

Abstract
Objective:
This study's aim was to evaluate whether the GCKR rs1260326 variant increases hepatic de novo lipogenesis (DNL).
Setting and Design:
To test this hypothesis, 14 adolescents, seven homozygous for the common allele (CC) and seven homozygous for the risk allele (TT), underwent measurement of hepatic DNL during the fasting state and after consumption of a carbohydrate (CHO) drink (75 g glucose and 25 g fructose). DNL was assessed through incorporation of deuterium in the palmitate contained in the very low-density lipoprotein.
Results:
Subjects with TT demonstrated higher fasting fractional DNL (P = .036) and a lower increase in fractional DNL after the CHO challenge (P = .016). With regard to absolute lipogenesis, TT subjects had both higher fasting rates (P = .015) and 44% greater area under the curve of absolute lipogenesis during the study (P = .016), compared to CC subjects. Furthermore, subjects carrying the TT genotype showed higher basal rates of glucose oxidation (P = .0028) and a lower ability than CC subjects to increase the rates of glucose oxidation after the CHO load (P = .054).
Conclusions:
This study reports for the first time rates of DNL in obese adolescents and suggests that the GCKR rs1260326 gene variant, which is associated with greater glycolysis, increases hepatic DNL. These data highlight the role of glycolytic carbon flux in liver lipid synthesis and hypertriglyceridemia in these youngsters.
Hepatic de novo lipogenesis (DNL), particularly under conditions of high carbohydrate (CHO) diets, represents one of the main sources of fatty acids for the liver, thereby contributing to an elevation in plasma and intrahepatic triglycerides (TGs) (1). Recently, the rs1260326 single nucleotide polymorphism (SNP) in the glucokinase regulatory protein (GCKR) gene has been associated with elevated concentrations of plasma large very low-density lipoprotein (VLDL) and TGs and an increased hepatic fat content in obese adolescents and adults (2–4). The GCKR encodes the GCKR, and the rs1260326 is a functionally relevant SNP consisting of a C to T substitution coding for a proline to leucine substitution at the codon 446 (P446L). The rs1260326 minor allele frequency has been estimated to be 0.446 in Caucasians, 0.129 in African Americans, and 0.355 in Hispanics (3). The physiological role of the GCKR, highly expressed in the hepatocytes, is to bind and to keep inactivated the glucokinase (GK) in the nucleus (5–7). In vitro studies have shown that the P446L mutation confers to the GCKR a lower affinity to bind the GK (8), possibly causing an increased release of GK into the cytoplasm. The higher cytoplasmatic concentrations of GK are predicted to lead to an enhanced glycolysis (8, 9), and we hypothesize that it would thus increase hepatic DNL. Building on our earlier observation of an association between the GCKR rs1260326 and high plasma and intrahepatic TGs in obese youth (3), in this study we aimed at assessing whether we could translate the evidence gained from previous in vitro studies (9) and demonstrate, in vivo, that the rs1260326 SNP minor allele is associated with an increased hepatic DNL.
Subjects and Methods
Adolescents (n = 14) were recruited from the Yale multiethnic cohort of the study entitled “Genetics of Pediatric Fatty Liver.” This cohort was started in 2009, and as of today approximately 500 obese children and adolescents have been enrolled and genotyped for polymorphisms found to be associated with fatty liver. The study was approved by the Yale University Human Investigation Committee. Written informed parental consent and written child assent were obtained from all participants. All clinical investigations have been conducted according to the principles expressed in the Declaration of Helsinki.
All subjects underwent an abdominal/liver magnetic resonance imaging (MRI) and a detailed assessment of glucose and lipid phenotypes (3, 10, 11). From this cohort we recruited subjects who were homozygotes for the common allele (C; n = 7) and homozygotes for the minor/risk allele (T; n = 7) of the rs1260326 SNP in the GCKR gene. The two groups of subjects were matched for age, gender, ethnicity, body mass index (BMI), and degree of insulin resistance. Moreover, given the strong relationship between hepatic fat content and DNL (1, 12), to test the sole effect of the gene variant and rule out the effect of fatty liver on DNL, the two groups were matched also for hepatic fat content. In particular, we attempted to recruit subjects without fatty liver, and when it was not possible, we enrolled pairs with similar hepatic fat content in the study (Table 1). Food intake was assessed by 3-day food record (including 2 weekdays and 1 weekend day), and food records were analyzed using the Nutrition Data System for Research (NDSR 2009).
