroxidation have been reported[78]. Recently, Heuer et al[79] studied pigs with acute intracranial hypertension. They reported higher scores of inflammation, edema and necrosis in the lung and other organs compared with control pigs without intracranial hypertension despite the absence of hypoperfusion and hypoxemia[79]. Previously, they compared 4 groups of pigs: control, with intracranial hypertension, with ARDS and with intracranial hypertension + ARDS. They analyzed lung CT-scans of each group. Intracranial hypertension alone increased lung density and exacerbated the increase in lung density in pigs with ARDS. Moreover, the gas-tissue ratio of the lung was decreased by intracranial hypertension in normal and injured lungs with an increase of poorly aerated and atelectatic lung areas. These lung CT-scan injuries were exacerbated by intracranial hypertension[74].
Figure 1.
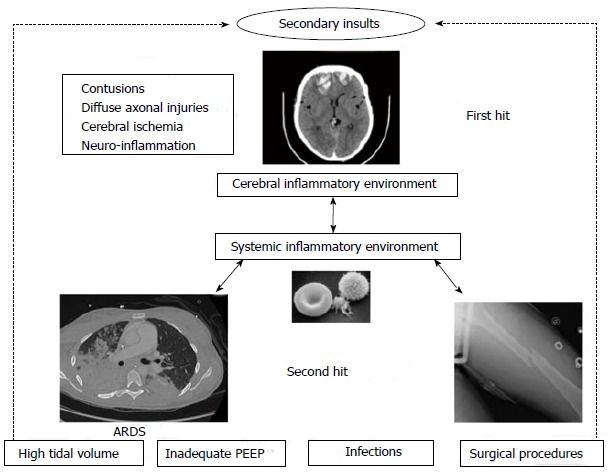
The double hit model in the context of brain injury. ARDS: Acute respiratory distress syndrome; PEEP: Positive end-expiratory pressure.
The catecholamine storm, in conjunction with the cerebral and systemic inflammatory reaction (first hit) creates an inflammatory environment leading to an increased susceptibility of the lung to further injurious events (second hit). This pathway could be the bed for lung injuries in patients with acute cerebral damage. However, this inflammatory cascade does not occur only in one way: from the brain to the lung, but also from the lung to the brain.
Hypothalamo-pituitary adrenal axis
Since several years, hypothalamo-pituitary adrenal axis [Hypothalamo-pituitary adrenal (HPA) axis] in brain injury has been explored in experimental and clinical studies and it could participate to lung dysfunction. Indeed, it has major effects on stress and systemic inflammatory response after trauma[80,81]. In the initial phase of trauma, inflammation mediators, such as IL-6, activate massively HAP axis to induce an initial hypercortisolism, main effector of compensatory anti-inflammatory response syndrome[80,82,83]. This hypercortisolism allow decreasing deleterious effects of inflammatory response, as its spread in organism and protect also other organs[81,84]. Moreover, endogenous glucocorticoids stimulate anti-infectious immunity[85] and HAP axis has major role in hemodynamic response and maintain of blood pressure[86,87].
After TBI, 25%-50% of patients present an acute secondary adrenal insufficiency[88-91]. These patients had worse outcomes and neurologic prognostic, lower arterial pressure, greater vasopressor use and higher mortality rate[88,89,92,93]. Moreover, trauma-induced adrenal insufficiency is correlated with systemic inflammatory response syndrome[94]. Patients with adrenal insufficiency have longer high plasma IL-6 levels than patients with normal adrenal response to stress[89,95]. In multiple-injured patients, persistence of high IL-6 plasma level at day 7 is associated with higher mortality rate and incidence of pneumonia[96]. Persistence of systemic inflammatory response syndrome seems to be predictive of nosocomial infection in trauma patients[97,98]. The principal theory is that secondary adrenal insufficiency exposes patients to deleterious effects of uncontrolled systemic inflammation with immunodepression, nosocomial infections, especially VAP and overwhelming inflammatory response[90,98,99]. So this HAP axis dysfunction could participate to weaken the lung after TBI.
A multicenter, randomized trial reported in 150 intubated patients with severe trauma and corticosteroid insufficiency, a decrease risk of hospital-acquired pneumonia with stress-dose of hydrocortisone, particularly in the sub-group of patients with severe TBI[100]. However, this result was not confirmed with recent trial in patients with severe TBI[101]. Stroke-induced immunodepression has been described with HAP axis-related abnormalities following acute ischemic stroke[102] and is probably implicated in high incidence of pneumonia[103].
Lung to brain pathway
A complex pathway throughout autonomic, neuro-inflammatory, neuro-endocrine and immunologic systems has been described. This pathway is involved in normal physiology to contribute to maintain homeostasis, but may lead to adverse effects[104]. Two components may be involved in this lung to brain pathway: lung injuries themselves, such as ARDS, and mechanical ventilation.
Lung injuries due to inadequate ventilator settings, could result in an inflammatory response, initially located in the lung parenchyma. But this could extend to the systemic circulation and then to other organs and the brain. Multi-organ failure can occur as a result of pulmonary injuries[105]. The main cause of mortality in patients with ARDS is multiple organ failure and not hypoxemia or pulmonary dysfunction[106]. It has been well described that ARDS survivors have cognitive deterioration including memory, language and cognitive decline[107-109] and that patients with a long duration of mechanical ventilation present neurologic impairment with memory and cognitive alteration[110]. The hippocampus, which is involved in learning and memory processes, is particularly vulnerable to hypoxia[111]. However, ARDS can lead to hippocampal injuries with memory defects, regardless of the degree of hypoxia[112]. ARDS, in the same way than septic shock, can induce neuronal damages. Nguyen et al[113] studied 170 patients with severe sepsis or septic shock in a prospective study. They found an increase in plasmatic marker of brain damages as S-100β protein and neuron-specific enolase (NSE) in respectively 42% and 53% of these patients[114]. High S-100β protein levels were reported in patients with decreased consciousness and encephalopathy. In pig models of ARDS (lavage model), S-100β protein levels were significantly higher than in pigs with hypoxemia induced by lavage than when hypoxia was induced by reducing the inspired oxygen fraction[115]. Moreover, histopathologic changes in the hippocampus occurred only in pigs with ARDS. The authors suggested that brain damage could only be observed in ARDS independently to hypoxemia. S-100β protein and NSE might represent cerebral injuries and BBB alterations in patients with ARDS[113]. Permeability of both the blood-brain and lung barriers can be altered by pathophysiologic situations and allows communication between the brain and the lung[116].
Lung injuries may aggravate the sensitivity of the brain to acute injuries. In their previous study, Heuer et al[74] reported brain damage in pigs with ARDS alone and reciprocal synergistic effects between the lung and brain with worsening of brain damage in the group with ARDS + intracranial hypertension[74]. Indeed, cerebral tissue oxygenation (PtiO2) and brain tissue density (reflecting cerebral edema) decreased in all animals (intracranial hypertension, ARDS and ARDS + intracranial hypertension) compared to the control group. NSE and S-100β protein levels increased significantly in all animals compared to the control group, but the most marked increase was in the group with ARDS, as for IL-1β and IL-6. So ARDS could exacerbate cerebral damage in acute cerebral hypertension. Hegeman et al[105] described, after injurious stress and strain in the lung, inflammation of the alveoli, recruitment of neutrophils and production of cytokines. Endothelial cells, activated by cytokines, secrete chemokines and express adhesion molecules on their surface, leading to enhanced leukocyte adhesiveness and transmigration of active immune cells across the endothelium[105]. This local inflammation can then spread into the systemic circulation. Lung inflammation could spread to the cerebral system through humoral, cellular and neural pathways[116].
Beyond pulmonary injuries, mechanical ventilation strategies, used daily in the ICU, could impair regional blood flow and brain oxygenation. Indeed, Bickenbach et al[117] studied PtiO2 and cerebral metabolism in a porcine model of ARDS over 8 h. Pigs were randomized in 2 groups: low tidal (LT) volume (6 mL/kg) and high tidal (HT) volume (12 mL/kg)[117]. No differences between the two groups were found in terms of PaO2, PaCO2 and pH. ARDS induced a significant decrease in PtiO2 in both groups, but the PtiO2 increased significantly at 4 and 8 h in the LT group compared to the HT group. Lactates in microdialysis were higher in the HT group at 2, 4 and 8 h. After 2 h, the plasmatic S-100 protein level decreased in the LT group, and IL-6 increased in the HT group. Therefore, LT volume ventilation improved cerebral tissue oxygenation compared to HT volume ventilation in ARDS. HT volume ventilation could increase the inflammatory response and could impair cerebral oxygenation and metabolism. Quilez et al[118] studied the effect of Vt on activation in areas of the brain in a rat model of MV with c-fos expression, a marker of neuronal activation. They randomized 3 groups of healthy-brain rats: basal (not submitted to mechanical ventilation), low Vt (8 mL/kg and positive end-expiratory pressure (PEEP) of 0 cmH2O) and high Vt (30 mL/kg and PEEP of 0 cmH2O). The inflammatory response (TNF-α) and c-fos expression in the retrosplenial cortex and thalamus were higher in the high Vt group than in the low Vt group[118]. So, setting of mechanical ventilation can directly affect the brain, most likely via inflammatory mediators. These data highlight the importance of the ventilator setting in patients undergoing mechanical ventilation and particularly in brain injured patients.
THE CONFLICT BETWEEN THE LUNG AND THE BRAIN
Mechanical ventilation allows the supply of oxygen and the removal of carbon dioxide (CO2) with tight control of the PaO2 and PaCO2, the goal is to prevent secondary cerebral ischemia and increase neurologic outcomes.
To prevent or limit Ventilation-Induced Lung Injury (VILI) the concept of protective ventilation has been developed using with low Vt, plateau pressure < 30 cmH2O and adequate PEEP levels[119]. VILI has been described as the results of 3 mechanisms: volotrauma, atelectrauma and biotrauma[120,121]. Volotrauma results from overdistension of the lung parenchyma with a high Vt. Atelectrauma results from the recruitment-derecruitment of collapsed alveoli due to an inadequate PEEP level. Biotrauma comes from a local inflammatory process due to overdistending tidal volumes and repetitive opening and closing lung units. However, most of the studies that have enhanced ventilation strategy in ARDS patients have excluded brain-injured patients[122-124]. The concept of “open the lung and keep it open” for ARDS with a low Vt, high PEEP and recruitment maneuvers, with permissive hypercapnia could have potential deleterious consequences on the brain, and intensivists are often fearful to use some parts of protective ventilation in patients with brain injury.
