

Abstract
A mounting body of evidence in cancer research suggests that the local microenvironment of tumor cells has a profound influence on cancer progression and metastasis. In vitro studies on the tumor microenvironment and its pharmacological modulation, however, are often hampered by the technical challenges associated with creating physiological cell culture environments that integrate cancer cells with the key components of their native niche such as neighboring cells and extracellular matrix (ECM) to mimic complex microarchitecture of cancerous tissue. Using early-stage breast cancer as a model disease, here we describe a biomimetic microengineering strategy to reconstitute three-dimensional (3D) structural organization and microenvironment of breast tumors in human cell-based in vitro models. Specifically, we developed a microsystem that enabled co-culture of breast tumor spheroids with human mammary ductal epithelial cells and mammary fibroblasts in a compartmentalized 3D microfluidic device to replicate microarchitecture of breast ductal carcinoma in situ (DCIS). We also explored the potential of this breast cancer-on-a-chip system as a drug screening platform by evaluating the efficacy and toxicity of an anticancer drug (paclitaxel). Our microengineered disease model represents the first critical step towards recapitulating pathophysiological complexity of breast cancer, and may serve as an enabling tool to systematically examine the contribution of the breast cancer microenvironment to the progression of DCIS to an invasive form of the disease.
Introduction
In the early stages of breast cancer, neoplastic epithelial cells accumulate in the lumen of the mammary duct and form a pre-invasive cancerous lesion known as ductal carcinoma in situ (DCIS) (Fig. 1A). Progression to invasive breast cancer occurs when tumor cells in DCIS acquire the ability to penetrate their basement membrane and invade the surrounding tissue.1, 2 This transition from DCIS to invasive ductal carcinoma (IDC) is accompanied by aberrant changes in various biological processes such as matrix remodeling,3 paracrine signaling,4 and immune responses5 that together contribute to increased invasion of cancer cells and their metastasis to distant organs. With the introduction of screening mammography, the rate at which DCIS is diagnosed has increased by more than tenfold over the past decades and as a result, DCIS now accounts for approximately 20% of all breast cancers6. However, it remains a formidable clinical challenge to identify DCIS patients with an increased likelihood of progression to invasive cancer. The most critical barrier to this type of predictive diagnosis has been a lack of fundamental understanding on the biological underpinnings of the malignant transformation of DCIS lesions to IDC.
Figure 1. A human breast cancer-on-a-chip.
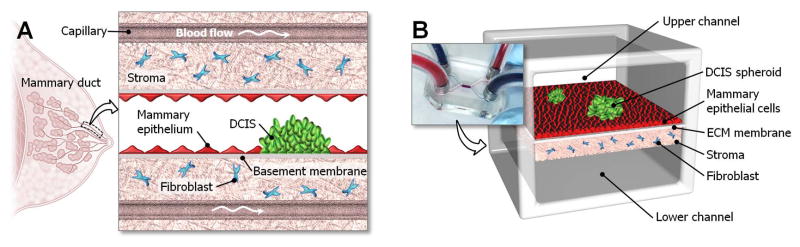
A. DCIS is embedded in a mammary duct consisting of the mammary epithelium and a basement membrane surrounded by stromal tissue that contains fibroblasts. B. The microarchitecture of DCIS and the surrounding tissue layers is reproduced in the breast cancer-on-a-chip microdevice comprised of the upper and lower cell culture chambers separated by an ECM-derived membrane that mimics a basement membrane in vivo. DCIS spheroids are embedded in the mammary epithelium formed in the upper channel, and the fibroblast-containing stromal layer is created on the other side of the membrane.
