

Abstract
Exacerbated mRNA translation during brain development has been linked to autism spectrum disorders (ASDs). Deletion of the eukaryotic initiation factor 4E (eIF4E)-binding protein 2 gene (Eif4ebp2), encoding the suppressor of mRNA translation initiation 4E-BP2, leads to an imbalance in excitatory-to-inhibitory neurotransmission and ASD-like behaviors. Inhibition of group I metabotropic glutamate receptors (mGluRs) mGluR1 and mGluR5 reverses the autistic phenotypes in several ASD mouse models. Importantly, these receptors control synaptic physiology via activation of mRNA translation. We investigated the potential reversal of autistic-like phenotypes in Eif4ebp2−/− mice by using antagonists of mGluR1 (JNJ16259685) or mGluR5 (fenobam). Augmented hippocampal mGluR-induced long-term depression (LTD; or chemically induced mGluR-LTD) in Eif4ebp2−/− mice was rescued by mGluR1 or mGluR5 antagonists. While rescue by mGluR5 inhibition occurs through the blockade of a protein synthesis-dependent component of LTD, normalization by mGluR1 antagonists requires the activation of protein synthesis. Synaptically induced LTD was deficient in Eif4ebp2−/− mice, and this deficit was not rescued by group I mGluR antagonists. Furthermore, a single dose of mGluR1 (0.3 mg/kg) or mGluR5 (3 mg/kg) antagonists in vivo reversed the deficits in social interaction and repetitive behaviors (marble burying) in Eif4ebp2−/− mice. Our results demonstrate that Eif4ebp2−/− mice serve as a relevant model to test potential therapies for ASD symptoms. In addition, we provide substantive evidence that the inhibition of mGluR1/mGluR5 is an effective treatment for physiological and behavioral alterations caused by exacerbated mRNA translation initiation.
SIGNIFICANCE STATEMENT Exacerbated mRNA translation during brain development is associated with several autism spectrum disorders (ASDs). We recently demonstrated that the deletion of a negative regulator of mRNA translation initiation, the eukaryotic initiation factor 4E-binding protein 2, leads to ASD-like behaviors and increased excitatory synaptic activity. Here we demonstrated that autistic behavioral and electrophysiological phenotypes can be treated in adult mice with antagonists of group I metabotropic glutamate receptors (mGluRs), which have been previously used in other ASD models (i.e., fragile X syndrome). These findings support the use of group I mGluR antagonists as a potential therapy that extends to autism models involving exacerbated mRNA translation initiation.
Keywords: autism spectrum disorders, group I mGluRs, long-term depression, repetitive behavior, social interaction, translation initiation
Introduction
Autism spectrum disorders (ASDs) are thought to be caused by an imbalance in the ratio of excitatory-to-inhibitory (E/I) neurotransmission due to impaired neurodevelopment (Kelleher and Bear, 2008). Several single-gene disorders, harboring mutations upstream of the mammalian target of rapamycin (mTOR) complex 1 (mTORC1), manifest high rates of autism and are characterized by increased cap-dependent mRNA translation of synaptic proteins (Kelleher and Bear, 2008). These disorders include tuberous sclerosis complex (Curatolo et al., 2010) and PTEN (phosphatase and tensin homolog) hamartoma tumor syndrome (Butler et al., 2005).
mTORC1 activates cap-dependent translation via phosphorylation and inactivation of the eukaryotic initiation factor 4E (eIF4E) binding proteins (4E-BPs; Gingras et al., 1999). eIF4E is the cap-binding component of the eIF4F complex, which also contains the RNA helicase, eIF4A, and the modular scaffolding protein eIF4G, which bridges the mRNA to the ribosome. 4E-BPs repress translation initiation by binding to eIF4E and preventing the eIF4E–eIF4G interaction, thereby inhibiting eIF4F complex formation. There are three 4E-BP isoforms, of which 4E-BP2 is the predominant form in the brain (Tsukiyama-Kohara et al., 2001; Banko et al., 2005).
Recently, we (Gkogkas et al., 2013) and Santini et al. (2013) demonstrated that excessive eIF4E activity promotes the development of ASD-like behaviors in mice. Genetic deletion of Eif4ebp2 (encoding 4E-BP2) or overexpression of Eif4e resulted in augmented translation of mRNAs encoding for neuroligins, engendering an imbalance in the E/I ratio, impaired social interaction, repetitive behaviors, and vocalization defects (Gkogkas et al., 2013; Santini et al., 2013).
Group I metabotropic glutamate receptors (mGluRs), comprising mGluR1 and mGluR5, have been explored as a therapeutic target for fragile X syndrome (FXS; Bear et al., 2004) and other disorders with autistic features (Tian et al., 2015). FXS is a genetic disorder with a high prevalence of autism caused by the loss of the negative regulator of mRNA translation FMR1 (Weiler et al., 1997). In Fmr1-null mice, mGluR1/mGluR5 activation induces exacerbated hippocampal long-term depression (mGluR-LTD; Huber et al., 2002; Hou et al., 2006; Nosyreva and Huber, 2006). mGluR-LTD is dependent on de novo protein synthesis (Weiler and Greenough, 1993; Huber et al., 2000), and involves the activation of mTORC1 and cap-dependent translation (Hou and Klann, 2004; Banko et al., 2006; Antion et al., 2008). Importantly, mGluR-dependent LTD is also exacerbated in Eif4ebp2-null mice (Banko et al., 2006); thus, inhibition of group I mGluRs could benefit ASD caused by dysregulated cap-dependent translation initiation. We sought to determine whether mice lacking 4E-BP2 could serve as a useful model to investigate therapies aimed to improve decreased social interaction and repetitive behaviors. Consequently, we determined whether mGluR1 or mGluR5 antagonists could rescue the synaptic and behavioral deficits in Eif4ebp2 knock-out mice.
Materials and Methods
Eif4ebp2 knock-out mice were previously described (Banko et al., 2005). Male littermates from heterozygote crossings were housed in temperature-controlled (21°C) and humidity-controlled (∼55%) rooms, with ad libitum food and water, and a 12 h light/dark cycle. All procedures were conducted in compliance with the Canadian Council on Animal Care guidelines and were approved by the McGill University and Université de Montréal Animal Care Committees.
