

Abstract
The inositol hexakisphosphate kinases (IP6Ks) are the principal enzymes that generate inositol pyrophosphates. There are three IP6Ks (IP6K1, 2, and 3). Functions of IP6K1 and IP6K2 have been substantially delineated, but little is known of IP6K3's role in normal physiology, especially in the brain. To elucidate functions of IP6K3, we generated mice with targeted deletion of IP6K3. We demonstrate that IP6K3 is highly concentrated in the brain in cerebellar Purkinje cells. IP6K3 physiologically binds to the cytoskeletal proteins adducin and spectrin, whose mutual interactions are perturbed in IP6K3-null mutants. Consequently, IP6K3 knock-out cerebella manifest abnormalities in Purkinje cell structure and synapse number, and the mutant mice display deficits in motor learning and coordination. Thus, IP6K3 is a major determinant of cytoskeletal disposition and function of cerebellar Purkinje cells.
SIGNIFICANCE STATEMENT We identified and cloned a family of three inositol hexakisphosphate kinases (IP6Ks) that generate the inositol pyrophosphates, most notably 5-diphosphoinositol pentakisphosphate (IP7). Of these, IP6K3 has been least characterized. In the present study we generated IP6K3 knock-out mice and show that IP6K3 is highly expressed in cerebellar Purkinje cells. IP6K3-deleted mice display defects of motor learning and coordination. IP6K3-null mice manifest aberrations of Purkinje cells with a diminished number of synapses. IP6K3 interacts with the cytoskeletal proteins spectrin and adducin whose altered disposition in IP6K3 knock-out mice may mediate phenotypic features of the mutant mice. These findings afford molecular/cytoskeletal mechanisms by which the inositol polyphosphate system impacts brain function.
Keywords: adducin, cerebellar synapse, IP6K, kinase independent, Purkinje cell, spectrin
Introduction
Inositol phosphates are major signaling molecules with inositol 1, 4, 5-trisphosphate (IP3) best known as a second messenger molecule releasing intracellular calcium. During the past decade higher inositol polyphosphates have been characterized, most notably diphosphoinositol-pentakisphosphate (PP-IP5), which contains an energetic pyrophosphate bond and is typically referred to as IP7 (Stephens et al., 1993). IP7 is synthesized by a family of three inositol hexakisphosphate kinases (IP6K;Voglmaier et al., 1996; Saiardi et al., 1999; Saiardi et al., 2001), and, to a lesser extent, by the VIP group of enzymes (Mulugu et al., 2007). The three IP6Ks generate the same isomer of IP7 with the pyrophosphate bond at C-5. However, they derive from three distinct genes and appear to mediate different functions (Saiardi et al., 2001; Chakraborty et al., 2010; Rao et al., 2014). Mice with targeted deletion of IP6K1 display male sterility, enhanced Akt signaling, and resistance to diabetes (Chakraborty et al., 2010). IP6K2 appears to be a mediator of cancer cell migration and tumor metastasis (Rao et al., 2015) and is required for p53-associated apoptosis (Koldobskiy et al., 2010). However, little is known about the function of IP6K3.
Adducins and spectrins are ubiquitously expressed cytoskeletal proteins. Adducin has three subtypes: α, β, and γ. α- and γ-Adducins are expressed similarly in most tissues, whereas β-adducin is most abundant in the brain (Joshi et al., 1991; Dong et al., 1995). All three adducin proteins contain an N-terminal globular head domain, a neck domain, and a C-terminal tail domain with a highly conserved MARCKS motif at the end, which is required for association with spectrin (Matsuoka et al., 2000). α- and β-Adducins form heterodimers in the brain.
Spectrins are comprised of α- and β-subunits, which associate to form heterotetramers. Human spectrin contains two α-subunits (α1 and α2) and five β-subunits (β1, β2, β3, β4, and β5; Bennett and Baines, 2001). α1/β1-Spectrins are expressed in erythrocytes, while α2/β2, α2/β3, and α2/β4 spectrins predominate in nonerythroid tissues. α-Spectrin incorporates triple helical repeat domains, an src homology domain 3 (SH3), and a C-terminal calmodulin-related domain. β-Spectrin contains an N-terminal calponin homology (CH) domain, which binds adducin, a triple helical repeat, and a C-terminal pleckstrin homology (PH) domain region (Bennett and Baines, 2001).
Adducin is localized to and binds spectrin-actin junctions, promoting the association of spectrin with actin (Gardner and Bennett, 1987). Binding of the two proteins is mediated by the C-terminal tail and MARCKS domain of adducin (Li et al., 1998) and the N-terminal CH domain of β-spectrin (Li and Bennett, 1996).
Morphologic alterations in neuronal dendritic spines underlie synaptic plasticity, processes that are especially well exemplified in Purkinje cells of the cerebellum. The cytoskeleton of dendritic spines is comprised of actin and actin-regulating proteins, including adducins and spectrins (Matsuoka et al., 1998; Gao et al., 2011; Nestor et al., 2011). The shape of Purkinje cell dendritic spines is determined by the arrangements and attachments of their cytoskeletal elements (Landis and Reese, 1983). β-Spectrins regulate the morphology and functional dynamics of dendritic spines (Gao et al., 2011; Nestor et al., 2011). Cells with deficiencies of spectrin/adducin interaction display disrupted morphology (Hu et al., 1995).
In the present study we report that IP6K3 is abundantly expressed in cerebellar Purkinje cells and physically binds adducin/spectrin. Mutual interactions of adducin and spectrin are severely disrupted in IP6K3-null mice cerebellum. Mice with targeted deletion of IP6K3 manifest abnormalities in cerebellar Purkinje cell structure as well as reduced numbers of synapses. IP6K3 mutant mice manifest disorders of motor learning and coordination. IP6K3 noncatalytically influences the disposition of the spectrin-adducin complex, which can account for its influences upon cerebellar Purkinje cells and motor behavior.
Materials and Methods
Reagents.
Chemicals were from Sigma-Aldrich. Anti-IP6K1, IP6K2, IP6K3, anti-GST (HRP conjugate), and anti-Flag-tag antibodies were from Sigma-Aldrich. Anti-GAD65, GAPDH, β-actin antibodies, and rabbit anti-calbindin antibody were from Cell Signaling Technology. Anti-VGlut-1 and anti-VGlut-2 antibodies were from Millipore. Anti-gephyrin, vesicular GABA transporter (VGAT), and Homer1 antibodies were from Synaptic Systems. Anti-α-adducin, β-adducin, α2-spectrin, normal rabbit IgG, and normal mouse IgG antibodies and mouse anti-calbindin antibody were from Santa Cruz Biotechnology. Anti-β2-spectrin antibody was from Bethyl Laboratories. Alexa 488 goat anti-rabbit and Alexa 568 goat anti-mouse IgG were from Life Technologies. IP6K3 shRNA and control shRNA plasmids were from Sigma-Aldrich.
Animals.
Eight-week-old male mice were used. The wild-type and IP6K3 KO animals were littermates from heterozygous parents. Animal breeding and procedures were conducted in strict accordance with the NIH Guide for Care and Use of Laboratory Animals.
Immunoblotting and immunoprecipitation.
Standard methods for cell lysis, immunoprecipitation, SDS-PAGE, and Western blot were as described previously (Zhu et al., 2011). Briefly, mouse brain tissue or cultured cells were lysed on ice in buffer containing 20 mm Tris-HCl, pH 7.5; 150 mm NaCl; 1% NP-40; 1% sodium deoxycholate; 1 mm Na2EDTA; 1 mm EGTA; protease inhibitors; and phosphatase inhibitors. Insoluble cell debris was removed by centrifugation for 10 min at 16,000 g. For immunoprecipitation, cell lysates were precleared by protein A/G beads for 1 h at 4°C, then treated with the primary antibody overnight at 4°C, followed by pull-down with protein A/G beads for 1 h at 4°C. Beads were washed six times with lysis buffer on ice.