Increasing recognition of the tumor microenvironment as a key regulator of cancer progression has led researchers to investigate its role in the transition of DCIS to malignancy. The native microenvironment of DCIS is composed of ductal epithelial cells, the basement membrane, and the underlying mesenchyme that contains ECM and various cell types such as mammary fibroblasts, adipocytes, and endothelial cells. Previous studies have suggested that biochemical and biophysical signals produced by these microenvironmental components may trigger and facilitate the invasive progression of DCIS. For example, researchers have shown that growth factors and matrix enzymes secreted by mammary fibroblasts in the stroma of DCIS lesions can increase tumor cell proliferation and promote their invasion7, 8. Similar studies also suggest that ECM stiffening due to abnormal matrix remodeling in DCIS-associated stroma may lead to increased tumor cell migration and invasion9. Despite emerging evidence, however, further research progress in this area has been greatly challenged by the limited ability of existing models to recapitulate the complexity of DCIS and its in vivo microenvironment. Specifically, mixed co-cultures of DCIS cells and mammary fibroblasts commonly employed in current in vitro models fail to reproduce physiological relative spatial arrangement of DCIS and its surrounding stroma, which has been suggested as an important determinant of cancer-stromal interactions and tumor invasiveness10, 11. Limitations of conventional in vitro approaches also make it challenging to reconstitute three-dimensionality of DCIS lesions and their association with surrounding normal epithelium and basement membrane that may affect dynamics of intercellular interactions leading to cancer progression and metastasis12–14. Although xenograft animal models have been used successfully in DCIS studies15, 16, they require complex experimental procedures for intraductal injection of DCIS cells and more importantly, suffer from the inability to precisely control and manipulate microenvironmental factors for mechanistic investigation of underlying disease processes. Therefore, a critical need remains for human-relevant disease models that better represent DCIS in vivo and enable precise spatiotemporal control over key biochemical and biomechanical components of its tumor microenvironment.
As the first step towards addressing this unmet need, here we describe a biomimetic microengineering approach to develop a human cell-based disease model that replicates 3D microarchitecture of DCIS lesions in tissue-specific microenvironment of the human mammary duct. This 3D microsystem enables microfluidic co-culture of multicellular DCIS spheroids with normal human mammary ductal epithelial cells in close apposition to human mammary fibroblasts embedded in a 3D ECM scaffold (Fig. 1B). Our work builds upon recent advances in organ-on-chip technology for breast cancer research and extends the capabilities of existing microfluidic breast cancer models10, 11, 17–20. We also demonstrate the potential use of our model as a screening platform for assessing the efficacy and toxicity of chemotherapeutic drugs.
Materials and Methods
Cell culture
Human mammary epithelial cell line (HMT-3522, also known as S1; Public Health England) was cultured in mammary epithelium growth medium (MEGM) (Lonza). Human primary mammary fibroblasts (HMF; ScienCell) were grown in fibroblast medium containing 2% fetal bovine serum (FBS), 1% fibroblast growth supplement, and 1% penicillin/streptomycin (ScienCell). The DCIS cell line (MCF10-DCIS.com; Asterand) was cultured in DMEM/F12 (50:50) supplemented with 2mM L-glutamine, 5% horse serum, and 1% penicillin/streptomycin (Invitrogen).
Generation of fluorescent protein expressing cells
To achieve endogenous fluorescence, lentiviruses expressing red fluorescent protein (RFP), cyan fluorescent protein (CFP), and green fluorescent protein (GFP) were transduced into the mammary epithelial cells (S1), mammary fibroblasts (HMF), and DCIS cells (DCIS.com), respectively. 106–107 transduction units/ml of lentivirus was used and incubated in the culture media for 6–10 hours in the presence of 8 μg/ml polybrene. Three days of transduction were followed by treatment of culture media with puromycin (10 ng/ml) for 2 weeks, and cells expressing the fluorescent proteins were selected.
Formation of DCIS spheroids
To generate DCIS spheroids, DCIS cells were seeded into a 96-well hanging drop plate (Perfecta 3D hanging drop plate, 3D Biomatrix) and cultured for three days. The average size of the spheroids was calculated by measuring the diameters of spheres in a well.
Microdevice design and fabrication
To mimic the 3D structural organization of the human mammary duct, we designed a compartmentalized microdevice consisting of upper and lower microchannels separated by a thin ECM-derived membrane that mimics the basement membrane (Fig. 1B). In this design, the upper microchannel recreates the ductal lumen in vivo and enables continuous flow of culture media required for growing and maintaining mammary epithelial cells and DCIS spheroids on the ECM membrane. A stromal layer impregnated with mammary fibroblasts is formed on the lower side of the ECM membrane and perfused with culture medium through the lower microchannel to mimic the vascular compartment of capillaries in mammary stroma in vivo.
