

Abstract
Context:
The contiguous gene deletion syndrome (CAH-X) was described in a subset (7%) of congenital adrenal hyperplasia (CAH) patients with a TNXA/TNXB chimera, resulting in deletions of CYP21A2, encoding 21-hydroxylase necessary for cortisol biosynthesis, and TNXB, encoding the extracellular matrix glycoprotein tenascin-X (TNX). This TNXA/TNXB chimera is characterized by a 120-bp deletion in exon 35 and results in TNXB haploinsufficiency, disrupted TGF-β signaling, and an Ehlers Danlos syndrome phenotype.
Objective:
The objective of the study was to determine the genetic status of TNXB and resulting protein defects in CAH patients with a CAH-X phenotype but not the previously described TNXA/TNXB chimera.
Design, Settings, Participants, and Intervention:
A total of 246 unrelated CAH patients were screened for TNXB defects. Genetic defects were investigated by Southern blotting, multiplex ligation-dependent probe amplification, Sanger, and next-generation sequencing. Dermal fibroblasts and tissue were used for immunoblotting, immunohistochemical, and coimmunoprecipitation experiments.
Main Outcome Measures:
The genetic and protein status of tenascin-X in phenotypic CAH-X patients was measured.
Results:
Seven families harbor a novel TNXB missense variant c.12174C>G (p.C4058W) and a clinical phenotype consistent with hypermobility-type Ehlers Danlos syndrome. Fourteen CAH probands carry previously described TNXA/TNXB chimeras, and seven unrelated patients carry the novel TNXB variant, resulting in a CAH-X prevalence of 8.5%. This highly conserved pseudogene-derived variant in the TNX fibrinogen-like domain is predicted to be deleterious and disulfide bonded, results in reduced dermal elastin and fibrillin-1 staining and altered TGF-β1 binding, and represents a novel TNXA/TNXB chimera. Tenascin-X protein expression was normal in dermal fibroblasts, suggesting a dominant-negative effect.
Conclusions:
CAH-X syndrome is commonly found in CAH due to 21-hydroxylase deficiency and may result from various etiological mechanisms.
A deficiency in 21-hydroxylase (encoded by CYP21A2) leads to congenital adrenal hyperplasia [CAH; online Mendelian inheritance in man (OMIM) 201910], an autosomal recessive disorder of the adrenal cortex characterized by cortisol deficiency, with or without aldosterone deficiency, and androgen excess, the severity of which depends on the degree of 21-hydroxylase impairment (1). The severe or classic form is a rare orphan disease occurring in 1 in 16 000 live births, whereas the mild or nonclassic form is estimated to occur in 1 in 1000 people (2). Flanking CYP21A2 is the TNXB gene encoding tenascin-X (TNX), an extracellular matrix (ECM) glycoprotein that is highly expressed in connective tissue. TNX plays a role in collagen fibrillogenesis and matrix maturation, although it functions in collagen fibril deposition independent of collagen synthesis (3). Normal collagen fibril deposition in connective tissue is essential for the collagenous matrix integrity, and defects lead to Ehlers Danlos syndrome (EDS), a hereditary connective tissue disorder (4). Complete TNX deficiency was first reported in a patient with CAH and EDS (5). Whereas autosomal recessive complete TNX deficiency is a cause of classical EDS (OMIM 130000) (6), TNXB haploinsufficiency is associated with hypermobile EDS (OMIM 130020) (7), and when present in patients with CAH, results in a contiguous gene deletion syndrome termed CAH-X (8).
The CYP21A2 (OMIM 201910) and the TNXB (OMIM 600985) genes reside in tandem on chromosome 6p23.1 within the human leukocyte antigen histocompatibility complex in a module characterized by highly homologous sequences between functional genes (CYP21A2 and TNXB) and their corresponding pseudogenes (CYP21A1P and TNXA), which leads to frequent homologous recombination. Chimeric genes generated by large gene deletion or gene conversion events, account for 20%–30% of the common CYP21A2 pathogenic variants in 21-hydroxylase-deficient patients (9–11). To date, nine CYP21A2/CYP21A1P (CH-1 to CH-9) and one TNXA/TNXB chimera (termed CH-1 here) have been described (5, 11). The previously described TNXA/TNXB CH-1 results in a contiguous CYP21A2 and TNXB deletion found in 7% of CAH patients and has been estimated to account for 13% of large CYP21A2 deletions (8). TNXA/TNXB CH-1 is characterized by a 120-bp deletion crossing exon 35 and intron 35 carried over from the TNXA pseudogene; this is the only well-documented discrepancy between TNXB and TNXA.