Cell culture and lentiviral transduction.
HEK293 and HEK293T cells were cultured under standard conditions. Lentiviral production and transduction were generated as previously reported (Fu et al., 2013). IP6K3 knock-down HEK293 cells were generated by lentiviral transduction and maintained in DMEM with 5 μg/ml puromycin.
In vitro protein expression.
The actin-binding (CH) domain (1–275 aa), repeat (RE) domain (303–525 aa), and PH domain (2197–2307 aa) of β2-spectrin (Li and Bennett, 1996) and the wild-type IP6K3 and kinase-dead mutant IP6K3 (K217A) were cloned into the pGEX-6P-2 vector (GE Healthcare Life Sciences). PreScission Protease was used to cleave the wild-type IP6K3 and kinase-dead mutant IP6K3 (K217A) from the fusion protein. The Flag-tag β-adducin tail and MARCKS domain (437–726 aa; Li et al., 1998) were cloned into pT7-MAT-1 for in vitro protein expression.
Immunofluorescence staining and electron microscopy.
Immunofluorescence staining and electron microscopy were performed as previously reported (Fu et al., 2010). The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the NIH. For immunofluorescence staining, animals were perfused with 4% paraformaldehyde and brain slices were cut in sagittal section and incubated with 10% goat serum and then with primary antibodies at 4°C overnight. The fluorescence-labeled secondary antibodies were incubated for 1 h at room temperature. Nuclei were counterstained with DAPI. Slices were mounted with ProLong Gold Antifade Mountant. Pictures were taken under confocal microscope (Zeiss LSM 700; NIH Award S10 OD016374). The range indicator on the microscope shows no “red” or no oversaturated pixels to prevent artifacts caused by overexposure. Synapses were identified by double staining of presynaptic and postsynaptic markers (VGAT and gephyrin for GABAergic synapses, VGlut1 and Homer1 for parallel fiber synapses, and VGlut2 and Homer1 for climbing fiber synapses). The primary antibodies were anti-VGAT (rabbit polyclonal; Synaptic Systems), anti-gephyrin (mouse monoclonal; Synaptic Systems), anti-VGlut1 (mouse monoclonal; Millipore), anti-VGlut2 (mouse monoclonal; Millipore), and anti-Homer1 (rabbit polyclonal; Synaptic Systems). The primary antibodies were diluted in PBS at a ratio of 1:100. The fluorescence-labeled secondary antibodies were Alexa 488 goat anti-rabbit IgG (Life Technology) and Alexa 568 goat anti-mouse IgG (Life Technology). The secondary antibodies were diluted in PBS at a ratio of 1:300. Volocity software (V6.3; PerkinElmer) was used to quantify the density of synapses, determined by colocalization of both presynaptic and postsynaptic markers. Synapse density was determined by counting the number of synapses per unit area. For electron microscopy, animals were perfused with 2% glutaraldehyde and 2% paraformaldehyde, and the samples were embedded into EPON resin. Slides were cut in sagittal and coronal sections with focus on the molecular layer of cerebellum, where the synapses on Purkinje cell dendrites predominate. Sections were stained with uranyl acetate and lead citrate and viewed by transmission electron microscopy. Synapses were counted from at least six different views of pictures.
Golgi stain and H&E stain.
The Golgi stain was performed using the FD Rapid GolgiStain Kit (FD Neurotechnologies). The H&E stain followed standard protocols. Mice were anesthetized and briefly perfused with PBS followed by 4% paraformaldehyde. For Golgi stain, mice brains were immersed in impregnation solution at room temperature for 3 weeks in the dark. Tissues were then transferred into Solution C (FD Rapid GolgiStain Kit) and stored in the dark at 4°C for 1 week. Sections (200 μm in thickness) were cut on a cryostat at −20°C. Sections were stained and, following dehydration and clearing, they were mounted with Permount Mounting Medium.
Mouse behavioral tests.
Mouse behavioral tests were conducted on 8-week-old males at the Johns Hopkins University Brain Science Institute Behavioral Core. Wild-type and IP6K3-null mice were littermates of heterozygous parents, 12 mice per group. Animals were housed in ventilated racks and given ad libitum access to food and water. Animals were moved to their final house location 2 weeks before testing. Starting 1 week before testing, animals were handled daily up until the first day of behavior testing. All testing occurred during the animals' normal light phase (7:30 A.M.–9:30 P.M.). The order of testing was as follows: open field, rotarod, and gait analysis with only one test done per week. All procedures were approved by The Johns Hopkins University Animal Care and Use Committee.
Open field.
General locomotor activity in IP6K3 knock-out mice was assessed by open field analysis (Photobeam Activity System; San Diego Instruments). Mice were placed in the center of an enclosed acrylic chamber and allowed to explore freely for 30 min. Photobeams tracked horizontal movements and rearing behavior in the x and y directions.
Rotarod.
Following open field studies, motor learning and coordination were examined in IP6K3-null mice by the rotarod test (Rotamex-5; Columbus Instruments). Testing was conducted over a 3 d period, where each mouse was given five trials per day and latency to fall off the rotarod was measured as it accelerated from 4 to 99 rpm over a 5 min period. Before the start of testing on the first 2 d, each mouse was given a habituation trial by being placed on the rotarod, which was rotating at a constant speed of 4 rpm for 10 min. The highest and lowest latencies of the trials were discarded and the average taken from those remaining.
Gait analysis.
The CatWalk XT system (Noldus Information Technologies) was used to study walking sequence and patterns of IP6K3 mutant mice. Before testing, mice were given four training days to acclimate to the testing apparatus. On the testing day, animals were placed on the glass plate until they completed three valid trials. A valid trial was one in which the mouse walked the length of the tunnel in <10 s with <60% variation in speed. Data were recorded using a high-speed camera fixed directly beneath the glass plate and were analyzed using CatWalk 9.0 XT software (Noldus Information Technologies). Sequence of movement was used to identify stride length, step width, and percentage of forepaw-hindpaw overlap. Averages were taken across valid trials on testing day. All trials marked as valid by the software were then reviewed manually to ensure that animals did not turn around or stop during the trial.
Statistical analysis.
Quantitative data are expressed as means with SEM. Data were analyzed by one-way ANOVA, unpaired Student's t test, and Pearson's correlation coefficient. Open field and rotarod data were analyzed by two-way repeated-measures ANOVA. The volume of Purkinje cells was quantified by using ImageJ. The Purkinje cell number was quantified by counting the cell number per unit length. P < 0.05 was considered statistically significant. For nonquantitative data, results were representative of at least three independent experiments.
Results
IP6K3 is highly expressed in cerebellar Purkinje cells, defects of motor learning, and coordination in IP6K3 knock-outs
To evaluate rigorously physiologic functions of IP6K3, we developed mice with targeted deletion of IP6K3 (Fig. 1A). IP6K3 knock-outs breed fairly normally, though they produce fewer progeny than wild-type mice (wild-type 7 ± 1 vs KO 5 ± 1; six pairs of breeding). IP6K3 immunostaining reveals substantial levels in cortex, hippocampus, thalamus, hypothalamus, and cerebellum with virtually total loss of the enzyme in null mice (Fig. 1B). Staining is somewhat patchy in midbrain and cortex. At this low level of resolution, the basis for the patchiness and cellular localizations cannot be discriminated. The IP6K3-null mice do not display gross abnormalities of brain structure (Fig. 1C–E). We explored motor behavior of mutant mice (Fig. 1F–H). Overall activity in an open field does not appear impaired in the knock-out mice (Fig. 1F); however, motor learning and coordination are disturbed, as is evident in rotarod performance (Fig. 1G). Moreover, gait analysis indicates defects in the overlap of hindpaws and forepaws but no significant difference in stride length and step width (Fig. 1H). Motor deficits manifested by the mutant mice resemble disabilities of mice with Purkinje cell dysfunction. Interestingly, in cerebellar molecular layer, IP6K3 is most highly expressed in Purkinje cells with substantial levels as well in interneurons (Fig. 1B,I,J).