The upper and lower microchannel layers were fabricated using soft lithography. Briefly, poly(dimethylsiloxane) (PDMS; Sylgard, Dow Corning) pre-polymer was mixed with a curing agent (10:1 w/w ratio of PDMS to curing agent) and cast against a mold containing photolithographically prepared microchannel features. The size of the cell culture chamber was 1 mm (width) × 3 mm (length) × 200 μm (height) (Supplementary Fig. 1A).
A vitrified collagen membrane was used as a biomimetic basement membrane that served to separate the upper and lower microchannels. Preparation of the vitrified collagen membrane began by mixing type I collagen (9.46 mg/ml; BD Biosciences) with 10X DMEM (Sigma-Aldrich), 1M HEPES (HEPES, Gibco), distilled water, and 1N NaOH. The final collagen concentration was 4.0 mg/ml, and pH was neutralized to 7.2–7.4. 14 μl of the mixture solution was then placed over the cell culture chamber of the lower PDMS layer and incubated at 37°C in 100% humidity for 1 hour to induce gelation (Supplementary Fig. 1B). Subsequently, the collagen hydrogel was dried at room temperature for at least 48 hours to produce a thin membrane. Since this resulting membrane was anchored to the large flat surface surrounding the recessed microchannel, it remained firmly attached to the lower PDMS slab. For bonding, a hand-held corona treater (Harrick plasma) was used to treat the surfaces of the upper and lower PDMS layers. For the lower layer containing the collagen membrane, care was taken to treat only the edges of the PDMS slab and to avoid direct exposure of the membrane to corona. Finally, the two layers were brought in contact and incubated at 60 °C for 3 hours to achieve permanent bonding (Supplementary Fig. 1B). After bonding, the membrane was visually inspected using a microscope to ensure its integrity without structural failure.
Formation of 3D mammary tissue structures
Prior to cell seeding, a fully assembled microdevice was sterilized by ultraviolet irradiation. To model the stromal tissue in the mammary duct, we formed a thin fibroblast-laden collagen gel layer attached to the vitrified membrane in the lower microchannel (Figs. 2B, 2C). As the first step to accomplish this, the lower microchannel was filled with 2% bovine serum albumin (BSA; Sigma-Aldrich) and incubated for 1 hour at 37°C to prevent binding of the collagen gel to the channel surfaces. Upon aspiration of BSA, a collagen precursor solution (3.0 mg/ml) mixed with human mammary fibroblasts (5×106 cells/ml) was injected into the lower microchannel and incubated for 2 hours to permit collagen gelation (Fig. 2B). For this step, type I collagen was used to represent the most abundant ECM component in the mammary stroma. Subsequently, HMF medium was perfused into the upper channel by a syringe pump (New Era Pump System) at a volumetric flow rate of 60 μl/hour. The fibroblast-embedded collagen gel was maintained for 3 days with continuous medium perfusion. During this period, traction forces generated by the fibroblasts resulted in contraction and subsequent detachment of the cell-laden gel from the bottom surface of the lower microchannel, creating a continuous gap along the channel length that permitted flow of culture media (Fig. 2C). Consistent gel contraction during this procedure was achieved by controlling parameters such as cell seeding density, collagen concentration, and polymerization conditions characterized by time, temperature, and humidity.
Figure 2. Formation of multilayered breast cancer tissue.
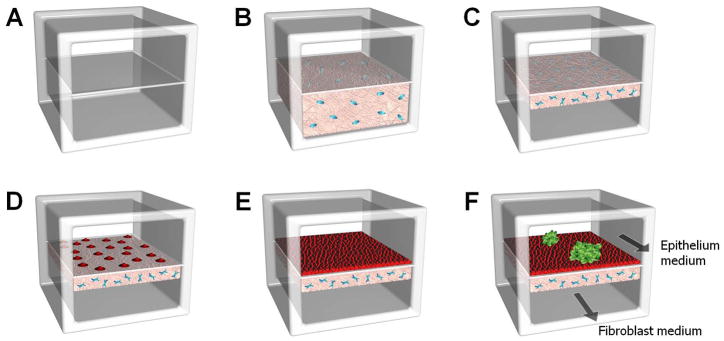
A. After channel bonding, the upper and lower cell culture chambers are separated by a thin vitrified collagen membrane. B. A human mammary fibroblast (HMF)-containing collagen solution is introduced into the BSA-coated lower chamber and polymerized. C. The fibroblasts in the collagen gel are cultured with continuous flow of HMF medium through the upper chamber, and this leads to gel contraction and resulting gap formation in the lower chamber. D. E. For the formation of the normal mammary epithelium, mammary epithelial cells are seeded onto the upper side of the Matrigel-coated basement membrane and cultured to confluence. F. Pre-formed DCIS spheroids are then injected into the upper chamber and allowed to attach to the epithelial cell layer.