Electrophysiology.
Transverse hippocampal slices (400 μm thickness), prepared from 30- to 35-d-old male mice, were allowed to recover for at least 2 h at 32°C submerged in oxygenated artificial CSF (ACSF; 124 mm NaCl, 5 mm KCl, 1.25 mm NaH2PO4, 2 mm MgSO4, 2 mm CaCl2, 26 mm NaHCO3, and 10 mm glucose) and were perfused with ACSF for an additional 30 min in a recording chamber at 27–28°C. Field EPSPs (fEPSPs) were recorded in CA1 stratum radiatum with glass electrodes (2–3 MΩ) filled with ASCF. Schaffer collateral fEPSPs were evoked with a concentric bipolar tungsten stimulating electrode placed in mid-stratum radiatum proximal to the CA3 region. Baseline stimulation was applied at 0.033 Hz by delivering 0.1 ms pulses, with intensity adjusted to evoke fEPSPs with 60% of maximal amplitude. For chemically induced mGluR-LTD, CA1 and CA3 hippocampal regions were isolated by a surgical cut before recovery, and group I mGluR agonist (S)-3,5-dihydroxyphenylglycine (DHPG; 50 μm; Tocris Bioscience) was added to ACSF for 10 min for induction. For synaptically induced LTD, paired-pulse low-frequency stimulation (PP-LFS) was performed as reported (Huber et al., 2000; Volk et al., 2006). PP-LFS stimulation (50 ms interstimulus interval) was administered at 1 Hz for 20 min in the presence of the NMDAR antagonist aminophosphonovalerate (50 μm). fEPSP slope measurements were performed on digitized analog recordings using the Clampfit analyze function. The slope was measured at between 10% and 90% of maximal fEPSP amplitude during an epoch defined by constant cursor placements, which excluded fiber volley and population spikes. Where indicated, we used anisomycin (40 μm; Tocris Bioscience) 20 min prior to, during, and for 20 min after LTD induction; and fenobam, an mGluR5 antagonist (0.9 μm; Tocris Bioscience), or JNJ16259685, an mGluR1 antagonist (6 nm; Tocris Bioscience) 10 min prior to, during, and for 5 min after LTD induction.
In vivo experiments.
To assess the concentration of fenobam or JNJ16259685 that had no effect on the social behavior of wild-type mice, 7-week-old wild-type C57BL/6J mice were treated with vehicle [30% DMSO in 0.9% NaCl; 8 ml/kg body weight (BW), i.p.], JNJ16259685 (0.3 or 1 mg/kg BW), or fenobam (3 or 10 mg/kg BW) and tested 30 min later. After determining the concentrations that spared social behavior in wild-type mice, a different cohort of wild-type and Eif4ebp2−/− mice was treated with the chosen doses of each antagonist (0.3 mg/kg for JNJ16259685; 3 mg/kg for fenobam) or vehicle, and 30 min or 24 h after, brains were collected, hippocampi were isolated, flash frozen, and used later for Western blotting.
In separate experiments, Eif4ebp2−/− mice and wild-type littermates were treated with vehicle and tested for social interaction 30 min after injection. One week after, mice were treated with either JNJ16259685 or fenobam and tested again for social interaction 30 min after injection. Twenty-four hours after treatment, mice were tested again without further injections. The social behavior of wild-type mice treated with JNJ16259285 or fenobam was indistinguishable, and data from both groups were pooled. For the marble-burying test, a different cohort of mice was treated with vehicle, JNJ16259685, and fenobam, and examined in the marble-burying test 30 min after injection. In all cases, the quantification of behavior was performed on recorded video by an experimenter who was blind to genotype and treatment.
Social preference test.
A three-chamber arena was used to assess social approach, as previously reported (Gkogkas et al., 2013), except that habituation and social preference sessions lasted 10 min.
Marble-burying task.
Mice were individually placed in Plexiglas cages containing 5-cm-deep fresh bedding with 20 black marbles prearranged in 5 × 4 evenly spaced rows for 20 min (Gkogkas et al., 2013). Marbles were considered to be buried if at least one-half was covered with bedding.
Western blotting and antibodies.
Tissues were dissociated in RIPA buffer. Western blotting was performed as previously described (Bidinosti et al., 2010). The antibodies used against indicated proteins were as follows: phospho-S6 ribosomal protein (pS6; Ser235/236, 1:1000; Cell Signaling Technology); total S6 ribosomal protein (tS6; 1:1000; Cell Signaling Technology); and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), coupled to horseradish peroxidase (1:5000; Abcam). The secondary antibodies used were anti-mouse and anti-rabbit (GE Healthcare). Quantification of immunoblots was performed using ImageJ (National Institutes of Health). Values were normalized to GAPDH and expressed as a ratio of pS6/tS6/GAPDH.
Statistical analysis.
All data are presented as the mean ± SEM. Electrophysiology, Western blotting, and marble-burying test data were analyzed using a two-way ANOVA (using genotype and treatment as between-subjects factors), except for mGluR-LTD data from Eif4ebp2−/− treated with both anisomycin and JNJ16259285 or fenobam, which were compared only to Eif4ebp2−/− treated with JNJ16259285 or fenobam using a two-tailed Student's t test. Social preference data in wild-type mice (see Fig. 3) were analyzed using repeated-measures ANOVA (using time in chambers as the within-subject factor) independently for each treatment. Social preference data (see Fig. 4) were analyzed using two-way ANOVA, with treatment used as the between-subjects factor and with chamber used as the within-subject factor; each time point (baseline, 0.5, and 24 h after treatment) were analyzed independently as no comparisons between these were intended. When the main factor or interaction of an ANOVA was significant, analysis was followed by Bonferroni-corrected pairwise comparisons. p values <0.05 were deemed significant.
Figure 3.