Table 1.
Clinical Features of the Study Subjects
| CC | TT | P Value | |
|---|---|---|---|
| n | 7 | 7 | |
| Age, y | 16.4 ± 0.62 | 16.2 ± 0.78 | .54 |
| Gender (males/females), n | 2/5 | 2/5 | 1.00 |
| Ethnicity (C/AA/H), n | 3/1/3 | 3/1/3 | 1.00 |
| BMI, kg/m2 | 37.7 ± 2.64 | 35.4 ± 2.54 | .15 |
| Waist to hip ratio | 0.93 ± 0.01 | 0.91 ± 0.02 | .38 |
| Whole body insulin sensitivity index | 1.45 ± 0.36 | 1.27 ± 0.33 | .49 |
| Fasting glucose, mg/dL | 84.7 ± 2.95 | 87.2 ± 1.90 | .51 |
| Fasting insulin, μU/mLa | 30.4 ± 2.93 | 30.4 ± 8.07 | .39 |
| Hepatic fat content, %a | 3.81 ± 1.83 | 4.45 ± 2.92 | .77 |
| Fasting triglycerides, mg/dLa | 69.0 ± 10.1 | 121.5 ± 15.4 | .02 |
Abbreviations: C, Caucasian; AA, African American; H, Hispanic. Data are expressed as mean ± SEM, unless stated otherwise.
Data are expressed as median ± SEM.
DNL studies
Using the deuterated water (D2O) method (1), hepatic DNL was assessed after an overnight fast by providing subjects with a CHO drink containing 75 g of glucose and 25 g of fructose. Subjects were asked to refrain from excessive physical activity 3 days before the study. As shown in Figure 1, subjects were admitted to the hospital research unit of the Yale New Haven Hospital at 5:30 pm of day 1, and at this time they consumed the first of three doses of D2O. Additional doses were given at 10 pm of day 1 and 3 am of day 2. The total D2O given (3 mL per kg of body water) was designed to raise body deuterium levels to 0.3% and was administrated in three doses to maintain the steady state until the end of the study (Figure 1). Each subject pair consumed the same dinner between 7 and 7:30 pm on day 1, and then subjects remained fasting until the beginning of the study on day 2. At 6 am on day 2, indirect calorimetry was performed in the fasting state. At 7:45 am, the subjects ingested the CHO drink and thereafter underwent hourly blood draws for 6 hours. Indirect calorimetry was again performed 1 hour after the drink (9 am) (Figure 1). The subjects stayed in bed throughout the study (except for bathroom breaks). By evaluating the incorporation of the D2O into palmitate, we were able to assess the DNL expressed as a percentage of DNL as previously described (1, 12, 13). Absolute lipogenesis was calculated by multiplying the percentage of DNL and the fasting TG concentration contained in the isolated VLDL. As shown previously, with careful control of the previous evening meal, fasting DNL is reproducible in subjects in the range of 0.5–1.0% (14).
Figure 1.
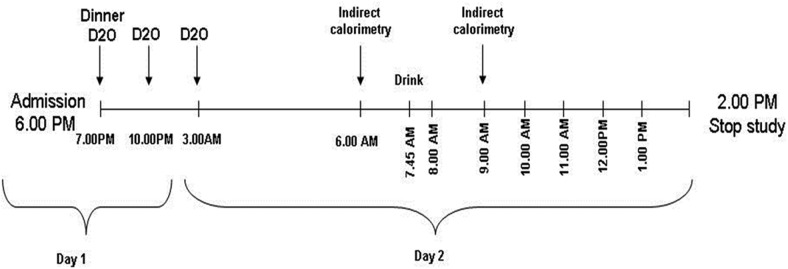
The design of the study to assess DNL.