After 3 days of culture, the upper side of the vitrified membrane was coated with a mixture of Matrigel (2 mg/ml) and fibronectin (0.5 mg/ml) for 2 hours. Matrigel, which contains laminin and type IV collagen, was advantageous for more faithfully recapitulating the key composition of the basement membrane of the mammary ductal epithelium in vivo. Human mammary epithelial cells (7×106 cells/ml) were then introduced into the upper microchannel and allowed to attach to the membrane surface for 2 hours. During microfluidic co-culture, the epithelial cells and fibroblasts were maintained by flowing MEGM and HMF culture media through the upper (40 μl/hour) and lower (30 μl/hour) microchannels, respectively (Fig. 2E). These flow rates were determined empirically by identifying the optimal rates of medium flow at which the cells remained viable for extended culture periods and no significant structural changes to the collagen stromal layer were induced by fluid flow. The flow rates used in our study were also within the physiological range of interstitial flow (2–70 μl/hour)21, which plays an important role in the survival of the mammary cells.
Introduction of DCIS spheroids
DCIS spheroids were collected from the 96-well hanging drop plate and centrifuged. The spheroids were then reconstituted in MEGM medium at 5 spheroids/μl and introduced into the upper microchannel. This step was followed by incubation of the microdevice without media perfusion for at least 8 hours to enable adhesion of the spheroids to the mammary epithelium (Fig. 2F). After firm attachment of the spheroids, flow of culture medium was resumed in the upper microchannel at a volumetric flow rate of 40 μl/hour.
Cell viability analysis
We used a commercially available colorimetric lactate dehydrogenase (LDH) cytotoxicity detection kit (Takara Biotechnology) to evaluate cell viability in our model. Perfused media from the upper and lower microchannels were collected separately and assayed using the detection kit to measure LDH release from the mammary epithelial cells and fibroblasts, respectively. The measured LDH levels were then converted to percent cytotoxicity according to the instruction provided by the manufacturer. Finally, percent cell viability was determined by subtracting the percent cytotoxicity from 100%.
Paclitaxel treatment and cytotoxicity evaluation
For drug testing studies, the cells were treated with paclitaxel (Sigma Aldrich) at a concentration of 20 nM, which was injected into the lower microchannel for 24 hours. This particular dose was chosen based on clinically relevant doses previously used in in vitro studies of paclitaxel efficacy22. Analysis of cytotoxicity was carried out using the aforementioned method based on a commercial LDH detection kit to calculate the percentage of cells that exhibited cytotoxic responses to paclitaxel. To assess the relative contribution of LDH production by DCIS cells and normal mammary epithelial cells, we analyzed the levels of paclitaxel-induced LDH release from normal epithelial cells in control microdevices that excluded DCIS spheroids from the upper microchannel while maintaining the lower stromal layer unchanged. The cytotoxic response of our DCIS-on-a-chip model to paclitaxel was also compared to that measured in another control group that did not receive paclitaxel treatment. Additionally, changes in the projected area of DCIS spheroids due to drug treatment were analyzed by using ImageJ (NIH) to evaluate the therapeutic effect of paclitaxel on the suppression of tumor growth.
Scanning electron microscopy
The microstructure of the vitrified collagen membrane was imaged using scanning electron microscopy (SEM). Prior to imaging, membrane samples were fixed with 2% formaldehyde for 10 minutes. SEM images were acquired using a JSM 7410F scanning electron microscope (JEOL Ltd.) with a Gatan cooled charge-coupled device camera.
