

Abstract
Neuron–astrocyte interactions are critical for proper CNS development and function. Astrocytes secrete factors that are pivotal for synaptic development and function, neuronal metabolism, and neuronal survival. Our understanding of this relationship, however, remains incomplete due to technical hurdles that have prevented the removal of astrocytes from neuronal circuits without changing other important conditions. Here we overcame this obstacle by growing solitary rat hippocampal neurons on microcultures that were comprised of either an astrocyte bed (+astrocyte) or a collagen bed (−astrocyte) within the same culture dish. −Astrocyte autaptic evoked EPSCs, but not IPSCs, displayed an altered temporal profile, which included increased synaptic delay, increased time to peak, and severe glutamate release asynchrony, distinct from previously described quantal asynchrony. Although we observed minimal alteration of the somatically recorded action potential waveform, action potential propagation was altered. We observed a longer latency between somatic initiation and arrival at distal locations, which likely explains asynchronous EPSC peaks, and we observed broadening of the axonal spike, which likely underlies changes to evoked EPSC onset. No apparent changes in axon structure were observed, suggesting altered axonal excitability. In conclusion, we propose that local astrocyte support has an unappreciated role in maintaining glutamate release synchrony by disturbing axonal signal propagation.
SIGNIFICANCE STATEMENT Certain glial cell types (oligodendrocytes, Schwann cells) facilitate the propagation of neuronal electrical signals, but a role for astrocytes has not been identified despite many other functions of astrocytes in supporting and modulating neuronal signaling. Under identical global conditions, we cultured neurons with or without local astrocyte support. Without local astrocytes, glutamate transmission was desynchronized by an alteration of the waveform and arrival time of axonal action potentials to synaptic terminals. GABA transmission was not disrupted. The disruption did not involve detectable morphological changes to axons of glutamate neurons. Our work identifies a developmental role for astrocytes in the temporal precision of excitatory signals.
Keywords: action potential, astrocyte, glutamate, synchrony
Introduction
Neurons send and receive synchronous synaptic inputs, which are key to the precision with which the nervous system functions. Astrocytes aid this precise signaling by supporting synaptic development, maturation, pruning, and metabolism (Hama et al., 2004; Allen and Barres, 2009; Perea et al., 2009; Pellerin and Magistretti, 2012; Clarke and Barres, 2013). However, our picture of astrocytes' role in neuronal development remains incomplete in part because of an inability to remove astrocytes while keeping other experimental conditions constant. In the present work, we lifted this barrier and uncovered an unanticipated role for local astrocyte cues in supporting the temporal precision of neuronal signaling.
Previous studies have investigated astrocyte effects on neuronal signaling by growing neurons sans astrocytes or by applying astrocyte-specific toxins. Although neurons can survive without astrocyte support, astrocyte-free neuronal survival requires special culture media composition (Brewer et al., 1993; Meyer-Franke et al., 1995). These components are likely to alter neuronal development directly, complicating efforts to specifically explore astrocyte contributions. Furthermore, adding back astrocyte cues in astrocyte-free cultures is restricted to global, soluble cues from astrocyte conditioned medium (Christopherson et al., 2005; Kucukdereli et al., 2011; Allen et al., 2012), leaving contact-mediated factors and other local cues obscure. Removal of astrocytes can also be achieved by astrocyte specific toxins such as l-α-aminoadipate (Khurgel et al., 1996) or fluorocitrate (Swanson and Graham, 1994). These can have unintended off-target effects (Fonnum et al., 1997), and this strategy is typically limited to exploring acute effects of astrocytes rather than developmental roles of astrocytes in neuronal function. These limitations leave our understanding of local astrocyte cues in neuron development and function incomplete.
In the present study we use a cell microculture preparation that allowed us to test the role of local astrocytic support on neuronal physiology. The microcultures permitted us to grow hippocampal neurons with (+astrocyte) or without (−astrocyte) a local supporting layer of astrocytes, interspersed on the same coverslip (Fig. 1A). Thus, neurons on both +astrocyte and −astrocyte microcultures experience identical global cell culture conditions. By comparing +astrocyte and −astrocyte neurons on neighboring microcultures, we assessed the impact of local astrocytic support on neuronal development and function.
Figure 1.
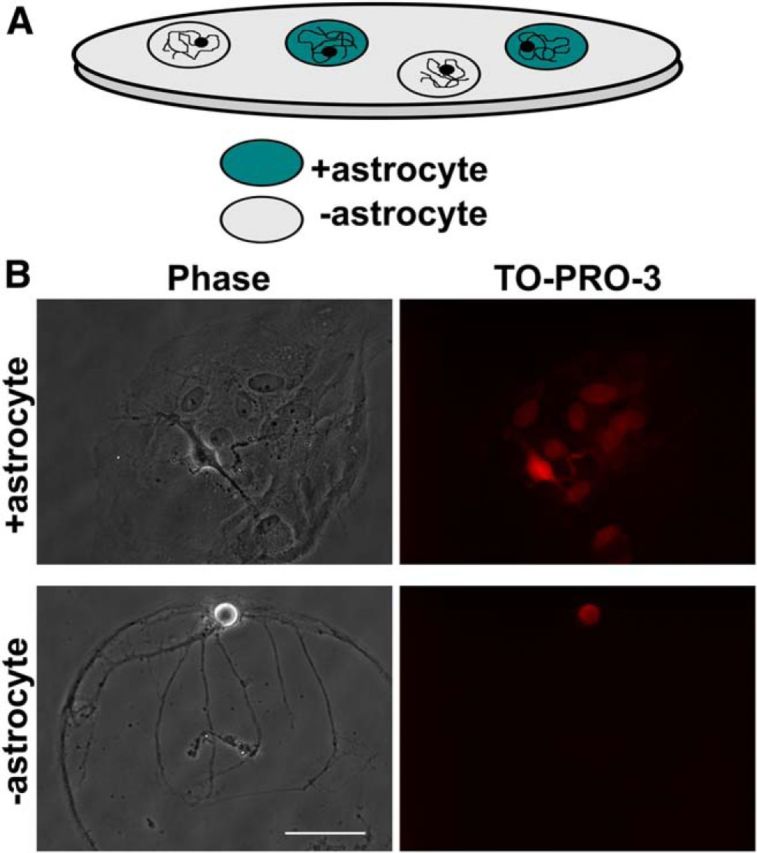
Preparation for studying local astrocyte effects on neuronal function. A, Schematic of cell culture system in which solitary neurons develop on either +astrocyte (blue circles) or −astrocyte (gray circles) microcultures. B, Examples of phase microscopy images (left) and TO-PRO-3 nuclear staining (right) of solitary neurons plated on +astrocyte (top row) and −astrocyte (bottom row) microcultures. Scale bar, 100 μm.
We found that glutamatergic, but not GABAergic, solitary neurons grown on −astrocyte microcultures displayed temporally distorted and asynchronous EPSCs. This previously uncharacterized form of asynchronous transmission is distinct from classical quantal asynchrony. Without astrocytes, neurons exhibited an unexpected change in action potential arrival to different points along the presumed axon, as well as alterations in the action potential shape at distal processes. Together, these changes likely underlie altered timing and synchrony of EPSC waveforms. Although oligodendrocytes are typically associated with proper action potential conduction of CNS neurons, our data suggest an unanticipated and distinct developmental effect of local astrocytic signals on signal propagation in unmyelinated axons.
Materials and Methods
Microculture coverslip preparation.
Our microculture system was prepared by merging previously published primary cell culture protocols (Mennerick and Zorumski, 1995; Moulder et al., 2007; Ricoult et al., 2012). Briefly, 25 mm glass coverslips were stamped with a polydimethylsiloxane microstamp coated with 0.5 mg/ml collagen to create 150–200 μm diameter microdots. The coverslips were next backfilled with a nonpermissive substrate poly-l-lysine grafted polyethylene glycol [PLL(20 kDa)-g[3.5]-PEG(2 kDa); Surface Solutions] at a concentration of 10 μg/ml in PBS for 1 h and then washed with 1× PBS. These microdots acted as the cell-permissive foundation for both +astrocyte and −astrocyte microcultures to form.
Hippocampal cell culture.
Following detailed preparations published previously (Mennerick and Zorumski, 1995; Moulder et al., 2007; Crawford et al., 2012), hippocampal (neurons) and cortical (astrocytes) tissue of postnatal day 1–4 Sprague Dawley female rat pups were harvested under protocols approved by the Washington University Animal Studies Committee, digested by 1 mg/ml papain, and mechanically dispersed. Astrocytes were first plated on collagen microcultures in Eagle's medium (Invitrogen) supplemented with 5% heat-inactivated horse serum, 5% fetal bovine serum, 17 mm d-glucose, 400 μm glutamine, 500 U/ml penicillin, and 50 μg/ml streptomycin and maintained at 37°C in a humidified incubator (5% CO2/95% air) before neuronal plating. Neurons were plated at a low density (∼100 cells/mm−2). Serendipitously, as the microcultures developed, astrocytes were evident on only a subset of collagen microcultures, thus creating the +astrocyte/−astrocyte microculture system schematized in Figure 1A. Fixed-astrocyte microcultures were prepared by fixing glial cells 7–10 d after plating with 4% paraformaldehyde/0.2% glutaraldehyde in PBS at room temperature for 5 min followed by three washes in PBS. Unless otherwise stated, experiments were performed between 9–14 d in vitro.
Electrophysiology.
Electrophysiological recordings were made at room temperature on the stage of an Eclipse TE2000-S inverted microscope. Whole-cell patch-clamp recording was performed on solitary neurons on either +astrocyte or −astrocyte microcultures unless noted otherwise. Data were collected using pClamp 10 software with a Multiclamp 700B amplifier and Digidata 1550 data acquisition board (Molecular Devices). Intracellular pipette solution for whole-cell recordings typically consisted of the following (in mm): 140 potassium gluconate, 40 NaCl, 10 HEPES, 5 EGTA, and 0.5 CaCl2. The pH was adjusted to 7.25 with KOH. Experiments in which both GABAergic and glutamatergic autaptic PSCs were studied, we used 140 mm potassium chloride instead of potassium gluconate to provide driving force for IPSCs near the resting potential. The extracellular solution during voltage-clamp recordings typically consisted of the following (in mm): 138 NaCl, 4 KCl, 10 HEPES, 10 glucose, 2 CaCl2, 1 MgCl2, and 0.01 d-2-amino-5-phosphonovalerate (d-APV; Tocris Bioscience), pH 7.25, adjusted with NaOH. Extracellular recording pipettes were filled with external saline solution with the addition of 1 μm 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX) and 25 μm d-APV. For current-clamp recordings, recording saline contained 50 μm d-APV and 1 μm NBQX. For experiments designed to examine synaptic delay, recordings were performed in both standard saline solution and saline containing 25 μm d-APV and 1 μm NBQX to isolate the PSC from capacitive and voltage-gated currents associated with stimulation. Miniature EPSCs were recorded in saline containing 1 μm tetrodotoxin (TTX) and 100 mm sucrose to promote vesicle release (Rosenmund and Stevens, 1996).
