

Abstract
Most synapses release neurotransmitters in two phases: (1) a fast synchronous phase lasting a few milliseconds; and (2) a delayed “asynchronous” phase lasting hundreds of milliseconds. Ca2+ triggers fast synchronous neurotransmitter release by binding to synaptotagmin-1, synaptotagmin-2, or synaptotagmin-9, but how Ca2+ triggers delayed asynchronous release has long remained enigmatic. Recent results suggested that consistent with the Ca2+-sensor function of synaptotagmin-7 in neuroendocrine exocytosis, synaptotagmin-7 also functions as a Ca2+ sensor for synaptic vesicle exocytosis but operates during delayed asynchronous release. Puzzlingly, a subsequent study postulated that synaptotagmin-7 is not a Ca2+ sensor for release but mediates Ca2+-dependent vesicle repriming after intense stimulation. To address these issues, we here analyzed synaptic transmission at rod bipolar neuron–AII amacrine cell synapses in acute mouse retina slices as a model system. Using paired recordings, we show that knock-out of synaptotagmin-7 selectively impairs delayed asynchronous release but not fast synchronous release. Delayed asynchronous release was blocked in wild-type synapses by intracellular addition of high concentrations of the slow Ca2+-chelator EGTA, but EGTA had no effect in synaptotagmin-7 knock-out neurons because delayed asynchronous release was already impaired. Moreover, direct measurements of vesicle repriming failed to uncover an effect of the synaptotagmin-7 knock-out on vesicle repriming. Our data demonstrate that synaptotagmin-7 is selectively essential for Ca2+-dependent delayed asynchronous release in retinal rod bipolar cell synapses, that its function can be blocked by simply introducing a slow Ca2+ buffer into the cells, and that synaptotagmin-7 is not required for normal vesicle repriming.
SIGNIFICANCE STATEMENT How Ca2+ triggers delayed asynchronous release has long remained enigmatic. Synaptotagmin-7 has been implicated recently as Ca2+ sensor in mediating delayed asynchronous release, or vesicle repriming, in cultured neurons. To test the precise function of synaptotagmin-7 in a physiologically important synapse in situ, we have used pair recordings to study the synaptic transmission between retinal rod bipolar cells and AII amacrine cells. Our data demonstrate that the knock-out of synaptotagmin-7 selectively impaired delayed asynchronous release but not synchronous release. In contrast, the readily releasable vesicles after depletion recover normally in knock-out mice. Therefore, our findings extend our knowledge of synaptotagmins as Ca2+ sensors in vesicle fusion and support the idea that synapses are governed universally by different synaptotagmin Ca2+ sensors mediating distinct release.
Keywords: active zone, calcium channel, ribbon synapse, vesicle fusion
Introduction
Most Ca2+-triggered neurotransmitter release proceeds in a primary fast and synchronous reaction lasting a few milliseconds, followed by a secondary delayed and asynchronous reaction lasting hundreds of milliseconds. Synchronous release is driven by local Ca2+ at release sites that activates fast synaptotagmin Ca2+ sensors [synaptotagmin-1 (Syt1), Syt2, and Syt9; Xu et al., 2007]. Delayed asynchronous release is a major contributor to release in some of synapses, for example, parallel fiber synapses (Atluri and Regehr, 1998), inhibitory synapses onto inferior olive neurons (Best and Regehr, 2009), and synapses formed by cholecystokinin-containing interneurons (Hefft and Jonas, 2005; Daw et al., 2009). Syt1, Syt2, and Syt9 also act as Ca2+ sensors in other forms of exocytosis, and other synaptotagmins have also been implicated in exocytosis. Among these, Syt7 is closely related to Syt1, Syt2, and Syt9 and, like them, functions as a Ca2+ sensor for exocytosis in neuroendocrine PC12 cells (Sugita et al., 2001), chromaffin cells (Schonn et al., 2008), and β cells (Gustavsson et al., 2009). Moreover, Syt7 was implicated as a Ca2+ sensor mediating delayed asynchronous release in zebrafish neuromuscular junctions (Wen et al., 2010) and cultured cortical neurons (Bacaj et al., 2013). Confusingly, however, it was suggested recently in cultured hippocampal neurons that Syt7 does not act as a Ca2+ sensor for exocytosis but does enable Ca2+-dependent vesicle repriming (Liu et al., 2014).
In the mammalian retina, rod bipolar cells (RBCs) form ribbon-type excitatory synapses on AII amacrine (AII) cells. These synapses play a central role in transmission of scotopic visual signals to the brain (Bloomfield and Dacheux, 2001). In response to sustained stimulation, RBC–AII cell synaptic transmission exhibits a fast transient component, followed by a sustained component that continues throughout the stimulus (Singer and Diamond, 2003). The transient component is thought to arise from fast exocytosis of the vesicles that are primed in the readily releasable pool (RRP), whereas the sustained component is thought to reflect the continuous Ca2+-triggered exocytosis of vesicles as they are being reprimed into the RRP (Oesch and Diamond, 2011). Increasing the duration of the depolarization or reducing the intracellular Ca2+ buffer can enhance and prolong the sustained component, such that release continues for seconds after the membrane repolarized, effectively constituting a form of delayed asynchronous release (Singer and Diamond, 2003). Dual recordings of paired RBCs and AII cells (Singer and Diamond, 2003; Veruki et al., 2003) offer an opportunity to study in a physiologically important synapse in situ the fast synchronous, the sustained, and the delayed asynchronous components of release.
Here, we investigated the functional role of Syt7 in delayed asynchronous release and/or vesicle replenishment at the RBC–AII cell synapse using paired whole-cell voltage-clamp recordings from RBCs and AII cells in retina slices from littermate wild-type (WT) and Syt7 knock-out (KO) mice. Our data suggest that Syt7 is selectively essential for Ca2+-triggered delayed asynchronous release but not for Ca2+-dependent vesicle replenishment, consistent with the notion that Syt7, like its closely related homologs Syt1, Syt2, and Syt9, acts in Ca2+ triggering of release but exhibits distinct properties (Bacaj et al., 2013).
Materials and Methods
Mouse husbandry.
All experiments were performed on adult male and female hybrid Bl6/CD1/SJ129 mice without identification of the genotype to the experimenter. All experiments were approved by Stanford Institutional Animal Care and Use Committee review.
Electrophysiology experiments.