Tidal volume
The use of low Vt decreases systemic and pulmonary inflammatory responses in patients with ARDS[124-126] but also in patients with inflammatory processes such as aspiration, sepsis, pneumonia or trauma[127,128]. Mascia et al[30] reported that the proportion of ARDS in patients with severe TBI increased with higher initial tidal volume (Vt) settings in a dose-response relationship[30]. The ventilator management of patients with severe TBI seems to be a key point of ARDS development. As we described before high Vt could affect the brain and could be an injurious event (second hit) in the lung that is particularly sensitive due to brain injury. There is no prospective study regarding the use of low Vt in TBI patients. However, recently, Krebs et al[129] reported in rats with massive brain damage that a low Vt (6 mL/kg) with open lung PEEP (set according to the minimal static elastance of the respiratory system) compared to a high Vt (12 mL/kg) and low PEEP improved oxygenation reduced lung damage according to histology, genome analysis and real-time quantitative polymerase chain reaction with a decrease of IL-6[129].
The protective mechanical ventilation for ARDS includes low Vt (6 mL/kg PBW) and then low minute ventilation, with consequently permissive hypercapnia. Cerebral effects of hypercapnia are well known (vasodilation) and should be avoided in case of intracranial hypertension[130]. Objectives for the management of severe TBI are maintaining the PaCO2 between 35 to 40 mmHg[131] but this goal is sometimes not possible when using protective mechanical ventilation. Individualized management with neuromonitoring could allow us, in specific difficult cases, to use higher values of PaCO2 and supervise its impact on brain homeostasis. A small retrospective study in 12 patients with SAH and ARDS reported no increase in ICP with lung protective ventilation and hypercapnia (50-60 mmHg)[132]. Recently, Westermaier et al[133] performed a gradual increase of PaCO2 to 40, 50 and 60 mmHg in patients with poor-grade SAH. Cerebral blood flow and brain tissue oxygen saturation (StiO2) reacted with sustained elevation without an increase in intracranial pressure[133].
PEEP
Application of PEEP is part of the protective mechanical ventilation to recruit collapsed alveoli, improve PaO2 and lung compliance[134]. However, the use of PEEP may alter the cerebral blood flow by CO2-mediated and hemodynamic repercussion[135,136]. Therefore, Pelosi et al[13] reported in a prospective observational multicenter study that more than 80% of neurologic patients in the ICU were ventilated with a PEEP ≤ 5 cmH2O[13]. PEEP is necessary to prevent collapse and/or recruit collapsed alveoli and thereby reduce atelectasis, especially when low Vt is used. Its application is also a key point of protective ventilation.
Some studies reported the effects of PEEP on cerebral hemodynamics. Mascia et al[137] randomly applied PEEP at 5 and 10 cmH2O in 12 brain-injured patients with ARDS. Patients who were responders had decreased elastance and increased PaO2, while patients who were non-responders had an increase of elastance and PaCO2. Intracranial pressure and jugular saturation were constant in recruiters but increased in non-recruiters suggesting deleterious effects in this group[137]. Therefore, the use of PEEP in brain-injured patients seems to be safe when patients are responders to the PEEP level (i.e., not creating overdistension, increase in dead space and in PaCO2)[138]. When PEEP induces lung recruitment, intracranial pressure and cerebral perfusion do not change, and PaO2 increases[1]. PEEP could be safety used and must probably be used in brain-injured patients if the optimal PEEP is searched and adapted individually, as for patients with ARDS and a healthy brain.
Muench et al[139] examined the influence of PEEP levels on intracranial pressure, PtiO2, cerebral blood flow and systemic hemodynamics in healthy pigs and patients with SAH[139]. High levels of PEEP did not influence cerebral parameters in pigs. In patients with SAH, changes in the regional cerebral blood flow were reported, resulting from arterial pressure changes and altered cerebral autoregulation. Normalization of systemic arterial pressure restored cerebral blood flow. Recently, Schramm et al[140] measured cerebral blood flow in 20 patients with ARDS. An increase in PEEP from 9 to 14 cmH2O did not influence blood flow velocity. Caricato et al[141] examined the effect of respiratory system compliance on the intracranial effects of PEEP. No impact on cerebral and systemic hemodynamics were reported with 0, 5, 8 or 12 cmH2O of PEEP[141]. The use of PEEP appears to be safe, if arterial blood pressure is maintained. Euvolemia is probably a condition that can minimize the effect of PEEP on arterial blood pressure[139,142,143].
Moreover, some authors recommend to optimize elevation of the head to enhance cerebro-venous drainage through the vertebral venous system, not subjected to intrathoracic pressure and to maintain PEEP lower than ICP to limit interference with venous outflow[1,144,145].
An accurate monitoring of macrohemodynamic, respiratory system and cerebral parameters is needed to optimize the use of PEEP in brain-injured patients.
Recruitment maneuvers
Several studies in patients with ARDS recommended recruitment maneuvers (RM) to recruit collapsed pulmonary alveoli and open the lung followed by appropriate PEEP to maintain recruitment of the lung leading to improvement of oxygenation and compliance of the respiratory system[146,147]. However, for the same reasons as PEEP, RM could decrease arterial blood pressure and increase ICP by interfering with venous blood return and causing an increase in intrathoracic pressure[137]. Bein et al[148] reported in 11 patients with severe cerebral lesions (traumatic and non-traumatic) and ARDS, the effects of RM, which included sustaining 60 cmH2O for 30 s[148]. They recorded an increase in ICP, a decrease in mean arterial pressure, cerebral perfusion pressure (< 65 mmHg) and jugular oxygen saturation (< 55%) at the end of the RM. The improvement of arterial oxygenation was reported just after the RM but was not maintained after. Therefore, the authors did not recommend this maneuver. The impact on cerebral blood flow and intracranial pressure depends on the hemodynamic tolerance of RM. Re-aeration of lung units depends not only on the inflating pressure but also on the duration of sustained pressure (inflating pressure-time product)[149-151]. Constantin et al[146] compared 2 RM: continuous airway pressure (CPAP) with 40 cmH2O for 40 s and extended sigh (eSigh) with PEEP maintained at 10 cmH2O above the lower inflection point for 15 min[146]. They reported that only eSigh increased recruited volume and that eSigh was hemodynamically better tolerated than CPAP and induced a greater and more prolonged increase in arterial oxygenation. Moreover, response to RM seems to depend on the lung morphology. Patients with diffuse loss of aeration are more responsive than patients with a focal loss of aeration[152]. These parameters have to be considered before using RM. Therefore, eSigh may be better adapted to patients with severe brain injuries due to its better hemodynamic tolerance. Nemer et al[153] compared 2 RM: CPAP at 35 cmH2O for 40 s and PEEP of 15 cmH2O and pressure control above PEEP of 35 cmH2O for 2 min in patients with SAH and ARDS[153]. CPAP recruitment leads to higher intracranial pressure (> 20 mmHg) and lower cerebral perfusion pressure (< 65 mmHg). In another study, 28 RMs were performed in 9 patients with ARDS and cerebral injury in a stepwise with 3 cmH2O increments and decrements of PEEP. No significant differences were found for mean arterial pressure, intracranial pressure and cerebral perfusion pressure after RMs compared with baseline values[154]. Therefore the use of RM may be safe and possible with strict monitoring of systemic and cerebral parameters and use of progressive and soft maneuvers.
Wolf et al[155] evaluated the feasibility of the “open lung approach” with low tidal volume, a high level of PEEP and RM in 13 patients with acute brain injury and ARDS[155]. They reported a decrease of FiO2 from 0.85 to 0.55, 24 h after the first RM with an increase of PaO2/ FiO2 from 142 to 257. In parallel, intracranial pressure, PaCO2 and PtiO2 remained stable. The authors concluded that protective ventilation is safe in neurosurgical patients and improves oxygenation without side effects.
Prone position
Prone position has been used for 30 years in patients with ARDS. It has been proven to increase oxygenation with different mechanisms such as net recruitment, more homogeneous distribution of alveolar inflation and protection of VILI. Benefits in terms of outcomes and mortality have been shown in severely hypoxemic ARDS if a sufficient duration of prone position is used[156-158]. This respiratory management has been sparsely studied in patients with cerebral injuries. Some authors reported cases or series of prone position[159-161]. Reinprecht et al[159] analyzed the effect of this position in 16 patients with severe SAH and ARDS. They reported a significant increase in PaO2 and PtiO2 with significant, but not deleterious, increases in intracranial pressure and decreases in cerebral perfusion pressure[159]. A case report of a patient with severe traumatic chest and brain injuries showed improvement of oxygenation with a moderate, but very transient, increase in intracranial pressure after 20 h of prone position[161].
The Table 1 summarizes the effects of different parts of protective ventilation on brain hemodynamic and metabolism.
Table 1.
Effects of protective ventilation on brain hemodynamic and metabolism
| CBF | ICP | CPP | PtiO2 | SjO2 | Lactates (microdialysis) | |
| High Vt In pigs with ARDS[117] | ↓ | ↑ | ||||
| Low Vt In pigs with ARDS[117] | ↑ | ↓ | ||||
| Permissive hypercapnia (PaCO2: 40-60 mmHg) in patients with SAH[132,133] | ↑ | = | = | ↑ | ||
| PEEP | = if MAP is maintained[140] | = if responder patient[137] | = if responder patient[137] | |||
| ↑ If non-responder patient[137] | ↓ If non-responder patient[137] | |||||
| RM | ↑ If MAP decreased[148] | ↓ If MAP decreased[148] | ↓ If MAP decreased[148] | |||
| Open lung approach (low Vt + high PEEP + RM) in patients with acute brain injury and ARDS[155] | = | = | = |
Responder patient to PEEP: Decrease in elastance and increased PaO2; Non-responder patient to PEEP: Increase in elastance and PaCO2. CBF: Cerebral blood flow; ICP: Intracranial pressure; CPP: Cerebral perfusion pressure; PtiO2: Cerebral tissue oxygenation; SjO2: Jugular vein oxygen saturation; Vt: Tidal volume; PEEP: Positive end-expiratory pressure; RM: Recruitment maneuvers; MAP: Mean arterial pressure; ARDS: Acute respiratory distress syndrome.