The TNX protein is divided into an N-terminal signal peptide, followed by TNX assembly, epidermal growth factor-like, fibronectin type III, and C-terminal fibrinogen-like domains (12). All but one previously reported TNX genetic defects are in the fibronectin type III domains (13–15). Although the exact mechanism behind TNX's ability to organize the ECM is unknown, the TNX fibrinogen-like domain activates the TGF-β pathway, a regulator of collagen production important in connective tissue dysplasias (16). Additionally, we recently showed aberrant TGF-β signaling in patients with CAH-X syndrome (17).
In this study, we evaluated the TNXB gene and the TNX protein in CAH patients with a CAH-X phenotype who did not have the previously described TNXA/TNXB CH-1. Because the junction site of a chimeric TNXA/TNXB gene theoretically can be anywhere between exons 32 and 44 (the homologous region), we hypothesized that novel TNXA/TNXB chimeras might exist. Our study provides functional evidence of a novel TNXA/TNXB chimera, resulting in CAH-X syndrome, thereby broadening the genetic and phenotypic spectrum of CAH due to 21-hydroxylase deficiency.
Materials and Methods
Study subjects
In a large prospective cohort of 246 unrelated patients (146 females, aged 1–65 y) with CAH due to 21-hydroxylase deficiency, we studied patients with a phenotype suspicious for CAH-X syndrome. Patients were enrolled in an ongoing prospective natural history study at the National Institutes of Health Clinical Center (Bethesda, Maryland) (Clinical Trials number NCT00250159), and approval was obtained from the Eunice Kennedy Shriver National Institute of Child Health and Human Development Institutional Review Board. Written informed consent was obtained from all participants and parents of participating children. All minors at least 8 years old gave assent. The diagnosis of 21-hydroxylase deficiency was determined by hormonal evaluation (17-hydroxyprogesterone > 1200 ng/dL) and CYP21A2 genotyping (18). Previously reported subjects with CAH-X syndrome due to TNXB haploinsufficiency (14 probands, 16 related patients) and CAH controls were used as comparison groups (8).
All patients were evaluated prior to genotyping for joint hypermobility using the Beighton 9-point scale and for major and minor criteria of classical and hypermobile EDS as previously described (8). Generalized hypermobility was determined by an individual's age and Beighton score and defined as 5 or greater for children and 4 or greater for postpubertal adolescents and adults (19). Skin characteristics, including extensibility, thinness, scar formation, and striae, were evaluated. Transthoracic two-dimensional echocardiography and magnetic resonance imaging of the heart (1.5 T scanner) were performed in patients with a CAH-X phenotype and their relatives when possible.
Genetic analysis
Sanger sequencing (CYP21A2 and TNXB genes) and next-generation sequencing (EDS panel: COL3A1, COL5A1, COL5A2 genes) were performed in a commercial diagnostics laboratory (PreventionGenetics, Marshfield, Wisconsin). The amino acid substitution prediction programs, Polymorphism Phenotyping version 2 and Sorting Intolerant From Tolerant, were used to predict missense variant pathogenicity (12, 13). Multiplex ligation-dependent probe amplification and Southern blotting were done as previously described (8, 20). Two hundred healthy subjects (Coriell Repository, http://ccr.coriell.org) served as controls for evaluating allele frequency in the general population. All new TNXB variants described were submitted to the ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar/).