Statistical analysis
Statistical significance was determined using a two-tailed Student’s t-test for at least three independent experiments and are presented as the mean ± standard deviation (std.). P-values less than 0.05 were considered to be statistically significant and indicated with asterisks (**).
Results and Discussion
Recapitulating microarchitecture of normal human mammary duct
Our microengineering techniques allowed for the production and prolonged maintenance of the human mammary ductal epithelium and a human mammary fibroblast-containing 3D collagen matrix separated by a thin biomimetic basement membrane (Fig. 3A). The collagen vitrification process conducted in this study produced thin ECM membranes with an average thickness of 20.9 μm that consisted of a dense network of collagen fibers (Fig. 3A). The vitrified membrane remained attached to the lower microchannel, and its structural integrity was maintained during corona treatment and subsequent channel bonding. When buffer solutions were flowed into the channels prior to cell seeding, the membrane was observed to become hydrated rapidly without exhibiting significant structural changes. The hydrated vitrified membrane served as a stable barrier during injection of collagen gel solution into the lower microchannel.
Figure 3. Microengineered normal human mammary duct.
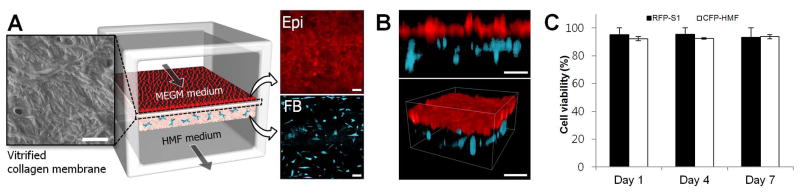
A. As visualized by scanning electron microscopy, the intervening vitrified membrane consists of a dense network of collagen fibers. This membrane supports adhesion and growth of mammary epithelial cells (shown in red; upper right inset) to a confluent monolayer, as well as 3D culture of fibroblasts (shown in cyan; lower right inset) within the collagen gel in the lower chamber. Epi and FB represent epithelial cells and fibroblasts, respectively. The micrographs were taken at day 7. Scale bars: 20 μm. B. A cross-sectional view (upper) and 3D rendered image (lower) show an intact normal mammary epithelium and fibroblasts embedded in the stromal layer of the lower chamber. Scale bars: 100 μm. C. In this microfluidic device, percent viability of the mammary epithelial cells (RFP-S1) and mammary fibroblasts (CFP-HMF) is maintained over 85% throughout the culture period (7 days). Data shown mean ± std.
In the formation of the membrane-bound stromal layer in the lower microchannel, BSA surface coating was critical for preventing unwanted adhesion of collagen matrix to the bottom channel floor during continuous contraction of the collagen gel. In the absence of BSA coating, this contraction led to the detachment of the collagen gel layer from the vitrified membrane and its shrinkage to the bottom surface of the lower microchannel (Supplementary Fig. 2). Three days of microfluidic culture and concomitant gel contraction produced a stromal layer with an average thickness of approximately 150 μm that remained attached to the vitrified membrane. This process also resulted in the formation of a gap between the contracted gel and the bottom floor of the lower microchannel that provided fluidic access for perfusion of fibroblast medium during co-culture with epithelial cells. Reduction in the concentration of serum in the fibroblast culture medium inhibited further shrinkage of the gel layer.
During the course of culture, mammary fibroblasts embedded in the collagen gel began to spread within 24 hours after seeding and gradually became elongated to exhibit their characteristic spindle-shaped morphology (Fig. 3A). Human mammary epithelial cells seeded into the upper microchannel were observed to establish firm attachment to the vitrified membrane surface and proliferate to form a confluent monolayer over a period of 24 hours. Cross-sectional images of the tissue constructs showed a dense mammary epithelium overlying the 3D stromal layer containing uniformly distributed fibroblasts (Fig. 3B). As shown in Fig. 3C, microfluidic culture conditions in our device permitted prolonged co-culture of the epithelial cells and fibroblasts without significant viability loss, with over 85% of the cells maintained alive throughout the 1-week culture period. Flow of culture medium through the gap in the lower channel created by gel contraction was effective for maintaining the fibroblasts embedded in the 3D matrix scaffold and did not induce undesirable changes in the structural integrity of the stromal tissue during long-term culture.