Alternative methods for tight CO2 control and refractory hypoxia such as high frequency oscillatory ventilation and extracorporeal lung support techniques (percutaneous extracorporeal lung assist and extracorporeal membrane oxygenation) have been poorly evaluated in patients with head injuries[145].
CLINICAL MANAGEMENT OF LUNG INJURIES IN BRAIN-INJURED PATIENTS
In clinical practice, there is actually no recommendation for ventilator strategy of brain-injured patients except for PaO2 and PaCO2 targets[131].
Treatment of VAP is not specific for patients with cerebral injuries but it is important to note that prevention seems to be a key point. Treatment of VAP has to be started quickly as VAP is associated with higher mortality rate and poor neurologic outcome. It may follow the guidelines for hospital-acquired and VAP[162]. Risk factors of VAP in brain-injured patients are numerous and prophylactic measures have to focus on these, including oral care[23,103,163]. The high rate of VAP in brain-injured patients is, in part, explained by long duration of mechanical ventilation[164]. So Roquilly et al[165] reported in a before/after evaluation of an extubation readiness bundle, a decrease of duration of mechanical ventilation in patients with brain injury[165]. The bundle components were 1/protective ventilation (Vt: 6-8 mL/kg PBW, PEEP > 3 cmH2O) 2/early enteral nutrition (initiation day 1 and 25 kCal/kg per day before day 3) 3/optimization of the probabilistic antibiotherapy for VAP and 4/a systematic approach of extubation (ventilator weaning and removal of tube if Glasgow Coma Scale ≥ 10 and cough). Despite a compliance with bundle elements of 21% in the intervention phase, they observed a reduction of duration of mechanical ventilation, rate of VAP and rate of unplanned extubation compared to the control observational phase. In acute stroke, the major measure is to avoid per os nutrition until swallowing is evaluated and validated[166-168]. No difference has been found between percutaneous gastrostomy or nasal feeding tube in terms of rate of pneumonia but percutaneous gastrostomy tube seems to be safer and more effective for feeding[169]. For TBI, in front of traumatic-induced adrenal insufficiency, the use of stress-dose steroids during initial management are still debated for prevention of VAP but literature doesn’t allow us to provide an answer[101].
Concerning NPE, few studies have reported specific treatment in humans. Some animal studies have focused on α-blockers treatment to limit massive sympathetic discharge after brain injuries[48,170]. Two cases of human NPE were published about use of adrenergic blocker (phentolamine or chlorpromazine) and successful treatment with improvement of hemodynamic instability and oxygenation[171,172]. Further studies are needed to explore this way. But the key point of NPE management is to treat the underlying cerebral injuries to decrease ICP, mitigate the sympathetic discharge and improve oxygenation[41,48].
Concerning ARDS, protective ventilation has been largely discussed in the previous section. An accurate monitoring of macrohemodynamic, respiratory and cerebral parameters are needed to optimize the management.
When a brain-injured patient presents hypoxia, all diagnoses evoked in this review could be discussed. The Figure 2 summarizes different steps of management and prevention of respiratory failure in brain-injured patient. The response of the cardiopulmonary system varies widely among patients with brain injury (direct myocardial injury, non-cardiogenic mechanisms, etc.). So first of all, it is important to evaluate cardiac function to adapt our management and initiate treatment of cardiogenic failure if necessary. Moreover, normalization of ICP is an important step to decrease sympathetic discharge and its consequences. Criteria of VAP, ARDS and NPE have to be researched and for some patients in which difference between NPE and ARDS could be difficult, measurement of serum catecholamines may be helpful[48].
Figure 2.
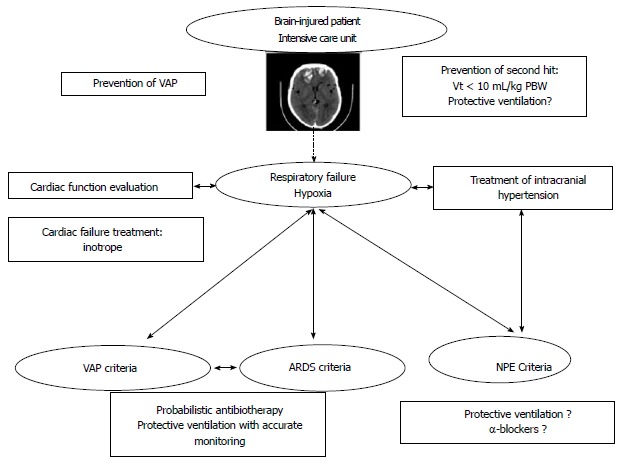
Algorithm approach for pulmonary dysfunction in brain-injured patient. ARDS: Acute respiratory distress syndrome; VAP: Ventilator-associated pneumonia; Vt: Tidal volume; PBW: Predictive body weight; NPE: Neurogenic pulmonary edema.
CONCLUSION
Brain and lung strongly interact via complex pathways. In cases of brain injury, therapeutic strategies should protect the brain but also the lung to avoid worsening of both brain and lung dysfunction. If correctly applied, mechanical ventilation could have beneficial effect on brain oxygenation, even if PEEP and recruitment maneuvers are used. Experimental and clinical studies are needed to explore pathophysiological processes and evaluate optimal ventilator setting in brain-injured patients with lung injuries. A strict monitoring of systemic, respiratory and cerebral parameters is probably required to optimize the management of these patients.
Footnotes
Conflict-of-interest statement: None.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: February 9, 2015
First decision: April 10, 2015
Article in press: May 28, 2015
P- Reviewer: Tanriverdi F, Tanabe S S- Editor: Ji FF L- Editor: A E- Editor: Wu HL
References
- 1.Mascia L. Acute lung injury in patients with severe brain injury: a double hit model. Neurocrit Care. 2009;11:417–426. doi: 10.1007/s12028-009-9242-8. [DOI] [PubMed] [Google Scholar]
- 2.Lee K, Rincon F. Pulmonary complications in patients with severe brain injury. Crit Care Res Pract. 2012;2012:207247. doi: 10.1155/2012/207247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Veeravagu A, Chen YR, Ludwig C, Rincon F, Maltenfort M, Jallo J, Choudhri O, Steinberg GK, Ratliff JK. Acute lung injury in patients with subarachnoid hemorrhage: a nationwide inpatient sample study. World Neurosurg. 2014;82:e235–e241. doi: 10.1016/j.wneu.2014.02.030. [DOI] [PubMed] [Google Scholar]
- 4.Rincon F, Maltenfort M, Dey S, Ghosh S, Vibbert M, Urtecho J, Jallo J, Ratliff JK, McBride JW, Bell R. The prevalence and impact of mortality of the acute respiratory distress syndrome on admissions of patients with ischemic stroke in the United States. J Intensive Care Med. 2014;29:357–364. doi: 10.1177/0885066613491919. [DOI] [PubMed] [Google Scholar]
- 5.Maramattom BV, Weigand S, Reinalda M, Wijdicks EF, Manno EM. Pulmonary complications after intracerebral hemorrhage. Neurocrit Care. 2006;5:115–119. doi: 10.1385/NCC:5:2:115. [DOI] [PubMed] [Google Scholar]
- 6.Zygun DA, Kortbeek JB, Fick GH, Laupland KB, Doig CJ. Non-neurologic organ dysfunction in severe traumatic brain injury. Crit Care Med. 2005;33:654–660. doi: 10.1097/01.ccm.0000155911.01844.54. [DOI] [PubMed] [Google Scholar]
- 7.Kahn JM, Caldwell EC, Deem S, Newell DW, Heckbert SR, Rubenfeld GD. Acute lung injury in patients with subarachnoid hemorrhage: incidence, risk factors, and outcome. Crit Care Med. 2006;34:196–202. doi: 10.1097/01.ccm.0000194540.44020.8e. [DOI] [PubMed] [Google Scholar]
- 8.Santoli F, De Jonghe B, Hayon J, Tran B, Piperaud M, Merrer J, Outin H. Mechanical ventilation in patients with acute ischemic stroke: survival and outcome at one year. Intensive Care Med. 2001;27:1141–1146. doi: 10.1007/s001340100998. [DOI] [PubMed] [Google Scholar]
- 9.Roch A, Michelet P, Jullien AC, Thirion X, Bregeon F, Papazian L, Roche P, Pellet W, Auffray JP. Long-term outcome in intensive care unit survivors after mechanical ventilation for intracerebral hemorrhage. Crit Care Med. 2003;31:2651–2656. doi: 10.1097/01.CCM.0000094222.57803.B4. [DOI] [PubMed] [Google Scholar]
- 10.Piek J, Chesnut RM, Marshall LF, van Berkum-Clark M, Klauber MR, Blunt BA, Eisenberg HM, Jane JA, Marmarou A, Foulkes MA. Extracranial complications of severe head injury. J Neurosurg. 1992;77:901–907. doi: 10.3171/jns.1992.77.6.0901. [DOI] [PubMed] [Google Scholar]
- 11.Solenski NJ, Haley EC, Kassell NF, Kongable G, Germanson T, Truskowski L, Torner JC. Medical complications of aneurysmal subarachnoid hemorrhage: a report of the multicenter, cooperative aneurysm study. Participants of the Multicenter Cooperative Aneurysm Study. Crit Care Med. 1995;23:1007–1017. doi: 10.1097/00003246-199506000-00004. [DOI] [PubMed] [Google Scholar]