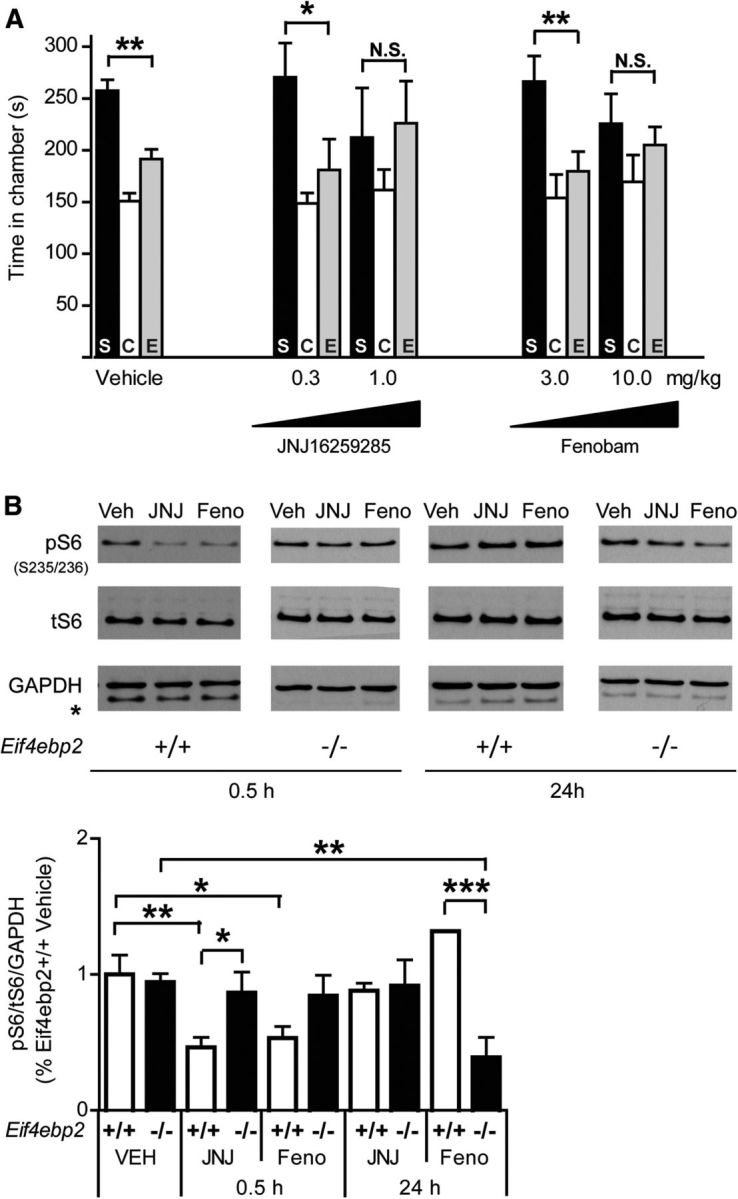
Effects of mGluR1 and mGluR5 antagonists on social interaction and mTORC1 signaling in wild-type mice. A, JNJ16259285 and fenobam impaired social preference at the higher doses (1 mg/kg for JNJ16259285 and 10 mg/kg for fenobam), but not at lower doses (0.3 mg/kg for JNJ16259285 and 3 mg/kg for fenobam; vehicle-treated mice, n = 20; for all other groups, n = 7. C, Center). B, At doses that did not affect the social preference, JNJ16259285 and fenobam were effective in reducing S6 ribosomal protein phosphorylation in the hippocampus of wild-type mice but not in Eif4ebp2-null mice. Twenty-four hours after injection, the short-term effect of JNJ16259285 and fenobam disappeared in wild-type mice, but fenobam had a delayed effect in reducing pS6 in 4E-BP2 knock-out mice; representative Western blots are presented in the top (asterisk indicates a residual signal of the anti-tS6 antibody when probing for GAPDH). Eif4ebp2+/+ plus vehicle, n = 10; Eif4ebp2+/+ plus JNJ16259285 at 0.5 h, n = 12; Eif4ebp2+/+ plus fenobam at 0.5 h, n = 11; Eif4ebp2+/+ plus JNJ16259285 at 24 h, n = 3; Eif4ebp2+/+ plus fenobam at 24 h, n = 3; Eif4ebp2−/− plus vehicle, n = 10; Eif4ebp2−/− plus JNJ16259285 at 0.5 h, n = 5; Eif4ebp2−/− plus fenobam at 0.5 h, n = 5; Eif4ebp2−/− plus JNJ16259285 at 24 h, n = 5; and Eif4ebp2−/− plus fenobam at 24 h, n = 8 mice per group. N.S., Not significant. *p < 0.05, **p < 0.01, and ***p < 0.001.
Figure 4.