Light-adapted WT or Syt7 KO mice (3–6 weeks old) were anesthetized with isoflurane and killed, and their retinas were isolated in oxygenated ACSF containing the following (in mm): 119 NaCl, 26 NaHCO3, 10 glucose, 1.25 NaH2PO4, 2.5 KCl, 2 CaCl2, 1 MgCl2, 2 Na-pyruvate, and 0.5 ascorbic acid, pH 7.4. Isolated retinas were embedded in low-melting agar (2% in ACSF with HEPES substituted for NaHCO3). Slices, 200 μm, from the midtemporal retina were cut using a Vibratome (VT1200s; Leica), incubated for 1 h at 32°C, and stored at room temperature for experiments.
Paired whole-cell patch-clamp recordings were made from synaptically connected RBCs and AIIs visually identified under infrared differential interference contrast (IR-DIC) video microscopy (Axioskop 2; Zeiss; Singer and Diamond, 2003; Veruki et al., 2003). Patch pipettes (resistance of 4–5 MΩ for postsynaptic recording and 5–6 MΩ for presynaptic recording) were pulled using borosilicate glass (WPI) on a two-stage vertical puller (Narishige). The input and series resistances of AII cells were ∼2 GΩ and ∼13–20 MΩ, respectively. Cells were discarded if the series resistance exceeded 40 MΩ or if the holding current changed abruptly.
RBC Ca2+ currents, which arise exclusively from the nerve terminal (Protti and Llano, 1998), were evoked by 500 ms step depolarizations from −70 to −10 mV and isolated using the following pipette internal solution (in mm): 120 Cs-gluconate, 20 tetraethylammonium-Cl, 20 HEPES, 1.3 BAPTA, 4 MgATP, 0.4 NaGTP, and 10 phosphocreatine. Presynaptic Ca2+ currents were leak subtracted using the P/4 protocol. AII cells were voltage clamped at −70 mV, and EPSCs were recorded in ACSF containing picrotoxin (100 μm), strychnine (0.5 μm), and tetrodotoxin (0.5 μm) to block GABAA receptors, glycine receptors, and voltage-gated Na+ channels, respectively.
For sucrose puffing, AII cells were patched and visualized by including Alexa Fluor 594 (Invitrogen) in the pipette solution. Sucrose at 2 m contained in a 2–3 MΩ pipette was puffed directly on the dendritic field of the patched AII cell. A puff pressure of ∼5 psi was applied using a Picospritzer system (Parker Hannifin).
Immunohistochemistry.
Retinas were fixed in 4% paraformaldehyde overnight and then embedded in low-melting agar (2% in ACSF), sectioned at 50 μm thickness using a Vibratome (VT1200s; Leica). The retina slices were pretreated with 0.5% Triton X-100 for 1 h at room temperature and incubated overnight at 4°C with primary antibodies in blocking solution (0.1% Triton X-100 and 5% goat serum in PBS). The slices were washed and incubated with fluorescence-conjugated secondary antibodies for 2 h at room temperature. The slices were washed and mounted with DAPI fluoromount (SouthernBiotech). Primary antibodies against Syt1 (V216, 1:1000), PKCα (1:500, rabbit; Santa Cruz Biotechnology), and VGluT1 (1:1000, guinea pig; Millipore) were used. Secondary antibodies were Alexa Fluor conjugates (1:500; Invitrogen). Images were acquired using Nikon A1RSi confocal microscope with a 60× oil-immersion objective (1.45 numerical aperture) and analyzed in Nikon Analysis software.
Data analyses and statistics.
Data were acquired using PatchMaster (HEKA Elektronik) and analyzed in Igor Pro (Wavemetrics) and MiniAnalysis (Synaptosoft). Statistics were performed using a Student's t test unless specifically stated otherwise.
Results
Delayed asynchronous neurotransmitter release at the RBC–AII cell synapses
AII cells were identified with IR-DIC microscopy by their unique morphology and position in the inner nuclear layer of the retina (Singer and Diamond, 2003; Fig. 1A). AII cells have an inverted pear-shaped soma that gives rise to a primary dendrite and are typically located at the border between the inner nuclear and inner plexiform layers. AII cells were patched first and usually displayed high rates of miniature EPSCs (mEPSCs; Singer and Diamond, 2003; Veruki et al., 2003; Fig. 1B). After 1–2 min of mEPSC recording, presynaptic RBCs, which are located above the recorded AII cell at the border between the inner nuclear and outer plexiform layers, were identified and patched. When both cells were patched successfully, the probability that these two cells were synaptically connected was relatively high (∼60–80%), and occasionally we sequentially patched two or more RBCs that were connected to a single AII cell, allowing us to record multiple RBC–AII cell pairs for the same postsynaptic AII cell.
Figure 1.
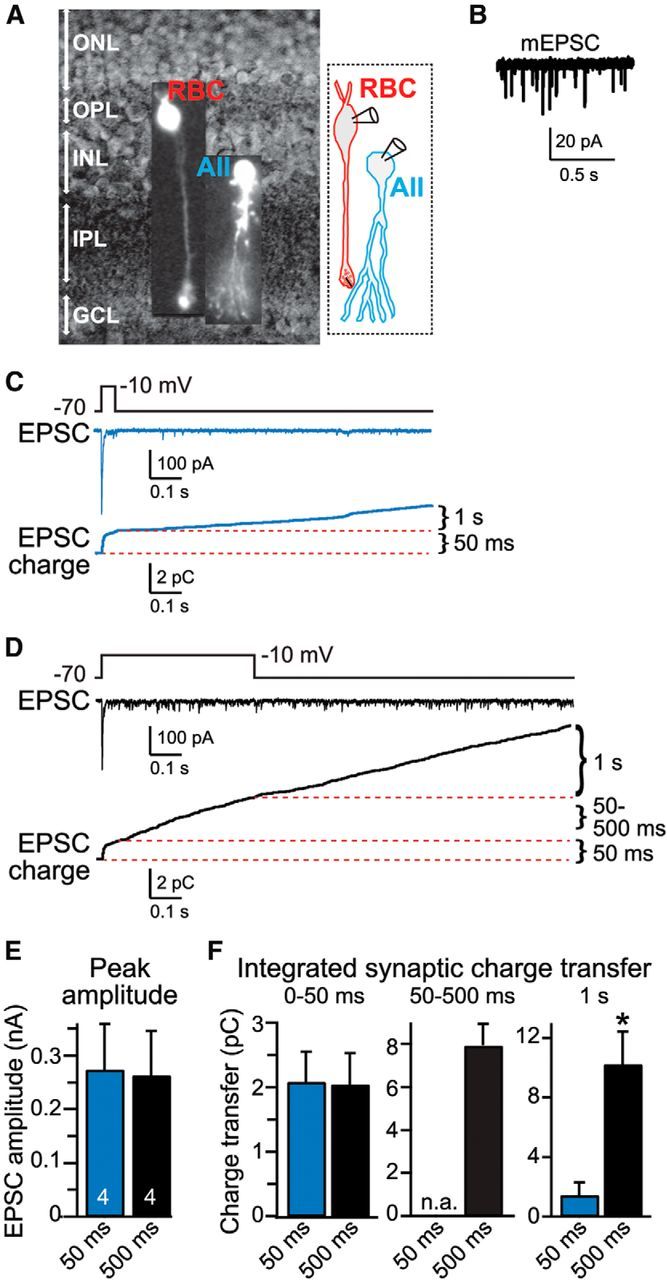
Paired recordings of synaptic transmission at RBC–AII cell synapses. A, Left, Retinal slice visualized with IR-DIC optics with superimposed images of an RBC and an AII cell that are labeled fluorescently by Alexa Fluor-594. Right, Schematic of a synaptic connection between an RBC and an AII cell. GCL, Ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer. B, Representative trace of spontaneous mEPSCs recorded from an AII cell. C, D, Representative traces of EPSCs recorded from AII cells as a function of 50 ms (C) or 500 ms (D) duration step depolarizations of the presynaptic RBC (top, presynaptic depolarization protocol; middle, postsynaptic EPSCs; bottom, plots of the cumulative EPSC charge transfer). The three time periods analyzed for quantifying release (50 ms, 50–500 ms, and 1 s after repolarization) are illustrated in the cumulative charge graphs. Recordings were