- 12.Mascia L, Sakr Y, Pasero D, Payen D, Reinhart K, Vincent JL. Extracranial complications in patients with acute brain injury: a post-hoc analysis of the SOAP study. Intensive Care Med. 2008;34:720–727. doi: 10.1007/s00134-007-0974-7. [DOI] [PubMed] [Google Scholar]
- 13.Pelosi P, Ferguson ND, Frutos-Vivar F, Anzueto A, Putensen C, Raymondos K, Apezteguia C, Desmery P, Hurtado J, Abroug F, et al. Management and outcome of mechanically ventilated neurologic patients. Crit Care Med. 2011;39:1482–1492. doi: 10.1097/CCM.0b013e31821209a8. [DOI] [PubMed] [Google Scholar]
- 14.Kollef MH, Morrow LE, Niederman MS, Leeper KV, Anzueto A, Benz-Scott L, Rodino FJ. Clinical characteristics and treatment patterns among patients with ventilator-associated pneumonia. Chest. 2006;129:1210–1218. doi: 10.1378/chest.129.5.1210. [DOI] [PubMed] [Google Scholar]
- 15.Bronchard R, Albaladejo P, Brezac G, Geffroy A, Seince PF, Morris W, Branger C, Marty J. Early onset pneumonia: risk factors and consequences in head trauma patients. Anesthesiology. 2004;100:234–239. doi: 10.1097/00000542-200402000-00009. [DOI] [PubMed] [Google Scholar]
- 16.American Thoracic Society, Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2005;171:388–416. doi: 10.1164/rccm.200405-644ST. [DOI] [PubMed] [Google Scholar]
- 17.Chamorro Á, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R. The immunology of acute stroke. Nat Rev Neurol. 2012;8:401–410. doi: 10.1038/nrneurol.2012.98. [DOI] [PubMed] [Google Scholar]
- 18.Dirnagl U, Klehmet J, Braun JS, Harms H, Meisel C, Ziemssen T, Prass K, Meisel A. Stroke-induced immunodepression: experimental evidence and clinical relevance. Stroke. 2007;38:770–773. doi: 10.1161/01.STR.0000251441.89665.bc. [DOI] [PubMed] [Google Scholar]
- 19.Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci. 2005;6:775–786. doi: 10.1038/nrn1765. [DOI] [PubMed] [Google Scholar]
- 20.Dziedzic T, Slowik A, Szczudlik A. Nosocomial infections and immunity: lesson from brain-injured patients. Crit Care. 2004;8:266–270. doi: 10.1186/cc2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woratyla SP, Morgan AS, Mackay L, Bernstein B, Barba C. Factors associated with early onset pneumonia in the severely brain-injured patient. Conn Med. 1995;59:643–647. [PubMed] [Google Scholar]
- 22.Lepelletier D, Roquilly A, Demeure dit latte D, Mahe PJ, Loutrel O, Champin P, Corvec S, Naux E, Pinaud M, Lejus C, et al. Retrospective analysis of the risk factors and pathogens associated with early-onset ventilator-associated pneumonia in surgical-ICU head-trauma patients. J Neurosurg Anesthesiol. 2010;22:32–37. doi: 10.1097/ANA.0b013e3181bdf52f. [DOI] [PubMed] [Google Scholar]
- 23.Fields LB. Oral care intervention to reduce incidence of ventilator-associated pneumonia in the neurologic intensive care unit. J Neurosci Nurs. 2008;40:291–298. doi: 10.1097/01376517-200810000-00007. [DOI] [PubMed] [Google Scholar]
- 24.Cinotti R, Dordonnat-Moynard A, Feuillet F, Roquilly A, Rondeau N, Lepelletier D, Caillon J, Asseray N, Blanloeil Y, Rozec B, et al. Risk factors and pathogens involved in early ventilator-acquired pneumonia in patients with severe subarachnoid hemorrhage. Eur J Clin Microbiol Infect Dis. 2014;33:823–830. doi: 10.1007/s10096-013-2020-8. [DOI] [PubMed] [Google Scholar]
- 25.Frontera JA, Fernandez A, Schmidt JM, Claassen J, Wartenberg KE, Badjatia N, Parra A, Connolly ES, Mayer SA. Impact of nosocomial infectious complications after subarachnoid hemorrhage. Neurosurgery. 2008;62:80–87; discussion 87. doi: 10.1227/01.NEU.0000311064.18368.EA. [DOI] [PubMed] [Google Scholar]
- 26.Kasuya Y, Hargett JL, Lenhardt R, Heine MF, Doufas AG, Remmel KS, Ramirez JA, Akça O. Ventilator-associated pneumonia in critically ill stroke patients: frequency, risk factors, and outcomes. J Crit Care. 2011;26:273–279. doi: 10.1016/j.jcrc.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 27.Hilker R, Poetter C, Findeisen N, Sobesky J, Jacobs A, Neveling M, Heiss WD. Nosocomial pneumonia after acute stroke: implications for neurological intensive care medicine. Stroke. 2003;34:975–981. doi: 10.1161/01.STR.0000063373.70993.CD. [DOI] [PubMed] [Google Scholar]
- 28.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, Legall JR, Morris A, Spragg R. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149:818–824. doi: 10.1164/ajrccm.149.3.7509706. [DOI] [PubMed] [Google Scholar]
- 29.Hoesch RE, Lin E, Young M, Gottesman RF, Altaweel L, Nyquist PA, Stevens RD. Acute lung injury in critical neurological illness. Crit Care Med. 2012;40:587–593. doi: 10.1097/CCM.0b013e3182329617. [DOI] [PubMed] [Google Scholar]
- 30.Mascia L, Zavala E, Bosma K, Pasero D, Decaroli D, Andrews P, Isnardi D, Davi A, Arguis MJ, Berardino M, et al. High tidal volume is associated with the development of acute lung injury after severe brain injury: an international observational study. Crit Care Med. 2007;35:1815–1820. doi: 10.1097/01.CCM.0000275269.77467.DF. [DOI] [PubMed] [Google Scholar]
- 31.Holland MC, Mackersie RC, Morabito D, Campbell AR, Kivett VA, Patel R, Erickson VR, Pittet JF. The development of acute lung injury is associated with worse neurologic outcome in patients with severe traumatic brain injury. J Trauma. 2003;55:106–111. doi: 10.1097/01.TA.0000071620.27375.BE. [DOI] [PubMed] [Google Scholar]
- 32.Rincon F, Ghosh S, Dey S, Maltenfort M, Vibbert M, Urtecho J, McBride W, Moussouttas M, Bell R, Ratliff JK, et al. Impact of acute lung injury and acute respiratory distress syndrome after traumatic brain injury in the United States. Neurosurgery. 2012;71:795–803. doi: 10.1227/NEU.0b013e3182672ae5. [DOI] [PubMed] [Google Scholar]
- 33.Wartenberg KE, Schmidt JM, Claassen J, Temes RE, Frontera JA, Ostapkovich N, Parra A, Connolly ES, Mayer SA. Impact of medical complications on outcome after subarachnoid hemorrhage. Crit Care Med. 2006;34:617–623; quiz 624. doi: 10.1097/01.ccm.0000201903.46435.35. [DOI] [PubMed] [Google Scholar]
- 34.Zhao JN, Liu Y, Li HC. Aspiration-related acute respiratory distress syndrome in acute stroke patient. PLoS One. 2015;10:e0118682. doi: 10.1371/journal.pone.0118682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Contant CF, Valadka AB, Gopinath SP, Hannay HJ, Robertson CS. Adult respiratory distress syndrome: a complication of induced hypertension after severe head injury. J Neurosurg. 2001;95:560–568. doi: 10.3171/jns.2001.95.4.0560. [DOI] [PubMed] [Google Scholar]
- 36.Bratton SL, Davis RL. Acute lung injury in isolated traumatic brain injury. Neurosurgery. 1997;40:707–712; discussion 712. doi: 10.1097/00006123-199704000-00009. [DOI] [PubMed] [Google Scholar]
- 37.Ghosh S, Dey SK, Maltenfort M, Vibbert M, Urtecho J, Jallo J. Epidemiological Trends of Adult Respiratory Distress Syndrome (ARDS) After Traumatic Brain Injury in the United States. American Academy of Neurology, New Orleans, La, USA. 2012. [Google Scholar]
- 38.Shanahan W. Acute pulmonary edema as a complication of epileptic seizures. NY Med J. 1908;37:54–56. [Google Scholar]
- 39.Simmons RL, Heisterkamp CA, Collins JA, Genslar S, Martin AM. Respiratory insufficiency in combat casualties. 3. Arterial hypoxemia after wounding. Ann Surg. 1969;170:45–52. doi: 10.1097/00000658-196907000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rogers FB, Shackford SR, Trevisani GT, Davis JW, Mackersie RC, Hoyt DB. Neurogenic pulmonary edema in fatal and nonfatal head injuries. J Trauma. 1995;39:860–866; discussion 866-868. doi: 10.1097/00005373-199511000-00009. [DOI] [PubMed] [Google Scholar]
- 41.Fontes RB, Aguiar PH, Zanetti MV, Andrade F, Mandel M, Teixeira MJ. Acute neurogenic pulmonary edema: case reports and literature review. J Neurosurg Anesthesiol. 2003;15:144–150. doi: 10.1097/00008506-200304000-00013. [DOI] [PubMed] [Google Scholar]
- 42.Baumann A, Audibert G, McDonnell J, Mertes PM. Neurogenic pulmonary edema. Acta Anaesthesiol Scand. 2007;51:447–455. doi: 10.1111/j.1399-6576.2007.01276.x. [DOI] [PubMed] [Google Scholar]
- 43.Friedman JA, Pichelmann MA, Piepgras DG, McIver JI, Toussaint LG, McClelland RL, Nichols DA, Meyer FB, Atkinson JL, Wijdicks EF. Pulmonary complications of aneurysmal subarachnoid hemorrhage. Neurosurgery. 2003;52:1025–1031; discussion 1031-1032. [PubMed] [Google Scholar]
- 44.Weir BK. Pulmonary edema following fatal aneurysm rupture. J Neurosurg. 1978;49:502–507. doi: 10.3171/jns.1978.49.4.0502. [DOI] [PubMed] [Google Scholar]