Rescue of social interaction deficits and repetitive behavior by mGluR1 and mGluR5 antagonists in Eif4ebp2−/− mice. A, Wild-type or Eif4ebp2−/− mice were tested after treatment with vehicle, JNJ16259285, or fenobam (0.5 and 24 h after). JNJ16259285 had a short-term effect, rescuing the social approach (time spent in chamber with a stranger mouse) in Eif4ebp2−/− mice at 0.5 h after treatment, but not 24 h later. Fenobam also rescued the social preference in Eif4ebp2−/− mutant mice, but 24 h after treatment. B, The effect on social preference was corroborated by the time spent by the mice sniffing the enclosure with the stranger mouse. JNJ16259285 and fenobam did not affect social preference in wild-type mice. Eif4ebp2+/+, n = 21, of which 10 were treated with JNJ16259285 and 10 with fenobam; Eif4ebp2−/−, n = 22, of which 11 were treated with JNJ16259285 and 11 with fenobam. C, In the marble-burying task, the elevated number of marbles buried by the Eif4ebp2−/− mice was reduced to control levels by JNJ16259285 and fenobam treatment. The antagonists did not affect marble burying in wild-type mice. Eif4ebp2+/+ plus vehicle, n = 12; Eif4ebp2+/+ plus JNJ16259285, n = 7; Eif4ebp2+/+ plus fenobam, n = 10; Eif4ebp2−/− plus vehicle, n = 16; Eif4ebp2−/− plus JNJ16259285, n = 9; and Eif4ebp2−/− plus fenobam, n = 13. N.S., Not significant. *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
Exacerbated group I mGluR-dependent LTD is rescued by inhibition of mGluR1/mGluR5
We first tested the effect of mGluR1 or mGluR5 antagonists on exacerbated mGluR-LTD in Eif4ebp2−/− mice. As previously reported (Banko et al., 2006), DHPG-induced LTD in the CA1 region of the hippocampus was significantly exacerbated in Eif4ebp2−/− mice (Eif4ebp2+/+ vs Eif4ebp2−/− vehicle; F(1,119) = 10.01, p = 0.002; Fig. 1A,E). In wild-type mice, hippocampal mGluR-LTD induction is dependent on the activation of both mGluR1 and mGluR5, and pharmacological inhibition of only one of these receptors during induction is not sufficient to block mGluR-LTD (Hou and Klann, 2004; Volk et al., 2006). Accordingly, incubation with either JNJ16259685 (mGluR1) or fenobam (mGluR5) in Eif4ebp2+/+ mice did not affect mGluR-LTD (F(3,119) = 5.31, p = 0.002, vehicle vs JNJ16259685 or fenobam, p = 1.0; Fig. 1B,C,E). In contrast, in Eif4ebp2−/− mice, JNJ16259685 and fenobam reduced mGluR-LTD (F(3,119) = 9.46, p = 0.0009; vehicle vs JNJ16259685, p = 0.00109; vehicle vs fenobam, p = 0.001; Fig. 1B,C,E) to control levels for JNJ16259685 or to a greater degree than control levels for fenobam. These results demonstrate a greater sensitivity of hippocampal mGluR-LTD in Eif4ebp2−/− mice to the inhibition of either mGluR1 or mGluR5, which results in the rescue of synaptic plasticity deficits caused by increased cap-dependent translation initiation.
Figure 1.

Enhanced LTD in Eif4ebp2−/− mice is normalized by mGluR1 and mGluR5 antagonists. A, DHPG induced enhanced LTD in Eif4ebp2−/− mice compared with Eif4ebp2+/+ mice. B, The mGluR1 antagonist JNJ16259285 abrogated the difference in LTD between Eif4ebp2+/+ and Eif4ebp2−/− without affecting wild-type mice. This effect was prevented when anisomycin was coadministered. C, Fenobam, an mGluR5 antagonist, also normalized mGluR-LTD in Eif4ebp2−/− mice without affecting Eif4ebp2+/+ mice. Anisomycin cotreatment did not further affect the normalized mGluR-LTD. D, In the presence of anisomycin, mGluR-LTD was completely blocked in wild-type mice, but only normalized in 4E-BP2 mutant mice. E, fEPSP slope at 50–60 min after DHPG application for Eif4ebp2+/+ (n = 23), Eif4ebp2+/+ plus JNJ16259285 (n = 12), Eif4ebp2+/+ plus fenobam (n = 11), Eif4ebp2+/+ plus anisomycin (n = 12), Eif4ebp2−/− (n = 33), Eif4ebp2−/− plus JNJ16259285 (n = 15), Eif4ebp2−/− plus fenobam (n = 11), Eif4ebp2−/− plus anisomycin (n = 12), Eif4ebp2−/− plus JNJ16259285 and anisomycin (n = 10), and Eif4ebp2−/− plus fenobam and anisomycin (n = 8). Insets in A–D are representative traces. Calibration: 50 ms and 0.5 mV. **p < 0.01 vs Eif4ebp2+/+ plus vehicle; $p < 0.05 vs Eif4ebp2+/+ plus fenobam; #p < 0.05, ##p < 0.01, and ###p < 0.001 vs Eif4ebp2−/− plus vehicle; &&&p < 0.001 vs Eif4ebp2−/− plus JNJ16259285; N.S. vs Eif4ebp2−/− plus fenobam.
To determine the relationship between the rescued mGluR-LTD in Eif4ebp2−/− mice and protein synthesis, we treated hippocampal slices with the protein synthesis inhibitor anisomycin, alone or in combination with either JNJ16259685 or fenobam (Fig. 1B–E). As expected, anisomycin completely inhibited hippocampal mGluR-LTD in wild-type mice (Eif4ebp2+/+ vehicle vs anisomycin, p = 0.002; Fig. 1D,E), while restoring the mGluR-LTD in Eif4ebp2−/− mice to control levels (Eif4ebp2−/− vehicle vs anisomycin; p = 0.014; Fig. 1D,E). This effect indicates that in Eif4ebp2-null mice LTD is still partially dependent on de novo protein synthesis and is similar to the normalization of CA1 LTP by anisomycin or cycloheximide in Eif4ebp2−/− mice (Banko et al., 2005). Cotreatment of hippocampal slices of Eif4ebp2-null mice with anisomycin and JNJ16259685 abrogated the ability of each drug to rescue mGluR-LTD when applied individually, resulting in mGluR-LTD levels that were significantly lower than those in the group of JNJ16259685-treated Eif4ebp2−/− mice (Fig. 1B,E; Eif4ebp2−/− plus JNJ16259685 vs Eif4ebp2−/− plus JNJ16259685 and anisomycin; t(22) = 5.75, p = 0.0009) and were comparable to those of DHPG-treated Eif4ebp2−/− mice. Thus, the rescue of exaggerated LTD by mGluR1 antagonism requires protein synthesis. In contrast, the rescued mGluR-LTD by fenobam was insensitive to anisomycin treatment (Fig. 1C,E; Eif4ebp2−/− plus fenobam vs Eif4ebp2−/− plus fenobam and anisomycin; t(17) = 0.06, p = 0.95). Given that both anisomycin and fenobam reduce the exaggerated mGluR-LTD in Eif4ebp2−/− mice when applied individually, the lack of an additive effect when coadministered suggests an occlusion of the rescue effect, and that fenobam acts by reducing the protein synthesis-sensitive component of the exaggerated mGluR-LTD in Eif4ebp2−/− mice.