Tenascin-X modeling and disulfide prediction
The primary sequence of the tenascin-X C-terminal fibrinogen-like domain, corresponding to residues 4023–4233, was submitted to the Robetta server (http://www.robetta.org) for domain (Ginzu) (21) and tertiary structure predictions (Rosetta) (22, 23). The disulfide bonding state of C4058 was analyzed by submitting the C-terminal fibrinogen-like domain to several university-based online disulfide bonding state predictors (http://bioserv.rpbs.jussieu.fr/cgi-bin/CysState) (24, 25).
Fibroblast cell culture
Primary skin fibroblasts were initiated from 3-mm punch biopsy explants from seven CAH patients with the novel TNXB variant and 10 age- and sex-matched CAH controls with a normal TNXB genotype. Fibroblasts from second through third passages were cultured to confluence in high-glucose DMEM (Invitrogen), 10% fetal bovine serum, penicillin, and streptomycin (Invitrogen) at 37°C in 5% CO2.
Western blot analysis
Protein expression was analyzed by SDS-PAGE and Western blot as previously described (17) with the following changes: 10 μg protein was loaded onto a 3%–8% NuPAGE Novex Tris-acetate precast gel (Invitrogen), and immunoblotting was done using a rabbit polyclonal antihuman tenascin-X (H-90) antibody (1:200; Santa Cruz Biotechnology) or rabbit polyclonal antihuman β-tubulin antibody (1:2000; Cell Signaling Technology) overnight at 4°C. Secondary antibody incubation, blot visualization, and quantification were done as previously described (17). All data were normalized to the loading control. Experiments were performed in triplicate.
Elastin and fibrillin-1 staining in dermal biopsies
Five-micrometer sections of paraffin-embedded skin biopsies were mounted on Superfrost Plus slides (Erie Scientific), and sections were stained with hematoxylin and eosin using a Leica CV5030 autostainer (Leica).
Elastin-Van Gieson staining of elastic fibers was done using an Artisan multistainer (DakoCytomation) and Dako Artisan staining reagents (Artisan elastin-Van Gieson kit; DakoCytomation). The epidermal layer served as an internal negative control for each skin biopsy.
Mouse antifibrillin-1 monoclonal antibody clone 26 (number MAB2502; EMD Millipore) was used at a 1:300 dilution. Automated Leica BOND-MAX and its detection kit (Leica Biosystems) were used for antifibrillin-1 immunohistochemical staining. Primary antibody was incubated for 30 minutes, polymer and postpolymer for 15 minutes, 3,3′-diaminobenzidine chromogen for 10 minutes, and hematoxylin for 5 minutes. Negative controls were done likewise but used mouse serum without primary antibody. Slides were viewed at ×200 magnification.
Coimmunoprecipitation
Coimmunoprecipitation experiments were conducted according to Alcaraz et al (16). Briefly, whole-cell lysate (equivalent volume of 10 μg protein according to a bicinchoninic assay assay) from dermal fibroblasts was lysed as above and subjected to immunoprecipitation using the Dynabeads Protein G immunoprecipitation kit (Invitrogen) according to the manufacturer's instructions. A purified rat antimouse, human, pig TGF-β1 antibody (BD Biosciences) was used to pull down and normal rat IgG (R&D Systems) antibody was used as a control for nonspecific binding. The entire eluates from the Dynabeads kit were loaded onto the gel and subjected to SDS-PAGE and Western blotting with an antitenascin-X antibody as described above.
Statistical analysis
Comparisons were made using the unpaired Student's t test (Microsoft Excel). P values are two tailed and considered significant when P ≤ .05. Data are represented as mean ± SEM.
Results
Novel TNXB Variant c.12174C>G associated with CAH-X
Ten phenotypic CAH-X patients from seven families were heterozygous for the novel TNXB missense variant in exon 40 defined as c.12174C>G (Figure 1A), which is predicted to result in the amino acid substitution p.C4058W within the TNX C-terminal fibrinogen-like domain. The prediction programs, Polymorphism Phenotyping version 2 and Sorting Intolerant From Tolerant, predicted that the p.C4058W change to be probably damaging and deleterious, respectively. This variant was not found in 200 healthy subjects. In our cohort, 14 CAH probands carry previously described TNXA/TNXB chimeras and seven unrelated patients carry the novel TNXB variant, resulting in a CAH-X prevalence of 8.5%.