- 45.Ochiai H, Yamakawa Y, Kubota E. Deformation of the ventrolateral medulla oblongata by subarachnoid hemorrhage from ruptured vertebral artery aneurysms causes neurogenic pulmonary edema. Neurol Med Chir (Tokyo) 2001;41:529–534; discussion 534-535. doi: 10.2176/nmc.41.529. [DOI] [PubMed] [Google Scholar]
- 46.Fein IA, Rackow EC. Neurogenic pulmonary edema. Chest. 1982;81:318–320. doi: 10.1378/chest.81.3.318. [DOI] [PubMed] [Google Scholar]
- 47.Mayer SA, Fink ME, Homma S, Sherman D, LiMandri G, Lennihan L, Solomon RA, Klebanoff LM, Beckford A, Raps EC. Cardiac injury associated with neurogenic pulmonary edema following subarachnoid hemorrhage. Neurology. 1994;44:815–820. doi: 10.1212/wnl.44.5.815. [DOI] [PubMed] [Google Scholar]
- 48.Davison DL, Terek M, Chawla LS. Neurogenic pulmonary edema. Crit Care. 2012;16:212. doi: 10.1186/cc11226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bahloul M, Chaari AN, Kallel H, Khabir A, Ayadi A, Charfeddine H, Hergafi L, Chaari AD, Chelly HE, Ben Hamida C, et al. Neurogenic pulmonary edema due to traumatic brain injury: evidence of cardiac dysfunction. Am J Crit Care. 2006;15:462–470. [PubMed] [Google Scholar]
- 50.Connor RC. Myocardial damage secondary to brain lesions. Am Heart J. 1969;78:145–148. doi: 10.1016/0002-8703(69)90001-5. [DOI] [PubMed] [Google Scholar]
- 51.Mayer SA, Lin J, Homma S, Solomon RA, Lennihan L, Sherman D, Fink ME, Beckford A, Klebanoff LM. Myocardial injury and left ventricular performance after subarachnoid hemorrhage. Stroke. 1999;30:780–786. doi: 10.1161/01.str.30.4.780. [DOI] [PubMed] [Google Scholar]
- 52.Zaroff JG, Rordorf GA, Ogilvy CS, Picard MH. Regional patterns of left ventricular systolic dysfunction after subarachnoid hemorrhage: evidence for neurally mediated cardiac injury. J Am Soc Echocardiogr. 2000;13:774–779. doi: 10.1067/mje.2000.105763. [DOI] [PubMed] [Google Scholar]
- 53.SARNOFF SJ, SARNOFF LC. Neurohemodynamics of pulmonary edema. II. The role of sympathetic pathways in the elevation of pulmonary and stemic vascular pressures following the intracisternal injection of fibrin. Circulation. 1952;6:51–62. doi: 10.1161/01.cir.6.1.51. [DOI] [PubMed] [Google Scholar]
- 54.Ducker TB, Simmons RL. Increased intracranial pressure and pulmonary edema. 2. The hemodynamic response of dogs and monkeys to increased intracranial pressure. J Neurosurg. 1968;28:118–123. doi: 10.3171/jns.1968.28.2.0118. [DOI] [PubMed] [Google Scholar]
- 55.Brashear RE, Ross JC. Hemodynamic effects of elevated cerebrospinal fluid pressure: alterations with adrenergic blockade. J Clin Invest. 1970;49:1324–1333. doi: 10.1172/JCI106348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Minnear FL, Kite C, Hill LA, van der Zee H. Endothelial injury and pulmonary congestion characterize neurogenic pulmonary edema in rabbits. J Appl Physiol (1985) 1987;63:335–341. doi: 10.1152/jappl.1987.63.1.335. [DOI] [PubMed] [Google Scholar]
- 57.van der Zee H, Malik AB, Lee BC, Hakim TS. Lung fluid and protein exchange during intracranial hypertension and role of sympathetic mechanisms. J Appl Physiol Respir Environ Exerc Physiol. 1980;48:273–280. doi: 10.1152/jappl.1980.48.2.273. [DOI] [PubMed] [Google Scholar]
- 58.Theodore J, Robin ED. Speculations on neurogenic pulmonary edema (NPE) Am Rev Respir Dis. 1976;113:405–411. doi: 10.1164/arrd.1976.113.4.405. [DOI] [PubMed] [Google Scholar]
- 59.Maron MB. Effect of elevated vascular pressure transients on protein permeability in the lung. J Appl Physiol (1985) 1989;67:305–310. doi: 10.1152/jappl.1989.67.1.305. [DOI] [PubMed] [Google Scholar]
- 60.Bosso FJ, Lang SA, Maron MB. Role of hemodynamics and vagus nerves in development of fibrin-induced pulmonary edema. J Appl Physiol (1985) 1990;69:2227–2232. doi: 10.1152/jappl.1990.69.6.2227. [DOI] [PubMed] [Google Scholar]
- 61.Bowers RE, McKeen CR, Park BE, Brigham KL. Increased pulmonary vascular permeability follows intracranial hypertension in sheep. Am Rev Respir Dis. 1979;119:637–641. doi: 10.1164/arrd.1979.119.4.637. [DOI] [PubMed] [Google Scholar]
- 62.Melon E, Bonnet F, Lepresle E, Fevrier MJ, Djindjian M, François Y, Gray F, Debras C. Altered capillary permeability in neurogenic pulmonary oedema. Intensive Care Med. 1985;11:323–325. doi: 10.1007/BF00273546. [DOI] [PubMed] [Google Scholar]
- 63.Keegan MT, Lanier WL. Pulmonary edema after resection of a fourth ventricle tumor: possible evidence for a medulla-mediated mechanism. Mayo Clin Proc. 1999;74:264–268. doi: 10.4065/74.3.264. [DOI] [PubMed] [Google Scholar]
- 64.Fein A, Grossman RF, Jones JG, Overland E, Pitts L, Murray JF, Staub NC. The value of edema fluid protein measurement in patients with pulmonary edema. Am J Med. 1979;67:32–38. doi: 10.1016/0002-9343(79)90066-4. [DOI] [PubMed] [Google Scholar]
- 65.Richardson JB. Innervation of the pulmonary circulation: an overview. The Pulmonary Circulation in Health and Disease. 1987:9–14. [Google Scholar]
- 66.McClellan MD, Dauber IM, Weil JV. Elevated intracranial pressure increases pulmonary vascular permeability to protein. J Appl Physiol (1985) 1989;67:1185–1191. doi: 10.1152/jappl.1989.67.3.1185. [DOI] [PubMed] [Google Scholar]
- 67.Peterson BT, Ross JC, Brigham KL. Effect of naloxone on the pulmonary vascular responses to graded levels of intracranial hypertension in anesthetized sheep. Am Rev Respir Dis. 1983;128:1024–1029. doi: 10.1164/arrd.1983.128.6.1024. [DOI] [PubMed] [Google Scholar]
- 68.Avlonitis VS, Fisher AJ, Kirby JA, Dark JH. Pulmonary transplantation: the role of brain death in donor lung injury. Transplantation. 2003;75:1928–1933. doi: 10.1097/01.TP.0000066351.87480.9E. [DOI] [PubMed] [Google Scholar]
- 69.Ott L, McClain CJ, Gillespie M, Young B. Cytokines and metabolic dysfunction after severe head injury. J Neurotrauma. 1994;11:447–472. doi: 10.1089/neu.1994.11.447. [DOI] [PubMed] [Google Scholar]
- 70.Habgood MD, Bye N, Dziegielewska KM, Ek CJ, Lane MA, Potter A, Morganti-Kossmann C, Saunders NR. Changes in blood-brain barrier permeability to large and small molecules following traumatic brain injury in mice. Eur J Neurosci. 2007;25:231–238. doi: 10.1111/j.1460-9568.2006.05275.x. [DOI] [PubMed] [Google Scholar]
- 71.Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr Opin Crit Care. 2002;8:101–105. doi: 10.1097/00075198-200204000-00002. [DOI] [PubMed] [Google Scholar]
- 72.McKeating EG, Andrews PJ, Signorini DF, Mascia L. Transcranial cytokine gradients in patients requiring intensive care after acute brain injury. Br J Anaesth. 1997;78:520–523. doi: 10.1093/bja/78.5.520. [DOI] [PubMed] [Google Scholar]
- 73.López-Aguilar J, Villagrá A, Bernabé F, Murias G, Piacentini E, Real J, Fernández-Segoviano P, Romero PV, Hotchkiss JR, Blanch L. Massive brain injury enhances lung damage in an isolated lung model of ventilator-induced lung injury. Crit Care Med. 2005;33:1077–1083. doi: 10.1097/01.ccm.0000162913.72479.f7. [DOI] [PubMed] [Google Scholar]
- 74.Heuer JF, Pelosi P, Hermann P, Perske C, Crozier TA, Brück W, Quintel M. Acute effects of intracranial hypertension and ARDS on pulmonary and neuronal damage: a randomized experimental study in pigs. Intensive Care Med. 2011;37:1182–1191. doi: 10.1007/s00134-011-2232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu S, Fang CX, Kim J, Ren J. Enhanced pulmonary inflammation following experimental intracerebral hemorrhage. Exp Neurol. 2006;200:245–249. doi: 10.1016/j.expneurol.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 76.Kalsotra A, Zhao J, Anakk S, Dash PK, Strobel HW. Brain trauma leads to enhanced lung inflammation and injury: evidence for role of P4504Fs in resolution. J Cereb Blood Flow Metab. 2007;27:963–974. doi: 10.1038/sj.jcbfm.9600396. [DOI] [PubMed] [Google Scholar]
- 77.Fisher AJ, Donnelly SC, Hirani N, Burdick MD, Strieter RM, Dark JH, Corris PA. Enhanced pulmonary inflammation in organ donors following fatal non-traumatic brain injury. Lancet. 1999;353:1412–1413. doi: 10.1016/S0140-6736(99)00494-8. [DOI] [PubMed] [Google Scholar]
- 78.Yildirim E, Solaroglu I, Okutan O, Ozisik K, Kaptanoglu E, Sargon MF, Sakinci U. Ultrastructural changes in tracheobronchial epithelia following experimental traumatic brain injury in rats: protective effect of erythropoietin. J Heart Lung Transplant. 2004;23:1423–1429. doi: 10.1016/j.healun.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 79.Heuer JF, Selke M, Crozier TA, Pelosi P, Herrmann P, Perske C, Quintel M. Effects of acute intracranial hypertension on extracerebral organs: a randomized experimental study in pigs. J Neurol Surg A Cent Eur Neurosurg. 2012;73:289–295. doi: 10.1055/s-0032-1304813. [DOI] [PubMed] [Google Scholar]