Indirect calorimetry-derived measures
Indirect calorimetry (Deltatrac; Datex-Ohmeda) was performed on day 2 during the fasted state at 6 am and after the ingestion of the sugar load at 9 am, when the glucose levels reached their peak. Resting energy expenditure (REE) was expressed in kilocalories per kilogram of body weight per 24 hours. Rates of glucose and lipid oxidation were calculated from the gas exchange measurements as previously reported (15). Previous studies have shown that the measures derived from the indirect calorimetry, including O2 consumption, CO2 production, respiratory quotient, and total energy production per 24 hours, show acceptable mean coefficients of intraindividual variability of 3.7, 4.6, 3.5, and 3.6%, respectively (16).
Analytical procedures
Plasma glucose was determined with an YSI 2700 Analyzer (YSI Inc). A colorimetric method was used to measure plasma concentrations of free fatty acids (NEFA-HR kit; Wako Diagnostics), glycerol (CMA microdialysis kit P000025; M Dialysis Inc), and lactate (kit LC2389; Randox) during the study. Plasma insulin concentrations were measured by RIA (Linco), and TG was measured on the Alera (Kit SA1023; Alfa Wassermann). To isolate VLDLs, 1.5 mL of plasma was spun in a Beckman Optima XL-100K ultracentrifuge in a fixed angle 50.3 Ti rotor at 40 000 rpm for 20 hours at 15°C. The upper fraction containing VLDL (∼1 mL) was removed, and lipids were extracted with 3.75 mL CHCl3/MeOH 1:2. VLDL-TG was separated by thin layer chromatography, and TG-fatty acids were prepared for gas chromatography-mass spectrometry as described previously (17). Gas chromatography-mass spectrometry was performed using a DB-225, 30-m column (250 mm inner diameter, and 0.25 μm film thickness; J&W, Chrom Tech) in an Agilent 6890 GC with helium as the carrier gas. Selected ion monitoring was used for ions with mass to charge ratios 270, 271, and 272, which were analyzed with an Agilent 5975 mass spectrometer. Newly made fatty acids from DNL were calculated by mass isotopomer distribution analysis (18). Deuterium enrichment in plasma was measured by cavity ring-down spectroscopy using a liquid water isotope analyzer with automated injection system, version 2 upgrade (Los Gatos Research) by Metabolic Solutions Inc.
Oral glucose tolerance test
A standard oral glucose tolerance test (1.75 g/kg body weight, up to 75 g) was performed in all subjects (19, 20). The whole body insulin sensitivity index was used as the index of insulin sensitivity, validated for use in obese children and adolescents (20).
Imaging studies
Abdominal MRI studies were performed on a GE or Siemens Sonata 1.5 Tesla system (21). Fast-MRI measurement of liver fat content was performed by MRI using the two-point Dixon method as modified by Fishbein et al (22). Using the MRIcro software program, five regions of interest were drawn on each image, and the mean pixel signal intensity level was recorded. The hepatic fat fraction (HFF%) was calculated in duplicate from the mean pixel signal intensity data using the following formula: [(Sin − Sout)/(2 × Sin)] × 100. The imaging parameters were: matrix size = 128 × 256, flip angle (α) = 30°; TR = 18 milliseconds, TEs = 2.38/4.76 milliseconds out-of-phase and in-phase, respectively; bandwidth = 420 Hz/pixel, six averages; slice thickness = 10 mm, one slice, 2.3 seconds/slice (for two points); and scan time = 14 seconds in a single breath-hold (22).
Genotyping
The GCKR rs1260326 was genotyped using the Sequenom Massarray as previously described (3, 10).
Calculations and statistics
The area under the curve for both fractional and absolute lipogenesis and for TG contained in the VLDL was calculated by using the trapezoid method (23). Changes of DNL and lactate levels during the study were assessed by calculating the delta between the fasting and peak data. Delta changes for the calorimetry-derived measures were calculated by subtracting the value of the second calorimetry from the value obtained during the fasting state. Differences between the two groups of genotype were assessed by using a two-sided paired t test. A χ2 test was used to assess the difference in prevalence. Data are expressed as mean or median ± SEM.