Incorporation of DCIS in the microengineered mammary duct
For optimal injection and handling of DCIS spheroids in the upper microchannel with a height of 200 μm, we examined the relationship between the number of seeded cells and the diameter of resulting spheroids in standard hanging drop culture after 3 days of culture (Supplementary Fig. 3). Spheroids having a diameter of approximately 150 μm were selected to mimic early-stage DCIS lesions. Under static conditions without the flow of culture medium, the spheroids injected into the upper channel successfully adhered to the epithelial cell surface within 10 hours after seeding. When flow was resumed, the DCIS spheroids remained adherent on the surface and gradually became integrated into the epithelium over the course of two days (Fig. 4A). Confocal analysis of the epithelium revealed that the spheroids became flattened and positioned themselves on the same horizontal plane as the epithelial cells, suggesting full incorporation of DCIS into the epithelial layer (Fig. 4B). Importantly, the vast majority of the DCIS cells remained in their original spheroids without invading the surrounding epithelial tissue and the vitrified membrane, and disintegration of the spheroids due to cell migration was not observed throughout the entire culture period. Prolonged culture in the microfluidic device led to the enlargement of the spheroids, indicating the ability of our model to support proliferation of the DCIS cells. In addition, DCIS spheroids did not induce any significant alterations in the morphology of the underlying fibroblasts (Fig. 4A).
Figure 4. DCIS-on-a-chip.
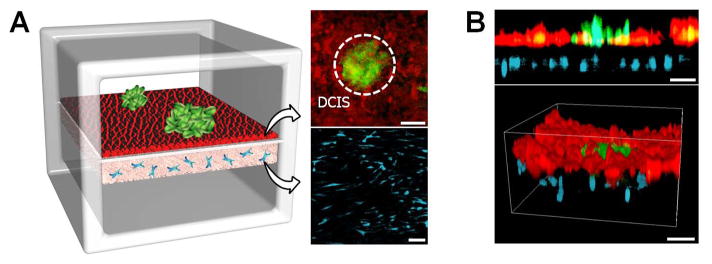
A. DCIS spheroids (green; shown with a dotted circle) introduced into the upper channel bind to the mammary epithelial cells (red) and become embedded in the epithelium over time. During this process, HMFs (cyan) in the stromal layer maintain their stretched morphology. The fluorescence micrographs show cells at Day 2. B. Cross-sectional (upper) and 3D rendered (lower) views of the multilayered tissue structure illustrating integration of DCIS spheroids in the mammary epithelium. Scale bars: 100 μm.
Paclitaxel treatment in DCIS-on-a-chip
Next, we evaluated the potential of our disease model as a drug screening platform using a clinical anticancer drug, paclitaxel. To simulate physiological delivery of intravenously administered paclitaxel from the vasculature in the mammary stroma to DCIS lesions, we generated a continuous flow of paclitaxel solution through the lower microchannel. As shown in Fig. 5A, during the course of drug treatment, cellular production of LDH, as represented by percent cytotoxicity, gradually increased and doubled at day 4 (DCIS+S1 Paclitaxel), indicating a paclitaxel-induced statistically significant increase in cytotoxicity compared to paclitaxel untreated group (DCIS+S1; p<0.05). Interestingly, when paclitaxel was introduced into control devices without DCIS spheroids (S1 Paclitaxel) at the same dose and duration, cytotoxicity did not increase and statistically significant difference between DCIS+S1 and S1 Paclitaxel was not observed (Fig. 5A). This is presumably due to substantially higher sensitivity of the cells in the DCIS spheroids to the drug designed to selectively inhibit the assembly of mitotic spindles and cell division in actively proliferating cancer cells.
Figure 5. Testing of anticancer drug in the breast cancer-on-a-chip.