Whole-cell and extracellular recording pipettes were pulled from borosilicate glass capillary tubes (World Precision Instruments) and had final open-tip resistances between 3 and 6 MΩ. Data were sampled at 20 kHz and filtered at 10 kHz for PSC recordings as well as action potential recordings. Access resistance was compensated to 90–95% for evoked autaptic PSC recordings. Miniature EPSCs were sampled at 5 kHz and filtered at 2 kHz; access resistance was not compensated. Unless stated otherwise, neurons in voltage-clamp mode were held at −70 mV. Evoked autaptic PSCs were elicited with a 1.5 ms depolarizing pulse to 0 mV. Neurons in current-clamp experiments were biased with an appropriate current injection to hold the membrane potential at approximately −65 mV. To measure action potential waveform and rheobase, a step current injection protocol was applied. Current pulses started at 0 pA and depolarized in increments of 1–5 pA for 1200 ms until a single action potential was evoked. For extracellular action potential recordings of distal processes, local currents were recorded in voltage-clamp mode at bath potential while action potentials were elicited at the soma by a second pipette in whole-cell current-clamp mode. When solution exchange or drug delivery was required, solutions were delivered to the target neuron by a gravity-driven local perfusion system from a common tip with an exchange time of ∼100 ms. Whenever possible, effects of experimental manipulations on PSCs were determined by averaging 2–5 interleaved control and test applications to help account for any time-dependent changes in EPSCs.
Immunohistochemistry and microscopy.
Cell cultures were fixed before immunostaining with 4% paraformaldehyde plus 0–0.2% glutaraldehyde at room temperature or with 100% methanol at −20°C for 10 min. Cultures were then washed with PBS and incubated in 4% normal goat serum and permeabilized with 0.1% Triton X-100 or by a freeze–thaw method (3 min in −70°C and 5 min in 37°C, five times). Primary antibodies against vesicular glutamate transporter 1 (vGluT1; 1:2000; Synaptic Systems), voltage-gated sodium channel 1 (pan-Nav; 1:250; Alomone Labs), voltage-gated potassium channel 1.2 (Kv1.2; 1:2000; Alomone Labs), ankyrin-G (ankG; 1:2000; Santa Cruz Biotechnology), and axonal neurofilament marker (SMI-312; 1:20,000; Abcam) were applied for 2–16 h at room temperature. Cells were subsequently washed with PBS and incubated with Alexa Fluor conjugates (Invitrogen). Some experiments included a nuclear stain (TO-PRO-3, 1:2000; Invitrogen), cell-viability marker, calcein-AM (AnaSpec), and/or a marker of free cholesterol (filipin, 50 μg/ml; Cayman Chemical).
Images for axon morphology analyses were collected from neurons previously characterized as glutamatergic by evoking EPSCs, followed by fixation and immunolabeling. Images of SMI-312/vGluT1/TO-PRO-3 labeling for axon structure analyses were acquired on an inverted Eclipse TE2000-S microscope with a 40×, 0.95 numerical aperture (NA) objective using a C1 scanning confocal laser (488, 543, and/or 633 nm). Z-stack images were collected with the gain and acquisition settings held constant for a given experiment. Images were projected into a 2D image and analyzed using MetaMorph 7 (Molecular Devices), MATLAB 2013 (MathWorks), and ImageJ (NIH). Pan-Nav, ankyrin-G, filipin, and TO-PRO-3 images were collected on an inverted Eclipse TE2000-S microscope with a 40×, 0.6 NA objective using a metal halide lamp and appropriate filters, and images were acquired with MetaMorph 7 software and a 12 bit CCD camera (QImaging).
Data analysis.
Data were analyzed and plotted using MetaMorph 7, Clampfit 10 (Molecular Devices), MiniAnalysis 6 (Synaptosoft), Excel 2011 (Microsoft), Prism 6 (GraphPad), ImageJ, and MATLAB. Unless stated otherwise, data in figures and text are given as mean ± SEM. Student's two-tailed unpaired t test was used to compare two groups unless noted otherwise. If more than two parameters were compared between two groups, a Bonferroni correction was applied. To test differences in variance between groups, an F test was conducted. Significance was defined as a corrected p value of <0.05. The reported n refers to the number of neurons in each group within a particular experiment.
When evoked PSCs were elicited, at least two sweeps were averaged in a given condition for each autaptic neuron. Clampfit 10 was used to measure parameters of peak amplitude, rise time, decay time, half-width, and charge transfer. Synaptic delay was measured by subtracting traces recorded in the presence of glutamate receptor blockers, NBQX (1 μm) and d-APV (25 μm), from control EPSC traces in the absence of blockers and measuring the time from presynaptic stimulation to onset of the postsynaptic response. Local peaks of an EPSC and their temporal location were identified using custom MATLAB code by normalizing EPSCs to the largest peak and detecting the local minima that were larger than a threshold, defined as 10 times the SD of the baseline, compared to the previous data point. This ensured that we detected supraquantal local minima regardless of the original size of the EPSC. In a small number of cases, false negatives (missed peaks) were detected by eye, but we did not perform any corrections of the analysis to retain an objective standard for comparisons among experimental conditions, and the false negatives were not evident disproportionately in any experimental condition. Local peak count was determined by summing the number of local peaks in a given EPSC. The number of local peaks and their corresponding temporal location in an EPSC is referred to as the local peak profile. Decay time constants were measured by fitting the decay from the last detected peak in the EPSC with a single exponential function.
To measure changes in the local peak profile during different experimental conditions, we measured the number of temporally locked common peaks shared between control and test conditions. Common peaks were defined as local minima in the test condition that occur within 1 ms before or after a local minimum in the control condition. The number of common peaks was then normalized to the control EPSC local peak count to measure the percentage of peaks conserved between test and control conditions. This ratio is referred to as the common peak ratio. If the common peak ratio was 1, then the local peak profile was unchanged between control and test conditions. If the common peak ratio was 0, then the local peak profile showed no temporal correspondence between control and test conditions.
For all immunostaining experiments, solitary glutamatergic neurons were identified via vGluT-1 immunoreactivity or by evoked EPSCs before fixation. The presence or absence of astrocytes on microcultures was predominantly defined with phase microscopy and confirmed in a subset of experiments by the nuclei marker, TO-PRO-3, or calcein-AM for fixed-astrocyte experiments. Neurons selected for filipin analyses required robust SMI-312 staining in a well-isolated axon with an obvious somatic origin. Axon initial segment (AIS) structure measurements were conducted with the help of ImageJ to set a constant threshold value between conditions, established by eye. For AIS length, a line was drawn between start of threshold to the end of threshold. For length between the soma and AIS, a line was drawn between the soma and the proximal edge of the threshold. Axon morphology analyses were performed by first evoking an EPSC to characterize the local peak profile of the +astrocyte or −astrocyte glutamatergic neurons and were chosen if their EPSCs displayed temporal distortion (−astrocyte) or were synchronous (+astrocyte; see Fig. 6C). After sampling and marking for later identification, the cells were immunostained, and the subsequent images were processed using the MATLAB-based program SynD (Schmitz et al., 2011). Axon detection was performed in SynD using steerable filters that thresholded the SMI-312 staining and then used the threshold to create an axon mask. This was used to measure the number of branch points as well as axon end points. The axon mask produced by SynD was then transferred to ImageJ, where the image was made binary and skeletonized with the skeletonize plug-in for ImageJ to measure axon length.
Figure 6.
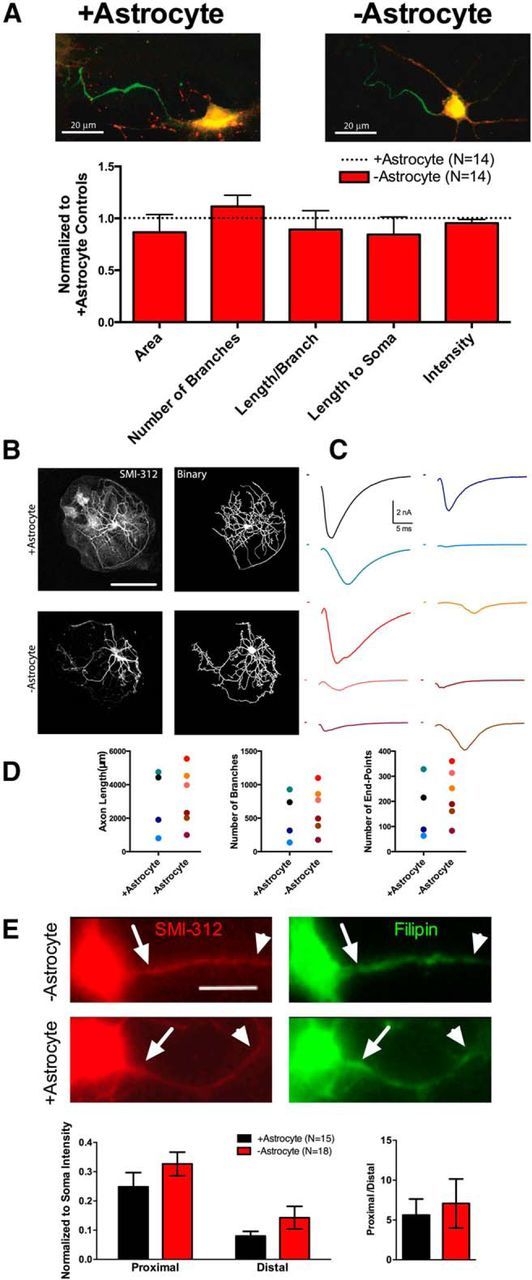
−Astrocyte and +astrocyte axons have similar physical properties. A, Example pan-Nav immunostaining (green; vGluT1, red) of a +astrocyte neuron (left) and −astrocyte neuron (right). The bar graph quantifies staining intensity, size, and location for −astrocyte neurons (red bars; n = 13) compared to +astrocyte controls (black dotted line; n = 14). −Astrocyte AIS immunostaining values were normalized to +astrocyte neuron immunostaining values. B, C, Distal axon morphology and EPSCs were compared between +astrocyte and −astrocyte glutamatergic neurons. B, Representative SMI-312 staining (left) of +astrocyte (top row) and −astrocyte (bottom row) glutamatergic neurons. Immunostaining was thresholded and binarized (right) for subsequent axon length and branching measurements. (For a description, see Materials and Methods.) C, EPSCs from +astrocyte neurons (n = 4; black and blue hues) and −astrocyte neurons (n = 6; red hues) before fixation and SMI-312 immunostaining. D, Plots of axon length (left), number of branch points (middle), and number of end points (right) for all +astrocyte (blue hue dots) and −astrocyte (red hue dots) neurons recorded and measured. The color of the dots corresponds with the color of EPSCs displayed in C. E, Representative SMI-312 (left) and filipin (right) stains for −astrocyte (top row) and +astrocyte (bottom row) glutamatergic neurons, verified in preceding whole-cell recordings. Proximal (arrows) and distal (arrowheads) axon regions of interest were defined on SMI-312 images and subsequently used to measure filipin intensity via background-subtracted line scan analyses. These analyses are quantified in the bar graphs (+astrocytes, n = 15; −astrocytes, n = 18). Filipin intensity was normalized to somatic filipin intensity to account for variations in staining intensity between biological replicates. Scale bars: A, E, 20 μm; B, 100 μm.