Results
The clinical features of the study population according to the genotype are shown in Table 1. The two genotype groups were of similar age (P = .54), gender (P = 1.0), ethnicity (P = 1.0), BMI (P = .15), whole body insulin sensitivity index (P = .49), and fasting concentrations of glucose (P = .51) and insulin (P = .39). Although we recruited subjects with matched liver fat content, the TT genotype subject mean hepatic-TG was slightly higher than the CC group, although not different statistically (P = .77). In each genotype group, two subjects showed fatty liver, as defined by an HFF% higher than 5.5 (24); the two pairs with high HFF% showed the following values: CC = 7.2 vs TT = 11.9, and CC = 12 vs TT = 10. By contrast, the TT subjects had higher fasting plasma TG compared to the subjects carrying the CC genotype (P = .020). Analysis of 3-day food records revealed no differences between the groups for intake of total daily energy (2658 ± 229 vs 2679 ± 249 kcal/d, for the CC vs TT, respectively; P = .66, data not shown), energy from CHO (48.9 ± 2.1% vs 50.2 ± 2.1%; P = .99), and intake of added sugars (86.7 ± 13.2 vs 90.7 ± 26.3 g/d; P = .89). In response to the glucose/fructose drink, the two genotype groups displayed similar changes in glucose, insulin, glycerol, and free fatty acid concentrations (Figure 2). The groups differed with respect to fasting lactate (P = .006) and the change in lactate after the CHO challenge (P = .027), with the TT showing a lower delta lactate than the CC (Figure 2).
Figure 2.

Changes in concentrations of plasma metabolites. Values represent mean ± SEM, showing changes in glucose (A), insulin (B), glycerol (C), free fatty acids (D), lactate (E), and delta lactate (F) associated with the CHO challenge (75 g glucose + 75 g fructose). The dashed lines represent the TT subjects, and each data point is shown as a closed square; the continuous lines represent the CC, and each data point is represented by an open circle. F, White bar represents the CC, and black bar represents the TT. The groups were compared by a paired t test. P values are given only for statistically significant comparisons.
DNL: effect of GCKR rs1260326
As shown in Figure 3, subjects homozygous for the T allele demonstrated higher fasting VLDL-TG (P = .041), fractional DNL (P = .036), and total absolute lipogenesis (P = .015). Two hours after the CHO load, fractional DNL increased in both groups; however, the magnitude of changes in fractional DNL was significantly different between the two groups. In particular, compared to the CC genotype, subjects carrying the TT genotype showed a lower increase in fractional DNL (P = .016), a smaller area under the curve of fractional DNL (P = .040), but significantly greater areas under the curve for VLDL-TG (P = .019) and absolute lipogenesis (P = .017).
Figure 3.

Changes in VLDL-TG concentration and fractional and absolute DNL. Values are mean ± SEM data for the plasma concentration of VLDL-TG (A), area under the curve (AUC) of VLDL-TG (B), fractional DNL (C), delta increase DNL (D), rates of absolute lipogenesis (E), and AUC of the rates of absolute lipogenesis (F) according to the GCKR rs1260326 genotypes. The dashed lines represent the TT, and each time point is shown as a closed square; the continuous lines represent the CC, and each time point is represented by an open circle. White bars represent the CC, and black bars represent the TT. The groups were compared by a paired t test. P values are given only for statistically significant comparisons.
Fuel oxidation during the CHO drink: effect of the GCKR rs1260326
In the fasting state, subjects carrying the TT genotype exhibited significantly higher respiratory quotients (RQs) (P = .017), higher REE (P = .032) (Figure 4), higher rates of glucose oxidation (P = .0028), and lower rates of lipid oxidation (P = .044) (Figure 5). After consumption of the CHO drink, the subjects homozygous for the risk allele showed a trend toward a lower increase of RQ (P = .077), REE (P = .063) (Figure 4), and rates of glucose oxidation (P = .054), as well as a trend toward a lower decrease of the rates of lipid oxidation (P = .125) (Figure 5).
Figure 4.

Changes in RQ and REE during the study. In the upper panels are described the basal RQ (A), the RQ after the glucose/fructose load (B), and the delta RQ (C). In the lower panels are shown the basal REE (D), the REE after the glucose/fructose load (E), and the delta REE (F). Values are expressed as mean ± SEM. The groups were compared by a paired t test. P values are given only for statistically significant comparisons.