Figure 1.
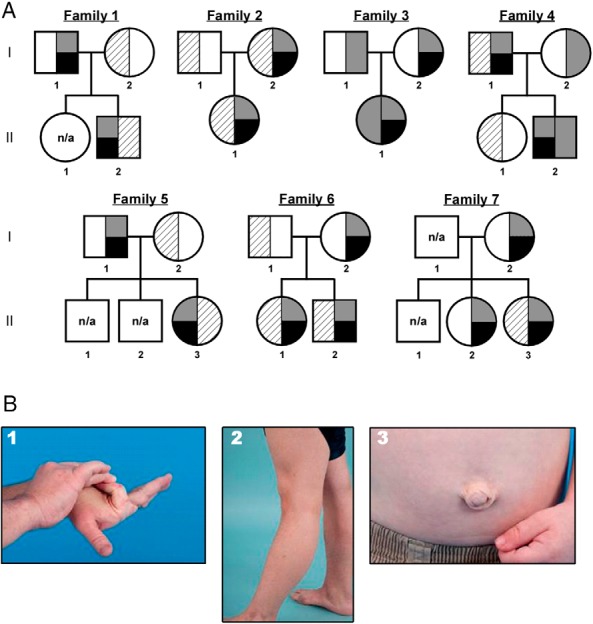
CAH-X pedigrees and clinical phenotypes. A, The pedigrees of seven novel CAH-X families. Black represents the TNXB C4058W variant. CYP21A2 variants are represented as gray (CYP21A2 deletion due to CYP21A2/CYP21A1P chimera) or striped (other CYP21A2 pathogenic variants). Most CAH patients are compound heterozygotes. Five CAH carrier parents and one CAH carrier sibling also have the C4058W mutation. DNA was not available from siblings in families 1 and 5. B, Clinical findings in patients with CAH-X syndrome due to a C4058W mutation in TNXB. Hypermobile small (panel 1) and large joints (panel 2), and umbilical hernia (panel 3) are shown. n/a, not available.
All 10 CAH patients with the c.12174C>G variant had EDS clinical features (Table 1). Generalized joint hypermobility, a major diagnostic criterion for classical EDS, was found in all patients. Hypermobility of small (Figure 1, B1) and large joints (Figure 1, B2) and hernias (Figure 1, B3) were found in four (40%), seven (70%), and three (30%) patients, respectively. Most adults (90%) had chronic arthralgia in four or more joints. Other characteristic CAH-X clinical findings included structural cardiac abnormalities, elongated uvula with midline crease, and piezogenic pedal papules. Unlike the previously reported CAH-X CH-1 patients, a subset of patients with the novel c.12174C>G variant had skin laxity (40%). The clinical phenotype of relatives with the c.12174C>G variant, but not CAH, was less severe, although the majority had hypermobile joints.
Table 1.
Clinical Findings in Patients With CAH-X Due to the Novel TNXB Variant C4058W
| Subject | Sex and Age, y | CAH Phenotype | Hypermobility Scorea | Other Joint Findings | Skin Findings | Cardiac Findings | Additional Clinical Features |
|---|---|---|---|---|---|---|---|
| Family 1: II-2 | M/18 | SV | 4 | Multiple subluxations; hypermobile shoulders and feet; hip laxity; chronic arthralgiab | Wide scars; striae; skin laxity | Normal | Elongated uvula with midline crease; strabismus; gastroesophageal reflux; hiatal hernia; irritable bowel syndrome; piezogenic papules; bunions; osteopenia |
| Family 1: I-1 | M/49 | Carrier | 3 | Hypermobile hands; chronic arthralgia | Skin laxity | Normal | Asthma; hypertension |
| Family 2: I-2 | F/20 | NC | 5 | Chronic arthralgia | Normal | Normal | Scoliosis; gastroesophageal reflux; piezogenic papules |
| Family 2: II-1 | F/45 | NC | 7 | Multiple subluxations; hypermobile shoulders; hip laxity; chronic arthralgia; chronic tendonitis of wrists and knees | Skin laxity | Normal | Umbilical hernia; piezogenic papules |
| Family 3: II-1 | F/6 | SW | 8 | Hip laxity | Normal | Patent foramen ovale until age 4 y | None |
| Family 3: I-2 | F/43 | Carrier | 1 | Hip laxity; torn rotator cuff | Normal | Normal | None |
| Family 4: II-2 | M/2 | SW | 9 | Hip laxity | Skin laxity; doughy skin | Atrial septal aneurysm with patent foramen ovale | Elongated uvula with midline crease; umbilical hernia |
| Family 4: I-1 | M/44 | NC | 7 | Hypermobile hands | Wide scars; congenital third nipple | Mildly enlarged aortic root | Elongated uvula with midline crease; pectus excavatum; varicose veins; chronic plantar fasciitis; pes planus |
| Family 5: II-3 | F/41 | SW | 6 | Shoulder subluxation and bursitis; chronic arthralgia | Striae; skin laxity | Normal | None |