- 80.Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. 1996;24:1125–1128. doi: 10.1097/00003246-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 81.Munford RS, Pugin J. Normal responses to injury prevent systemic inflammation and can be immunosuppressive. Am J Respir Crit Care Med. 2001;163:316–321. doi: 10.1164/ajrccm.163.2.2007102. [DOI] [PubMed] [Google Scholar]
- 82.Offner PJ, Moore EE, Ciesla D. The adrenal response after severe trauma. Am J Surg. 2002;184:649–653; discussion 653-654. doi: 10.1016/s0002-9610(02)01101-7. [DOI] [PubMed] [Google Scholar]
- 83.Chrousos GP. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med. 1995;332:1351–1362. doi: 10.1056/NEJM199505183322008. [DOI] [PubMed] [Google Scholar]
- 84.Moore FA, Sauaia A, Moore EE, Haenel JB, Burch JM, Lezotte DC. Postinjury multiple organ failure: a bimodal phenomenon. J Trauma. 1996;40:501–510; discussion 510-512. doi: 10.1097/00005373-199604000-00001. [DOI] [PubMed] [Google Scholar]
- 85.Webster JI, Tonelli L, Sternberg EM. Neuroendocrine regulation of immunity. Annu Rev Immunol. 2002;20:125–163. doi: 10.1146/annurev.immunol.20.082401.104914. [DOI] [PubMed] [Google Scholar]
- 86.Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. N Engl J Med. 2005;353:1711–1723. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 87.Prigent H, Maxime V, Annane D. Clinical review: corticotherapy in sepsis. Crit Care. 2004;8:122–129. doi: 10.1186/cc2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cohan P, Wang C, McArthur DL, Cook SW, Dusick JR, Armin B, Swerdloff R, Vespa P, Muizelaar JP, Cryer HG, et al. Acute secondary adrenal insufficiency after traumatic brain injury: a prospective study. Crit Care Med. 2005;33:2358–2366. doi: 10.1097/01.ccm.0000181735.51183.a7. [DOI] [PubMed] [Google Scholar]
- 89.Dimopoulou I, Tsagarakis S, Kouyialis AT, Roussou P, Assithianakis G, Christoforaki M, Ilias I, Sakas DE, Thalassinos N, Roussos C. Hypothalamic-pituitary-adrenal axis dysfunction in critically ill patients with traumatic brain injury: incidence, pathophysiology, and relationship to vasopressor dependence and peripheral interleukin-6 levels. Crit Care Med. 2004;32:404–408. doi: 10.1097/01.CCM.0000108885.37811.CA. [DOI] [PubMed] [Google Scholar]
- 90.Dimopoulou I, Tsagarakis S, Theodorakopoulou M, Douka E, Zervou M, Kouyialis AT, Thalassinos N, Roussos C. Endocrine abnormalities in critical care patients with moderate-to-severe head trauma: incidence, pattern and predisposing factors. Intensive Care Med. 2004;30:1051–1057. doi: 10.1007/s00134-004-2257-x. [DOI] [PubMed] [Google Scholar]
- 91.Llompart-Pou JA, Raurich JM, Pérez-Bárcena J, Barceló A, Ibáñez J, Ayestarán JI. Acute Hypothalamic-pituitary-adrenal response in traumatic brain injury with and without extracerebral trauma. Neurocrit Care. 2008;9:230–236. doi: 10.1007/s12028-008-9115-6. [DOI] [PubMed] [Google Scholar]
- 92.Mesotten D, Vanhorebeek I, Van den Berghe G. The altered adrenal axis and treatment with glucocorticoids during critical illness. Nat Clin Pract Endocrinol Metab. 2008;4:496–505. doi: 10.1038/ncpendmet0921. [DOI] [PubMed] [Google Scholar]
- 93.Agha A, Rogers B, Mylotte D, Taleb F, Tormey W, Phillips J, Thompson CJ. Neuroendocrine dysfunction in the acute phase of traumatic brain injury. Clin Endocrinol (Oxf) 2004;60:584–591. doi: 10.1111/j.1365-2265.2004.02023.x. [DOI] [PubMed] [Google Scholar]
- 94.Hoen S, Asehnoune K, Brailly-Tabard S, Mazoit JX, Benhamou D, Moine P, Edouard AR. Cortisol response to corticotropin stimulation in trauma patients: influence of hemorrhagic shock. Anesthesiology. 2002;97:807–813. doi: 10.1097/00000542-200210000-00010. [DOI] [PubMed] [Google Scholar]
- 95.Papanicolaou DA, Tsigos C, Oldfield EH, Chrousos GP. Acute glucocorticoid deficiency is associated with plasma elevations of interleukin-6: does the latter participate in the symptomatology of the steroid withdrawal syndrome and adrenal insufficiency? J Clin Endocrinol Metab. 1996;81:2303–2306. doi: 10.1210/jcem.81.6.8964868. [DOI] [PubMed] [Google Scholar]
- 96.Gebhard F, Pfetsch H, Steinbach G, Strecker W, Kinzl L, Brückner UB. Is interleukin 6 an early marker of injury severity following major trauma in humans? Arch Surg. 2000;135:291–295. doi: 10.1001/archsurg.135.3.291. [DOI] [PubMed] [Google Scholar]
- 97.Bochicchio GV, Napolitano LM, Joshi M, McCarter RJ, Scalea TM. Systemic inflammatory response syndrome score at admission independently predicts infection in blunt trauma patients. J Trauma. 2001;50:817–820. doi: 10.1097/00005373-200105000-00007. [DOI] [PubMed] [Google Scholar]
- 98.Hoover L, Bochicchio GV, Napolitano LM, Joshi M, Bochicchio K, Meyer W, Scalea TM. Systemic inflammatory response syndrome and nosocomial infection in trauma. J Trauma. 2006;61:310–316; discussion 316-317. doi: 10.1097/01.ta.0000229052.75460.c2. [DOI] [PubMed] [Google Scholar]
- 99.Giannoudis PV. Current concepts of the inflammatory response after major trauma: an update. Injury. 2003;34:397–404. doi: 10.1016/s0020-1383(02)00416-3. [DOI] [PubMed] [Google Scholar]
- 100.Roquilly A, Mahe PJ, Seguin P, Guitton C, Floch H, Tellier AC, Merson L, Renard B, Malledant Y, Flet L, et al. Hydrocortisone therapy for patients with multiple trauma: the randomized controlled HYPOLYTE study. JAMA. 2011;305:1201–1209. doi: 10.1001/jama.2011.360. [DOI] [PubMed] [Google Scholar]
- 101.Asehnoune K, Seguin P, Allary J, Feuillet F, Lasocki S, Cook F, Floch H, Chabanne R, Geeraerts T, Roger C, et al. Hydrocortisone and fludrocortisone for prevention of hospital-acquired pneumonia in patients with severe traumatic brain injury (Corti-TC): a double-blind, multicentre phase 3, randomised placebo-controlled trial. Lancet Respir Med. 2014;2:706–716. doi: 10.1016/S2213-2600(14)70144-4. [DOI] [PubMed] [Google Scholar]
- 102.Marklund N, Peltonen M, Nilsson TK, Olsson T. Low and high circulating cortisol levels predict mortality and cognitive dysfunction early after stroke. J Intern Med. 2004;256:15–21. doi: 10.1111/j.1365-2796.2004.01334.x. [DOI] [PubMed] [Google Scholar]
- 103.Hannawi Y, Hannawi B, Rao CP, Suarez JI, Bershad EM. Stroke-associated pneumonia: major advances and obstacles. Cerebrovasc Dis. 2013;35:430–443. doi: 10.1159/000350199. [DOI] [PubMed] [Google Scholar]
- 104.Stevens RD, Puybasset L. The brain-lung-brain axis. Intensive Care Med. 2011;37:1054–1056. doi: 10.1007/s00134-011-2233-1. [DOI] [PubMed] [Google Scholar]
- 105.Hegeman MA, Hennus MP, Heijnen CJ, Specht PA, Lachmann B, Jansen NJ, van Vught AJ, Cobelens PM. Ventilator-induced endothelial activation and inflammation in the lung and distal organs. Crit Care. 2009;13:R182. doi: 10.1186/cc8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Slutsky AS, Tremblay LN. Multiple system organ failure. Is mechanical ventilation a contributing factor? Am J Respir Crit Care Med. 1998;157:1721–1725. doi: 10.1164/ajrccm.157.6.9709092. [DOI] [PubMed] [Google Scholar]
- 107.Hopkins RO, Brett S. Chronic neurocognitive effects of critical illness. Curr Opin Crit Care. 2005;11:369–375. doi: 10.1097/01.ccx.0000166399.88635.a5. [DOI] [PubMed] [Google Scholar]
- 108.Milbrandt EB, Angus DC. Potential mechanisms and markers of critical illness-associated cognitive dysfunction. Curr Opin Crit Care. 2005;11:355–359. doi: 10.1097/01.ccx.0000170508.63067.04. [DOI] [PubMed] [Google Scholar]
- 109.Hopkins RO, Jackson JC. Long-term neurocognitive function after critical illness. Chest. 2006;130:869–878. doi: 10.1378/chest.130.3.869. [DOI] [PubMed] [Google Scholar]
- 110.Pustavoitau A, Stevens RD. Mechanisms of neurologic failure in critical illness. Crit Care Clin. 2008;24:1–24, vii. doi: 10.1016/j.ccc.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 111.Neves G, Cooke SF, Bliss TV. Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat Rev Neurosci. 2008;9:65–75. doi: 10.1038/nrn2303. [DOI] [PubMed] [Google Scholar]