Figure 5.

Rates of glucose and lipid oxidation during the study. Values are expressed as mean ± SEM. Upper panels describe the rates of glucose oxidation during the fasting state (A) and after the glucose/fructose load (B), as well as the absolute changes in the rates of glucose oxidation (C). In the lower panels are shown the fasting rates of lipid oxidation (D), the rates of lipid oxidation after the glucose/fructose load (E), and the change in rates of lipid oxidation (F). The groups were compared by a paired t test. P values are given only for statistically significant comparisons.
Discussion
This study demonstrates that the GCKR rs1260326 genotype is a strong modulator of glycolysis and of DNL in obese adolescents. In particular, we observed that subjects homozygous for the minor allele of the rs1260326 variant (TT), compared to the subjects homozygous for the non-risk allele (CC), exhibit a higher basal REE, higher fasting rates of glucose oxidation, and fractional DNL. Elevated fasting DNL is evidence of an increased flux through the hepatic glycolysis pathway because the carbon source used for fatty acid synthesis likely arises from glycolysis. Furthermore, the TT subjects failed to increase glucose oxidation after the CHO load, which is consistent with their inability to significantly increase DNL after consumption of sugars. Both of these findings could be interpreted as reflecting an already high fasting flux of carbons down through glycolysis and through fatty acid synthesis. Although it has been hypothesized that a high glycolytic carbon flux can result in elevated lipogenesis, this is the first study to utilize a genetic variant to test this hypothesis. The sum of these observations is likely the consequence of the low affinity between GCKR and GK in subjects homozygous for the rs1260326 variant. The GK, which catalyzes the conversion of glucose in glucose-6-phosphate, is regulated by the adaptive interaction with a GCKR (7), which sequesters GK in the nucleus in the fasted state, allowing rapid translocation to the cytoplasm (activation) during postprandial hyperglycemia (25). In vitro studies have shown that a leucine to proline substitution at position 446 is characterized by a lower affinity between the GCKR and GK (9), which may cause an increase of the GK activity in the liver and therefore an increased conversion of glucose to glucose-6-phosphate, which represents the substrate needed to begin glycolysis. The increased glycolytic rate in subjects homozygous for the rs1260326 minor allele is supported by their higher lactate concentrations in the fasting state. Indeed, although glycolysis ends with the synthesis of pyruvate, some of these carbons may be converted into lactate, which in turn is exported into the hepatic venous effluent or stored as glycogen via the gluconeogenic pathway (26). The molecules of pyruvate not converted into lactate can be converted into acetyl-coenzyme A and, eventually, malonyl-coenzyme A, which represents the link between glycolysis and DNL. Together, these events provide the metabolic mechanisms leading to the increased production of TG observed in subjects carrying the rs1260326 minor allele (27).
Because an increased intrahepatic fat content is associated with an increased DNL (12), in this study, to rule out the effect of intrahepatic fat accumulation on DNL, we tried to recruit subjects without fatty liver, and when this was not possible, we matched the hepatic fat content, which is why the two groups of genotypes in this study show similar hepatic fat content. Nevertheless, subjects carrying the risk genotype showed higher VLDL-TG, which suggested that the VLDL export compensated for the increased lipogenesis. Lastly, this is the first study to directly measure DNL in a pediatric population and the first human study showing that a common gene variant might lead to hyperlipidemia by increasing the liver's ability to convert CHO into lipids. Moreover, our findings raise two questions. First, would subjects homozygous for the rs1260326 variant benefit from a reduced dietary intake of glucose and/or fructose? Second, what would be the impact of the GK activators on the metabolism of subjects with this variant? The latter question is timely and needs to be answered because clinical trials using GK activators seem to demonstrate that these compounds will lose effectiveness after a few months and cause hyperlipidemia and hypertension (28). However, no data exist showing how and if the rs1260326 variant in the GCKR gene might impact the response to GK activators.