Synaptically elicited LTD is deficient in Eif4ebpb2−/− and not rescued by group I mGluR antagonists
Protein synthesis-dependent LTD can be induced in CA1 by using PP-LFS of Schaffer collaterals (Huber et al., 2000). This form of plasticity requires the activation of muscarinic acetylcholine receptors, in addition to group I mGluRs (Volk et al., 2007). We determined whether the induction of PP-LFS LTD was altered in Eif4ebp2−/− mice and found that it was impaired (effect of genotype: F(1,48) = 14.42, p = 0.0009; Fig. 2A,D), suggesting a differential role of 4E-BP2 in chemically and synaptically induced LTD in the hippocampus.
Figure 2.

Synaptically induced LTD is impaired in Eif4ebp2−/− mice and unresponsive to group I mGluR antagonists. A, PP-LFS induced a stable LTD in wild-type mice, while it was completely absent in Eif4ebp2−/− mice. B, PP-LFS LTD was not affected by incubation with JNJ16259285 in wild-type mice. Similarly, the impairment in this form of LTD in Eif4ebp2−/− mice was not changed by the mGluR1 antagonist. C, The mGluR5 antagonist fenobam did not affect the response to PP-LFS in either wild-type or Eif4ebp2−/− mice. D, fEPSP slope at 70–80 min after PP-LFS onset for Eif4ebp2+/+ (n = 12), Eif4ebp2+/+ plus JNJ16259285 (n = 7), Eif4ebp2+/+ plus fenobam (n = 7), Eif4ebp2−/− (n = 10), Eif4ebp2−/− plus JNJ16259285 (n = 8), and Eif4ebp2−/− plus fenobam (n = 12). Insets in A–C are representative traces. Calibration: 50 ms and 0.5 mV. ***p < 0.001.
We then examined whether mGluR1 or mGluR5 antagonists could rescue the impaired LTD in Eif4ebp2−/− mice. JNJ16259685 or fenobam, at the doses that rescued chemically induced mGluR-LTD, failed to normalize the deficit in LTD (effect of treatment: F(2,48) = 0.68, p = 0.51; Fig. 2B,C) and did not affect PP-LFS LTD in controls (Fig. 2B–D). These results are not surprising, since the role of group I mGluRs in LTD induced by PP-LFS is complex, seemingly secondary to muscarinic receptor activation in the hippocampus (Volk et al., 2006, 2007).
JNJ16259685 and fenobam rescue social interaction and repetitive behaviors
To investigate the effect of JNJ16259685 and fenobam on social interaction and repetitive behaviors in Eif4ebp2−/− mice, we first established the maximal tolerable dose of each antagonist that does not impair social behavior in wild-type mice. JNJ16259285 and fenobam had no effect at the lower dose [Stranger (S) vs Empty (E) chamber: 0.3 mg/kg JNJ16259285, p = 0.03; 3 mg/kg fenobam, p = 0.007; Fig. 3A] but impaired social preference at the higher dose (S vs E: p = 1.0 for 1 and 10 mg/kg, respectively; Fig. 3A). We verified that the antagonists reduced signaling downstream of group I mGluRs at the lowest doses used, using pS6 (at the Ser235/236) as the readout for mTORC1 and MAPK/ERK activation (Thomas, 2002). Both antagonists reduced pS6 in the hippocampus of wild-type mice 30 min after their administration (F(4,60) = 7.46, p = 0.0009; vehicle vs JNJ16259285 at 0.5 h, p = 0.003; vehicle vs fenobam at 0.5 h, p = 0.012; Fig. 3B). This reduction was transient, as levels returned to baseline 24 h after the injection of either antagonist (vehicle vs JNJ16259285 or fenobam at 24 h, p = 1.0; Fig. 3B). S6 phosphorylation occurs in parallel with that of the 4E-BPs, downstream of mTOR (Gingras et al., 1999); thus, it may not necessarily correlate with increased translation in Eif4ebp2−/− mice. Indeed, in Eif4ebp2−/− mice pS6 levels were comparable to those of wild-type mice (F(1,60) = 0.15, p = 0.70; Fig. 3B), but JNJ16259285 and fenobam failed to significantly reduce the levels of pS6, 30 min after injection (F(4,60) = 3.91, p = 0.007; vehicle vs JNJ16259285 or fenobam at 0.5 h, p = 1.0; Fig. 3B). One potential reason for this discrepancy is that increased levels of network activity in baseline conditions in Eif4ebp2-null mice (Bidinosti et al., 2010; Gkogkas et al., 2013) could mask the contribution of mGluR1 and R5 on S6 phosphorylation in vivo. Intriguingly, despite the lack of a short-term effect, fenobam, but not JNJ16259285, had a delayed influence in mice lacking 4E-BP2, resulting in reduced S6 phosphorylation levels 24 h after injection (Eif4ebp2−/− vehicle vs Eif4ebp2−/− fenobam at 24 h, p = 0.006). These results suggest that, although phosphorylation of S6 may reflect group I mGluR activity in wild-type mice, this may not necessarily be the case in Eif4ebp2-null mice.