Figure 1.
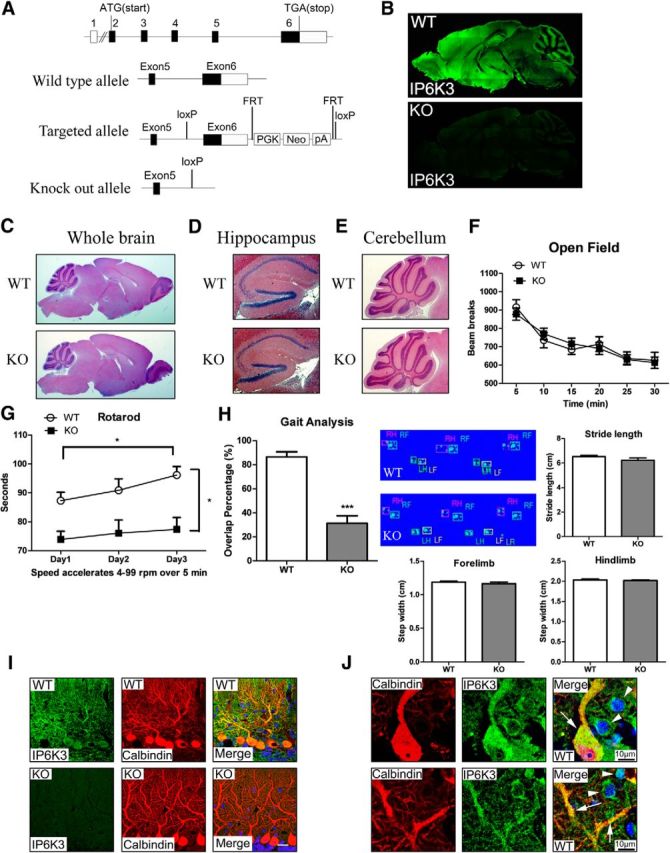
IP6K3 is highly expressed in cerebellar Purkinje cells, defects of motor learning, and coordination in IP6K3 knock-outs. A, Schematic representation of the mouse IP6K3 gene with coding regions shaded black, noncoding regions in white, and the exon number indicated on top. A close-up view of exons 5 and 6 in the wild-type, targeted, and knock-out alleles is shown. The knock-out allele loses the splice site, coding region, and the 3UTR of exon 6. B, Immunostaining of IP6K3 in sagittal sections of wild-type and IP6K3 KO mice brain (top) reveals abundant levels of IP6K3 in the cortex and cerebellar molecular layer. C–E, H&E staining on wild-type and IP6K3 KO mice brain sagittal section. C, Whole brain. D, Hippocampus. E, Cerebellum. F, Open field behavioral analysis reveals negligible alterations in IP6K3 KO mice; y-axis, total activity (beams breaks); x-axis, 5 min intervals. G, Rotarod tests reveal reduced motor learning and coordination in IP6K3 KO mice; y-axis, the latency to falling in second; x-axis, days of testing. H, Gait analysis shows significantly decreased overlap of hindpaws and forepaws in IP6K3 KO mice; y-axis, the percentage of forepaw-hindpaw overlap, with the averages taken across trials on testing day. The stride length and step width do not display notable differences. I, Immunostaining of IP6K3 and calbindin in cerebellar molecular layer from wild-type and IP6K3 KO mice. IP6K3 is expressed mainly in Purkinje cells. Scale bar, 20 μm. J, Immunostaining of IP6K3 and calbindin in wild-type cerebellar molecular layer. IP6K3 is most highly expressed in Purkinje cells (arrow) with substantial levels as well in interneurons (top arrowhead, basket cell; bottom arrowhead, stellate cell). Data are represented as mean ± SEM, *p < 0.05, ***p < 0.001, 8-week-old male mice. B–E, I, J, n = 5; F–H, n = 12.
IP6K3 binds spectrin and adducin in mouse cerebellum
Much less is known about protein–protein interactions of IP6K3 compared with IP6K1 or IP6K2. To gain insight into functions of IP6K3, we sought potential interacting proteins. Accordingly, we immunoprecipitated IP6K3 and explored proteins that coprecipitate. Mass spectrometric analysis of the most prominently interacting proteins identifies spectrins and adducins as major binding partners for IP6K3 (Fig. 2A).
Figure 2.
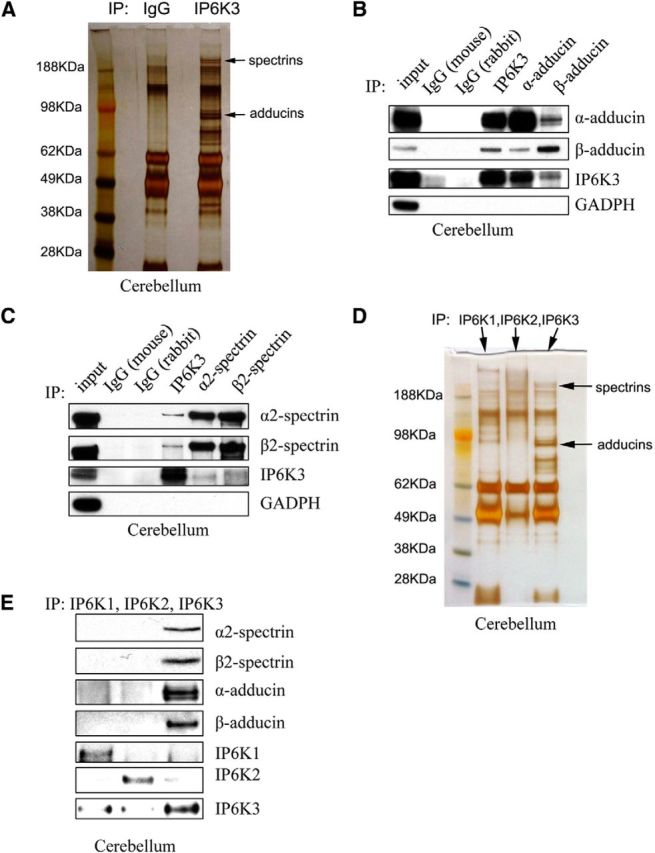
IP6K3 binds spectrins and adducins in the cerebellum. A, Immunoprecipitation (IP) of IP6K3 from wild-type mice cerebellum with rabbit IgG as a control. Silver staining and mass spectrometric analysis identify spectrins and adducins pulled down by IP6K3. B, Immunoprecipitation of IP6K3, α-adducin, and β-adducin from wild-type mice cerebellum with mouse IgG and rabbit IgG as controls. Western blot indicates that IP6K3 binds to α-adducin and β-adducin. C, Immunoprecipitation of IP6K3, α2-spectrin, and β2-spectrin from wild-type mice cerebellum with mouse IgG and rabbit IgG as controls. Western blot shows IP6K3 binding to α2-spectrin and β2-spectrin. D, Immunoprecipitation of IP6K1, IP6K2, and IP6K3 from wild-type mice cerebellum. Silver staining and mass spectrometric analysis indicates pull-down of spectrins and adducins by IP6K3 but not IP6K1 or IP6K2. E, Immunoprecipitation of IP6K1, IP6K2, and IP6K3 from wild-type mice cerebellum. Western blot shows that IP6K3 but not IP6K1 or IP6K2 binds to α-adducin, β-adducin, α2-spectrin, and β2-spectrin.