performed in standard bath solution containing 2 mm Ca2+ and 1 mm Mg2+ with an internal pipette solution containing 1.3 mm BAPTA to mimic the endogenous Ca2+-buffering capacity of RBCs. E, Summary graph of the peak EPSC amplitude during the initial transient EPSC phase shows that this amplitude is independent of the length of the depolarization pulse. F, Summary graphs of the EPSC charge transfer during the initial 50 ms of depolarization (fast release), the 50–500 ms depolarization period for the longer pulse (sustained release), and the 1 s after the depolarization (delayed asynchronous release). Note that delayed asynchronous release is approximately fivefold larger after the 500 ms pulse than after the 50 ms pulse. All data are means ± SEMs (n = 4). Statistical analyses were performed by Student's t test comparing the 50 and 500 ms depolarization pulses. *p < 0.05.
To induce release at the RBC–AII cell synapse, we depolarized the presynaptic RBC for 50 or 500 ms from a holding membrane potential of −70 to −10 mV, whereas the voltage-clamped postsynaptic AII cell was held at −70 mV (Fig. 1C,D). We found that both depolarization pulses elicited a fast synchronous response that exhibited the same amplitude independent of the depolarization pulse duration (Fig. 1E). When integrated over 50 ms, the fast synchronous response also mediated the same synaptic charge transfer (Fig. 1F). During the longer 500 ms depolarization, we observed a sustained release component as described previously (Singer and Diamond, 2003). After the depolarization pulses ended, both depolarization pulses were followed by an extended phase of delayed asynchronous release. Delayed asynchronous release was much larger after the 500 ms depolarization than after the 50 ms depolarization (approximately sevenfold; Fig. 1F), leading us to adopt the 500 ms depolarization pulse as the standard experimental protocol. Note that quantitatively, delayed asynchronous release accounts for nearly half of synaptic signaling when an RBC is stimulated by the 500 ms depolarization, suggesting that delayed asynchronous release is of potential physiological importance.
Syt7 KO selectively impairs Ca2+-triggered delayed asynchronous release
To examine the effect of the Syt7 KO on neurotransmitter release from RBC–AII cell synapses, we performed simultaneous recordings from synaptically paired RBCs and AII cells in retinas from littermate WT and Syt7 KO mice, using the recording configuration described in Figure 1. We monitored postsynaptic EPSCs in response to a presynaptic 500 ms depolarization and simultaneously recorded presynaptic Ca2+ currents (Fig. 2A). We observed no difference between WT and Syt7 KO cells in Ca2+ currents or Ca2+ fluxes induced by the step depolarization (Fig. 2B), suggesting that the Syt7 KO has no effect on the function of neuronal Ca2+ channels.
Figure 2.

Syt7 KO selectively impairs delayed asynchronous release at RBC–AII cell synapses. A, Experimental design (top), representative presynaptic Ca2+ current traces (ICa; middle), and postsynaptic EPSC traces (bottom) recorded from connected RBCs and AII cells in littermate WT (left) and Syt7 KO (right) mice. The insets show the initial 50 ms of the EPSC at an expanded timescale. Note that these measurements were performed under standard conditions using an internal pipette solution with 1.3 mm BAPTA. B, Summary graph of the Ca2+ current amplitude (left) and integrated Ca2+ charge transfer (right) shows that the Syt7 KO has no effect on Ca2+ influx. C, Plot of the cumulative EPSC charge transfer during and after the 500 ms depolarization reveals selective impairment in Syt7 KO synapses of delayed asynchronous release after the end of the depolarization. D, Summary graph of peak EPSC amplitudes (corresponding to fast synchronous release) measured in WT and Syt7 KO mice. E, Summary graphs of the charge transfer integrated over the initial 50 ms of depolarization (fast synchronous release), the remaining depolarization period lasting from 50 to 500 ms (sustained release), and the 1 s period after the depolarization (0.5–1.5 s, delayed asynchronous release). Only delayed asynchronous release is significantly different between WT and KO mice. F–H, Analysis of spontaneous mEPSCs recorded in AII cells from WT and Syt7 KO mice (F, representative traces; G, summary graphs of mEPSC frequency and amplitude; H, summary graphs of mEPSC rise and decay times). All data are means ± SEMs (n = 10–12). Statistical analyses were performed by Student's t test comparing Syt7 KO with WT littermates, except an ANOVA followed by the post hoc Bonferroni's test was used in C and E. **p < 0.01.
We then quantified the three phases of the EPSCs described in Figure 1. We found that the Syt7 KO did not significantly affect the fast initial phase of release or sustained release during the depolarization phase, although there was a trend toward a decrease in the latter (Fig. 2C–E). However, the Syt7 KO nearly completely blocked delayed asynchronous release (approximately threefold), suggesting that Syt7 is selectively essential for delayed asynchronous release. This finding is consistent with the notion that Syt7 functions as a Ca2+ sensor for delayed asynchronous release (Bacaj et al., 2013). We also measured spontaneous mEPSCs but observed no difference between WT and Syt7 KO synapses in any parameter evaluated (Fig. 2F–H).