Next, we examined whether the group I mGluR antagonists could improve social interaction in Eif4ebp2−/− mice. As previously reported (Gkogkas et al., 2013), Eif4ebp2−/− mice display impaired social preference, measured either as time spent in the chamber with the stranger mouse (baseline: F(2,44) = 5.42, p = 0.008; Eif4ebp2+/+ S vs E chamber, p = 0.0009; Eif4ebp2+/+ S vs Eif4ebp2−/− S chamber, p = 0.019; Eif4ebp2−/− S vs E chamber, p = 1.0; Fig. 4A) or as the time spent sniffing the enclosure of the stranger mouse (baseline: F(1,37) = 7.05, p = 0.012; Eif4ebp2+/+ S vs E chamber, p = 0.0009; Eif4ebp2+/+ S vs Eif4ebp2−/− S chamber, p = 0.036; Eif4ebp2−/− S vs E chamber, p = 0.25; Fig. 4B). We then administered JNJ16259285 or fenobam to Eif4ebp2−/− or wild-type littermates. As reported above (Fig. 3A), neither of these drugs had a significant effect on the social preference exhibited by Eif4ebp2+/+ mice [0.5 h: F(2,33) = 20.76, p = 0.0009; S vs E chamber, p = 0.045 (Fig. 4A); F(1,34) = 36.46, p = 0.0009; S vs E chamber, p = 0.0009 (Fig. 4B)]. Importantly, JNJ16259285 rescued the deficit in social approach of Eif4ebp2−/− mice [0.5 h: S vs E chamber, p = 0.011 (Fig. 4A); S vs E cage, p = 0.0009 (Fig. 4B)], albeit transiently, because the rescue was not observed 24 h after JNJ16259285 application [24 h: F(2,33) = 37.48, p = 0.0009; S vs E chamber, p = 1.0 (Fig. 4A); F(1,37) = 20.02, p = 0.0009; S vs E chamber, p = 0.09 (Fig. 4B)]. Fenobam did not affect the impaired social interaction in Ei4ebp2−/− mice 0.5 h after injection (S vs E chamber: p = 1.0, Fig. 4A; p = 0.2, Fig. 4B). However, Ei4ebp2−/− mice exhibited a significant improvement in social approach 24 h after fenobam treatment (S vs E chamber, p = 0.023; Fig. 4A,B). The latter result parallels our finding that fenobam caused a significant reduction in pS6 levels 24 h after fenobam administration (but not at 0.5 h; Fig. 3B). Thus, despite differences in the time course, inhibition of either mGluR1 or mGluR5 rescues the impaired social interaction caused deficient 4E-BP2.
We also investigated whether JNJ16259285 and fenobam could reverse the augmented marble-burying behavior in Eif4ebp2−/− mice. In wild-type mice, JNJ16259285 and fenobam did not affect the number of marbles buried (genotype × drug interaction: F(2,63), p = 0.012; Eif4ebp2+/+ vehicle vs fenobam, p = 1.0; or vehicle vs JNJ16259285, p = 0.85; Fig. 4C). In the Eif4ebp2−/− mice, the increased number of marbles buried (Eif4ebp+/+ vehicle vs Eif4ebp2−/− vehicle, p = 0.0009) was reversed to control levels by treatment with JNJ16259285 or fenobam (Eif4ebp2−/− JNJ16259285 or Eif4ebp2−/− fenobam vs Eif4ebp2−/− vehicle, p = 0.0009). Thus, the inhibition of either mGluR1 or mGluR5 rescues the enhanced repetitive behavior phenotype caused by enhanced cap-dependent mRNA translation.
Discussion
Here, we demonstrated that in Eif4ebp2−/− mice, the inhibition of mGluR1 or mGluR5 rescued exaggerated mGluR-LTD (Fig. 1), as well as impaired social interaction and repetitive marble burying (Fig. 4), while leaving unaltered the impaired PP-LFS LTD (Fig. 2). Importantly, the doses required to rescue synaptic physiology and behavior in Eif4ebp2-null mice had no effect in wild-type mice (Figs. 1, 2, 3, 4). These results establish the Eif4ebp2-null mouse as a tool for the testing of mGluR1/mGluR5 antagonists as candidate drugs to treat ASD and cognitive impairment.
Genetic or pharmacological mGluR5 inhibition rescues autistic and cognitive phenotypes in mouse and Drosophila FXS models (Aschrafi et al., 2005; Chuang et al., 2005; McBride et al., 2005; Yan et al., 2005; Dölen et al., 2007; Osterweil et al., 2010; Levenga et al., 2011). mGluR5 inhibition is also effective in other ASD models, including BTBR mice (Silverman et al., 2010, 2012), which model a microdeletion on the human chromosome 16p11.2 (Tian et al., 2015), and in ASD-like alterations caused by prenatal exposure to the anticonvulsant drug valproic acid (Mehta et al., 2011). These successful preclinical studies led to the implementation of clinical trials with mGluR5 antagonists in FXS. An early open-label clinical trial with fenobam showed an improvement in the general anxiety level and in prepulse inhibition, without safety concerns in FXS (Berry-Kravis et al., 2009), but this was not followed up by further trials (Scharf et al., 2015). Other mGluR5 antagonists (AFQ056, Novartis; STX107, Seaside Therapeutics; RO4917523, Roche) also had initial successes, yet more recently failed to show significant symptomatic improvement compared with placebo in patients (Healy et al., 2011; Jacquemont et al., 2011; Gross et al., 2012; Scharf et al., 2015). The compound AFQ056 appeared to improve the behavioral outcome in FXS patients with fully methylated alleles (and no detectable FMR1 mRNA), but not in those with partial methylation (Jacquemont et al., 2011), suggesting that the underlying genetic variability may be important for the responsiveness to treatment.
Our current study shows that the therapeutic effect of mGluR5 inhibition with fenobam is also evident in a model where cap-dependent translation initiation is augmented. Additionally, we demonstrated that mGluR1 is also a promising drug target. In this regard, both fenobam and JNJ16259285 were equally potent in rescuing exacerbated chemically induced mGluR-LTD, and increased marble burying, although they differed in the time course of their beneficial effect on impaired social interaction and on phosphorylation levels of hippocampal S6. The reason for the delayed effect of fenobam is unclear, as antagonists of mGluR5 (MPEP or GRN-529) have a short-term effect on social interaction in other ASD models (Silverman et al., 2010, 2012; Mehta et al., 2011).
Eif4ebp2−/− mice are more sensitive, at the electrophysiological and behavioral level, than their wild-type littermates to the effects of mGluR1/mGluR5 antagonists. The mechanism for this increased sensitivity is not straightforward, but may involve MAPK/ERK signaling (Banko et al., 2006). Given that altered hippocampal plasticity in Eif4ebp2−/− mice still depends, at least partly, on translation (Fig. 1D; Banko et al., 2005, 2006), the ERK-MNK1/2-eIF4E pathway may be important in the therapeutic effect of mGluR1/mGluR5 antagonists. In addition, our results showed that mGluR5 inhibition exerted its rescue in Eif4ebp2−/− mice via the regulation of protein synthesis, downstream of the initiation step. Accordingly, group I mGluRs can regulate elongation (Park et al., 2008) and reactivation of stalled ribosomes (Graber et al., 2013). In contrast, the rescue of mGluR-LTD by mGluR1 antagonist JNJ16259285 requires protein synthesis activation. One potential explanation for the differential requirement of protein synthesis by mGluR1 and mGluR5 is that each receptor may involve different cellular substrates. For example, mGluR1 is enriched in CA1 oriens-alveus interneurons (Baude et al., 1993; Ran et al., 2009). Therefore, inhibition by JNJ16259285 may preferentially affect this subpopulation of interneurons and indirectly impact pyramidal cell function. In contrast, mGluR5 appears to be localized on the neurites of pyramidal neurons in the hippocampus (López-Bendito et al., 2002) and therefore directly impact their function.