We examined in more detail interactions of IP6K3, adducins, and spectrins (Fig. 2B,C). IP6K3 coprecipitates with α- and β-adducin, while precipitation of the two adducins leads to coprecipitation of IP6K3 and the complementary adducin isoforms (Fig. 2B). Similarly, precipitation of IP6K3 leads to coprecipitation of α2- and β2-spectrin, while each of these spectrins coprecipitates with IP6K3 as well as the other spectrin isoform (Fig. 2C). Thus, IP6K3 occurs in a complex with the adducins and spectrins.
We wondered whether interactions with spectrins and adducins are selective for IP6K3 or might be evident with other IP6 kinase isoforms. Immunoprecipitation with antibodies to IP6K3 coprecipitates spectrins and adducins, while these proteins do not coprecipitate with IP6K1 or IP6K2 (Fig. 2D). In the same experiments we conducted Western blots focusing on subtypes of spectrins and adducins that bind IP6 kinase isoforms (Fig. 2E). IP6K3 coprecipitates with α2- and β2-spectrin and α- and β-adducin (Fig. 2E). Spectrins and adducins do not coprecipitate with IP6K1 or IP6K2.
Altered adducin/spectrin interactions in IP6K3-deleted cells
Spectrin and adducin are well known to interact biochemically and to colocalize in cells (Li et al., 1998). We wondered whether IP6K3 impacts such interactions. Accordingly, we deleted IP6K3 by RNA interference in HEK293 cells (Fig. 3A). In the IP6K3-deleted cells coprecipitation of α2- and β2-spectrin with α-adducin is markedly diminished (Fig. 3B). Similarly coprecipitation of α- and β-adducin with β2-spectrin is much lower in the mutants (Fig. 3B). Thus, physiologic interactions of spectrins and adducins are substantially impaired with loss of IP6K3, indicating an important role for IP6K3 in maintaining the spectrin-adducin complexes.
Figure 3.

Impaired adducin/spectrin interaction in IP6K3-deleted cells. A, IP6K3 is deleted in HEK293 cells by shRNA lentiviral transduction. B, Knockdown of IP6K3 in HEK293 cells reduces adducin/spectrin binding. C, Immunostaining in IP6K3-deleted HEK293 cells of β-adducin and β2-spectrin (top two lanes) as well as F-actin and β2-spectrin (bottom two lanes). Colocalization of β-adducin with β2-spectrin is significantly reduced in IP6K3 knockdown cells, as is colocalization of F-actin with β2-spectrin. Data are presented as mean ± SEM, ***p < 0.001. IP, immunoprecipitation; Ctrl, control.
Similarly, colocalizations of β-adducin and β2-spectrin monitored microscopically depend upon IP6K3 (Fig. 3C). In control HEK293 cells β-adducin, β2-spectrin, and F-actin are similarly localized in the vicinity of the plasma membrane. Colocalization of β-adducin and β2-spectrin is reduced almost 60% in IP6K3 knock-outs with a similar reduction in the colocalization of β2-spectrin and F-actin.
Reduced adducin/spectrin binding in IP6K3 knock-out mice cerebellum
Using IP6K3 knock-out mice, we confirm the importance of IP6K3 for the disposition of spectrins and adducins (Fig. 4). IP6K3 deletion does not change cerebellar protein levels of spectrins and adducins (Fig. 4A) but markedly alters their interaction (Fig. 4D). Coprecipitation of spectrins and adducins with IP6K3 is verified (Fig. 4B,C). Coprecipitation of β2-spectrin with α-adducin or β-adducin is greatly diminished in IP6K3 knock-outs. Similarly, coprecipitation of α- and β-adducin with β2-spectrin is virtually abolished in IP6K3 mutants (Fig. 4D).
Figure 4.

Adducin/spectrin binding is substantially reduced in IP6K3-null mice cerebellum. A, Expression of α-adducin, β-adducin, α2-spectrin, and β2-spectrin is normal in IP6K3 KO mice cerebellum. B, C, Immunoprecipitation (IP) verifies IP6K3 binding to α-adducin, β-adducin, α2-spectrin, and β2-spectrin in mouse cerebellum. D, Immunoprecipitation of α-adducin, β-adducin, α2-spectrin, and β2-spectrin from cerebellum of both wild-type and IP6K3 KO mice. Binding of α-adducin and β-adducin with β2-spectrin is reduced in IP6K3 KO mice cerebellum.
Spectrin/adducin binding is substantially increased by IP6K3, independent of the enzyme's kinase activity
To explore molecular mechanisms for IP6K3/adducin/spectrin interactions, we obtained bacterially purified forms of these proteins (Li and Bennett, 1996; Li et al., 1998; Fig. 5A). Utilizing purified proteins, we show that IP6K3 binds to β-adducin and to β2- spectrin's CH domain directly but not to the repeat or PH domains of spectrin (Fig. 5B,C). Binding of spectrin to adducin is substantially enhanced in the presence of IP6K3 (Fig. 5D). We wondered whether catalytic activity of IP6K3 is required for interactions with spectrin/adducin. Catalytically inactive IP6K3 (K217A) binds spectrin/adducin similarly to the wild-type enzyme (Fig. 5E–G). Moreover, physiological concentrations of IP6 or IP7 fail to influence spectrin-adducin binding, consistent with catalytic activity of IP6K3 not being required for spectrin/adducin interactions (Fig. 5H).
Figure 5.
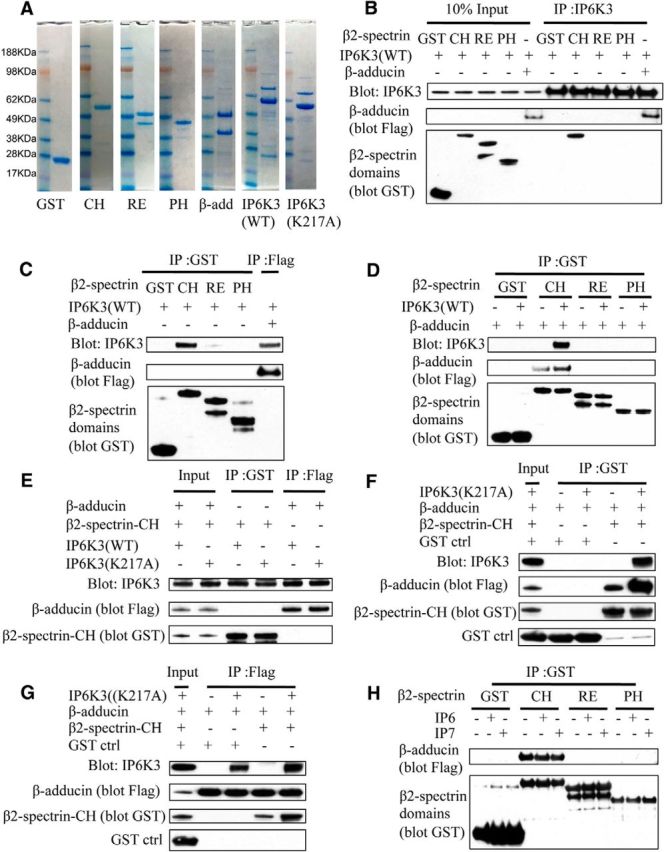