Materials.
D-APV, NBQX, TTX, and 10,10-bis(4-Pyridinylmethyl)-9(10H)-anthracenone dihydrochloride (XE-991) were obtained from Tocris Bioscience. Retigabine was obtained from Alomone Labs. All materials without identified suppliers above were obtained from Sigma-Aldrich.
Results
Loss of local astrocytic support has no effect on passive neuronal properties
We tested the consequences of removing local astrocytic support from neurons by growing hippocampal neurons on microcultures that were either astrocyte rich (+astrocyte) or devoid of astrocytes (−astrocyte). Figure 1A schematizes the preparation used. The presence or absence of astrocytes was normally identified with phase contrast microscopy and confirmed in a subset of experiments using TO-PRO-3 nuclear staining (Fig. 1B). The system allowed us to investigate the influence of local astrocytic support on neurotransmission without altering other cell culture conditions, given the random interspersion of +astrocyte and −astrocyte microcultures across a common coverslip. This setup permitted us to exclude contributions of global factors and to attribute differences to local astrocytic cues mediated by contact and/or by local soluble cues. No differences were detected in membrane surface area or in background leak channels as measured by cell capacitance (+astrocyte = 69.86 ± 8.40 pF, n = 12; −astrocyte = 79.29 ± 12.06 pF, n = 14), input resistance (+astrocyte = 208.33 ± 26.03 MΩ, n = 12; −astrocyte = 296.42 ± 51.43 MΩ; n = 14), or resting membrane potential (+astrocyte = −57.67 ± 2.10 mV, n = 12; −astrocyte = −55.75 ± 1.55 mV, n = 12; p > 0.05, Bonferroni corrected Student's unpaired t test for all conditions) between glutamatergic neurons grown on +astrocyte or −astrocyte microcultures.
Postsynaptic currents of glutamatergic, but not GABAergic, neurons are desynchronized on −astrocyte microcultures
To broadly assess neuronal function, we chose to examine evoked synaptic transmission, which encompasses membrane excitability, signal propagation, excitation–secretion coupling (Ca2+ influx, transmitter packaging, vesicle fusion), and postsynaptic receptor localization and function. Our lab and others have studied autaptic transmission in mixed astrocyte–neuron microcultures and have found that many salient features of both evoked and spontaneous transmission, including action potential initiation near the soma, axonal propagation, transmitter release timing, quantal size, and postsynaptic receptor properties, are preserved (Bekkers and Stevens, 1991; Mennerick and Zorumski, 1995), although some aspects of synaptic transmission may differ between autaptic and dissociated cell cultures, such as frequency of spontaneous release events and release probability (Liu et al., 2009). Autaptic PSCs were evoked with a brief depolarization of cells in voltage-clamp recording mode (Bekkers and Stevens, 1991). We found that evoked GABAergic IPSCs displayed no waveform differences when grown on either +astrocyte or −astrocyte microcultures (Fig. 2A, gray bars) and were indistinguishable from IPSCs from previous work (Bekkers and Stevens, 1991; Mennerick and Zorumski, 1995; Prakriya and Mennerick, 2000; Vecchia et al., 2014).
Figure 2.
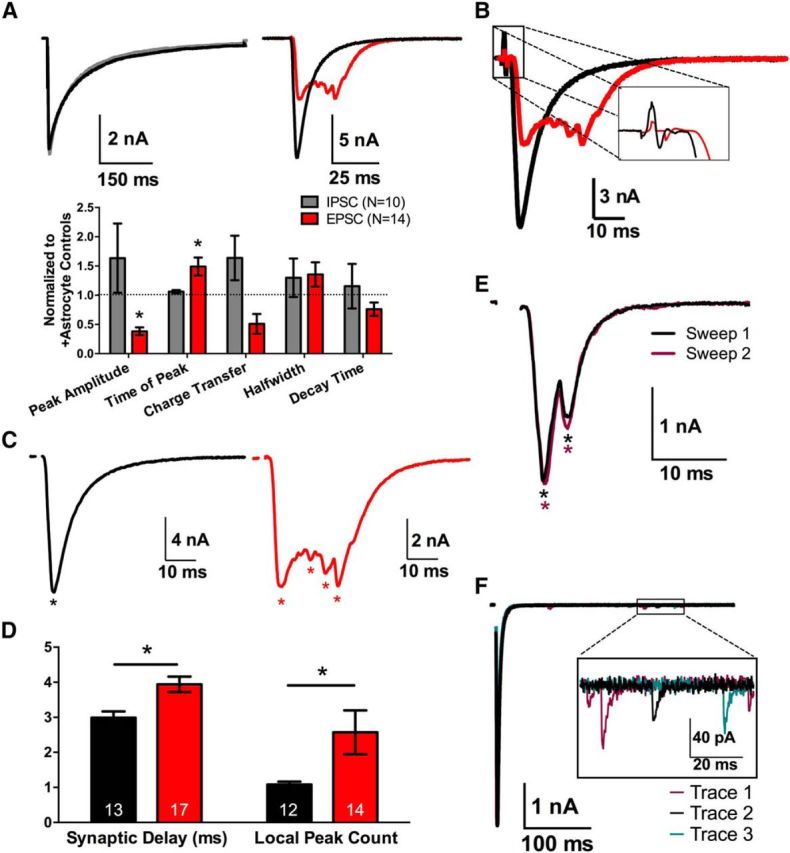
Evoked −astrocyte EPSCs, but not IPSCs, are desynchronized. A, Representative PSCs of GABAergic (left) and glutamatergic autaptic neurons (right) grown on +astrocyte microcultures (black traces, n = 9 and 12, respectively) or −astrocyte microcultures (gray IPSCs, n = 10; red EPSCs, n = 14). The bar graph summarizes differences in PSC waveforms between +astrocyte (black dotted line) and −astrocyte (gray and red bars, respectively) PSCs. −Astrocyte PSCs are normalized to +astrocyte PSC values. B, Representative traces of +astrocyte (black trace, n = 13) and −astrocyte (red trace, n = 17) EPSCs, focusing on synaptic delay between presynaptic stimulation and postsynaptic response (inset). C, Representative EPSCs from A, with asterisks denoting local minima in each trace (see Materials and Methods). D, Averaged local peak count and synaptic delay for +astrocyte and −astrocyte conditions displayed in bar graph (+astrocytes, n = 12; −astrocytes, n = 13). E, Two sequential evoked EPSCs from a −astrocyte glutamatergic neuron, separated by 25 s. Asterisks indicate local peaks detected via local peak count analysis, with color matched to trace color. F, Three evoked EPSCs recorded from +astrocyte glutamatergic neuron with 25 s between each stimulation. The boxed inset magnifies examples of quantal asynchrony occurring after the evoked response. *p < 0.05.
Glutamatergic neurons, however, displayed a marked change in the waveform of EPSCs when grown on −astrocyte microcultures compared to +astrocyte counterparts (Fig. 2A, red bars). The −astrocyte EPSCs had decreased peak amplitudes and increased time to peak (Fig. 2A, red bars; p < 0.05, Bonferroni corrected Student's unpaired t test) and increased synaptic delays (Fig. 2B,D; p < 0.05, Student's unpaired t test). The most obvious effect, however, was large-scale asynchrony of −astrocyte glutamatergic neuron EPSCs. This effect was quantified using local peak count (see Materials and Methods), which summed the number of local minima in the EPSC. −Astrocyte EPSCs exhibited ∼2.5 times the number of local peaks of +astrocyte EPSCs (Fig. 2C,D; p < 0.05, Student's unpaired t test).
This large-scale asynchrony was intriguing, in part because of fundamental differences from classical quantal synaptic asynchrony. Classical quantal asynchrony is defined by stochastic delayed release of a few vesicles after presynaptic stimulation, attributable to persisting presynaptic Ca2+ (Katz and Miledi, 1968; Barrett and Stevens, 1972; Rahamimoff and Yaari, 1973; Cummings et al., 1996). Large-scale asynchrony typically yielded late PSC peaks hundreds of picoamperes to nanoamperes in amplitude, compared with quantal amplitudes of ∼10 picoamperes (Fig. 2F). Furthermore, the pattern of large-scale asynchrony was largely invariant and stereotyped from trial to trial in a given cell (Fig. 2E), unlike the stochastic release characterizing quantal asynchrony (Fig. 2F). The asterisks in Figure 2E identify local peaks in each of the sweeps (asterisks are color-matched to their respective sweeps), and it is clear that the local peaks remained largely typecast between sweeps. This was quantified by measuring the common peak ratio between sweeps (see Materials and Methods). Indeed the common peak ratio for −astrocyte glutamatergic neurons was 0.82 ± 0.14 and was not significantly different compared to +astrocyte counterparts with a common peak ratio of 0.94 ± 0.06 (n = 14, 9 respectively; p > 0.05, Student's unpaired t test). In the absence of stimulation, we found no difference in spontaneous quantal vesicle release or waveform between +astrocyte and −astrocyte glutamatergic neurons recorded as miniature EPSCs (mEPSCs; frequency, +astrocyte = 2.72 ± 0.58 s−1, −astrocyte = 8.09 ± 3.02 s−1; mEPSC amplitude, +astrocyte = 29.74 ± 3.18 pA, −astrocyte = 32.89 ± 4.80 pA; 10–90% rise time, +astrocyte = 1.43 ± 0.15 ms, −astrocyte = 1.43 ± 0.12 ms; decay time, +astrocyte = 3.71 ± 0.28 ms, −astrocyte = 3.42 ± 0.30; n = 10 neurons; p > 0.05, Bonferroni corrected Student's unpaired t test for all parameters). In summary, our data suggest that the loss of local astrocytic support alters the EPSC waveform and glutamate release synchrony through an atypical mechanism of desynchronization.
Large-scale glutamate release asynchrony does not result from polysynaptic excitatory transmission
Large-scale glutamate release asynchrony could be explained by the presence of hidden polysynaptic connections from neighboring neurons (Fig. 3A). Although processes entering or exiting the presumed single-neuron −astrocyte microcultures were not evident, small neurites escaping detection are possible. If large-scale asynchrony from −astrocyte neurons results from polysynaptic connections, diminishing the stimulated excitatory input onto neighboring neurons should increase the likelihood of recurrent transmission delays or failure and alter the temporal location of peaks in the local peak profile, thus decreasing the common peak ratio.
Figure 3.