Syt7 KO does not cause major changes in the synaptic connectivity of the retina
Our previous studies in cultured neurons have shown that knockdown or KO of Syt7 does not alter synaptic density or size (Bacaj et al., 2013). However, loss of Syt7 may cause changes in synaptic connections in the retina and therefore result in specific blockade of delayed asynchronous release. To test this possibility, we examined the synaptic connections in the retina by immunostaining of retina sections for synaptic vesicle markers, including VGluT1 and Syt1. As shown in Figure 3, immunostaining for VGluT1 in vertical retina sections revealed normal synaptic connections in both outer and inner plexiform layers of Syt7 KO mice compared with WT littermates. Labeling RBCs with PKCα antibodies also revealed an apparently normal morphology and bouton size of RBCs. Furthermore, we found no evidence for the upregulation of Syt1 in Syt7 KO mice (Fig. 3B), consistent with our electrophysiological analysis that there is no change in the amplitude and kinetics of fast and sustained components of EPSC. We conclude that Syt7 is not required for synapse formation or maintenance in the retina.
Figure 3.

Synaptic connectivity is not changed in Syt7 KO retina. A, RBCs and synaptic boutons were labeled with antibodies against PKCα and VGluT1, respectively. Similar synaptic layering patterns and overall morphology of RBCs were found between WT (top) and KO (bottom) mice. GCL, Ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer. B, Syt1 was not upregulated in Syt7 KO mice. Scale bars, 20 μm.
High concentrations of EGTA block delayed asynchronous release and resemble the Syt7 KO phenotype
Delayed asynchronous release is thought to depend on the buildup of residual Ca2+ during extensive stimulation, such as that used in our recording configuration (Fig. 1B; Atluri and Regehr, 1998; Maximov and Südhof, 2005). Ca2+-triggered delayed asynchronous release is probably slower than fast synchronous release because the Ca2+ sensor involved (i.e., Syt7) is not as close to the Ca2+ channels mediating Ca2+-influx as the Ca2+ sensors mediating fast synchronous release (Syt1, Syt2, and Syt9; Xu et al., 2007). Indeed, Syt7 is absent from synaptic vesicles (Sugita et al., 2001; Takamori et al., 2006) and may thus be farther away from the site of Ca2+ influx than Syt1, Syt2, and Syt9, which are present on synaptic vesicles. At the same time, delayed asynchronous release is activated at lower Ca2+ concentrations (Sun et al., 2007), suggesting that the responsible Ca2+ sensor is not only farther away from the site of Ca2+ entry (the mouth of the Ca2+ channel) but also exhibits a higher apparent Ca2+ affinity, which is again consistent with Syt7 as the Ca2+ sensor for delayed asynchronous release (Sugita et al., 2001, 2002).
To further test the role of residual Ca2+ and Syt7 in delayed asynchronous release at the RBC–AII cell synapse, we included 10 mm EGTA in the internal presynaptic pipette solution (Fig. 4). The regular recording conditions at this synapse involve the addition of 1.3 mm BAPTA in the internal solution, which is a faster Ca2+ buffer than EGTA and is thought to mimic the endogenous Ca2+ buffer in these cells (Burrone et al., 2002). Because of its slow Ca2+-binding kinetics, EGTA presumably affects only global Ca2+ in the terminal but not local Ca2+ near the mouth of presynaptic Ca2+ channels at the release sites (Atluri and Regehr, 1998). Thus, if the hypotheses about delayed asynchronous release enunciated above are correct, EGTA would be expected to selectively suppress delayed asynchronous release induced by residual global Ca2+ but not fast release induced by acute Ca2+ influx. To ensure that the results obtained with 10 mm EGTA in the internal solution were not confounded by the absence or presence of the standard 1.3 mm BAPTA in the pipette solution, we performed the 10 mm EGTA experiments in both the absence and presence of 1.3 mm BAPTA in the internal pipette solution (Fig. 4).
Figure 4.
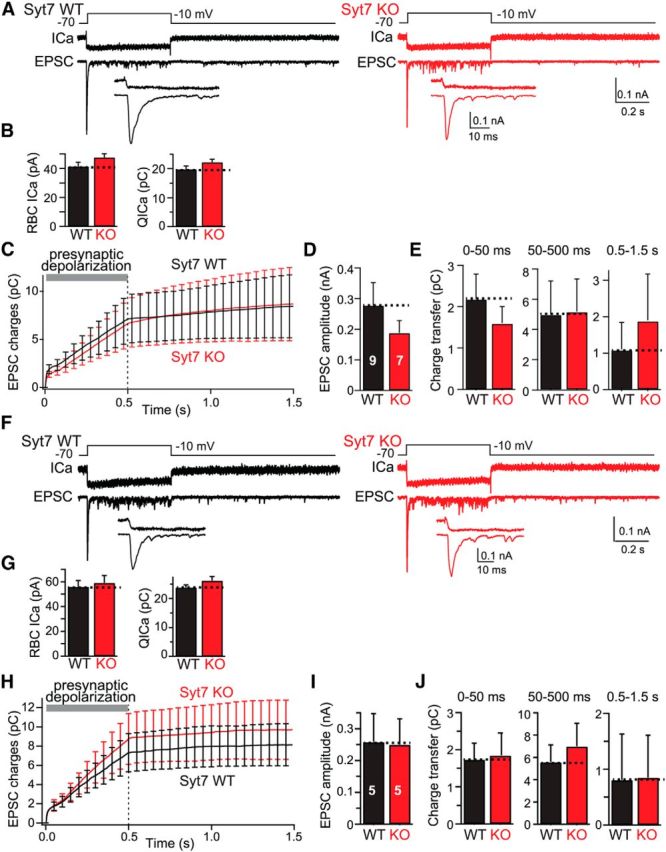
Introduction of a slow Ca2+ buffer (10 mm EGTA) into presynaptic RBCs blocks delayed asynchronous release and mimics the Syt7 KO phenotype. A, Experimental design (top), representative presynaptic Ca2+ current traces (ICa; middle), and postsynaptic EPSC traces (bottom) recorded from connected RBCs and AII cells from littermate WT (left) and Syt7 KO (right) mice. Insets (bottom) show expanded traces of the initial EPSCs. Experiments are as in Figure 2, except that the internal solution contained 10 mm EGTA instead of 1.3 mm BAPTA. B, Summary graph of the Ca2+ current amplitude (left) and integrated Ca2+ charge transfer (right) shows that the Syt7 KO has no effect on Ca2+ influx. C, Plot of the cumulative EPSC charge transfer during and after the 500 ms depolarization reveals that addition of 10 mm EGTA to the internal solution dramatically suppresses delayed asynchronous release in WT synapses but has no effect on Syt7 KO synapses, rendering delayed asynchronous release identical in WT and Syt7 KO synapses. D, Summary graph of peak EPSC amplitudes in WT and Syt7 KO mice. E, Summary graphs of the integrated charge transfer for fast synchronous release, sustained release, and delayed asynchronous release. Note that delayed asynchronous release in WT synapses is suppressed by the internal 10 mm EGTA to those of Syt7 KO synapses. F–J, Same as in A–E, except that all recordings were performed with an internal pipette solution containing both 1.3 mm BAPTA and 10 mm EGTA. All data are means ± SEMs (n = 5–9). Statistical analyses were performed by Student's t test comparing Syt7 KO with WT littermates, except an ANOVA followed by the post hoc Bonferroni's test was used in C, E, H, and J, but no statistically significant differences were noted.