IP6K3 facilitates the binding of adducins with spectrins; kinase activity of IP6K3 is not required. A, In vitro expression and purification of proteins. Coomassie blue staining of the control GST, β2-spectrin CH domain (1–275 aa), β2-spectrin repeat domain (303–525 aa), β2-spectrin PH domain (2197–2307 aa), β-adducin (437–726 aa), wild-type IP6K3, and kinase-dead mutant IP6K3 (K217A). GST, GST control; CH, β2-spectrin CH domain (1–275 aa); RE, β2-spectrin repeat domain (303–525 aa); PH, β2-spectrin PH domain (2197–2307 aa). B, In vitro binding of IP6K3 with β2-spectrin domains and β-adducin (437–726 aa). Immunoprecipitation (IP) of IP6K3, which binds to β2-spectrin CH domain and β-adducin (437–726 aa). The upper band for RE is the full-length band. C, In vitro binding of β2-spectrin domains and β-adducin (437–726 aa) with IP6K3. Immunoprecipitation of β2-spectrin domains (pull-down GST) and β-adducin (437–726 aa, pull-down flag tag). Western blot shows that IP6K3 binds to β2-spectrin CH domain and β-adducin (437–726 aa). D, In vitro binding of β2-spectrin domains with β-adducin (437–726 aa). Immunoprecipitation of β2-spectrin domains (pull-down GST) reveals that β-adducin (437–726 aa) binds only to β2-spectrin CH domain, and the binding is significantly increased by IP6K3. E, In vitro binding of wild-type IP6K3 and kinase-dead mutant IP6K3 (K217A) with β2-spectrin CH domain and β-adducin (437–726 aa). Immunoprecipitation of β2-spectrin CH domain (pull-down GST) and β-adducin (437–726 aa, pull-down flag-tag); Western blot shows kinase-dead mutant IP6K3 (K217A) binds with β-adducin (437–726 aa) and β2-spectrin CH domain similarly to wild-type IP6K3. F, In vitro binding of kinase-dead mutant IP6K3 (K217A) with β-adducin (437–726 aa) and β2-spectrin CH domain. Immunoprecipitation of β2-spectrin CH domain (pull-down GST) indicates that kinase-dead mutant IP6K3 (K217A) binding to β2-spectrin CH domain facilitates the binding of β2-spectrin CH domain with β-adducin (437–726 aa). G, In vitro binding of β-adducin (437–726 aa) and β2-spectrin CH domain with kinase-dead mutant IP6K3 (K217A). Immunoprecipitation of β-adducin (437–726 aa, pull-down flag tag). Western blot shows that binding of kinase-dead mutant IP6K3 (K217A) to β-adducin (437–726 aa) facilitates the binding of β2-spectrin CH domain with β-adducin (437–726 aa). H, In vitro binding of β-adducin (437–726 aa) with β2-spectrin domains. Immunoprecipitation of β2-spectrin domains (pull-down GST); Western blot shows that physiological concentrations of IP6 (20 μm) or IP7 (5 μm) do not affect the binding of β-adducin with β2-spectrin.
Abnormalities of cerebellar Purkinje cell morphology in IP6K3 knock-outs
We sought potential alterations of overall cerebellar structure in the mutant mice. In mutants, the width of the molecular layer is diminished, whereas width of the granule cell layer is normal as are the number of Purkinje cells (Fig. 6A). Examination of the IP6K3 mutants at high-power magnification reveals alterations in Purkinje cell structure with withered dendritic trees (Fig. 6B,D). The morphologic abnormalities of the mutant mice appear similar in 8-week-old and 12-month-old animals, suggesting that the disorder is developmental rather than degenerative. This conclusion is supported by our failure to detect any signs of inflammation in the mutant mouse brains. The cell size and spine density of mutant Purkinje cells are also decreased (Fig. 6B,C). In contrast, the morphology of granule cells appears normal. Golgi cells, basket cells, and stellate cells of the mutants possess modestly shorter dendrites but overall normal morphology (Fig. 6E).
Figure 6.
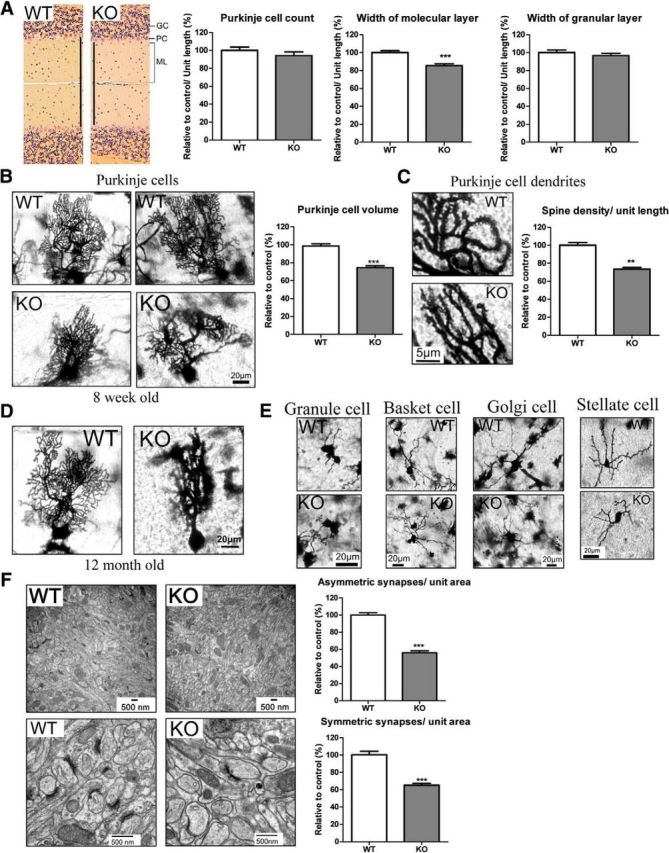
IP6K3 KO mutant mice manifest altered Purkinje cell structure and reduced synapses. A, H&E staining in cerebellum from wild-type and IP6K3 KO mice (8 weeks old). Width of the molecular layer is decreased in IP6K3 KO mice. The Purkinje cell number and the width of granule layer do not show any significant difference. B, Golgi staining reveals withered dendritic trees of Purkinje cells from 8-week-old IP6K3 KO mice cerebellum. Cell volume is significantly decreased in IP6K3 KO Purkinje cells. C, Golgi staining for the Purkinje cell dendrites from wild-type and IP6K3 KO mice cerebellum (8 weeks old). Spine number is significantly decreased in IP6K3 KO Purkinje cells. D, Golgi staining reveals withered dendritic trees of Purkinje cells from 12-month-old IP6K3 KO mice cerebellum. E, Golgi staining of granule cells, basket cells, Golgi cells, and stellate cells reveals shorter dendritic length (22 ± 5% less for basket cell, 15 ± 4% less for Golgi cell, and 25 ± 8% less for stellate cell) but no significant morphological change in the IP6K3 KOs (8 weeks old). We quantified the length of an average of five dendrites per cell, six cells per mouse. F, Electron microscopy of the cerebellar molecular layer from wild-type and IP6K3 KO mice (8 weeks old). Densities of both symmetric and asymmetric synapses are markedly decreased in IP6K3 KOs. The ultrastructure of synapses is relatively normal in the KOs. Data are represented as mean ± SEM, three mice per group, **p < 0.01, ***p < 0.001.
Diminished cerebellar synapses in IP6K3 mutants
Excitatory synapses contain asymmetric synaptic elements, whereas inhibitory synapses are symmetric (Harris and Weinberg, 2012). At the electron microscopic level we quantified numbers of symmetric and asymmetric synapses in the cerebellar molecular layer of IP6K3 knock-outs (Fig. 6F). Both symmetric and asymmetric synapses are reduced ∼40–50% in the mutant mice. The synaptic size, presynaptic vesicle number, and synaptic cleft thickness, however, do not show any notable differences.
We stained for markers of GABA and glutamate containing neurons (Fig. 7). IP6K3 knock-out cerebella display reduced levels of GAD65, the GABA synthesizing enzyme and also a marker for GABAergic synapses (Fig. 7A). Staining for VGlut1, a marker for parallel fibers, is also diminished in the mutants (Fig. 7B) as is VGlut2, marking climbing fibers (Fig. 7C).