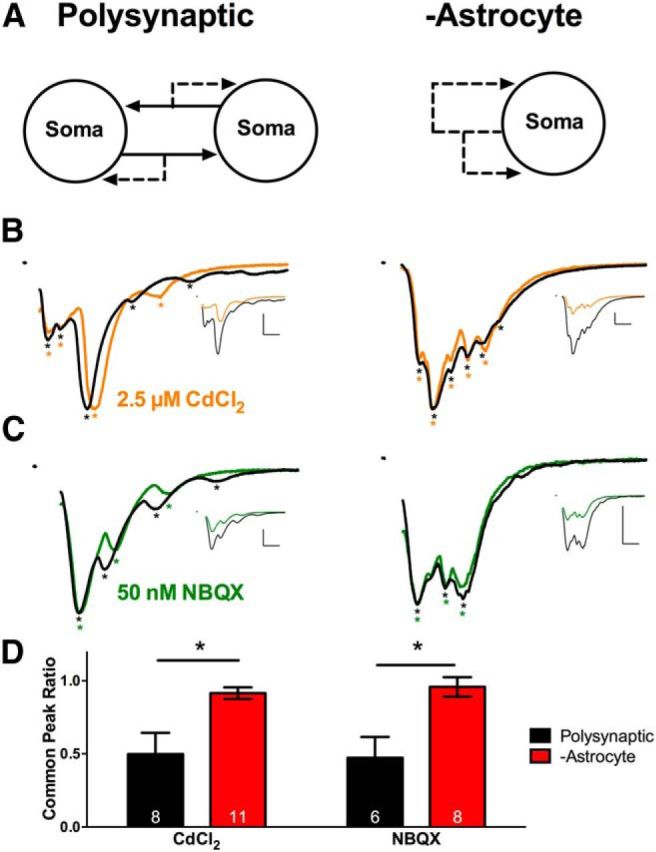
Large-scale asynchrony does not result from polysynaptic transmission. A, Schematic of a microculture containing multiple neurons with polysynaptic connections (left) and an autaptic −astrocyte microculture (right). B, C, EPSCs evoked by stimulating a neuron on a multineuron +astrocyte microculture (left) or a solitary neuron on a −astrocyte microculture (right). Asterisks denote local peaks. All EPSCs are normalized to the peak, and control and drug EPSCs are superimposed. The unscaled traces are displayed in the insets. Calibrations: 10 ms, 2 nA. EPSCs were evoked in control solution (black) or in 2.5 μm CdCl2 (orange; B). EPSCs in baseline solution (black) or in 50 nm NBQX (green; C). D, Common peak ratio (for a description of analysis, see Materials and Methods) was calculated for polysynaptic neurons (black bars) and −astrocyte neurons (red bars) in the CdCl2 experiment (left; n = 8 and 11, respectively) and NBQX experiment (right; n = 6 and 8, respectively). *p < 0.05.
To test this, we reduced excitatory neurotransmission in two distinct ways: (1) with a subsaturating concentration of Cd2+ (2.5 μm) to decrease synaptic glutamate release or (2) with NBQX (50 nm) to dampen postsynaptic responsiveness. The concentrations of NBQX and Cd2+ strongly reduced the peak amplitude (Cd2+, 46.39 ± 3.64%, n = 19 neurons; NBQX, 47.47 ± 1.65%, n = 14 neurons), ensuring initiation delays or failures would occur if recruitment of other cells underlies large-scale synaptic asynchrony. Glutamatergic neurons on multicell microcultures served as positive controls (Fig. 3A, left). On multicell microcultures, both Cd2+ and NBQX decreased the common peak ratio (Fig. 3B,C, left, D, black bars). However the common peak ratio of −astrocyte solitary neurons (Fig. 3A, right) remained largely unchanged by Cd2+ or by NBQX (Fig. 3B,C, right, D, red bars). These data suggest that large-scale asynchrony observed in −astrocyte glutamatergic neurons does not result from polysynaptic excitatory transmission.
Aberrant glutamate transmission is partially conserved in fixed-astrocyte microcultures
It is unclear whether the changes in the EPSC waveform of −astrocyte neurons result from loss of contact-mediated cues or loss of local soluble cues from astrocytes. We have previously addressed questions of local versus global astrocytic influence on neuronal physiology using neurons grown on a bed of lightly fixed astrocytes (Crawford et al., 2012). This fixed-astrocyte/live-astrocyte microculture system shares many similarities to the system described in the present work, but neurons presumably retain contact with astrocytic protein signals, albeit after fixation. Comparing these two systems allowed us to investigate whether alterations in glutamate neurotransmission likely results from contact mediated cues.
Evoked EPSCs elicited from fixed-astrocyte glutamatergic neurons (Fig. 4A, gray trace) displayed temporally altered EPSC waveforms compared to live-astrocyte controls (Fig. 4A, green trace), with an increase in the half-width and time to peak (Fig. 4C, gray bars; p < 0.05, Bonferroni corrected Student's unpaired t test). The increased time to peak in fixed-astrocyte EPSCs was similar to that in −astrocyte EPSCs, suggesting that some of the synchrony changes are recapitulated when contact with fixed astrocytes is maintained. Also, neither fixed-astrocyte nor −astrocyte glutamatergic neurons displayed differences in the charge transfer (p > 0.05, Bonferroni corrected Student's unpaired t test) nor decay time (p > 0.05, Bonferroni corrected Student's unpaired t test) compared with control +astrocyte EPSCs from culture mates in the respective conditions (Fig. 4C). This suggests that local astrocyte cues do not govern all aspects of synaptic efficacy in our context. However, the characteristic large-scale asynchrony was not observed in fixed-astrocyte EPSCs, with a local peak count of 1.00 ± 0.00 (n = 9), suggesting that soluble global or local cues were not responsible for glutamate release synchrony. Instead, this suggests that physical contact with an astrocyte, even if fixed, is sufficient to maintain synchrony.
Figure 4.

Temporal abnormalities are partially conserved in fixed-astrocyte microcultures. A, Example EPSCs from solitary glutamatergic neurons in microcultures containing live astrocytes (green trace) or lightly fixed astrocytes (gray trace), obtained from the same coverslip and superimposed. B, Similar EPSCs, but from +astrocyte (black trace) and −astrocyte neurons (red trace). C, The bar graph displays EPSC waveform parameters of fixed-astrocyte EPSCs (gray bars; n = 14) and −astrocyte EPSCs (red bars; n = 14) normalized to their respective sibling live-astrocyte EPSCs (green and black dotted line; n = 11 and 12, respectively). *p < 0.05, Bonferroni corrected.
Somatic action potential waveforms are mildly altered in −astrocyte glutamatergic neurons
We next queried possible neuronal deficits associated with the aberrant EPSC waveforms. We reasoned that large-scale asynchrony is most likely associated with disruptions of excitability and/or action potential propagation in the presynaptic cell. To test for changes in excitability near the site of action potential initiation, we first assessed action potential initiation through somatic current-clamp action potential recordings. The minimum current injection required for action potential initiation (rheobase) in −astrocyte neurons and +astrocyte neurons did not differ significantly (Fig. 5A; +astrocyte = 57.36 ± 8.50 pA, −astrocyte = 36.62 ± 5.16 pA; n = 12 neurons; p > 0.05, Bonferroni corrected Student's unpaired t test), suggesting that action potential initiation threshold cannot explain large-scale asynchrony. However, we did find subtle, yet significant differences in somatic action potential waveform between +astrocyte and −astrocyte glutamatergic neurons. Peak amplitude and maximum rise rate of −astrocyte action potentials were decreased (Fig. 5B,C; p < 0.05, Bonferroni corrected Student's unpaired t test). The small alterations in somatically recorded action potential waveform may hint at a source for altered EPSC waveforms, but such subtle changes are unlikely to directly orchestrate large-scale asynchrony. Rather, we hypothesize that subtle changes in somatic action potentials more likely reflect a larger difference in axonal action potential propagation, electrically remote from the soma.
Figure 5.
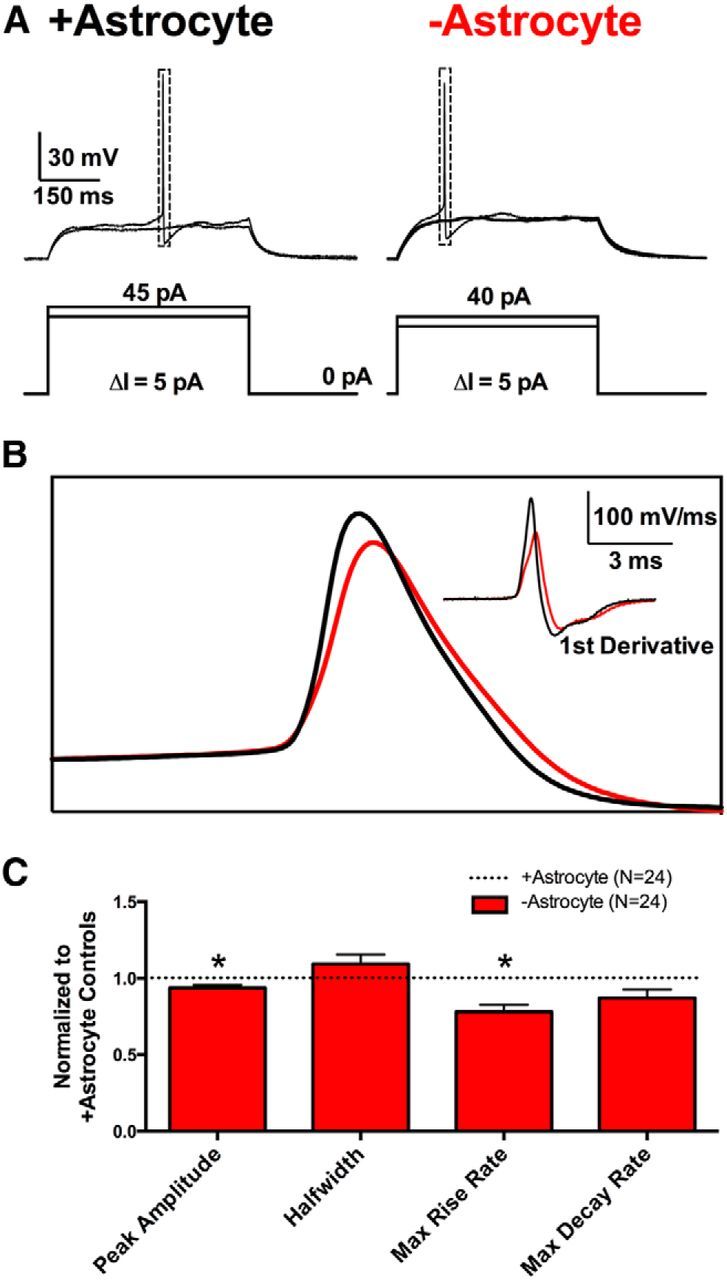
Somatic action potential waveforms are mildly affected by loss of local astrocytic support. A, Representative action potential initiation recordings from +astrocyte glutamatergic neurons (left) and −astrocyte glutamatergic neurons (right). The current-injection protocols displayed beneath the traces show current injections incremented (ΔI) in 5 pA steps until a single action potential was evoked. B, Superimposed +astrocyte (black) and −astrocyte (red) action potentials from A are displayed along with the first derivative of the action potentials (inset). C, Action potential waveform parameters are quantified in the bar graph below. −Astrocyte action potential values (red bars; n = 24) were normalized to +astrocyte controls (black dotted line; n = 24). *p < 0.05, Bonferroni corrected.