This study has some limitations, including the small sample size. However, metabolic studies in children are very demanding because they require a long fasting period and an overnight stay in the research unit; moreover, children undergoing these studies lose a day of school, and the parents lose a day of work. Moreover, the study design, in particular the matching by degree of insulin resistance and hepatic fat content, supports the notion that the differences observed among these metabolic tests were real and would be reproducible. In fact, one study strength is that the subjects were pair-matched for age, gender, ethnicity, BMI, insulin sensitivity, and hepatic fat content, and this allowed us to rule out the effect of many confounders that might affect DNL and therefore to dissect the effect of the genotype. Despite that, we acknowledge that other characteristics may have also influenced the findings. Two strong determinants of DNL were controlled for, including habitual intake of dietary sugars (dietary intake of sugars was similar between the CC and TT groups) and acute energy balance (subjects were asked to refrain from strenuous physical activity 3 d before the study, and during the inpatient measurements, subjects were sedentary). Strengths of our study are: 1) the young age of the studied population, which is free from many critical metabolic stressors (such as alcohol and age); 2) the detailed phenotype of the study participants; and 3) the use of state-of-the-art techniques to characterize their metabolic phenotype (eg, DNL measured by using stable isotopes).
In conclusion, this study demonstrated in adolescents that the GCKR rs1260326 gene variant is associated with an increased hepatic DNL, but future longitudinal studies are needed to understand the tempo of progression toward the fatty liver of subjects carrying the GCKR risk genotype.
Acknowledgments
The authors are grateful to the patients and their families, as well as to the Yale Center for Genome Analyses, the Yale Center for Clinical Investigation (YCCI), and Hospital Research Unit personnel.
This work was made possible by the American Heart Association through the 13SDG14640038 and 11CRP5620013 awards (to N.S.) and by the Yale Center for Clinical Investigation (2012 YCCI scholar award and YCCI just-in-time grant, to N.S.). S.C. is funded by the National Institutes of Health (NIH) (Grants R01-HD-40787 and R01-HD-28016) and the American Diabetes Association (Distinguished Clinical Scientist Award DK-49230). This work was also made possible by NIH Grant DK045735 to the Yale Diabetes Research Center, and by Clinical and Translational Science Awards Grant UL1-RR-024139 from the National Center for Advancing Translational Sciences, a component of the NIH and NIH Roadmap for Medical Research.
Author Contributions: N.S. and S.C. conceived the study and wrote the first draft of the manuscript. E.J.P. measured the DNL and provided data interpretation and manuscript writing. B.P., M.S., and M.V.N. helped recruit the patients. All of the authors have read and edited the manuscript. This paper's contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH. N.S. is the guarantor of this work and as such had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the analyses.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- BMI
- body mass index
- CHO
- carbohydrate
- DNL
- de novo lipogenesis
- D2O
- deuterated water
- GCKR
- glucokinase regulatory protein
- GK
- glucokinase
- HFF%
- hepatic fat fraction
- MRI
- magnetic resonance imaging
- REE
- resting energy expenditure
- RQ
- respiratory quotient
- SNP
- single nucleotide polymorphism
- TG
- triglyceride
- VLDL
- very low-density lipoprotein.