A. When the cells are treated with paclitaxel from the basal side to simulate intravenous administration of the drug, percent cytotoxicity as measured by the production of LDH increases over time in the DCIS-on-a-chip model (open triangle). Paclitaxel has negligible cytotoxic effects on normal epithelial cells (open circle), and percent cytotoxicity in this control group did not show any statistically significant difference from that in the untreated DCIS-on-a-chip (closed square) (p>0.05). B. Paclitaxel treatment prevents growth of DCIS spheroids in this model (white). In the absence of the drug, continuous proliferation of cancer cells in the tumor spheroids leads to a three-fold increase in the size of the spheroids. C. Fluorescence micrographs of DCIS spheroids at day 0 (leftmost), day 3 without paclitaxel (middle), and 3 days with paclitaxel treatment. Dotted lines show the outline of the spheroids. Scale bars: 100 μm. ** p<0.05
To quantitatively assess the anti-proliferative effects of paclitaxel, we measured changes in the projected area of the DCIS spheroids during drug treatment. In the untreated group, continuous proliferation of DCIS cells led to significant enlargement of the spheroids (p<0.05; Fig. 5B) and a nearly threefold increase in their size (Fig. 5C). Although mixed populations of DCIS cells and normal mammary epithelial cells were observed along the immediate periphery of the spheroids, it appeared that the proliferating DCIS cells continuously displaced their surrounding epithelial cells and managed to maintain the boundary between the two distinct populations during expansion of the spheroids. However, this growth was confined in the epithelial compartment and did not result in tumor cell invasion into the underlying stroma. In contrast, the diameter of the DCIS spheroids exposed to paclitaxel remained unchanged or slightly decreased, demonstrating the efficacy of paclitaxel in arresting tumor cell proliferation and preventing the growth of DCIS lesions (Fig. 5A&C). As was the case with the untreated group, the stromal layer remained intact without DCIS cell invasion during and following paclitaxel treatment.
Conclusion
In this study, we developed a microengineered cell culture platform to reproduce the physiological 3D architecture and microenvironment of early-stage breast cancer. This microphysiological human disease model provides new possibilities to simulate and probe structural and functional association of tumor cells with other cell types in the mammary duct and stromal compartment that play a critical role in the progression and metastasis of breast cancer. The ability to directly observe and visualize biological responses, combined with the precise parametric control of cell type, spatial cellular distribution, and microenvironmental cues in this model makes our breast cancer-on-a-chip approach well-suited for mechanistic investigation on malignant progression of DCIS. The paclitaxel study described in this paper demonstrates the feasibility of using our platform to develop new preclinical cancer models for evaluation and prediction of human-relevant responses to anticancer drugs.
Despite these advantages, further studies are necessary to improve the physiological relevance of this model. Our current system is an incomplete representation of complex cancerous mammary tissue, due in part to the absence of other important cell types such as adipocytes and endothelial cells. However, we envision inclusion of additional cell types to be readily achievable by using similar microengineering approaches. For example, the controlled gel contraction method could generate an additional ECM gel layer containing adipocytes that immediately neighbors the fibroblast-laden layer in the lower microchannel. It may also be possible to endothelialize the bottom surface of the ECM gel in the lower channel to recreate the stromal-vascular interface. The ability to tune stiffness and composition of the ECM gel layer may permit the development of more physiological systems that model characteristic changes in the stromal layer (e.g., stiffening) during the progression of breast cancer.23 Another important possibility is to create in vivo-like biochemical microenvironment by flowing soluble factors through the lower channel to form physiological gradients of chemokines and cytokines implicated in breast cancer across the mammary tissue in our model. These types of models would greatly facilitate mechanistic studies to delineate how biomechanical and biochemical microenvironmental cues contribute to malignant disease progression. We believe that our breast cancer-on-a-chip platform holds potential as an enabling approach to these critical future investigations in breast cancer research that may lead to increased understanding of key disease processes and identification of new therapeutic targets.
Supplementary Material
Acknowledgments
This work was supported by funding from the National Research Foundation of Korea (2012M3A7B4035286 and 2013R1A2A2A04013379), National Institutes of Health (NIH) (1DP2HL127720-01), and the University of Pennsylvania. D.H. is a recipient of the NIH Director’s New Innovator Award (1DP2HL127720-01).