- 112.Janz DR, Abel TW, Jackson JC, Gunther ML, Heckers S, Ely EW. Brain autopsy findings in intensive care unit patients previously suffering from delirium: a pilot study. J Crit Care. 2010;25:538.e7–538.12. doi: 10.1016/j.jcrc.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nguyen DN, Spapen H, Su F, Schiettecatte J, Shi L, Hachimi-Idrissi S, Huyghens L. Elevated serum levels of S-100beta protein and neuron-specific enolase are associated with brain injury in patients with severe sepsis and septic shock. Crit Care Med. 2006;34:1967–1974. doi: 10.1097/01.CCM.0000217218.51381.49. [DOI] [PubMed] [Google Scholar]
- 114.Mrozek S, Dumurgier J, Citerio G, Mebazaa A, Geeraerts T. Biomarkers and acute brain injuries: interest and limits. Crit Care. 2014;18:220. doi: 10.1186/cc13841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fries M, Bickenbach J, Henzler D, Beckers S, Dembinski R, Sellhaus B, Rossaint R, Kuhlen R. S-100 protein and neurohistopathologic changes in a porcine model of acute lung injury. Anesthesiology. 2005;102:761–767. doi: 10.1097/00000542-200504000-00011. [DOI] [PubMed] [Google Scholar]
- 116.López-Aguilar J, Fernández-Gonzalo MS, Turon M, Quílez ME, Gómez-Simón V, Jódar MM, Blanch L. Lung-brain interaction in the mechanically ventilated patient. Med Intensiva. 2013;37:485–492. doi: 10.1016/j.medin.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 117.Bickenbach J, Zoremba N, Fries M, Dembinski R, Doering R, Ogawa E, Rossaint R, Kuhlen R. Low tidal volume ventilation in a porcine model of acute lung injury improves cerebral tissue oxygenation. Anesth Analg. 2009;109:847–855. doi: 10.1213/ane.0b013e3181ad5769. [DOI] [PubMed] [Google Scholar]
- 118.Quilez ME, Fuster G, Villar J, Flores C, Martí-Sistac O, Blanch L, López-Aguilar J. Injurious mechanical ventilation affects neuronal activation in ventilated rats. Crit Care. 2011;15:R124. doi: 10.1186/cc10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Slutsky AS. Lung injury caused by mechanical ventilation. Chest. 1999;116:9S–15S. doi: 10.1378/chest.116.suppl_1.9s-a. [DOI] [PubMed] [Google Scholar]
- 120.Gattinoni L, Protti A, Caironi P, Carlesso E. Ventilator-induced lung injury: the anatomical and physiological framework. Crit Care Med. 2010;38:S539–S548. doi: 10.1097/CCM.0b013e3181f1fcf7. [DOI] [PubMed] [Google Scholar]
- 121.Tremblay LN, Slutsky AS. Ventilator-induced injury: from barotrauma to biotrauma. Proc Assoc Am Physicians. 1998;110:482–488. [PubMed] [Google Scholar]
- 122.Mercat A, Richard JC, Vielle B, Jaber S, Osman D, Diehl JL, Lefrant JY, Prat G, Richecoeur J, Nieszkowska A, et al. Positive end-expiratory pressure setting in adults with acute lung injury and acute respiratory distress syndrome: a randomized controlled trial. JAMA. 2008;299:646–655. doi: 10.1001/jama.299.6.646. [DOI] [PubMed] [Google Scholar]
- 123.Meade MO, Cook DJ, Guyatt GH, Slutsky AS, Arabi YM, Cooper DJ, Davies AR, Hand LE, Zhou Q, Thabane L, et al. Ventilation strategy using low tidal volumes, recruitment maneuvers, and high positive end-expiratory pressure for acute lung injury and acute respiratory distress syndrome: a randomized controlled trial. JAMA. 2008;299:637–645. doi: 10.1001/jama.299.6.637. [DOI] [PubMed] [Google Scholar]
- 124.Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 125.Ranieri VM, Suter PM, Tortorella C, De Tullio R, Dayer JM, Brienza A, Bruno F, Slutsky AS. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA. 1999;282:54–61. doi: 10.1001/jama.282.1.54. [DOI] [PubMed] [Google Scholar]
- 126.Ranieri VM, Giunta F, Suter PM, Slutsky AS. Mechanical ventilation as a mediator of multisystem organ failure in acute respiratory distress syndrome. JAMA. 2000;284:43–44. doi: 10.1001/jama.284.1.43. [DOI] [PubMed] [Google Scholar]
- 127.Gajic O, Dara SI, Mendez JL, Adesanya AO, Festic E, Caples SM, Rana R, St Sauver JL, Lymp JF, Afessa B, et al. Ventilator-associated lung injury in patients without acute lung injury at the onset of mechanical ventilation. Crit Care Med. 2004;32:1817–1824. doi: 10.1097/01.ccm.0000133019.52531.30. [DOI] [PubMed] [Google Scholar]
- 128.Gajic O, Frutos-Vivar F, Esteban A, Hubmayr RD, Anzueto A. Ventilator settings as a risk factor for acute respiratory distress syndrome in mechanically ventilated patients. Intensive Care Med. 2005;31:922–926. doi: 10.1007/s00134-005-2625-1. [DOI] [PubMed] [Google Scholar]
- 129.Krebs J, Tsagogiorgas C, Pelosi P, Rocco PR, Hottenrott M, Sticht C, Yard B, Luecke T. Open lung approach with low tidal volume mechanical ventilation attenuates lung injury in rats with massive brain damage. Crit Care. 2014;18:R59. doi: 10.1186/cc13813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ainslie PN, Duffin J. Integration of cerebrovascular CO2 reactivity and chemoreflex control of breathing: mechanisms of regulation, measurement, and interpretation. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1473–R1495. doi: 10.1152/ajpregu.91008.2008. [DOI] [PubMed] [Google Scholar]
- 131.Brain Trauma Foundation, American Association of Neurological Surgeons, Congress of Neurological Surgeons. Guidelines for the management of severe traumatic brain injury. J Neurotrauma. 2007;24 Suppl 1:S1–106. doi: 10.1089/neu.2007.9999. [DOI] [PubMed] [Google Scholar]
- 132.Petridis AK, Doukas A, Kienke S, Maslehaty H, Mahvash M, Barth H, Mehdorn HM. The effect of lung-protective permissive hypercapnia in intracerebral pressure in patients with subarachnoid haemorrhage and ARDS. A retrospective study. Acta Neurochir (Wien) 2010;152:2143–2145. doi: 10.1007/s00701-010-0761-z. [DOI] [PubMed] [Google Scholar]
- 133.Westermaier T, Stetter C, Kunze E, Willner N, Holzmeier J, Kilgenstein C, Lee JY, Ernestus RI, Roewer N, Muellenbach RM. Controlled transient hypercapnia: a novel approach for the treatment of delayed cerebral ischemia after subarachnoid hemorrhage? J Neurosurg. 2014;121:1056–1062. doi: 10.3171/2014.7.JNS132611. [DOI] [PubMed] [Google Scholar]
- 134.Ranieri VM, Eissa NT, Corbeil C, Chassé M, Braidy J, Matar N, Milic-Emili J. Effects of positive end-expiratory pressure on alveolar recruitment and gas exchange in patients with the adult respiratory distress syndrome. Am Rev Respir Dis. 1991;144:544–551. doi: 10.1164/ajrccm/144.3_Pt_1.544. [DOI] [PubMed] [Google Scholar]
- 135.Blanch L, Fernández R, Benito S, Mancebo J, Net A. Effect of PEEP on the arterial minus end-tidal carbon dioxide gradient. Chest. 1987;92:451–454. doi: 10.1378/chest.92.3.451. [DOI] [PubMed] [Google Scholar]
- 136.Doblar DD, Santiago TV, Kahn AU, Edelman NH. The effect of positive end-expiratory pressure ventilation (PEEP) on cerebral blood flow and cerebrospinal fluid pressure in goats. Anesthesiology. 1981;55:244–250. doi: 10.1097/00000542-198109000-00010. [DOI] [PubMed] [Google Scholar]
- 137.Mascia L, Grasso S, Fiore T, Bruno F, Berardino M, Ducati A. Cerebro-pulmonary interactions during the application of low levels of positive end-expiratory pressure. Intensive Care Med. 2005;31:373–379. doi: 10.1007/s00134-004-2491-2. [DOI] [PubMed] [Google Scholar]
- 138.Lou M, Xue F, Chen L, Xue Y, Wang K. Is high PEEP ventilation strategy safe for acute respiratory distress syndrome after severe traumatic brain injury? Brain Inj. 2012;26:887–890. doi: 10.3109/02699052.2012.660514. [DOI] [PubMed] [Google Scholar]
- 139.Muench E, Bauhuf C, Roth H, Horn P, Phillips M, Marquetant N, Quintel M, Vajkoczy P. Effects of positive end-expiratory pressure on regional cerebral blood flow, intracranial pressure, and brain tissue oxygenation. Crit Care Med. 2005;33:2367–2372. doi: 10.1097/01.ccm.0000181732.37319.df. [DOI] [PubMed] [Google Scholar]
- 140.Schramm P, Closhen D, Felkel M, Berres M, Klein KU, David M, Werner C, Engelhard K. Influence of PEEP on cerebral blood flow and cerebrovascular autoregulation in patients with acute respiratory distress syndrome. J Neurosurg Anesthesiol. 2013;25:162–167. doi: 10.1097/ANA.0b013e31827c2f46. [DOI] [PubMed] [Google Scholar]
- 141.Caricato A, Conti G, Della Corte F, Mancino A, Santilli F, Sandroni C, Proietti R, Antonelli M. Effects of PEEP on the intracranial system of patients with head injury and subarachnoid hemorrhage: the role of respiratory system compliance. J Trauma. 2005;58:571–576. doi: 10.1097/01.ta.0000152806.19198.db. [DOI] [PubMed] [Google Scholar]
- 142.Georgiadis D, Schwarz S, Baumgartner RW, Veltkamp R, Schwab S. Influence of positive end-expiratory pressure on intracranial pressure and cerebral perfusion pressure in patients with acute stroke. Stroke. 2001;32:2088–2092. doi: 10.1161/hs0901.095406. [DOI] [PubMed] [Google Scholar]