We found that 10 mm EGTA did not alter Ca2+ currents or Ca2+ fluxes and had no major effect on fast synchronous release or on sustained release triggered during the depolarization phase, independent of whether BAPTA was present (Fig. 4A–J). However, 10 mm EGTA dramatically decreased the amount of delayed “asynchronous” release in WT synapses, such that WT and Syt7 KO synapses now exhibited identical amounts of synaptic transmission (Fig. 4E,J). This result was obtained identically without and with 1.3 mm BAPTA in the pipette and mimics precisely what we observed previously for cultured hippocampal neurons in which EGTA also selectively blocked delayed asynchronous release (Maximov and Südhof, 2005). Thus, this result confirms the hypothesis that Syt7 is selectively essential for delayed asynchronous release.
Syt7 KO does not impair the priming rate in RBC–AII synapses
Although our data described above argue strongly for the role of Syt7 mediating Ca2+-triggered delayed asynchronous release, defects in vesicle repriming may conceivably contribute to the phenotype in delayed asynchronous release that we observed in Syt7 KO synapses. Repriming of synaptic vesicles after exocytosis replenishes vesicles in the RRP for subsequent release and thus plays a crucial role in maintaining synaptic strength during extensive neuronal firing. Activity-dependent Ca2+ accumulation is known to facilitate vesicle replenishment in both conventional synapses and ribbon synapses (Dittman and Regehr, 1998; Wang and Kaczmarek, 1998; Gomis et al., 1999; Hosoi et al., 2007; Cho et al., 2011; Schnee et al., 2011), and calmodulin, a ubiquitous Ca2+-binding protein, has been implicated in vesicle replenishment (Sakaba and Neher, 2001; Hosoi et al., 2007). Because Syt7 was proposed to function in the Ca2+-dependent repriming of vesicles (Liu et al., 2014), we tested directly whether Syt7 contributes to vesicle repriming at RBC–AII cell synapses and whether an impairment in repriming could account, at least in part, for the phenotype in delayed asynchronous release we observed in the Syt7 KO synapses.
We used a paired depolarization pulse protocol to examine the recovery time course after depletion of primed vesicles in the RRP. We applied a 50 ms depolarization pulse to completely deplete the RRP (Singer and Diamond, 2006; Oesch and Diamond, 2011; Fig. 1C), and applied a second 500 ms depolarization pulse after a variable recovery interval (Fig. 5A). To quantify vesicle repriming, we calculated the ratio of the peak EPSC amplitudes (second EPSC/first EPSC). We fitted the recovery time course with a double-exponential function as described previously (Sakaba and Neher, 2001), with the two time constants corresponding to a fast and slow phase of recovery. Moreover, to assess how much of the repriming process was dependent on global residual Ca2+ that is essential for delayed asynchronous release (Fig. 4), we performed the measurements of the repriming rates of RBC–AII cell synapses in both the absence and presence of 10 mm EGTA in the presynaptic pipette solution (Fig. 5).
Figure 5.

Syt7 KO has no effect on rapid Ca2+-dependent vesicle priming as measured by the RRP replenishment rate. A, Experimental design. As illustrated above the representative traces, paired recordings were used to depolarize the presynaptic neuron for 50 ms to deplete the RRP, followed by a variable repolarization period to allow recovery of the RRP, and a second depolarization to measure the extent of RRP recovery. Two representative experiments with 0.2 and 10 s intervals are depicted. To monitor possible Ca2+-dependent effects on the pool recovery rate that may differ between WT and Syt7 KO neurons, experiments were performed with internal pipette solutions containing 1.3 mm BAPTA (standard) or 10 mm EGTA as a slow Ca2+ buffer (EGTA). B, RRP recovery at RBC–AII cell synapses is extremely rapid and Ca2+ dependent but does not require Syt7. Plots show RRP recovery rates (measured as the ratio of the EPSC amplitudes of the second vs first stimulus) as a function of recovery time (interval between stimuli) for all four experimental conditions (WT vs Syt7 KO; 1.3 mm BAPTA vs 10 mm EGTA). The RRP replenishment rates can be fitted with a double exponential; time constants are 0.13 and 3.4 s in WT and 0.18 and 1.7 s in Syt7 KO synapses, respectively. No significant difference is found between WT and Syt7 KO mice. All data are means ± SEMs (n = 3–9). Statistical analyses were performed by Student's t test comparing Syt7 KO with WT littermates.
We found that the Syt7 KO did not change the replenishment kinetics under any condition. Under standard conditions without EGTA, the fast component (WT, τf = 0.13 ± 0.03 s; Syt7 KO, τf = 0.18 ± 0.04 s) and the slow component (WT, τs = 3.4 ± 0.8 s; Syt7 KO, τs = 1.7 ± 1.1 s) each accounted for approximately half of the total repriming reaction and were indistinguishable between WT and Syt7 KO synapses (Fig. 5). EGTA decreased selectively the amplitude and kinetics of the slow component but again had identical effects on WT and Syt7 KO synapses (Fig. 5B; WT, τf = 0.16 ± 0.03 s, τs = 12.6 ± 1.4 s; Syt7 KO, τf = 0.15 ± 0.04 s, τs = 11 ± 1.8 s). Our data thus demonstrate that, at the RBC–AII cell synapse, Syt7 is not required for repriming of vesicles. Moreover, these results suggest that vesicle repriming at the RBC–AII synapse is a mechanistically heterogeneous process that can be extremely fast and partly requires residual Ca2+ (Dittman and Regehr, 1998; Wang and Kaczmarek, 1998; Hosoi et al., 2007).