Synaptically induced LTD was completely absent in Eif4ebp2−/− mice, indicating that the control of cap-dependent mRNA translation through 4E-BP2 is necessary for its induction. In this regard, the absence of 4E-BP2 may result in increased levels of an endogenous inhibitor of synaptically induced LTD, or an occlusion of induction due to the saturation of endogenous mechanisms. Our findings indicate a critical, yet distinct, involvement of 4E-BP2 regulation of cap-dependent mRNA translation in synaptically and chemically induced LTD.
Our results clearly indicate that enhanced signaling through group I mGluR plays a role in the pathophysiology induced by the absence of 4E-BP2, a key repressor of cap-dependent translation initiation in the brain. Consequently, antagonists of mGluR1 or mGluR5 may be candidates for drugs to correct deficits in synaptic plasticity and behavior relevant for ASD due to dysregulated eIF4E-dependent translation.
Footnotes
This work was supported by Canadian Institutes of Health Research (CIHR) operating Grants MOP-114994 (N.S.), MOP-125985 (J.-C.L.), and MOP-136864 (N.S. and J.-C.L.); by a Fonds de la Recherche du Québec-Santé (FRQS) grant to J.-C.L. (Groupe de Recherche sur le Système Nerveux Central). A.A-V. was a recipient of an FRQS fellowship and, currently, a CIHR fellowship. J.-C.L. is the recipient of the Canada Research Chair in Cellular and Molecular Neurophysiology. N.S. is a senior international research scholar of the Howard Hughes Medical Institute. We thank I. Riebe, G. Ling, H. Tsui, A. Sylvestre, S. Perreault, I. Harvey, K. Gamache, and I. Laplante for their technical assistance; and C. Rouya for critical reading of the manuscript.
The authors declare no competing financial interests.
References
- Antion MD, Hou L, Wong H, Hoeffer CA, Klann E. mGluR-dependent long-term depression is associated with increased phosphorylation of S6 and synthesis of elongation factor 1A but remains expressed in S6K-deficient mice. Mol Cell Biol. 2008;28:2996–3007. doi: 10.1128/MCB.00201-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschrafi A, Cunningham BA, Edelman GM, Vanderklish PW. The fragile X mental retardation protein and group I metabotropic glutamate receptors regulate levels of mRNA granules in brain. Proc Natl Acad Sci U S A. 2005;102:2180–2185. doi: 10.1073/pnas.0409803102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banko JL, Poulin F, Hou L, DeMaria CT, Sonenberg N, Klann E. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J Neurosci. 2005;25:9581–9590. doi: 10.1523/JNEUROSCI.2423-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banko JL, Hou L, Poulin F, Sonenberg N, Klann E. Regulation of eukaryotic initiation factor 4E by converging signaling pathways during metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2006;26:2167–2173. doi: 10.1523/JNEUROSCI.5196-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baude A, Nusser Z, Roberts JD, Mulvihill E, McIlhinney RA, Somogyi P. The metabotropic glutamate receptor (mGluR1 alpha) is concentrated at perisynaptic membrane of neuronal subpopulations as detected by immunogold reaction. Neuron. 1993;11:771–787. doi: 10.1016/0896-6273(93)90086-7. [DOI] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Hessl D, Coffey S, Hervey C, Schneider A, Yuhas J, Hutchison J, Snape M, Tranfaglia M, Nguyen DV, Hagerman R. A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J Med Genet. 2009;46:266–271. doi: 10.1136/jmg.2008.063701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidinosti M, Ran I, Sanchez-Carbente MR, Martineau Y, Gingras AC, Gkogkas C, Raught B, Bramham CR, Sossin WS, Costa-Mattioli M, DesGroseillers L, Lacaille JC, Sonenberg N. Postnatal deamidation of 4E-BP2 in brain enhances its association with raptor and alters kinetics of excitatory synaptic transmission. Mol Cell. 2010;37:797–808. doi: 10.1016/j.molcel.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, Eng C. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42:318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang SC, Zhao W, Bauchwitz R, Yan Q, Bianchi R, Wong RK. Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J Neurosci. 2005;25:8048–8055. doi: 10.1523/JNEUROSCI.1777-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curatolo P, Napolioni V, Moavero R. Autism spectrum disorders in tuberous sclerosis: pathogenetic pathways and implications for treatment. J Child Neurol. 2010;25:873–880. doi: 10.1177/0883073810361789. [DOI] [PubMed] [Google Scholar]
- Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem. 1999;68:913–963. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, Vasuta C, Yee S, Truitt M, Dallaire P, Major F, Lasko P, Ruggero D, Nader K, Lacaille JC, Sonenberg N. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature. 2013;493:371–377. doi: 10.1038/nature11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber TE, Hébert-Seropian S, Khoutorsky A, David A, Yewdell JW, Lacaille JC, Sossin WS. Reactivation of stalled polyribosomes in synaptic plasticity. Proc Natl Acad Sci U S A. 2013;110:16205–16210. doi: 10.1073/pnas.1307747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Berry-Kravis EM, Bassell GJ. Therapeutic strategies in fragile X syndrome: dysregulated mGluR signaling and beyond. Neuropsychopharmacology. 2012;37:178–195. doi: 10.1038/npp.2011.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy A, Rush R, Ocain T. Fragile X syndrome: an update on developing treatment modalities. ACS Chem Neurosci. 