- 143.McGuire G, Crossley D, Richards J, Wong D. Effects of varying levels of positive end-expiratory pressure on intracranial pressure and cerebral perfusion pressure. Crit Care Med. 1997;25:1059–1062. doi: 10.1097/00003246-199706000-00025. [DOI] [PubMed] [Google Scholar]
- 144.Toung TJ, Aizawa H, Traystman RJ. Effects of positive end-expiratory pressure ventilation on cerebral venous pressure with head elevation in dogs. J Appl Physiol (1985) 2000;88:655–661. doi: 10.1152/jappl.2000.88.2.655. [DOI] [PubMed] [Google Scholar]
- 145.Mazzeo AT, Fanelli V, Mascia L. Brain-lung crosstalk in critical care: how protective mechanical ventilation can affect the brain homeostasis. Minerva Anestesiol. 2013;79:299–309. [PubMed] [Google Scholar]
- 146.Constantin JM, Jaber S, Futier E, Cayot-Constantin S, Verny-Pic M, Jung B, Bailly A, Guerin R, Bazin JE. Respiratory effects of different recruitment maneuvers in acute respiratory distress syndrome. Crit Care. 2008;12:R50. doi: 10.1186/cc6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Badet M, Bayle F, Richard JC, Guérin C. Comparison of optimal positive end-expiratory pressure and recruitment maneuvers during lung-protective mechanical ventilation in patients with acute lung injury/acute respiratory distress syndrome. Respir Care. 2009;54:847–854. doi: 10.4187/002013209793800448. [DOI] [PubMed] [Google Scholar]
- 148.Bein T, Kuhr LP, Bele S, Ploner F, Keyl C, Taeger K. Lung recruitment maneuver in patients with cerebral injury: effects on intracranial pressure and cerebral metabolism. Intensive Care Med. 2002;28:554–558. doi: 10.1007/s00134-002-1273-y. [DOI] [PubMed] [Google Scholar]
- 149.Marini JJ. A lung-protective approach to ventilating ARDS. Respir Care Clin N Am. 1998;4:633–663, viii. [PubMed] [Google Scholar]
- 150.Sydow M, Burchardi H, Ephraim E, Zielmann S, Crozier TA. Long-term effects of two different ventilatory modes on oxygenation in acute lung injury. Comparison of airway pressure release ventilation and volume-controlled inverse ratio ventilation. Am J Respir Crit Care Med. 1994;149:1550–1556. doi: 10.1164/ajrccm.149.6.8004312. [DOI] [PubMed] [Google Scholar]
- 151.Neumann P, Berglund JE, Mondéjar EF, Magnusson A, Hedenstierna G. Effect of different pressure levels on the dynamics of lung collapse and recruitment in oleic-acid-induced lung injury. Am J Respir Crit Care Med. 1998;158:1636–1643. doi: 10.1164/ajrccm.158.5.9711095. [DOI] [PubMed] [Google Scholar]
- 152.Constantin JM, Cayot-Constantin S, Roszyk L, Futier E, Sapin V, Dastugue B, Bazin JE, Rouby JJ. Response to recruitment maneuver influences net alveolar fluid clearance in acute respiratory distress syndrome. Anesthesiology. 2007;106:944–951. doi: 10.1097/01.anes.0000265153.17062.64. [DOI] [PubMed] [Google Scholar]
- 153.Nemer SN, Caldeira JB, Azeredo LM, Garcia JM, Silva RT, Prado D, Santos RG, Guimarães BS, Ramos RA, Noé RA, et al. Alveolar recruitment maneuver in patients with subarachnoid hemorrhage and acute respiratory distress syndrome: a comparison of 2 approaches. J Crit Care. 2011;26:22–27. doi: 10.1016/j.jcrc.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 154.Zhang XY, Yang ZJ, Wang QX, Fan HR. Impact of positive end-expiratory pressure on cerebral injury patients with hypoxemia. Am J Emerg Med. 2011;29:699–703. doi: 10.1016/j.ajem.2010.01.042. [DOI] [PubMed] [Google Scholar]
- 155.Wolf S, Plev DV, Trost HA, Lumenta CB. Open lung ventilation in neurosurgery: an update on brain tissue oxygenation. Acta Neurochir Suppl. 2005;95:103–105. doi: 10.1007/3-211-32318-x_22. [DOI] [PubMed] [Google Scholar]
- 156.Gattinoni L, Carlesso E, Taccone P, Polli F, Guérin C, Mancebo J. Prone positioning improves survival in severe ARDS: a pathophysiologic review and individual patient meta-analysis. Minerva Anestesiol. 2010;76:448–454. [PubMed] [Google Scholar]
- 157.Guerin C, Baboi L, Richard JC. Mechanisms of the effects of prone positioning in acute respiratory distress syndrome. Intensive Care Med. 2014;40:1634–1642. doi: 10.1007/s00134-014-3500-8. [DOI] [PubMed] [Google Scholar]
- 158.Lee JM, Bae W, Lee YJ, Cho YJ. The efficacy and safety of prone positional ventilation in acute respiratory distress syndrome: updated study-level meta-analysis of 11 randomized controlled trials. Crit Care Med. 2014;42:1252–1262. doi: 10.1097/CCM.0000000000000122. [DOI] [PubMed] [Google Scholar]
- 159.Reinprecht A, Greher M, Wolfsberger S, Dietrich W, Illievich UM, Gruber A. Prone position in subarachnoid hemorrhage patients with acute respiratory distress syndrome: effects on cerebral tissue oxygenation and intracranial pressure. Crit Care Med. 2003;31:1831–1838. doi: 10.1097/01.CCM.0000063453.93855.0A. [DOI] [PubMed] [Google Scholar]
- 160.Gritti P, Lanterna LA, Re M, Martchenko S, Olivotto P, Brembilla C, Agostinis C, Paganoni G, Lorini FL. The use of inhaled nitric oxide and prone position in an ARDS patient with severe traumatic brain injury during spine stabilization. J Anesth. 2013;27:293–297. doi: 10.1007/s00540-012-1495-2. [DOI] [PubMed] [Google Scholar]
- 161.Ashton-Cleary DT, Duffy MR. Prone ventilation for refractory hypoxaemia in a patient with severe chest wall disruption and traumatic brain injury. Br J Anaesth. 2011;107:1009–1010. doi: 10.1093/bja/aer374. [DOI] [PubMed] [Google Scholar]
- 162.Torres A, Ferrer M, Badia JR. Treatment guidelines and outcomes of hospital-acquired and ventilator-associated pneumonia. Clin Infect Dis. 2010;51 Suppl 1:S48–S53. doi: 10.1086/653049. [DOI] [PubMed] [Google Scholar]
- 163.O’Grady NP, Murray PR, Ames N. Preventing ventilator-associated pneumonia: does the evidence support the practice? JAMA. 2012;307:2534–2539. doi: 10.1001/jama.2012.6445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Esteban A, Anzueto A, Frutos F, Alía I, Brochard L, Stewart TE, Benito S, Epstein SK, Apezteguía C, Nightingale P, et al. Characteristics and outcomes in adult patients receiving mechanical ventilation: a 28-day international study. JAMA. 2002;287:345–355. doi: 10.1001/jama.287.3.345. [DOI] [PubMed] [Google Scholar]
- 165.Roquilly A, Cinotti R, Jaber S, Vourc’h M, Pengam F, Mahe PJ, Lakhal K, Demeure Dit Latte D, Rondeau N, Loutrel O, et al. Implementation of an evidence-based extubation readiness bundle in 499 brain-injured patients. a before-after evaluation of a quality improvement project. Am J Respir Crit Care Med. 2013;188:958–966. doi: 10.1164/rccm.201301-0116OC. [DOI] [PubMed] [Google Scholar]
- 166.Adams HP, del Zoppo G, Alberts MJ, Bhatt DL, Brass L, Furlan A, Grubb RL, Higashida RT, Jauch EC, Kidwell C, et al. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: The American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Circulation. 2007;115:e478–e534. doi: 10.1161/CIRCULATIONAHA.107.181486. [DOI] [PubMed] [Google Scholar]
- 167.Ickenstein GW, Riecker A, Höhlig C, Müller R, Becker U, Reichmann H, Prosiegel M. Pneumonia and in-hospital mortality in the context of neurogenic oropharyngeal dysphagia (NOD) in stroke and a new NOD step-wise concept. J Neurol. 2010;257:1492–1499. doi: 10.1007/s00415-010-5558-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hinchey JA, Shephard T, Furie K, Smith D, Wang D, Tonn S. Formal dysphagia screening protocols prevent pneumonia. Stroke. 2005;36:1972–1976. doi: 10.1161/01.STR.0000177529.86868.8d. [DOI] [PubMed] [Google Scholar]
- 169.Gomes CA, Lustosa SA, Matos D, Andriolo RB, Waisberg DR, Waisberg J. Percutaneous endoscopic gastrostomy versus nasogastric tube feeding for adults with swallowing disturbances. Cochrane Database Syst Rev. 2012;3:CD008096. doi: 10.1002/14651858.CD008096.pub3. [DOI] [PubMed] [Google Scholar]
- 170.Lu WH, Hsieh KS, Lu PJ, Wu YS, Ho WY, Cheng PW, Lai CC, Hsiao M, Tseng CJ. Different impacts of α- and β-blockers in neurogenic hypertension produced by brainstem lesions in rat. Anesthesiology. 2014;120:1192–1204. doi: 10.1097/ALN.0000000000000218. [DOI] [PubMed] [Google Scholar]
- 171.Davison DL, Chawla LS, Selassie L, Tevar R, Junker C, Seneff MG. Neurogenic pulmonary edema: successful treatment with IV phentolamine. Chest. 2012;141:793–795. doi: 10.1378/chest.11-0789. [DOI] [PubMed] [Google Scholar]
- 172.Wohns RN, Tamas L, Pierce KR, Howe JF. Chlorpromazine treatment for neurogenic pulmonary edema. Crit Care Med. 1985;13:210–211. doi: 10.1097/00003246-198503000-00016. [DOI] [PubMed] [Google Scholar]