An alternative hypothesis to account for our observations that might be proposed is that delayed asynchronous release derives from a separate pool of synaptic vesicles other than the standard RRP and that these vesicles may specifically require Syt7 for priming. To test this possibility, we applied hypertonic sucrose that is thought to trigger fusion of all primed vesicles (Rosenmund and Stevens, 1996). As shown in Figure 6A, the dendrites of AII cells were visualized by including Alexa Fluor-594 in the patch pipette, and the Alexa Fluor-594 fluorescence was used for guiding a puffing pipette containing 2 m sucrose. Sucrose was puffed onto the dendrites with a pressure of 5 psi, which should deplete all primed vesicles in RBC boutons located within the puffed area. The repriming of the refilled vesicles was accessed by applying the second sucrose puff with a 10 s interval. Our results showed that neither the RRP size of all primed vesicles nor the replenishment rate after depletion of the RRP changes in Syt7 KO mice compared with the control group (Fig. 6B–D). Thus, Syt7 is not required for the repriming of synaptic vesicles mediating delayed asynchronous release. However, we should note that, although hypertonic sucrose application is the only method we are aware of that can trigger exocytosis of all primed vesicles, the tissue distortions induced by the osmotic changes in acute slice preparations limit the precision and resolution of the recordings.
Figure 6.

Syt7 KO has no effect on the size of the RRP or RRP repriming as measured by the EPSCs induced by two sequential applications of hypertonic sucrose. A, Representative AII cell imaged after injecting Alexa Fluor-594 into the cell. The dashed circle indicates approximately the dendritic area targeted by puffing of hypertonic sucrose. B, Representative traces of EPSCs evoked by two 5 s pulses of 2 m sucrose delivered with a 10 s interval. The first puff was to measure and deplete the total RRP, and the second puff was to measure the repriming rate of synaptic vesicles after RRP depletion. C, The total pool size of releasable vesicles evoked by sucrose and the replenished vesicles are similar between WT and KO mice. Bar diagram depicts the mean RRP size measured as the charge transfer induced by hypertonic sucrose. D, The ratio of integrated charge of total release evoked by consequent sucrose puffs at 10 s intervals are not significantly different between WT and KO mice. Bar diagram displays the ratio of the size of the total charge transfer induced by the second sucrose application divided by that induced by the first sucrose application. Data in C and D are means ± SEMs. There was no statistically significant difference between control and KO data as probed with Student's t test.
Discussion
We found that Syt7 is selectively essential for delayed asynchronous release at RBC–AII cell ribbon synapses but not for transient synchronous or sustained release. Moreover, we showed that replenishment of readily releasable vesicles at these synapses exhibits two phases: (1) a fast phase that is independent of residual global Ca2+; and (2) a slow phase that depends on global Ca2+ but that both phases of vesicle replenishment do not require Syt7. Thus, our data are consistent with the notion that Syt7 functions as an essential Ca2+ sensor for delayed asynchronous release mediated by slow global Ca2+ increases, whereas “fast” synaptotagmins (Syt1, Syt2, and Syt9) function as Ca2+ sensors for synchronous release mediated by rapid local Ca2+ increases at the mouth of Ca2+ channels.
Neurotransmitter release at RBC ribbon synapses
RBC ribbon synapses release neurotransmitters with kinetically distinct components, including transient and prolonged or sustained release, which have been proposed to encode contrast and luminance of visual stimuli, respectively (Oesch and Diamond, 2011). Differences in release kinetics may derive from differences between (1) the Ca2+ sensors triggering exocytosis, (2) the coupling distance between vesicles and Ca2+ channels, or (3) the cell biological mechanisms of exocytosis (or a combination thereof because, for example, different Ca2+ sensors likely have distinct distances to the Ca2+ channels; Jarsky et al., 2010; Mehta et al., 2014). Based on previous studies (Mennerick and Matthews, 1996; Singer and Diamond, 2003; Oesch and Diamond, 2011), we conceptually divided release induced by 500 ms depolarizations into fast synchronous release (<50 ms), sustained release (50–500 ms during the depolarization), and delayed asynchronous release (>500 ms after the depolarization; Fig. 1). Our results indicate that transient and sustained release at RBC ribbon synapses are both primarily triggered by localized Ca2+ increase and probably both use the same fast Ca2+ sensor because they are not affected by high concentrations of EGTA or deletion of Syt7 (Figs. 2, 4). Because the transient fast EPSC component likely depletes the RRP, the sustained component during the depolarization derives from the replenishment of the RRP, which is thought to be accelerated by the specialized function of synaptic ribbons (Oesch and Diamond, 2011). Consistent with the notion that, at most synapses, delayed asynchronous release is primarily evoked by high-frequency action potential trains, delayed asynchronous release at ribbon synapses only manifests after strong depolarizations that induce accumulation of global Ca2+ in the nerve terminal (Mehta et al., 2014). Most importantly, our data show that Syt7 plays a major role in triggering delayed asynchronous release at RBC ribbon synapse, suggesting that Syt7 represents a universal Ca2+ sensor for delayed asynchronous release in various types of synapses.
We are proposing that the three phases of Ca2+-triggered release at the RBC–AII cell synapse proceed as follows. When the terminals are depolarized, Ca2+ channels open and the inflowing Ca2+ at the tip of the ribbons triggers exocytosis of the entire RRP. This initial release reaction is analogous to synchronous release at standard synapses and likely uses Syt1 as a Ca2+ sensor because Syt1 is concentrated in these synapses (Fox and Sanes, 2007). After depletion of the RRP, continuous depolarization produces continuous Ca2+ influx into the terminal, and the local Ca2+ influx triggers sustained release of vesicles as they are being reprimed rapidly (Figs. 1, 5). This sustained release continues to be triggered primarily by Ca2+ binding to Syt1 and not Syt7 because it is mediated by local Ca2+ influx and Ca2+ binding to Syt1 acts faster than Ca2+ binding to Syt7. In sustained release, Syt1 outcompetes Syt7 because it is triggered by Ca2+ that flows into the terminal via Ca2+ channels that are tethered adjacent to Syt1 or Syt2 on synaptic vesicles (Kaeser et al., 2011). However, when the depolarization ends, Ca2+ channels close and the high local Ca2+ concentration dissipates. As a result, Syt7 is now preferentially activated by Ca2+ binding, although Syt7 is slower than Syt1 because Syt7 is activated at lower Ca2+ concentrations than Syt1. As a result, delayed asynchronous release is dependent selectively on Syt7.
The cell biological basis and computational significance of delayed asynchronous release are only slowly beginning to emerge. Delayed release may be mediated by a non-ribbon-dependent mechanism (Mehta et al., 2014), but it is unknown whether the vesicles involved undergo exocytosis at the active zone or elsewhere in the terminal. The presence of distinct pathways of exocytosis has been observed similarly for α-latrotoxin triggered release (Deák et al., 2009) and spontaneous release (Ramirez et al., 2012). It is tempting to speculate that such different pathways of release perform distinct computational roles that may become manifest when more complex visual tasks are compared between WT and Syt7 KO mice.