| Family 5: I-1 | M/72 | Carrier | n/a | Hypermobile shoulders; hip laxity; two knee and left shoulder arthroplasties | Normal | Mild RV and LA enlargement; atrial septum aneurysm with patent foramen ovale | Osteoarthritis; spinal stenosis; prostate cancer; epilepsy |
| Family 6: I-2 | F/40 | Carrier | 2 | Torn anterior cruciate ligament | Normal | n/a | None |
| Family 6: II-1 | F/13 | SW | 4 | Scoliosis | Normal | Mild partial fusion of the commissure between left and right aortic cusps; mildly dilated aortic root | Turner syndrome; chronic otitis media |
| Family 6: II-2 | M/9 | SW | 8 | Hypermobile hands | Normal | Normal | Chronic urticarial; chronic abdominal pain |
| Family 7: I-2 | F/45 | Carrier | 5 | None | Striae | n/a | None |
| Family 7: II-2 | F/17 | Carrier | 6 | None | Striae; cystic acne | n/a | None |
| Family 7: II-3 | F/10 | NC | 5 | Rib and elbow subluxations; hypermobile shoulders and hands; hip laxity | Striae | n/a | None |
Abbreviations: LA, left atrium; LV, left ventricle; n/a, not available; NC, nonclassic; RA, right atrium; RV, right ventricle; SV, classic simple virilizing; SW, classic salt wasting.
Hypermobility score was assessed by the Beighton scale (19).
Arthralgia is at least 3 months' duration.
To rule out possible pathogenic variants in other known EDS-associated genes, we sequenced the COL3A1, COL5A1, and COL5A2 genes in the 10 novel CAH-X patients and found no pathogenic or likely pathogenic variants. Two missense variants of uncertain significance were found in the COL3A1 gene in patient I-1 from family 1 and patient II-1 from family 3. However, these two COL3A1 variants did not segregate with phenotypes between the parents and the probands, indicating that they are unlikely to be the primary cause of the phenotypes.
c.12174C>G as a marker of a novel TNXA/TNXB chimeric gene
At the locus of CYP21A2 and TNXB, the homology sequence between the pseudo- and active TNX genes spans the CYP21A2 5′ upstream region to intron 31 of TNXB; thus, the junction site of a chimeric gene resulting from an unequal crossover event can vary (Figure 2, A–F). The reference sequence of the pseudogene TNXA (NR_001284.2) at the corresponding c.12174 position is G, suggesting pseudogenic origin of this variation. To test this hypothesis, we sequenced TNXB exons 35–44 in 11 patients who were previously identified as heterozygous for TNXA/TNXB CH-1 (120-bp deletion) because the fragment spanning the exon 35/intron 35 boundary through exon 44 in the TNXA/TNXB CH-1 chimera represents the TNXA-specific sequence. All 11 patients were heterozygous for c.12174C>G in exon 40; analysis of seven available parents showed that the 120-bp deletion and the c.12174C>G reside on the same allele (in cis). In addition to c.12174C>G in exon 40, a cluster of three heterozygous variants (c.12218G>A, exon 41; c.12514G>A; and c.12524G>A, exon 43) was found in three individuals from two families: family 3: II-1; family 4: I-1 and family 4: II-2 (Figures 1A and 2G). Absence of c.12174C>G and these three variants in 100 healthy controls (a subgroup of the 200 controls mentioned above) strongly supports that they are TNXA specific, although only c.12174C>G is likely present in every copy of TNXA. Thus, c.12174C>G represents a novel distinguishable site between TNXA and TNXB.