+Astrocyte and −astrocyte glutamatergic neurons have similar axon structures
We first entertained the possibility that sites of ectopic initiation in distal axon may underlie large-scale asynchrony. A change in the size, position, or number of AISs could account for desynchronized EPSCs. We used immunohistochemistry to detect initiation sites using a broad-spectrum antibody against voltage-gated sodium channels (pan-Nav antibody). The antibody yields intense labeling at the AIS in many neuronal types (Kole et al., 2008; Kress et al., 2010; Lorincz and Nusser, 2010). We found that both +astrocyte neurons and −astrocyte solitary neurons had clearly defined proximal patches of strong pan-Nav labeling, but there were no differences in the area or normalized intensity of the immunofluorescence. There also was no difference in the number of pan-Nav-positive segments per neuron, the length of staining per segment, or length from soma to AIS staining onset (Fig. 6A; p > 0.05 Bonferroni corrected Student's t test; see Materials and Methods). We obtained similar findings when we stained for ankyrin-G, another marker of AIS (data not shown). Thus, there are no apparent alterations in AIS structure for −astrocyte glutamatergic neurons compared to +astrocyte controls. Because Navs and ankyrin-G immunoreactivity mark the site of action potential initiation in many cell types (Jenkins and Bennett, 2001; Hedstrom et al., 2008; Kole et al., 2008), it is unlikely that ectopic or abnormal sites of initiation explain large-scale asynchrony of −astrocyte EPSCs.
Although the AIS was comparable between +astrocyte and −astrocyte glutamatergic neurons, differences in the structure of the distal axon could underlie glutamate release asynchrony. For example, longer, more complex axons with more branch points could underlie action potential propagation and arrival time changes that could result in asynchronous glutamate release. We combined electrophysiological and morphological methods to correlate large-scale glutamate release asynchrony with distal axon structure. We first measured EPSCs from +astrocyte and −astrocyte neurons (Fig. 6C) to compare physiology with subsequent immunolabeling. We then performed immunocytochemistry with the axonal neurofilament antibody SMI-312, confocal microscopy, and image processing (see Materials and Methods) to create a binary image of the axon (Fig. 6B). We measured the number of branches, the number of endpoints, and the length of the axon and found no detectable difference in axon length or shape (p > 0.05, Bonferroni corrected Student's unpaired t test), suggesting that the structure of the distal axon is not responsible for large-scale glutamate release asynchrony (Fig. 6D).
We considered the possibility that altered lipid composition of the axon could underlie synchrony differences in the presence or absence of astrocytes. In particular, cholesterol content has been suggested to underlie excitability differences and to control release synchrony (Zamir and Charlton, 2006; Guo et al., 2008), and astrocytes synthesize and deliver cholesterol to mature hippocampal neurons (Mauch et al., 2001; Fünfschilling et al., 2012). We hypothesized that alterations in action potential propagation underlying large-scale asynchrony may stem from differences in membrane cholesterol content proximally versus distally along the axons of −astrocyte neurons compared to +astrocyte controls. We measured the free cholesterol content in the axons of +astrocyte and −astrocyte glutamatergic neurons using filipin staining (Yeagle, 1985). We first quantified filipin staining intensity proximally (Fig. 6E, arrow) and distally (arrowhead) and found no significant difference in staining intensity between +astrocyte and −astrocyte glutamatergic neurons (proximal, +astrocyte = 51.84 ± 13.14 A.U., −astrocyte = 50.42 ± 14.55 A.U., p > 0.05; distal, +astrocyte = 14.26 ± 3.91 A.U., −astrocyte = 15.70 ± 5.51 A.U., p > 0.05, Bonferroni corrected Student's unpaired t test). To reduce the possibility that proximal versus distal differences in axonal filipin staining intensity were masked by cell-to-cell variations in free-cholesterol content, we normalized proximal and distal axonal filipin intensity to somatic filipin intensity. We found no differences in proximal or distal intensity when normalized to somatic filipin intensity (Fig. 6E, left graphs). Because we hypothesized that large-scale asynchrony resulted from axonal propagation but not initiation deficits, we next asked whether there was a difference in the ratio of proximal to distal axonal free cholesterol. We found no difference in the ratio of proximal to distal fluorescence intensity (Fig. 6E, right graph). This suggests that the free cholesterol content does not account for aberrant EPSCs observed in −astrocyte glutamatergic neurons. Together, our data suggest that temporal disruptions of transmission are unlikely to stem from gross structural changes to axons.
Broader action potential waveforms may partially underlie EPSC waveform disruption
Neurotransmitter release is tightly coupled to presynaptic action potentials, and changes in action potential waveform can have a significant effect on synchronous release and synaptic delay of release (Borst and Sakmann, 1999; Hefft and Jonas, 2005; Boudkkazi et al., 2011). We next wanted to test whether aberrant EPSCs from −astrocyte glutamatergic neurons could be explained by broader action potentials. To test this, we broadened action potentials of +astrocyte glutamatergic neurons using voltage-gated potassium channels blockers tetraethylammonium (TEA), 4-aminopyradine (4-AP), and α-dendrotoxin (α-DTX). The Kv1 blocker α-DTX was prioritized because axonal Kv1 channels have been shown to increase the synaptic delay at cortical synapses by increasing action potential width (Boudkkazi et al., 2011).
α-DTX (100 nm) produced only a slight, trend-level increase in time to EPSC peak (Fig. 7A; p = 0.052, Student's two-tailed, paired t test). We also immunostained for one dendrotoxin-sensitive Kv subunit (Kv1.2) that is present in axons of excitatory principal cells (Wang et al., 1994; Gu et al., 2003; Lorincz and Nusser, 2008). In excitatory neurons identified by vGLUT1 immunoreactivity, we found no difference in Kv1.2 staining intensity in +astrocyte versus −astrocyte SMI-312-labeled axons (Fig. 7B; p > 0.05, Student's unpaired t test). Although certainly not exhaustive, the results of Figure 7A,B suggest that dendrotoxin-sensitive Kv1 channels are unlikely to drive differences between excitatory neurons reared with and without local astrocytes.
Figure 7.

Action potential broadening may partially account for the disturbed −astrocyte EPSC waveform. A, C–D, Evoked baseline (control) and experimental (indicated drug) EPSCs from solitary neurons on +astrocyte microcultures are superimposed. A, EPSCs evoked in control saline (black) or in the presence of 100 nm α-dendrotoxin (teal). The right plot shows the time to EPSC peak for baseline (black symbols) and during drug application (teal symbols; n = 9 neurons). B, Immunostaining for Kv1.2 in −astrocyte (n = 7) and +astrocyte (n = 8) axons. Cells were triple labeled with anti-vGLUT1 to identify glutamatergic neurons, SMI-312 to identify axons, and anti-Kv1.2. The right panel shows that Kv1.2 immunofluorescence intensity normalized to corresponding SMI-312 immunofluorescence intensity did not differ between +astrocyte and −astrocyte axons from three independent stains. C, EPSCs evoked in control saline (black) or in the presence of 10 mm TEA (green). The right plot shows the change in time to peak when the bath (black) or drug (green) was applied (n = 10 neurons). D, EPSCs evoked in control saline (black) or in the presence of 200 μm 4-AP (purple). The right plot shows the change in time to peak when the bath (black) or drug (purple) was applied (n = 10 neurons). E, A train of 12 EPSCs at 20 Hz was evoked in −astrocyte neurons before (black traces) and after (red) 5 min of 20 μm EGTA-AM application. Control (black solid line) and EGTA-AM incubated (red solid line) EPSCs from the first stimulation are magnified with the decay of the 12th EPSCs (gray and red dotted lines, respectively) superimposed. The 12th EPSC peaks are scaled to 1st EPSC peaks for comparison. Asterisks under EPSCs denote identified local peaks. The bar graph quantifies the decay time constant of the 1st and 12th EPSCs for control (black bars) and EGTA-AM incubated (red bars) EPSCs. F, Local peak count of control EPSCs (black bars) and drug-treated EPSCs (colored bars) across all experiments. For summary data in C–E, *p < 0.05; **p < 0.01.
The time to peak of +astrocyte evoked EPSCs was increased by nonspecific K+-channel blockers TEA (20 mm; Fig. 7C; p < 0.001 Student's paired t test) and 4-AP (200 μm; Fig. 7D; p < 0.05 Student's paired t test). Importantly, K+-channel blockers did not induce large-scale glutamate release asynchrony in +astrocyte glutamatergic neurons (Fig. 7F; p > 0.05 Student's paired t test).
If broad action potentials and resulting prolonged presynaptic Ca2+ elevation underlie −astrocyte EPSC asynchrony, chelation of presynaptic Ca2+ may abrogate the asynchrony. Thus, we tested the ability of EGTA-AM incubation to reduce large-scale asynchrony. To verify EGTA-AM effectiveness, we performed repetitive stimulation, which increases quantal asynchrony from primary hippocampal synaptic terminals, resulting in prolonged late EPSCs (Hagler and Goda, 2001; Chang and Mennerick, 2010). As expected, EGTA-AM treatment significantly decreased the asynchronous component of −astrocyte EPSCs after 12 pulses at 20 Hz, as quantified by the decreased decay time constant of the final evoked EPSC (Fig. 7E; p < 0.05 Student's paired t test). However, EGTA-AM incubation had no significant effect on the local peak count of isolated EPSCs evoked from these neurons (Fig. 7F). Interestingly, these experiments revealed that repetitive stimulation altered the local peak profile of late EPSCs in the train in an EGTA-sensitive manner (n = 4 of 5 neurons; Fig. 7E, top). This suggests that Ca2+ accumulation may interact with the mechanisms of asynchrony, but does not directly cause the asynchrony. Together, results of Figure 7 suggest that broader action potentials may contribute to the EPSC waveform differences between +astrocyte and −astrocyte neurons, but action potential broadening does not underlie the most prominent effect: large-scale glutamate release asynchrony.
Large-scale glutamate release asynchrony involves propagation deficits
Although −astrocyte glutamatergic neurons do not display altered axon structure, differences in axonal membrane excitability may participate in glutamate release asynchrony. We reasoned that if axon propagation fidelity is insecure, −astrocyte EPSCs may be more sensitive to acute disruption of excitability and propagation. We decreased membrane excitability by either applying a subsaturating concentration of TTX (10 nm) or increasing surface charge screening (Frankenhaeuser and Hodgkin, 1957; Hille, 2001) with a high concentration of extracellular divalent cations (4 mm Ca2+ and 3 mm Mg2+; Fig. 8A,B).
Figure 8.