Vesicle replenishment at the ribbon synapse
When the RRP is depleted at a synapse, it is refilled by repriming of vesicles. Depletion of the RRP may play a major role in short-term depression of synapses during high-frequency action potential trains. Repriming of vesicles into the RRP does not require Ca2+, but Ca2+ enhances the priming rate significantly (Neher and Sakaba, 2008). Consistent with these conclusions, we find that the fast phase of vesicle repriming is global Ca2+ independent, whereas the slow phase of vesicle repriming is strongly accelerated by increases in global Ca2+ concentration during depolarization (Fig. 5). The molecular identity of the Ca2+ sensors mediating Ca2+-dependent vesicle replenishment remains enigmatic, although the Ca2+-dependent properties of Munc13 are likely to be crucial (Shin et al., 2010), and calmodulin may additionally contribute (Sakaba and Neher, 2001; Lipstein et al., 2013). A recent study has proposed that Syt7 may act as a Ca2+ sensor for vesicle replenishment at cultured hippocampal neurons by interacting with CaM (Liu et al., 2014). However, by measuring directly the recovery of the RRP after complete depletion, we found that neither the fast nor the slow phase of RRP replenishment is altered by the Syt7 KO, arguing against a role of Syt7 as a Ca2+ sensor in vesicle replenishment. In addition, we found that application of hypertonic sucrose depletes a similar total pool of primed vesicles in Syt7 KO and WT mice and that this pool recovers similarly after depletion in these mice, suggesting that Syt7 is not required for vesicle priming (Fig. 6). It is possible that ribbon synapses use distinct molecular mechanisms in vesicle replenishment, because synaptic ribbons have been proposed to facilitate vesicle delivery to the presynaptic membrane and therefore RRP replenishment (Sterling and Matthews, 2005; Snellman et al., 2011). However, resupply of vesicles at ribbon synapses is also known to be Ca2+ dependent (Mennerick and Matthews, 1996; Gomis et al., 1999; Singer and Diamond, 2006; Babai et al., 2010; Wan et al., 2010; Cho et al., 2011; Schnee et al., 2011; Fig. 5).
Comparison with other synapses
Can these findings at RBC–AII cell synapses be applied to other synapses? Clearly synapses differ, but our results are broadly extending previous studies characterizing synaptotagmins as Ca2+ sensors in exocytosis in general and suggesting that synapses are universally governed by different synaptotagmin Ca2+ sensors that mediate distinct phases of release (for review, see Südhof, 2013).
Footnotes
This study was supported by National Institutes of Health Grant R01 NS077906.
References
- Atluri PP, Regehr WG. Delayed release of neurotransmitter from cerebellar granule cells. J Neurosci. 1998;18:8214–8227. doi: 10.1523/JNEUROSCI.18-20-08214.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babai N, Bartoletti TM, Thoreson WB. Calcium regulates vesicle replenishment at the cone ribbon synapse. J Neurosci. 2010;30:15866–15877. doi: 10.1523/JNEUROSCI.2891-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacaj T, Wu D, Yang X, Morishita W, Zhou P, Xu W, Malenka RC, Südhof TC. Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron. 2013;80:947–959. doi: 10.1016/j.neuron.2013.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best AR, Regehr WG. Inhibitory regulation of electrically coupled neurons in the inferior olive is mediated by asynchronous release of GABA. Neuron. 2009;62:555–565. doi: 10.1016/j.neuron.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomfield SA, Dacheux RF. Rod vision: pathways and processing in the mammalian retina. Prog Retin Eye Res. 2001;20:351–384. doi: 10.1016/S1350-9462(00)00031-8. [DOI] [PubMed] [Google Scholar]
- Burrone J, Neves G, Gomis A, Cooke A, Lagnado L. Endogenous calcium buffers regulate fast exocytosis in the synaptic terminal of retinal bipolar cells. Neuron. 2002;33:101–112. doi: 10.1016/S0896-6273(01)00565-7. [DOI] [PubMed] [Google Scholar]
- Cho S, Li GL, von Gersdorff H. Recovery from short-term depression and facilitation is ultrafast and Ca2+ dependent at auditory hair cell synapses. J Neurosci. 2011;31:5682–5692. doi: 10.1523/JNEUROSCI.5453-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw MI, Tricoire L, Erdelyi F, Szabo G, McBain CJ. Asynchronous transmitter release from cholecystokinin-containing inhibitory interneurons is widespread and target-cell independent. J Neurosci. 2009;29:11112–11122. doi: 10.1523/JNEUROSCI.5760-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deák F, Liu X, Khvotchev M, Li G, Kavalali ET, Sugita S, Südhof TC. Alpha-latrotoxin stimulates a novel pathway of Ca2+-dependent synaptic exocytosis independent of the classical synaptic fusion machinery. J Neurosci. 2009;29:8639–8648. doi: 10.1523/JNEUROSCI.0898-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG. Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to Purkinje cell synapse. J Neurosci. 1998;18:6147–6162. doi: 10.1523/JNEUROSCI.18-16-06147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MA, Sanes JR. Synaptotagmin I and II are present in distinct subsets of central synapses. J Comp Neurol. 2007;503:280–296. doi: 10.1002/cne.21381. [DOI] [PubMed] [Google Scholar]
- Gomis A, Burrone J, Lagnado L. Two actions of calcium regulate the supply of releasable vesicles at the ribbon synapse of retinal bipolar cells. J Neurosci. 1999;19:6309–6317. doi: 10.1523/JNEUROSCI.19-15-06309.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson N, Wei SH, Hoang DN, Lao Y, Zhang Q, Radda GK, Rorsman P, Südhof TC, Han W. Synaptotagmin-7 is a principal Ca2+ sensor for Ca2+ -induced glucagon exocytosis in pancreas. J Physiol. 2009;587:1169–1178. doi: 10.1113/jphysiol.2008.168005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefft S, Jonas P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci. 2005;8:1319–1328. doi: 10.1038/nn1542. [DOI] [PubMed] [Google Scholar]