References
- 1. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chambers JC, Zhang W, Sehmi J, et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet. 2011;43:1131–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Santoro N, Zhang CK, Zhao H, et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology. 2012;55:781–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Speliotes EK, Yerges-Armstrong LM, Wu J, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7:e1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brown KS, Kalinowski SS, Megill JR, Durham SK, Mookhtiar KA. Glucokinase regulatory protein may interact with glucokinase in the hepatocyte nucleus. Diabetes. 1997;46:179–186. [DOI] [PubMed] [Google Scholar]
- 6. Agius L, Peak M. Binding and translocation of glucokinase in hepatocytes. Biochem Soc Trans. 1997;25:145–150. [DOI] [PubMed] [Google Scholar]
- 7. de la Iglesia N, Mukhtar M, Seoane J, Guinovart JJ, Agius L. The role of the regulatory protein of glucokinase in the glucose sensory mechanism of the hepatocyte. J Biol Chem. 2000;275:10597–10603. [DOI] [PubMed] [Google Scholar]
- 8. Rees MG, Wincovitch S, Schultz J, et al. Cellular characterisation of the GCKR P446L variant associated with type 2 diabetes risk. Diabetologia. 2012;55:114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beer NL, Tribble ND, McCulloch LJ, et al. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum Mol Genet. 2009;18:4081–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Santoro N, Feldstein AE, Enoksson E, et al. The association between hepatic fat content and liver injury in obese children and adolescents: effects of ethnicity, insulin resistance, and common gene variants. Diabetes Care. 2013;36:1353–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Santoro N, Caprio S, Giannini C, et al. Oxidized fatty acids: a potential pathogenic link between fatty liver and type 2 diabetes in obese adolescents? Antioxid Redox Signal. 2014;20:383–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146:726–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Donnelly KL, Margosian MR, Sheth SS, Lusis AJ, Parks EJ. Increased lipogenesis and fatty acid reesterification contribute to hepatic triacylglycerol stores in hyperlipidemic Txnip−/− mice. J Nutr. 2004;134:1475–1480. [DOI] [PubMed] [Google Scholar]
- 14. Parks EJ, Skokan LE, Timlin MT, Dingfelder CS. Dietary sugars stimulate fatty acid synthesis in adults. J Nutr. 2008;138:1039–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Flannery C, Dufour S, Rabøl R, Shulman GI, Petersen KF. Skeletal muscle insulin resistance promotes increased hepatic de novo lipogenesis, hyperlipidemia, and hepatic steatosis in the elderly. Diabetes. 2012;61:2711–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gasic S, Schneider B, Waldhäusl W. Indirect calorimetry: variability of consecutive baseline determinations of carbohydrate and fat utilization from gas exchange measurements. Horm Metab Res. 1997;29:12–15. [DOI] [PubMed] [Google Scholar]
- 17. Barrows BR, Parks EJ. Contributions of different fatty acid sources to very low-density lipoprotein-triacylglycerol in the fasted and fed states. J Clin Endocrinol Metab. 2006;91:1446–1452. [DOI] [PubMed] [Google Scholar]
- 18. Hellerstein MK, Neese RA. Mass isotopomer distribution analysis at eight years: theoretical, analytic, and experimental considerations. Am J Physiol. 1999;276:E1146–E1170. [DOI] [PubMed] [Google Scholar]
- 19. D'Adamo E, Cali AM, Weiss R, et al. Central role of fatty liver in the pathogenesis of insulin resistance in obese adolescents. Diabetes Care. 2010;33:1817–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yeckel CW, Weiss R, Dziura J, et al. Validation of insulin sensitivity indices from oral glucose tolerance test parameters in obese children and adolescents. J Clin Endocrinol Metab. 2004;89:1096–1101. [DOI] [PubMed] [Google Scholar]
- 21. Cali AM, De Oliveira AM, Kim H, et al. Glucose dysregulation and hepatic steatosis in obese adolescents: is there a link? Hepatology. 2009;49:1896–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fishbein MH, Gardner KG, Potter CJ, Schmalbrock P, Smith MA. Introduction of fast MR imaging in the assessment of hepatic steatosis. Magn Reson Imaging. 1997;15:287–293. [DOI] [PubMed] [Google Scholar]
- 23. Le Floch JP, Escuyer P, Baudin E, Baudon D, Perlemuter L. Blood glucose area under the curve. Methodological aspects. Diabetes Care. 1990;13:172–175. [DOI] [PubMed] [Google Scholar]
- 24. Browning JD, Szczepaniak LS, Dobbins R, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. [DOI] [PubMed] [Google Scholar]
- 25. Arden C, Petrie JL, Tudhope SJ, et al. Elevated glucose represses liver glucokinase and induces its regulatory protein to safeguard hepatic phosphate homeostasis. Diabetes. 2011;60:3110–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stefanovski D, Youn JH, Rees M, et al. Estimating hepatic glucokinase activity using a simple model of lactate kinetics. Diabetes Care. 2012;35:1015–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van de Bunt M, Gloyn AL. From genetic association to molecular mechanism. Curr Diab Rep. 2010;10:452–466. [DOI] [PubMed] [Google Scholar]
- 28. Meininger GE, Scott R, Alba M, et al. Effects of MK-0941, a novel glucokinase activator, on glycemic control in insulin-treated patients with type 2 diabetes. Diabetes Care. 2011;34:2560–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]