References
- 1.Place AE, Jin Huh S, Polyak K. Breast cancer research: BCR. 2011;13:227. doi: 10.1186/bcr2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Espina V, Liotta LA. Nature reviews Cancer. 2011;11:68–75. doi: 10.1038/nrc2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lyons TR, O’Brien J, Borges VF, Conklin MW, Keely PJ, Eliceiri KW, Marusyk A, Tan AC, Schedin P. Nature medicine. 2011;17:1109–1115. doi: 10.1038/nm.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khurana A, McKean H, Kim H, Kim SH, mcguire J, Roberts LR, Goetz MP, Shridhar V. Breast cancer research: BCR. 2012;14:R43. doi: 10.1186/bcr3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soria G, Ofri-Shahak M, Haas I, Yaal-Hahoshen N, Leider-Trejo L, Leibovich-Rivkin T, Weitzenfeld P, Meshel T, Shabtai E, Gutman M, Ben-Baruch A. BMC cancer. 2011;11:130. doi: 10.1186/1471-2407-11-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.A. C. Socitey. Atalanta: American Cancer Society; 2013. [Google Scholar]
- 7.Zhao Y, Xiao A, Park HI, Newcomer RG, Yan M, Man Y, Heffelfinger SC, SQA Cancer research. 2015;64:590–598. doi: 10.1158/0008-5472.can-03-1932. [DOI] [PubMed] [Google Scholar]
- 8.Jedeszko C, Victor BC, Podgorski I, Sloane BF. Cancer research. 2009;69:9148–9155. doi: 10.1158/0008-5472.CAN-09-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sung KE, Yang N, Pehlke C, Keely PJ, Eliceiri KW, Friedl A, Beebe DJ. Integrative biology: quantitative biosciences from nano to macro. 2011;3:439–450. doi: 10.1039/c0ib00063a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bischel LL, Beebe DJ, Sung KE. BMC cancer. 2015;15:12. doi: 10.1186/s12885-015-1007-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim JB, Stein R, O’Hare MJ. Breast Cancer Res Treat. 2004;85:281–291. doi: 10.1023/B:BREA.0000025418.88785.2b. [DOI] [PubMed] [Google Scholar]
- 13.Campbell JJ, Davidenko N, Caffarel MM, Cameron RE, Watson CJ. PloS one. 2011;6:e25661. doi: 10.1371/journal.pone.0025661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmeichel KL, Bissell MJ. J Cell Sci. 2003;116:2377–2388. doi: 10.1242/jcs.00503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Behbod F, Kittrell FS, LaMarca H, Edwards D, Kerbawy S, Heestand JC, Young E, Mukhopadhyay P, Yeh HW, Allred DC, Hu M, Polyak K, Rosen JM, Medina D. Breast cancer research: BCR. 2009;11:R66. doi: 10.1186/bcr2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu M, Yao J, Carroll DK, Weremowicz S, Chen H, Carrasco D, Richardson A, Violette S, Nikolskaya T, Nikolsky Y, Bauerlein EL, Hahn WC, Gelman RS, Allred C, Bissell MJ, Schnitt S, Polyak K. Cancer cell. 2008;13:394–406. doi: 10.1016/j.ccr.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vidi PA, Leary JF, Lelievre SA. Integrative biology: quantitative biosciences from nano to macro. 2013;5:1110–1118. doi: 10.1039/c3ib40053k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grafton MM, Wang L, Vidi PA, Leary J, Lelievre SA. Integrative biology: quantitative biosciences from nano to macro. 2011;3:451–459. doi: 10.1039/c0ib00132e. [DOI] [PubMed] [Google Scholar]
- 19.Shin Y, Kim H, Han S, Won J, Jeong HE, Lee ES, Kamm RD, Kim JH, Chung S. Advanced healthcare materials. 2013;2:790–794. doi: 10.1002/adhm.201200320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esch EW, Bahinski A, Huh D. Nature reviews Drug discovery. 2015;14:248–260. doi: 10.1038/nrd4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heldin CH, Rubin K, Pietras K, Ostman A. Nature reviews Cancer. 2004;4:806–813. doi: 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- 22.Liebmann JE, Cook JA, Lipschultz C, Teague D, Fisher F, Mitchell JB. British Journal of Cancer. 1993;68:1104–1109. doi: 10.1038/bjc.1993.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Butcher DT, Alliston T, Weaver VM. Nature reviews Cancer. 2009;9:108–122. doi: 10.1038/nrc2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.