- Hosoi N, Sakaba T, Neher E. Quantitative analysis of calcium-dependent vesicle recruitment and its functional role at the calyx of Held synapse. J Neurosci. 2007;27:14286–14298. doi: 10.1523/JNEUROSCI.4122-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarsky T, Tian M, Singer JH. Nanodomain control of exocytosis is responsible for the signaling capability of a retinal ribbon synapse. J Neurosci. 2010;30:11885–11895. doi: 10.1523/JNEUROSCI.1415-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, Rizo J, Südhof TC. RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell. 2011;144:282–295. doi: 10.1016/j.cell.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipstein N, Sakaba T, Cooper BH, Lin KH, Strenzke N, Ashery U, Rhee JS, Taschenberger H, Neher E, Brose N. Dynamic control of synaptic vesicle replenishment and short-term plasticity by Ca(2+)-calmodulin-Munc13-1 signaling. Neuron. 2013;10:82–96. doi: 10.1016/j.neuron.2013.05.011. [DOI] [PubMed] [Google Scholar]
- Liu H, Bai H, Hui E, Yang L, Evans CS, Wang Z, Kwon SE, Chapman ER. Synaptotagmin 7 functions as a Ca2+-sensor for synaptic vesicle replenishment. Elife. 2014;3:e01524. doi: 10.7554/eLife.01524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Südhof TC. Autonomous function of synaptotagmin 1 in triggering synchronous release independent of asynchronous release. Neuron. 2005;48:547–554. doi: 10.1016/j.neuron.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Mehta B, Ke JB, Zhang L, Baden AD, Markowitz AL, Nayak S, Briggman KL, Zenisek D, Singer JH. Global Ca2+ signaling drives ribbon-independent synaptic transmission at rod bipolar cell synapses. J Neurosci. 2014;34:6233–6244. doi: 10.1523/JNEUROSCI.5324-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Matthews G. Ultrafast exocytosis elicited by calcium current in synaptic terminals of retinal bipolar neurons. Neuron. 1996;17:1241–1249. doi: 10.1016/S0896-6273(00)80254-8. [DOI] [PubMed] [Google Scholar]
- Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59:861–872. doi: 10.1016/j.neuron.2008.08.019. [DOI] [PubMed] [Google Scholar]
- Oesch NW, Diamond JS. Ribbon synapses compute temporal contrast and encode luminance in retinal rod bipolar cells. Nat Neurosci. 2011;14:1555–1561. doi: 10.1038/nn.2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protti DA, Llano I. Calcium currents and calcium signaling in rod bipolar cells of rat retinal slices. J Neurosci. 1998;18:3715–3724. doi: 10.1523/JNEUROSCI.18-10-03715.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DM, Khvotchev M, Trauterman B, Kavalali ET. Vti1a identifies a vesicle pool that preferentially recycles at rest and maintains spontaneous neurotransmission. Neuron. 2012;73:121–134. doi: 10.1016/j.neuron.2011.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/S0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron. 2001;32:1119–1131. doi: 10.1016/S0896-6273(01)00543-8. [DOI] [PubMed] [Google Scholar]
- Schnee ME, Santos-Sacchi J, Castellano-Muñoz M, Kong JH, Ricci AJ. Calcium-dependent synaptic vesicle trafficking underlies indefatigable release at the hair cell afferent fiber synapse. Neuron. 2011;70:326–338. doi: 10.1016/j.neuron.2011.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonn JS, Maximov A, Lao Y, Südhof TC, Sørensen JB. Synaptotagmin-1 and -7 are functionally overlapping Ca2+ sensors for exocytosis in adrenal chromaffin cells. Proc Natl Acad Sci U S A. 2008;105:3998–4003. doi: 10.1073/pnas.0712373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin OH, Lu J, Rhee JS, Tomchick DR, Pang ZP, Wojcik SM, Camacho-Perez M, Brose N, Machius M, Rizo J, Rosenmund C, Südhof TC. Munc13 C2B domain is an activity-dependent Ca2+ regulator of synaptic exocytosis. Nat Struct Mol Biol. 2010;17:280–288. doi: 10.1038/nsmb.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer JH, Diamond JS. Sustained Ca2+ entry elicits transient postsynaptic currents at a retinal ribbon synapse. J Neurosci. 2003;23:10923–10933. doi: 10.1523/JNEUROSCI.23-34-10923.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer JH, Diamond JS. Vesicle depletion and synaptic depression at a mammalian ribbon synapse. J Neurophysiol. 2006;95:3191–3198. doi: 10.1152/jn.01309.2005. [DOI] [PubMed] [Google Scholar]
- Snellman J, Mehta B, Babai N, Bartoletti TM, Akmentin W, Francis A, Matthews G, Thoreson W, Zenisek D. Acute destruction of the synaptic ribbon reveals a role for the ribbon in vesicle priming. Nat Neurosci. 2011;14:1135–1141. doi: 10.1038/nn.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling P, Matthews G. Structure and function of ribbon synapses. Trends Neurosci. 2005;28:20–29. doi: 10.1016/j.tins.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Südhof TC. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron. 2013;80:675–690. doi: 10.1016/j.neuron.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita S, Han W, Butz S, Liu X, Fernández-Chacón R, Lao Y, Südhof TC. Synaptotagmin VII as a plasma membrane Ca(2+) sensor in exocytosis. Neuron. 2001;30:459–473. doi: 10.1016/S0896-6273(01)00290-2. [DOI] [PubMed] [Google Scholar]
- Sugita S, Shin OH, Han W, Lao Y, Südhof TC. Synaptotagmins form a hierarchy of exocytotic Ca(2+) sensors with distinct Ca(2+) affinities. EMBO J. 2002;21:270–280. doi: 10.1093/emboj/21.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Pang ZP, Qin D, Fahim AT, Adachi R, Südhof TC. A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature. 2007;450:676–682. doi: 10.1038/nature06308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, Müller SA, Rammner B, Gräter F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmüller H, Heuser J, Wieland F, Jahn R. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Veruki ML, Mørkve SH, Hartveit E. Functional properties of spontaneous EPSCs and non-NMDA receptors in rod amacrine (AII) cells in the rat retina. J Physiol. 2003;549:759–774. doi: 10.1113/jphysiol.2003.039982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan QF, Zhou ZY, Thakur P, Vila A, Sherry DM, Janz R, Heidelberger R. SV2 acts via presynaptic calcium to regulate neurotransmitter release. Neuron. 2010;66:884–895. doi: 10.1016/j.neuron.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- Wen H, Linhoff MW, McGinley MJ, Li GL, Corson GM, Mandel G, Brehm P. Distinct roles for two synaptotagmin isoforms in synchronous and asynchronous transmitter release at zebrafish neuromuscular junction. Proc Natl Acad Sci U S A. 2010;107:13906–13911. doi: 10.1073/pnas.1008598107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Mashimo T, Südhof TC. Synaptotagmin-1, -2, and -9: Ca(2+) sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron. 2007;54:567–581. doi: 10.1016/j.neuron.2007.05.004. [DOI] [PubMed] [Google Scholar]