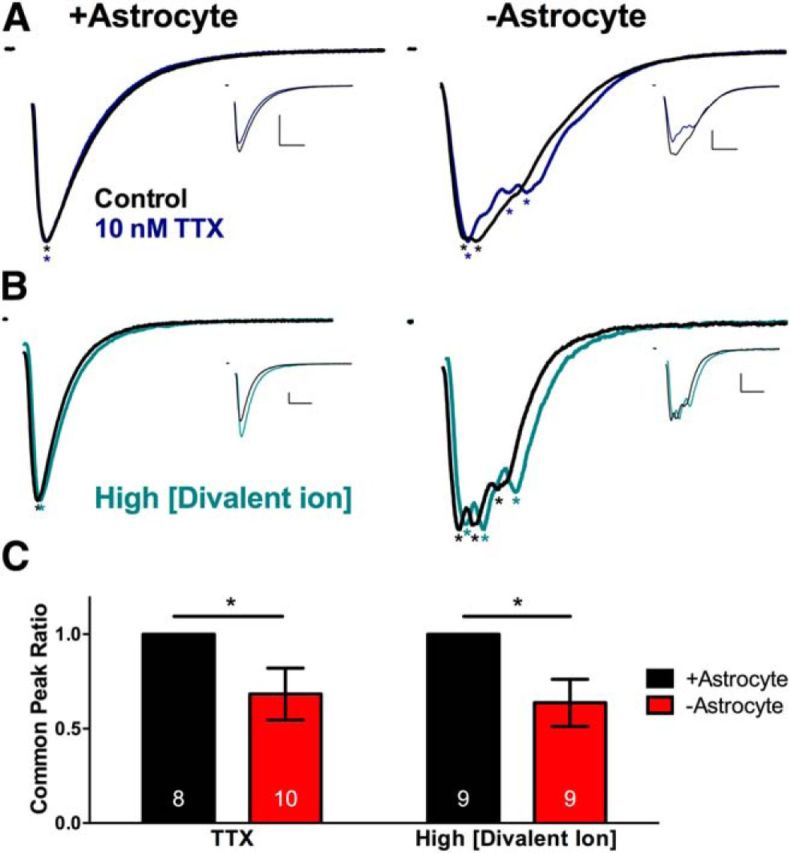
Decreasing membrane excitability alters temporal profile of −astrocyte EPSCs but not +astrocyte EPSCs. A, B, Evoked EPSCs from solitary neurons on +astrocyte microcultures (left) and −astrocyte microcultures (right). Local peaks are denoted by asterisks. All EPSCs are normalized to the largest peak, and control and experimental EPSCs are superimposed. Unscaled traces are displayed as insets. A, EPSCs evoked in control (black) or in 10 nm TTX (blue). Calibrations: 10 ms, 2 nA. B, EPSCs evoked in control (black) or in high extracellular divalent ion concentration (4 mm Ca2+, 3 mm Mg2+; teal). Calibrations: 10 ms, 500 pA. C, Bar graphs comparing common peak ratio between +astrocyte (black bars) and −astrocyte (red bars) EPSCs in the TTX condition (left; n = 8 and 10, respectively) and high extracellular divalent ion concentration (right; n = 9 and 9, respectively). *p < 0.05.
TTX decreased both +astrocyte and −astrocyte glutamatergic neuron EPSC charge transfer equally compared to control sweeps (+astrocyte, 17.35 ± 5.89% decrease, −astrocyte: 22.71 ± 6.42% decrease). However, TTX decreased the common peak ratio in −astrocyte but not +astrocyte EPSCs (Fig. 8A,C, left). The lack of effect on +astrocyte EPSCs is consistent with high propagation security under normal conditions (Allen and Stevens, 1994; Mackenzie et al., 1996; Mackenzie and Murphy, 1998; Meeks et al., 2005). Manipulation of surface charge screening also decreased the −astrocyte EPSC common peak ratio compared to +astrocyte counterparts, which showed no change (Fig. 8B,C, right). Thus, manipulating membrane excitability altered the local peak profile, which was typically invariant at baseline, as observed in Figure 2D. Figure 8 suggests that large-scale glutamate release asynchrony is associated with altered action potential propagation along the axon.
To explain axonal propagation deficits, we considered that altered axonal K+ channel expression or function along −astrocyte axons may underlie large-scale glutamate release asynchrony (Fig. 7). K+ channels are responsible for much of the variability among neurons in spiking patterns, and thus represent good candidates (Storm, 1990; Debanne et al., 2011). Kv7.2/3 channels in particular are enriched in axons of hippocampal pyramidal neurons but not interneurons (Chung et al., 2006; Vervaeke et al., 2006; Shah et al., 2008, 2011), thereby potentially explaining altered EPSCs, but not IPSCs (Fig. 2A). We elicited EPSCs in the presence of either Kv7 channel antagonist XE-991 (10 μm) or the Kv7 agonist retigabine (10 μm). Both +astrocyte and −astrocyte neurons responded similarly to XE-991 and retigabine applications, with no significant change in the local peak count between control and drug applications of −astrocyte EPSCs (XE-991 common peak ratio, 0.84 ± 0.09; retigabine common peak ratio, 0.84 ± 0.09) compared to +astrocyte counterparts (XE-991 and retigabine common peak ratio, 1.00 ± 0.00; p > 0.05, Bonferroni corrected unpaired Student's t test). These data suggest that differences in axon-specific Kv7 channels do not underlie large-scale asynchrony.
Extracellular axonal recordings demonstrate dispersed axonal arrival times
Although manipulations of excitability decreased EPSC common peak ratios of −astrocyte neurons, we pursued direct evidence that action potential propagation deficits underlie large-scale asynchrony, and we sought information about the nature of propagation changes. Is the action potential waveform altered, or does the action potential arrive at different axonal locations at different times?
We directly tested these two possibilities by simultaneously recording action potential arrival times at the soma and distal processes in +astrocyte and −astrocyte neurons. Dual recordings provide both the spatial and temporal resolution required to address these questions. We whole-cell patch-clamped the soma of +astrocyte or −astrocyte glutamatergic neurons and performed a loose-seal extracellular recording from the presumed axon (Fig. 9A). We evoked an action potential at the soma and measured action potential latency from the soma (whole-cell recording) to the distal site (loose-seal extracellular recording). Action potential latency was measured as the time between the peak of the first derivative of the somatic action potential and the peak of the extracellularly recorded action potential at the distal site (see Materials and Methods).
Figure 9.
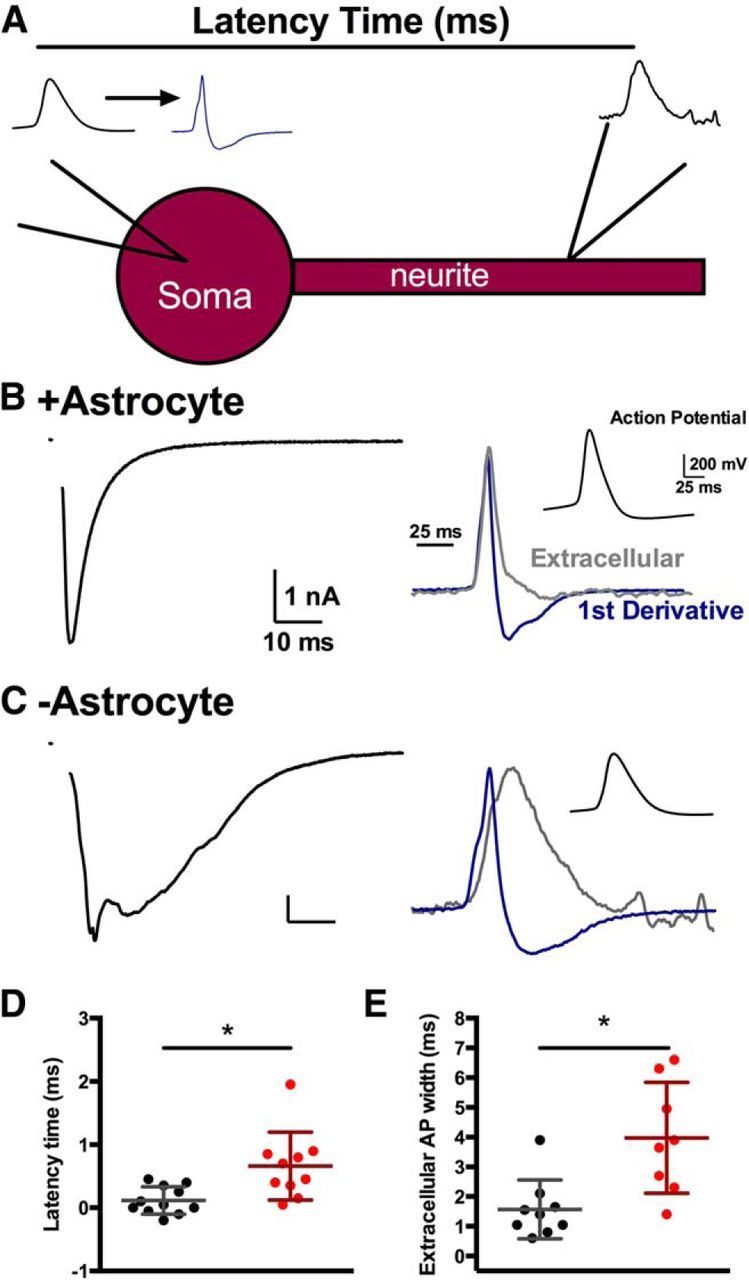
Longer, more variable action potential propagation for −astrocyte neurons. A, Schematic of the dual recording protocol performed on +astrocyte and −astrocyte glutamatergic neurons. A whole-cell patch pipette was placed at the soma, and a loose-seal extracellular pipette is placed along a distal thin neurite, the presumed axon. An action potential was evoked and recorded in the somatic whole-cell recording pipette, and the mainly capacitive current at the distal process was recorded with the loose-seal pipette. The extracellular action potential recording was compared to the first derivative of the somatic action potential, and the latency time was measured as the time between the peak of the somatic first derivative and the peak of the extracellular recording. B, C, Representative recordings from +astrocyte neurons (B) and −astrocyte neurons (C). EPSCs were evoked to assess large-scale asynchrony (left). The action potential measured at the distal neurite and the first derivative of the action potential are superimposed (right) and scaled to match baseline and maximum peak. The somatic action potential is displayed in the right inset. D, E, Plots of latency of action potential propagation (D) and extracellular action potential width (E). Dots represent individual +astrocyte neurons (black; n = 11 and 9, respectively) and −astrocyte neurons (red; n = 10 and 8, respectively). Values are mean ± SD. *p < 0.05.