Figure 7.
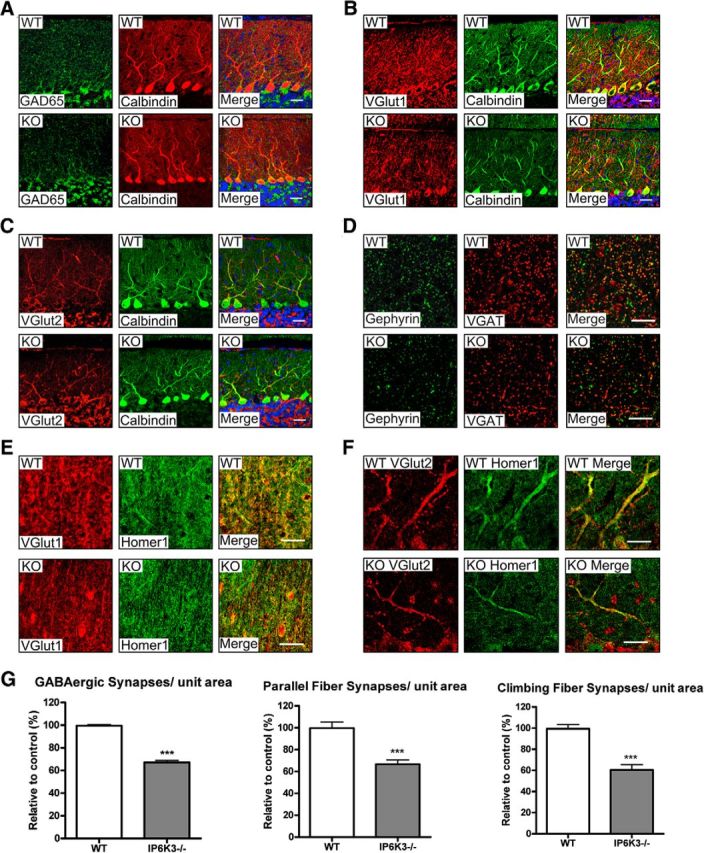
IP6K3 KO mice display significantly fewer synapses on cerebellar Purkinje cells. A, Immunostaining of GAD65 and calbindin in cerebellar Purkinje cells from wild-type and IP6K3 KO mice. The density of GAD65 is substantially less in IP6K3 KOs. B, Immunostaining of VGlut1 and calbindin in cerebellar Purkinje cells from wild-type and IP6K3 KO mice. The density of VGlut1 is markedly reduced in IP6K3 KOs. C, Immunostaining of VGlut2 and calbindin in cerebellar Purkinje cells from wild-type and IP6K3 KO mice. The density of VGlut2 is notably diminished in IP6K3 KOs. D, Immunostaining of gephyrin and VGAT in cerebellar Purkinje cells from wild-type and IP6K3 KO mice. The density of double-stained GABAergic synapses is significantly reduced in IP6K3 KOs. E, Immunostaining of VGlut1 and Homer1 in cerebellar Purkinje cells from wild-type and IP6K3 KO mice. The density of double-stained parallel fiber synapses is significantly reduced in IP6K3 KOs. F, Immunostaining of VGlut2 and Homer1 in cerebellar Purkinje cells from wild-type and IP6K3 KO mice. The density of double-stained climbing fiber synapses is much less in IP6K3 KOs. G, Statistical analysis from D–F indicates that GABAergic, parallel fiber, and climbing fiber synapses are significantly reduced in IP6K3 KOs. Scale bar, 20 μm. Data are presented as means ± SEM, ***p < 0.001. Mice were 8-week-old males, n = 5.
We explored properties of synapses that might be altered by diminution in these GABA and glutamate markers. We assessed GABA synapses in terms of the juxtaposition of VGAT, reflecting presynaptic GABA neurons, and gephyrin, marking postsynaptic elements of GABA synapses (Tyagarajan and Fritschy, 2014). The IP6K3 knock-outs display a 40% reduction in unit area for GABA synapses (Fig. 7D,G). We examined parallel fiber synapses by costaining for VGlut1, indicating parallel fibers, and Homer1, reflecting postsynaptic elements of excitatory synapses. Numbers of parallel fiber synapses are diminished by ∼40% in the mutant mice (Fig. 7E,G). We assessed climbing fiber synapses by monitoring colocalizations of VGlut2, marking climbing fiber processes, and Homer1, which labels postsynaptic elements. The density of climbing fiber synapses appears reduced ∼50% in IP6K3 knock-outs (Fig. 7F,G). In summary, the principal types of synapses in the cerebellum are substantially diminished in IP6K3 knock-out mice.
Discussion
In the present study we demonstrate that IP6K3 is highly expressed in cortex and cerebellum. There is no gross difference of brain structure in the mutant mice. The IP6K3 KO mice display impaired motor learning and coordination in rotarod and gait analysis but relative normal performance in open field study. The mutant mice may compensate for gait/rotarod deficits leading to normal open field performance, which incorporates a wide range of motor skills. Motor deficits manifested by the mutant mice are reminiscent of disabilities of mice with Purkinje cell dysfunction. Intriguingly, IP6K3 is most highly expressed in Purkinje cells in cerebellum. IP6K3 acting noncatalytically physiologically regulates interactions of spectrin and adducin. Spectrins and adducins appear to be the principal interactors of IP6K3, which is required for their physiologic association. Deletion of IP6K3 abrogates spectrin-adducin binding and impacts the morphology of cerebellar Purkinje cells as well as their synaptic disposition and motor activity. The dendritic trees of the mutant mice are withered, and the size and spine density of Purkinje cells are reduced along with a substantial decrease in the major forms of cerebellar synapses. Loss of Purkinje dendritic branches and decreased numbers of dendritic spines have been reported to cause decreased motor learning and coordination (Clark et al., 1997). Morphologic changes of Purkinje dendritic trees result in motor defects (Donald et al., 2008). The IP6K3-null Purkinje cells also have disordered dendritic trees, which may mediate the deficits of motor learning and coordination.
The physical binding of IP6K3 with adducins and spectrins to influence their function affords a molecular mechanism to account for cytoskeletal alterations of Purkinje cells in the IP6K3 mutants. Abundant literature establishes a role for β-adducin in the assembly of synapses in the brain and regulation of memory in mammals (Bednarek and Caroni, 2011) and in Drosophila (Pielage et al., 2011). In Drosophila adducin mediates assembly of synapses at the neuromuscular junction (Pielage et al., 2011), and mutations of spectrin impact presynaptic neurotransmitter release (Featherstone et al., 2001). There are multiple subtypes of spectrins. Our study focused upon β2-spectrins, which are robustly coimmunoprecipitated by IP6K3. The β1 subtype of spectrin also regulates the morphological and functional dynamics of dendritic spines (Nestor et al., 2011). The β3 subtype of spectrin has been implicated in synaptogenesis with mutations leading to ataxia (Stankewich et al., 2010). In the cerebellum β3-spectrin mediates development of Purkinje cell dendritic trees and morphogenesis of spines (Gao et al., 2011). Assembly of the spectrin-actin membrane skeleton is regulated by adducin/spectrin interactions (Gardner and Bennett, 1987), whose deficiency disrupts cell morphology (Hu et al., 1995). The disruption of adducin/spectrin interactions in IP6K3 KOs may mediate the morphologic alteration as well as the decreased synapse formation of Purkinje cells. Decreased synapses in the IP6K3 KO mice may also affect electrophysiological properties.