2011;2:402–410. doi: 10.1021/cn200019z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2004;24:6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–454. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S, Curie A, des Portes V, Torrioli MG, Berry-Kravis E, Hagerman RJ, Ramos FJ, Cornish K, He Y, Paulding C, Neri G, Chen F, Hadjikhani N, Martinet D, Meyer J, Beckmann JS, Delange K, Brun A, Bussy G, Gasparini F, et al. Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci Transl Med. 2011;3:64ra1. doi: 10.1126/scitranslmed.3001708. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135:401–406. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- Levenga J, Hayashi S, de Vrij FM, Koekkoek SK, van der Linde HC, Nieuwenhuizen I, Song C, Buijsen RA, Pop AS, Gomezmancilla B, Nelson DL, Willemsen R, Gasparini F, Oostra BA. AFQ056, a new mGluR5 antagonist for treatment of fragile X syndrome. Neurobiol Dis. 2011;42:311–317. doi: 10.1016/j.nbd.2011.01.022. [DOI] [PubMed] [Google Scholar]
- López-Bendito G, Shigemoto R, Fairén A, Luján R. Differential distribution of group I metabotropic glutamate receptors during rat cortical development. Cereb Cortex. 2002;12:625–638. doi: 10.1093/cercor/12.6.625. [DOI] [PubMed] [Google Scholar]
- McBride SM, Choi CH, Wang Y, Liebelt D, Braunstein E, Ferreiro D, Sehgal A, Siwicki KK, Dockendorff TC, Nguyen HT, McDonald TV, Jongens TA. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron. 2005;45:753–764. doi: 10.1016/j.neuron.2005.01.038. [DOI] [PubMed] [Google Scholar]
- Mehta MV, Gandal MJ, Siegel SJ. mGluR5-antagonist mediated reversal of elevated stereotyped, repetitive behaviors in the VPA model of autism. PLoS One. 2011;6:e26077. doi: 10.1371/journal.pone.0026077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol. 2006;95:3291–3295. doi: 10.1152/jn.01316.2005. [DOI] [PubMed] [Google Scholar]
- Osterweil EK, Krueger DD, Reinhold K, Bear MF. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci. 2010;30:15616–15627. doi: 10.1523/JNEUROSCI.3888-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Park JM, Kim S, Kim JA, Shepherd JD, Smith-Hicks CL, Chowdhury S, Kaufmann W, Kuhl D, Ryazanov AG, Huganir RL, Linden DJ, Worley PF. Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron. 2008;59:70–83. doi: 10.1016/j.neuron.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran I, Laplante I, Bourgeois C, Pépin J, Lacaille P, Costa-Mattioli M, Pelletier J, Sonenberg N, Lacaille JC. Persistent transcription- and translation-dependent long-term potentiation induced by mGluR1 in hippocampal interneurons. J Neurosci. 2009;29:5605–5615. doi: 10.1523/JNEUROSCI.5355-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D, Kaphzan H, Klann E. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature. 2013;493:411–415. doi: 10.1038/nature11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharf SH, Jaeschke G, Wettstein JG, Lindemann L. Metabotropic glutamate receptor 5 as drug target for Fragile X syndrome. Curr Opin Pharmacol. 2015;20:124–134. doi: 10.1016/j.coph.2014.11.004. [DOI] [PubMed] [Google Scholar]
- Silverman JL, Tolu SS, Barkan CL, Crawley JN. Repetitive self-grooming behavior in the BTBR mouse model of autism is blocked by the mGluR5 antagonist MPEP. Neuropsychopharmacology. 2010;35:976–989. doi: 10.1038/npp.2009.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JL, Smith DG, Rizzo SJ, Karras MN, Turner SM, Tolu SS, Bryce DK, Smith DL, Fonseca K, Ring RH, Crawley JN. Negative allosteric modulation of the mGluR5 receptor reduces repetitive behaviors and rescues social deficits in mouse models of autism. Sci Transl Med. 2012;4:131ra51. doi: 10.1126/scitranslmed.3003501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G. The S6 kinase signaling pathway in the control of development and growth. Biol Res. 2002;35:305–313. doi: 10.4067/s0716-97602002000200022. [DOI] [PubMed] [Google Scholar]
- Tian D, Stoppel LJ, Heynen AJ, Lindemann L, Jaeschke G, Mills AA, Bear MF. Contribution of mGluR5 to pathophysiology in a mouse model of human chromosome 16p11.2 microdeletion. Nat Neurosci. 2015;18:182–184. doi: 10.1038/nn.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukiyama-Kohara K, Poulin F, Kohara M, DeMaria CT, Cheng A, Wu Z, Gingras AC, Katsume A, Elchebly M, Spiegelman BM, Harper ME, Tremblay ML, Sonenberg N. Adipose tissue reduction in mice lacking the translational inhibitor 4E-BP1. Nat Med. 2001;7:1128–1132. doi: 10.1038/nm1001-1128. [DOI] [PubMed] [Google Scholar]
- Volk LJ, Daly CA, Huber KM. Differential roles for group 1 mGluR subtypes in induction and expression of chemically induced hippocampal long-term depression. J Neurophysiol. 2006;95:2427–2438. doi: 10.1152/jn.00383.2005. [DOI] [PubMed] [Google Scholar]
- Volk LJ, Pfeiffer BE, Gibson JR, Huber KM. Multiple Gq-coupled receptors converge on a common protein synthesis-dependent long-term depression that is affected in fragile X syndrome mental retardation. J Neurosci. 2007;27:11624–11634. doi: 10.1523/JNEUROSCI.2266-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler IJ, Greenough WT. Metabotropic glutamate receptors trigger postsynaptic protein synthesis. Proc Natl Acad Sci U S A. 1993;90:7168–7171. doi: 10.1073/pnas.90.15.7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K, Comery TA, Patel B, Eberwine J, Greenough WT. Fragile X mental retardation protein is translated near synapses. Proc Natl Acad Sci U S A. 1997;94:5395–5400. doi: 10.1073/pnas.94.10.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005;49:1053–1066. doi: 10.1016/j.neuropharm.2005.06.004. [DOI] [PubMed] [Google Scholar]