We found −astrocyte glutamatergic neurons had a significantly increased latency between somatic and neurite action potentials (Fig. 9C,D) compared to +astrocyte controls (Fig. 9B,D; p < 0.05, Bonferroni corrected Student's unpaired t test), and we also found that −astrocyte neurons showed more variance in their latency values than +astrocyte counterparts (p < 0.05, F test). The increased variance in action potential propagation supports the hypothesis that large-scale synaptic asynchrony may result from varied action potential arrival times to different parts of the axon and axon branches. For −astrocyte neurons, we also found an increase in time from the maximum peak to the minimum/baseline (Fig. 9C,E) compared to +astrocyte controls (Fig. 9B,E; p < 0.05, Bonferroni corrected Student's t test). Because extracellular local recordings approximate the first derivative of the intracellular voltage signal (Johnston and Wu, 1995; Meeks et al., 2005), we equate this measurement to the width of the action potential. This suggests that the width of the action potential also increases at more distal processes for −astrocyte neurons than +astrocyte neurons. Overall, neurons displayed broader action potentials and more varied and increased action potential propagation times throughout the presumed axon when local astrocytic support was lost.
Discussion
Here, we investigated the effects of removing local astrocytic support from developing hippocampal neurons. Glutamatergic neurons grown on −astrocyte microcultures displayed an altered EPSC waveform, with a significantly smaller peak amplitude, increased synaptic delay, and previously undescribed large-scale glutamate release asynchrony. This asynchrony was distinct from classical quantal asynchrony due to its invariance from stimulus to stimulus, the supraquantal size of the local peaks (Fig. 2), and the insensitivity to EGTA (Fig. 7). It was also not evident in GABAergic IPSCs. Large-scale asynchrony did not result from polysynaptic connections from neighboring neurons or from gross alterations in the structure of axons. Although the aberrant EPSC waveform appears to stem from changes to the waveform and propagation of axonal action potentials, action potential initiation appeared nearly unaltered. Thus, we have uncovered an axon-selective alteration in excitability of principal neurons that is under astrocytic control.
The role we have discovered for astrocytes appears to be fundamentally developmental in nature. Astrocytes have been implicated previously in the acute modulation of axonal excitability and action potential propagation through potassium buffering and gliotransmission (Sasaki et al., 2011; Bay and Butt, 2012). These acute forms of neuronal signal regulation cannot explain the effects reported here in part because the effects we observed are stimulus/frequency independent. For instance, potassium buildup (Bay and Butt, 2012) cannot explain the EPSC changes we report since we observed synchrony effects with the very first stimulus applied to neurons, and repeated stimuli were separated by >20 s. Similarly, gliotransmitter-induced action potential broadening (Sasaki et al., 2011) cannot explain our results, since we observed action potential broadening in the absence rather than the presence of astrocytes.
Our analysis suggests that glutamate but not GABA release exhibits large-scale asynchrony in the absence of local astrocytes. This differential effect suggests that local astrocytic cues are unnecessary for proper synchronous action potential propagation through the axons of interneurons. It seems most likely that interneuron propagation synchrony is established through molecular mechanisms distinct from those governing principal-cell propagation synchrony. We and others have suggested previously that even in the presence of local astrocyte support, action potential transmission in principal-cell axons is not as secure as interneuron transmission (Prakriya and Mennerick, 2000; He et al., 2002; Meeks and Mennerick, 2004; Hu and Jonas, 2014). Therefore, an alternative possibility is that principal-cell axons and interneuron axons respond similarly to astrocyte cues, but interneuron axons have a larger propagation safety factor and therefore better tolerate the disruption resulting from astrocyte loss.
A key conclusion from our work is that local astrocytic cues are required for the establishment of correct propagation synchrony. This conclusion is warranted because the global cues governing development of −astrocyte neurons and +astrocyte neurons are identical in our preparation, as the two classes are interspersed on the same coverslip. Furthermore, we found no significant effects on overall quantal content, frequency, or quantal size of EPSCs, all of which may be affected by astrocyte activity (Kucukdereli et al., 2011; Perea et al., 2014). Nevertheless, a fair question is, how local are these local astrocytic cues? Our results do not exclude a role for very local, diffusible signals that dilute to inconsequential concentrations distal to the site of astrocyte release. We found that culturing neurons on a fixed astrocyte bed reconstituted some temporal abnormalities, but contact with fixed astrocytes was sufficient to eliminate the large-scale asynchronous EPSC peaks. This suggests that contact mediated cues, which can be recapitulated even in fixed astrocytes, may be required to preserve glutamate release synchrony. Alternatively, desynchronization cues may come from direct neuronal contact with the collagen substrate. Other temporal disturbances, including increases time to peak, could involve local soluble cues since these changes are observed in the complete absence of underlying astrocytes and on fixed astrocytes. In summary, the ability to experimentally separate time-to-peak/synaptic delay alterations from large-scale asynchronous peaks could suggest that multiple cues contribute to synchrony disruptions, with at least some being contact mediated.
Our analysis of axonal action potentials revealed two propagation alterations in −astrocyte neurons: a wider distribution of action potential arrivals and a broader action potential waveform. We have shown that the former alteration likely contributes to large-scale asynchrony. Although the latency distribution from −astrocyte neurons did not approach latencies necessary to explain the latest EPSC peaks observed (>10 ms after stimulus), it seems likely that these late EPSC peaks arose from the most distal axon branches, whereas our extracellular recordings were likely biased toward proximal segments, where arrival delays are minimized. By contrast, the waveform broadening likely explains the increased synaptic delay and time to first peak, observed in both −astrocyte and fixed-astrocyte neurons. These changes were also recapitulated in +astrocyte EPSCs when the action potential was broadened by K+ channel block. Specific Kv1 block with α-DTX produced minimal alteration of EPSCs. Less selective blockers produced stronger effects (Fig. 7). Thus, if potassium channel alterations underlie differences between −astrocyte and +astrocyte axons, it appears that multiple classes of channels may be affected. Broadening of the action potential is well suited to increase synaptic delay, because most calcium entry into the presynaptic terminal occurs on the repolarization phase of the action potential (Llinás et al., 1981; Sabatini and Regehr, 1996; Boudkkazi et al., 2011). It remains unclear whether the broader spike waveform is causally related to the alteration in arrival time. Superficially, a broader action potential might be expected to result in more secure propagation, at least for single evoked responses. Furthermore, K+ channel block of +astrocyte EPSCs did not recapitulate the large-scale asynchrony (multiple EPSC peaks). Thus, it is tempting to speculate that action potential broadening and arrival times are dissociable, as are aspects of the EPSC asynchrony, as described above.
The alterations described herein represent an important foundation for future inquiry. Key outstanding issues include the specific local astrocytic signals and the downstream neuronal targets. Several initial candidates for downstream targets failed our tests of involvement. Although contact mediated cues are important for axon outgrowth and branching (for review, see Polleux and Snider, 2010; Raper and Mason, 2010), we found no evidence for structural alterations in the AIS, branching pattern, axonal length, or cholesterol content that can readily explain the strong propagation changes. Furthermore, although axon-specific potassium channels are prime candidates to explain the severe disruption of axonal signal propagation, we found no evidence for alterations in a putative axon-specific, principal-neuron potassium channel, Kv7.2/3. It is possible that a survey-based approach, such as a transcriptome analysis, might prove more efficient than a candidate-based approach in identifying altered targets in astrocyte-deficient neurons.
In conclusion, we found that the loss of local astrocytic support has a profound effect on evoked EPSCs, with decreased peak amplitudes, increased synaptic delays, and profound large-scale glutamate release asynchrony. We also found that −astrocyte glutamatergic neurons had aberrant action potential propagation, with increased and more variable action potential latency periods to distal processes, as well as increased action potential widths at the distal processes compared to +astrocyte neurons. We conclude that astrocytes have an unappreciated role in maintaining glutamate release synchrony by controlling proper maturation of unmyelinated principal-cell axons.
Footnotes
This work was supported by NIH Grants R21MH099658, R01MH101874, T32NS073547, T32GM08151, F31NS066611, and R01MH078823. We thank members of the Mennerick and Zorumski groups, Joe Henry Steinbach, Colin Nichols, and Andrew Yoo for advice and comments, and Ann Benz and Amanda Taylor for technical help with cultures.
The authors declare no competing financial interests.