IP6K3 physically binds to adducin's C-terminal domain and to β-spectrin's CH domain, which are the binding sites for adducin/spectrin interactions (Li and Bennett, 1996; Li et al., 1998). Adducin, protein 4.1, and dematin localize at the spectrin–actin junctional complex (Derick et al., 1992) and associate with spectrin to facilitate the spectrin–actin interaction (Correas et al., 1986; Gardner and Bennett, 1987; Koshino et al., 2012). Dematin and protein band 3 act as scaffolds to link adducin with spectrin (Drenckhahn et al., 1985; Khan et al., 2008; Anong et al., 2009; Koshino et al., 2012). Our study provides evidence that IP6K3 serves as a scaffold to link adducin and spectrin, a function that does not require IP6K3's catalytic activity. In the inositol phosphate kinase family there is precedence for noncatalytic actions. Inositol polyphosphate multikinase (IPMK) physiologically generates IP5 but also is a PI3 kinase that forms phosphatidylinositol (3,4,5)-trisphosphate (Resnick et al., 2005). Additionally, acting noncatalytically, IPMK stabilizes the mTOR complex (Kim et al., 2011) and acts as a transcriptional coactivator for several genes (Xu et al., 2013a, b). Our present findings are the first to establish noncatalytic functions for an IP6 kinase.
The ability of IP6K3 to regulate synaptic disposition noncatalytically via influences upon specific cytoskeletal proteins provides a new functional dimension for the IP6 kinases. This action, together with the noncatalytic activities of IPMK (Xu et al., 2013a, b), indicates that the class of inositol phosphate kinases may display myriad cellular activities not mediated by catalytic formation of inositol phosphates. Other classes of enzymes also manifest important noncatalytic activities. Known examples include kinase-dead Braf and oncogenic Ras, which cooperate to induce melanomas (Heidorn et al., 2010). The USP13 enzyme regulates Siah2 ligase stability and activity via noncatalytic ubiquitin-binding domains (Scortegagna et al., 2011). Moreover, a triosephosphate isomerase noncatalytically influences behavior and longevity (Roland et al., 2013).
The most obvious deficit showed by IP6K3 KO mice is the reduction in motor learning and coordination, which indicates cerebellar dysfunction. In the current study, we focus on these phenotypes and show that IP6K3 regulates the morphology and synaptic formation of cerebellar Purkinje cells via direct binding to spectrin/adducin. IP6K3 has also been found in several other regions of the brain, which may mediate diverse functions.
Footnotes
This work was supported by United States Public Health Service Grant DA000266 to S.H.S. We thank members of the Snyder laboratory for reagents and discussions.
The authors declare no competing financial interests.
References
- Anong WA, Franco T, Chu H, Weis TL, Devlin EE, Bodine DM, An X, Mohandas N, Low PS. Adducin forms a bridge between the erythrocyte membrane and its cytoskeleton and regulates membrane cohesion. Blood. 2009;114:1904–1912. doi: 10.1182/blood-2009-02-203216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarek E, Caroni P. beta-Adducin is required for stable assembly of new synapses and improved memory upon environmental enrichment. Neuron. 2011;69:1132–1146. doi: 10.1016/j.neuron.2011.02.034. [DOI] [PubMed] [Google Scholar]
- Bennett V, Baines AJ. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev. 2001;81:1353–1392. doi: 10.1152/physrev.2001.81.3.1353. [DOI] [PubMed] [Google Scholar]
- Chakraborty A, Koldobskiy MA, Bello NT, Maxwell M, Potter JJ, Juluri KR, Maag D, Kim S, Huang AS, Dailey MJ, Saleh M, Snowman AM, Moran TH, Mezey E, Snyder SH. Inositol pyrophosphates inhibit Akt signaling, thereby regulating insulin sensitivity and weight gain. Cell. 2010;143:897–910. doi: 10.1016/j.cell.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark HB, Burright EN, Yunis WS, Larson S, Wilcox C, Hartman B, Matilla A, Zoghbi HY, Orr HT. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci. 1997;17:7385–7395. doi: 10.1523/JNEUROSCI.17-19-07385.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correas I, Leto TL, Speicher DW, Marchesi VT. Identification of the functional site of erythrocyte protein 4.1 involved in spectrin-actin associations. J Biol Chem. 1986;261:3310–3315. [PubMed] [Google Scholar]
- Derick LH, Liu SC, Chishti AH, Palek J. Protein immunolocalization in the spread erythrocyte membrane skeleton. Eur J Cell Biol. 1992;57:317–320. [PubMed] [Google Scholar]
- Donald S, Humby T, Fyfe I, Segonds-Pichon A, Walker SA, Andrews SR, Coadwell WJ, Emson P, Wilkinson LS, Welch HC. P-Rex2 regulates Purkinje cell dendrite morphology and motor coordination. Proc Natl Acad Sci U S A. 2008;105:4483–4488. doi: 10.1073/pnas.0712324105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L, Chapline C, Mousseau B, Fowler L, Ramsay K, Stevens JL, Jaken S. 35H, a sequence isolated as a protein kinase C binding protein, is a novel member of the adducin family. J Biol Chem. 1995;270:25534–25540. doi: 10.1074/jbc.270.43.25534. [DOI] [PubMed] [Google Scholar]
- Drenckhahn D, Schlüter K, Allen DP, Bennett V. Colocalization of band 3 with ankyrin and spectrin at the basal membrane of intercalated cells in the rat kidney. Science. 1985;230:1287–1289. doi: 10.1126/science.2933809. [DOI] [PubMed] [Google Scholar]
- Featherstone DE, Davis WS, Dubreuil RR, Broadie K. Drosophila alpha- and beta-spectrin mutations disrupt presynaptic neurotransmitter release. J Neurosci. 2001;21:4215–4224. doi: 10.1523/JNEUROSCI.21-12-04215.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu C, He J, Li C, Shyy JY, Zhu Y. Cholesterol increases adhesion of monocytes to endothelium by moving adhesion molecules out of caveolae. Biochim Biophys Acta. 2010;1801:702–710. doi: 10.1016/j.bbalip.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Fu C, van der Zwan A, Gerber S, Van Den Berg S, No E, Wang WC, Sheibani N, Carducci MA, Kachhap S, Hammers HJ. Screening assay for blood vessel maturation inhibitors. Biochem Biophys Res Commun. 2013;438:364–369. doi: 10.1016/j.bbrc.2013.07.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Perkins EM, Clarkson YL, Tobia S, Lyndon AR, Jackson M, Rothstein JD. beta-III spectrin is critical for development of Purkinje cell dendritic tree and spine morphogenesis. J Neurosci. 2011;31:16581–16590. doi: 10.1523/JNEUROSCI.3332-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner K, Bennett V. Modulation of spectrin-actin assembly by erythrocyte adducin. Nature. 1987;328:359–362. doi: 10.1038/328359a0. [DOI] [PubMed] [Google Scholar]