Figure 2.

Schematic diagram of CYP21A1P/CYP21A2 and TNXA/TNXB chimera genes. A, Schematic showing exons (rectangles) of the CYP21A2 and TNXB genes. CYP21A2 encodes the active 21-hydroxylase enzyme (blue). TNXB encodes active tenascin-X (red). The sizes of the exons/introns are scaled, with the exception of introns with a slash (eg, CYP21A2 exon 9 is ∼100 bp). The homology sequence spans the CYP21A2 5′ upstream region to intron 31 of TNXB. The solid triangle at the boundary of TNXB exon 35 and intron 35 denotes the 120-bp fragment absent in TNXA. B, Unequal crossover during meiosis can occur at the CYP21A2 (blue cross) or TNXB (red cross) locus, resulting in a CYP21A1P/CYP21A2 or TNXA/TNXB chimera. Pseudogenes CYP21A1P and TNXA are in gray and are framed with the color of the corresponding functional gene. RP1 encodes a serine/threonine nuclear kinase (gray), and RP2 is the corresponding truncated pseudogene (gray). C4 encodes the fourth component of serum complement (gray). The open triangle at the boundary of exon 35 and intron 35 denotes the 120-bp deletion in TNXA. C, The CYP21A1P/CYP21A2 and TNXA/TNXB chimeric genes result from unequal crossover. D, Schematic showing exons (rectangles) of a representative CYP21A1P/CYP21A2 chimeric gene (with a junction site in exon 8) and the intact TNXB gene. CYP21A1P/CYP21A2 chimeric genes have been classified into nine types (CH-1 to CH-9) based on the junction site location. E, Schematic showing exons (rectangles) of the classic TNXA/TNXB chimeric gene with a 120-bp deletion at the boundary of exon 35 and intron 35 (CAHX-CH1). The CYP21A2 gene is completely deleted and replaced by the CYP21A1P pseudogene. F, Schematic showing exons (rectangles) of the newly identified TNXA/TNXB chimeric gene tagged by the c.12174C>G (p.Cys4058Trp) variant in exon 40 (CAHX-CH2). The exon 35 region is intact, whereas the CYP21A2 gene is completely deleted and replaced by the CYP21A1P pseudogene. G, Sanger sequencing chromatogram of the four variants: c.12174C>G (p.Cys4058Trp), c.12218G>A (p.Arg4073His), c.12514G>A (p.Asp4172Asn), and c.12524G>A (p.Ser4175Asn). The green bar is the DNA reference sequence with the encoded amino acids above. The gray bar is the representative patient's DNA sequence with the encoded amino acids above.
All of our patients with c.12174C>G had a CYP21A2 deletion. Most patients (all except patient II-3 from family 5) were found to have a CYP21A1P/CYP21A2 chimera with an undefined junction site downstream of exon 8 (11). Therefore, the junction site of these CYP21A1P/CYP21A2 chimeras could be located anywhere between intron 8 of CYP21A2 and intron 35 of TNXB, representing a contiguous gene deletion of CYP21A2 and part of TNXB. Thus, c.12174C>G can be used as a marker of a novel TNXA/TNXB chimeric gene. In keeping with the chimeric CYP21A1P/CYP21A2 gene-naming system (11), we termed the previously identified (120-bp deletion crossing exon 35 and intron 35) and the new (c.12174C>G) TNXA/TNXB chimeric genes CAH-X CH-1 and CAH-X CH-2, respectively) (Figure 2, E and F). Alternatively, the c. 12174C>G variant could be carried over from TNXA through a concurrent microgene conversion event instead of a contiguous gene deletion. This likely occurred in patient II-3 from family 5 (Table 1), who is compound heterozygous for the CYP21A1P/CYP21A2 CH-5 chimera (with V281L) and R484P in CYP21A2.