References
- Allen C, Stevens CF. An evaluation of causes for unreliability of synaptic transmission. Proc Natl Acad Sci U S A. 1994;91:10380–10383. doi: 10.1073/pnas.91.22.10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen NJ, Barres BA. Neuroscience: glia–more than just brain glue. Nature. 2009;457:675–677. doi: 10.1038/457675a. [DOI] [PubMed] [Google Scholar]
- Allen NJ, Bennett ML, Foo LC, Wang GX, Chakraborty C, Smith SJ, Barres BA. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature. 2012;486:410–414. doi: 10.1038/nature11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett EF, Stevens CF. Quantal independence and uniformity of presynaptic release kinetics at the frog neuromuscular junction. J Physiol. 1972;227:665–689. doi: 10.1113/jphysiol.1972.sp010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bay V, Butt AM. Relationship between glial potassium regulation and axon excitability: a role for glial Kir4.1 channels. Glia. 2012;60:651–660. doi: 10.1002/glia.22299. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc Natl Acad Sci U S A. 1991;88:7834–7838. doi: 10.1073/pnas.88.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Sakmann B. Effect of changes in action potential shape on calcium currents and transmitter release in a calyx-type synapse of the rat auditory brainstem. Philos Trans R Soc Lond B Biol Sci. 1999;354:347–355. doi: 10.1098/rstb.1999.0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudkkazi S, Fronzaroli-Molinieres L, Debanne D. Presynaptic action potential waveform determines cortical synaptic latency. J Physiol. 2011;589:1117–1131. doi: 10.1113/jphysiol.2010.199653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Chang CY, Mennerick S. Dynamic modulation of phasic and asynchronous glutamate release in hippocampal synapses. J Neurophysiol. 2010;103:392–401. doi: 10.1152/jn.00683.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Jan YN, Jan LY. Polarized axonal surface expression of neuronal KCNQ channels is mediated by multiple signals in the KCNQ2 and KCNQ3 C-terminal domains. Proc Natl Acad Sci U S A. 2006;103:8870–8875. doi: 10.1073/pnas.0603376103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LE, Barres BA. Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci. 2013;14:311–321. doi: 10.1038/nrn3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford DC, Jiang X, Taylor A, Mennerick S. Astrocyte-derived thrombospondins mediate the development of hippocampal presynaptic plasticity in vitro. J Neurosci. 2012;32:13100–13110. doi: 10.1523/JNEUROSCI.2604-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings DD, Wilcox KS, Dichter MA. Calcium-Dependent Paired-Pulse Facilitation of Miniature EPSC Frequency Accompanies Depression of EPSCs at Hippocampal Synapses in Culture. J Neurosci. 1996;16:5312–5323. doi: 10.1523/JNEUROSCI.16-17-05312.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Campanac E, Bialowas A, Carlier E, Alcaraz G. Axon physiology. Physiol Rev. 2011;91:555–602. doi: 10.1152/physrev.00048.2009. [DOI] [PubMed] [Google Scholar]
- Fonnum F, Johnsen A, Hassel B. Use of fluorocitrate and fluoroacetate in the study of brain metabolism. Glia. 1997;21:106–113. doi: 10.1002/(SICI)1098-1136(199709)21:1<106::AID-GLIA12>3.0.CO%3B2-W. [DOI] [PubMed] [Google Scholar]
- Frankenhaeuser B, Hodgkin AL. The action of calcium on the electrical properties of squid axons. J Physiol. 1957;137:218–244. doi: 10.1113/jphysiol.1957.sp005808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fünfschilling U, Jockusch WJ, Sivakumar N, Möbius W, Corthals K, Li S, Quintes S, Kim Y, Schaap IA, Rhee JS, Nave KA, Saher G. Critical time window of neuronal cholesterol synthesis during neurite outgrowth. J Neurosci. 2012;32:7632–7645. doi: 10.1523/JNEUROSCI.1352-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Jan YN, Jan LY. A conserved domain in axonal targeting of Kv1 (Shaker) voltage-gated potassium channels. Science. 2003;301:646–649. doi: 10.1126/science.1086998. [DOI] [PubMed] [Google Scholar]
- Guo J, Chi S, Xu H, Jin G, Qi Z. Effects of cholesterol levels on the excitability of rat hippocampal neurons. Mol Membr Biol. 2008;25:216–223. doi: 10.1080/09687680701805541. [DOI] [PubMed] [Google Scholar]
- Hagler DJ, Jr, Goda Y. Properties of synchronous and asynchronous release during pulse train depression in cultured hippocampal neurons. J Neurophysiol. 2001;85:2324–2334. doi: 10.1152/jn.2001.85.6.2324. [DOI] [PubMed] [Google Scholar]
- Hama H, Hara C, Yamaguchi K, Miyawaki A. PKC signaling mediates global enhancement of excitatory synaptogenesis in neurons triggered by local contact with astrocytes. Neuron. 2004;41:405–415. doi: 10.1016/S0896-6273(04)00007-8. [DOI] [PubMed] [Google Scholar]
- He Y, Zorumski CF, Mennerick S. Contribution of presynaptic Na(+) channel inactivation to paired-pulse synaptic depression in cultured hippocampal neurons. J Neurophysiol. 2002;87:925–936. doi: 10.1152/jn.00225.2001. [DOI] [PubMed] [Google Scholar]
- Hedstrom KL, Ogawa Y, Rasband MN. AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J Cell Biol. 2008;183:635–640. doi: 10.1083/jcb.200806112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefft S, Jonas P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci. 2005;8:1319–1328. doi: 10.1038/nn1542. [DOI] [PubMed] [Google Scholar]
- Hille B. Ion channels of excitable membranes. Ed 3. Sunderland, MA: Sinauer; 2001. [Google Scholar]
- Hu H, Jonas P. A supercritical density of Na(+) channels ensures fast signaling in GABAergic interneuron axons. Nat Neurosci. 2014;17:686–693. doi: 10.1038/nn.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol. 2001;155:739–746. doi: 10.1083/jcb.200109026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston D, Wu SM. Foundations of cellular neurophysiology. Cambridge, MA: MIT; 1995. [Google Scholar]
- Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol. 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurgel M, Koo AC, Ivy GO. Selective ablation of astrocytes by intracerebral injections of alpha-aminoadipate. Glia. 1996;16:351–358. doi: 10.1002/(SICI)1098-1136(199604)16:4<351::AID-GLIA7>3.0.CO%3B2-2. [DOI] [PubMed] [Google Scholar]
- Kole MH, Ilschner SU, Kampa BM, Williams SR, Ruben PC, Stuart GJ. Action potential generation requires a high sodium channel density in the axon initial segment. Nat Neurosci. 2008;11:178–186. doi: 10.1038/nn2040. [DOI] [PubMed] [Google Scholar]
- Kress GJ, Dowling MJ, Eisenman LN, Mennerick S. Axonal sodium channel distribution shapes the depolarized action potential threshold of dentate granule neurons. Hippocampus. 2010;20:558–571. doi: 10.1002/hipo.20667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, Chakraborty C, Workman G, Weaver M, Sage EH, Barres BA, Eroglu C. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci U S A. 2011;108:E440–E449. doi: 10.1073/pnas.1104977108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Dean C, Arthur CP, Dong M, Chapman ER. Autapses and networks of hippocampal neurons exhibit distinct synaptic transmission phenotypes in the absence of synaptotagmin I. J Neurosci. 2009;29:7395–7403. doi: 10.1523/JNEUROSCI.1341-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, Steinberg IZ, Walton K. Relationship between presynaptic calcium current and postsynaptic potential in squid giant synapse. Biophys J. 1981;33:323–351. doi: 10.1016/S0006-3495(81)84899-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz A, Nusser Z. Cell-type-dependent molecular composition of the axon initial segment. J Neurosci. 2008;28:14329–14340. doi: 10.1523/JNEUROSCI.4833-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz A, Nusser Z. Molecular identity of dendritic voltage-gated sodium channels. Science. 2010;328:906–909. doi: 10.1126/science.1187958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie PJ, Murphy TH. High safety factor for action potential conduction along axons but not dendrites of cultured hippocampal and cortical neurons. J Neurophysiol. 1998;80:2089–2101. doi: 10.1152/jn.1998.80.4.2089. [DOI] [PubMed] [Google Scholar]
- Mackenzie PJ, Umemiya MM, Murphy TH. Ca imaging of CNS axons in culture indicates reliable coupling Between single action potentials and distal functional release sites. Neuron. 1996;16:783–795. doi: 10.1016/S0896-6273(00)80098-7. [DOI] [PubMed] [Google Scholar]
- Mauch DH, Nägler K, Schumacher S, Göritz C, Müller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- Meeks JP, Mennerick S. Selective effects of potassium elevations on glutamate signaling and action potential conduction in hippocampus. J Neurosci. 2004;24:197–206. doi: 10.1523/JNEUROSCI.4845-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeks JP, Jiang X, Mennerick S. Action potential fidelity during normal and epileptiform activity in paired soma-axon recordings from rat hippocampus. J Physiol. 2005;566:425–441. doi: 10.1113/jphysiol.2005.089086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Zorumski CF. Paired-pulse modulation of fast excitatory synaptic currents in microcultures of rat hippocampal neurons. J Physiol. 1995;488:85–101. doi: 10.1113/jphysiol.1995.sp020948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron. 1995;15:805–819. doi: 10.1016/0896-6273(95)90172-8. [DOI] [PubMed] [Google Scholar]
- Moulder KL, Jiang X, Taylor AA, Shin W, Gillis KD, Mennerick S. Vesicle pool heterogeneity at hippocampal glutamate and GABA synapses. J Neurosci. 2007;27:9846–9854. doi: 10.1523/JNEUROSCI.2803-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Sweet sixteen for ANLS. J Cereb Blood Flow Metab. 2012;32:1152–1166. doi: 10.1038/jcbfm.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Navarrete M, Araque A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 2009;32:421–431. doi: 10.1016/j.tins.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Perea G, Yang A, Boyden ES, Sur M. Optogenetic astrocyte activation modulates response selectivity of visual cortex neurons in vivo. Nat Commun. 2014;5:3262. doi: 10.1038/ncomms4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polleux F, Snider W. Initiating and growing an axon. Cold Spring Harb Perspect Biol. 2010;2:a001925. doi: 10.1101/cshperspect.a001925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, Mennerick S. Selective Depression of Low–Release Probability Excitatory Synapses by Sodium Channel Blockers. Neuron. 2000;26:671–682. doi: 10.1016/S0896-6273(00)81203-9. [DOI] [PubMed] [Google Scholar]
- Rahamimoff R, Yaari Y. Delayed release of transmitter at the frog neuromuscular junction. J Physiol. 1973;228:241–257. doi: 10.1113/jphysiol.1973.sp010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raper J, Mason C. Cellular strategies of axonal pathfinding. Cold Spring Harb Perspect Biol. 2010;2:a001933. doi: 10.1101/cshperspect.a001933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricoult SG, Goldman JS, Stellwagen D, Juncker D, Kennedy TE. Generation of microisland cultures using microcontact printing to pattern protein substrates. J Neurosci Methods. 2012;208:10–17. doi: 10.1016/j.jneumeth.2012.04.016. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/S0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Timing of neurotransmission at fast synapses in the mammalian brain. Nature. 1996;384:170–172. doi: 10.1038/384170a0. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Matsuki N, Ikegaya Y. Action-potential modulation during axonal conduction. Science. 2011;331:599–601. doi: 10.1126/science.1197598. [DOI] [PubMed] [Google Scholar]
- Schmitz SK, Hjorth JJ, Joemai RM, Wijntjes R, Eijgenraam S, de Bruijn P, Georgiou C, de Jong AP, van Ooyen A, Verhage M, Cornelisse LN, Toonen RF, Veldkamp WJ, Veldkamp W. Automated analysis of neuronal morphology, synapse number and synaptic recruitment. J Neurosci Methods. 2011;195:185–193. doi: 10.1016/j.jneumeth.2010.12.011. [DOI] [PubMed] [Google Scholar]
- Shah MM, Migliore M, Valencia I, Cooper EC, Brown DA. Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 2008;105:7869–7874. doi: 10.1073/pnas.0802805105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Migliore M, Brown DA. Differential effects of Kv7 (M-) channels on synaptic integration in distinct subcellular compartments of rat hippocampal pyramidal neurons. J Physiol. 2011;589:6029–6038. doi: 10.1113/jphysiol.2011.220913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. Potassium currents in hippocampal pyramidal cells. Prog Brain Res. 1990;83:161–187. doi: 10.1016/S0079-6123(08)61248-0. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Graham SH. Fluorocitrate and fluoroacetate effects on astrocyte metabolism in vitro. Brain Res. 1994;664:94–100. doi: 10.1016/0006-8993(94)91958-5. [DOI] [PubMed] [Google Scholar]
- Vecchia D, Tottene A, van den Maagdenberg AM, Pietrobon D. Mechanism underlying unaltered cortical inhibitory synaptic transmission in contrast with enhanced excitatory transmission in CaV2.1 knockin migraine mice. Neurobiol Dis. 2014;69:225–234. doi: 10.1016/j.nbd.2014.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervaeke K, Gu N, Agdestein C, Hu H, Storm JF. Kv7/KCNQ/M-channels in rat glutamatergic hippocampal axons and their role in regulation of excitability and transmitter release. J Physiol. 2006;576:235–256. doi: 10.1113/jphysiol.2006.111336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Kunkel DD, Schwartzkroin PA, Tempel BL. Localization of Kv1.1 and Kv1.2, two K channel proteins, to synaptic terminals, somata, and dendrites in the mouse brain. J Neurosci. 1994;14:4588–4599. doi: 10.1523/JNEUROSCI.14-08-04588.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeagle PL. Cholesterol and the cell membrane. Biochim Biophys Acta. 1985;822:267–287. doi: 10.1016/0304-4157(85)90011-5. [DOI] [PubMed] [Google Scholar]
- Zamir O, Charlton MP. Cholesterol and synaptic transmitter release at crayfish neuromuscular junctions. J Physiol. 2006;571:83–99. doi: 10.1113/jphysiol.2005.098319. [DOI] [PMC free article] [PubMed] [Google Scholar]