- Harris KM, Weinberg RJ. Ultrastructure of synapses in the mammalian brain. Cold Spring Harb Perspect Biol. 2012;4:a005587. doi: 10.1101/cshperspect.a005587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, Marais R. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu RJ, Moorthy S, Bennett V. Expression of functional domains of beta G-spectrin disrupts epithelial morphology in cultured cells. J Cell Biol. 1995;128:1069–1080. doi: 10.1083/jcb.128.6.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi R, Gilligan DM, Otto E, McLaughlin T, Bennett V. Primary structure and domain organization of human alpha and beta adducin. J Cell Biol. 1991;115:665–675. doi: 10.1083/jcb.115.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AA, Hanada T, Mohseni M, Jeong JJ, Zeng L, Gaetani M, Li D, Reed BC, Speicher DW, Chishti AH. Dematin and adducin provide a novel link between the spectrin cytoskeleton and human erythrocyte membrane by directly interacting with glucose transporter-1. J Biol Chem. 2008;283:14600–14609. doi: 10.1074/jbc.M707818200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim SF, Maag D, Maxwell MJ, Resnick AC, Juluri KR, Chakraborty A, Koldobskiy MA, Cha SH, Barrow R, Snowman AM, Snyder SH. Amino acid signaling to mTOR mediated by inositol polyphosphate multikinase. Cell Metab. 2011;13:215–221. doi: 10.1016/j.cmet.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldobskiy MA, Chakraborty A, Werner JK, Jr, Snowman AM, Juluri KR, Vandiver MS, Kim S, Heletz S, Snyder SH. p53-mediated apoptosis requires inositol hexakisphosphate kinase-2. Proc Natl Acad Sci U S A. 2010;107:20947–20951. doi: 10.1073/pnas.1015671107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshino I, Mohandas N, Takakuwa Y. Identification of a novel role for dematin in regulating red cell membrane function by modulating spectrin-actin interaction. J Biol Chem. 2012;287:35244–35250. doi: 10.1074/jbc.M111.305441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis DM, Reese TS. Cytoplasmic organization in cerebellar dendritic spines. J Cell Biol. 1983;97:1169–1178. doi: 10.1083/jcb.97.4.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Bennett V. Identification of the spectrin subunit and domains required for formation of spectrin/adducin/actin complexes. J Biol Chem. 1996;271:15695–15702. doi: 10.1074/jbc.271.26.15695. [DOI] [PubMed] [Google Scholar]
- Li X, Matsuoka Y, Bennett V. Adducin preferentially recruits spectrin to the fast growing ends of actin filaments in a complex requiring the MARCKS-related domain and a newly defined oligomerization domain. J Biol Chem. 1998;273:19329–19338. doi: 10.1074/jbc.273.30.19329. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Li X, Bennett V. Adducin is an in vivo substrate for protein kinase C: phosphorylation in the MARCKS-related domain inhibits activity in promoting spectrin-actin complexes and occurs in many cells, including dendritic spines of neurons. J Cell Biol. 1998;142:485–497. doi: 10.1083/jcb.142.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka Y, Li X, Bennett V. Adducin: structure, function and regulation. Cell Mol Life Sci. 2000;57:884–895. doi: 10.1007/PL00000731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulugu S, Bai W, Fridy PC, Bastidas RJ, Otto JC, Dollins DE, Haystead TA, Ribeiro AA, York JD. A conserved family of enzymes that phosphorylate inositol hexakisphosphate. Science. 2007;316:106–109. doi: 10.1126/science.1139099. [DOI] [PubMed] [Google Scholar]
- Nestor MW, Cai X, Stone MR, Bloch RJ, Thompson SM. The actin binding domain of betaI-spectrin regulates the morphological and functional dynamics of dendritic spines. PLoS One. 2011;6:e16197. doi: 10.1371/journal.pone.0016197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielage J, Bulat V, Zuchero JB, Fetter RD, Davis GW. Hts/Adducin controls synaptic elaboration and elimination. Neuron. 2011;69:1114–1131. doi: 10.1016/j.neuron.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao F, Cha J, Xu J, Xu R, Vandiver MS, Tyagi R, Tokhunts R, Koldobskiy MA, Fu C, Barrow R, Wu M, Fiedler D, Barrow JC, Snyder SH. Inositol pyrophosphates mediate the DNA-PK/ATM-p53 cell death pathway by regulating CK2 phosphorylation of Tti1/Tel2. Mol Cell. 2014;54:119–132. doi: 10.1016/j.molcel.2014.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao F, Xu J, Fu C, Cha JY, Gadalla MM, Xu R, Barrow JC, Snyder SH. Inositol pyrophosphates promote tumor growth and metastasis by antagonizing liver kinase B1. Proc Natl Acad Sci U S A. 2015;112:1773–1778. doi: 10.1073/pnas.1424642112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick AC, Snowman AM, Kang BN, Hurt KJ, Snyder SH, Saiardi A. Inositol polyphosphate multikinase is a nuclear PI3-kinase with transcriptional regulatory activity. Proc Natl Acad Sci U S A. 2005;102:12783–12788. doi: 10.1073/pnas.0506184102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland BP, Stuchul KA, Larsen SB, Amrich CG, Vandemark AP, Celotto AM, Palladino MJ. Evidence of a triosephosphate isomerase non-catalytic function crucial to behavior and longevity. J Cell Sci. 2013;126:3151–3158. doi: 10.1242/jcs.124586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiardi A, Erdjument-Bromage H, Snowman AM, Tempst P, Snyder SH. Synthesis of diphosphoinositol pentakisphosphate by a newly identified family of higher inositol polyphosphate kinases. Curr Biol. 1999;9:1323–1326. doi: 10.1016/S0960-9822(00)80055-X. [DOI] [PubMed] [Google Scholar]
- Saiardi A, Nagata E, Luo HR, Snowman AM, Snyder SH. Identification and characterization of a novel inositol hexakisphosphate kinase. J Biol Chem. 2001;276:39179–39185. doi: 10.1074/jbc.M106842200. [DOI] [PubMed] [Google Scholar]
- Scortegagna M, Subtil T, Qi J, Kim H, Zhao W, Gu W, Kluger H, Ronai ZA. USP13 enzyme regulates Siah2 ligase stability and activity via noncatalytic ubiquitin-binding domains. J Biol Chem. 2011;286:27333–27341. doi: 10.1074/jbc.M111.218214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankewich MC, Gwynn B, Ardito T, Ji L, Kim J, Robledo RF, Lux SE, Peters LL, Morrow JS. Targeted deletion of betaIII spectrin impairs synaptogenesis and generates ataxic and seizure phenotypes. Proc Natl Acad Sci U S A. 2010;107:6022–6027. doi: 10.1073/pnas.1001522107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens L, Radenberg T, Thiel U, Vogel G, Khoo KH, Dell A, Jackson TR, Hawkins PT, Mayr GW. The detection, purification, structural characterization, and metabolism of diphosphoinositol pentakisphosphate(s) and bisdiphosphoinositol tetrakisphosphate(s) J Biol Chem. 1993;268:4009–4015. [PubMed] [Google Scholar]
- Tyagarajan SK, Fritschy JM. Gephyrin: a master regulator of neuronal function? Nat Rev Neurosci. 2014;15:141–156. doi: 10.1038/nrn3670. [DOI] [PubMed] [Google Scholar]
- Voglmaier SM, Bembenek ME, Kaplin AI, Dormán G, Olszewski JD, Prestwich GD, Snyder SH. Purified inositol hexakisphosphate kinase is an ATP synthase: diphosphoinositol pentakisphosphate as a high-energy phosphate donor. Proc Natl Acad Sci U S A. 1996;93:4305–4310. doi: 10.1073/pnas.93.9.4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Sen N, Paul BD, Snowman AM, Rao F, Vandiver MS, Xu J, Snyder SH. Inositol polyphosphate multikinase is a coactivator of p53-mediated transcription and cell death. Sci Signal. 2013a;6:ra22. doi: 10.1126/scisignal.2003405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Paul BD, Smith DR, Tyagi R, Rao F, Khan AB, Blech DJ, Vandiver MS, Harraz MM, Guha P, Ahmed I, Sen N, Gallagher M, Snyder SH. Inositol polyphosphate multikinase is a transcriptional coactivator required for immediate early gene induction. Proc Natl Acad Sci U S A. 2013b;110:16181–16186. doi: 10.1073/pnas.1315551110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Fu C, Li X, Song Y, Li C, Zou M, Guan Y, Zhu Y. Prostaglandin E2 promotes endothelial differentiation from bone marrow-derived cells through AMPK activation. PLoS One. 2011;6:e23554. doi: 10.1371/journal.pone.0023554. [DOI] [PMC free article] [PubMed] [Google Scholar]