The TNXB variants identified are located in the fibrinogen, α/β/γ-chain and C-terminal globular domain of the TNX protein. Because c.12174C>G (p.C4058W) was the only variant shared among all probands and affected family members, it is likely pathogenic and is suspected to be the primary cause of the phenotypes in these patients.
C4058W likely leads to partial TNX protein unfolding
A TNX primary amino acid sequence alignment showed that C4058 is highly conserved within the C-terminal fibrinogen-like domain across mammalian species (Figure 3A).
Figure 3.

Effects of C4058W in the TNX fibrinogen-like domain. A, Primary protein sequence alignment of C4058 in TNX. B, Robetta model of the TNX C-terminal fibrinogen-like domain. Left, Structural overlay of a TNX C-terminal fibrinogen domain model (pink) with the known crystal structure of the highly homologous ficolin-2 C-terminal fibrinogen domain [turquoise, PDB: 2J61 (26)]. Right, C4058 is located within probable disulfide bonding distance of C4028 according to disulfide bond state predictor algorithms. C, Representative Western blot of TNX expression in CAHX-CH2 patient skin fibroblasts (n = 7) compared with CAH controls (n = 10). Quantification graph includes all patients and controls. TNX expression was normalized to β-tubulin and experiments were done in triplicate. P ≤ .05 was considered significant. D, Representative elastin and fibrillin-1 staining in dermal tissue. Van Gieson's staining of elastic fibers was reduced (yellow arrows) and immunohistochemical staining showed disorganized fibrillin-1 fibers (black arrows) in both CAHX-CH1 haploinsufficient and CAHX-CH2 patients compared with normal controls. Hematoxylin and eosin were used to counterstain, and sections were viewed at ×200 magnification. E, Coimmunoprecipitation of the TNX C-terminal fibrinogen domain with TGF-β1. Immunoprecipitations were done using an antihuman TGF-β1 (α-TGF-β1) antibody or control IgG in whole-cell lysate from CAHX-CH2 and CAH control fibroblasts. A Western blot of the TNX C-terminal fibrinogen domain fragment that was pulled down is shown. Experiments were done in triplicate.
Because a full-length TNX tertiary structure was not available, the TNX C-terminal fibrinogen-like domain (residues 4023–4233) was submitted to the Robetta server for ab initio and comparative modeling. The primary protein sequence was first parsed into putative domains (Ginzu) (21), and then structural homologs for each domain were identified from protein structural databases and used to build structural models from homology modeling or ab initio calculations to predict protein structure (Rosetta) (22, 23).
The Ginzu prediction provided one domain for this region. Rosetta then modeled the domain with high confidence using homology and the known crystal structure for human ficolin-2, a carbohydrate-binding protein with a C-terminal fibrinogen-like domain (PDB: 2J61) (26). The ficolin-2 C-terminal fibrinogen-like domain crystal structure was overlaid with the TNX model (Figure 3B, left). The two structures nicely aligned, supporting the fidelity of this model. A neighboring cysteine in the tertiary structure (position 4028) was predicted to be within disulfide bonding distance of the highly conserved cysteine at position 4058 by three independent university-based online disulfide bonding state predictors (http://bioserv.rpbs.jussieu.fr/cgi-bin/CysState) (24, 25). This predicted disulfide bond is located at the interface between a small segment containing two β-sheets and an α-helix at the top of the structure and the remaining larger portion of the domain at the bottom, possibly being released and unfolding upon mutation to a tryptophan, resulting in a partially unfolded TNX fibrinogen-like domain (Figure 3B, right).
C4058W disrupts TNX function but not expression
In contrast to prior reports of CAH-X CH-1 (8), CAH-X CH-2 dermal fibroblasts did not have altered TNX expression in whole-cell lysate compared with CAH controls by Western blot (Figure 3C). However, CAH-X CH-2 patients had reduced Elastin-Van Gieson histology staining for elastic fibers in dermal biopsies compared with normal controls. Elastin staining in CAH-X CH-1 patients more closely resembled the controls, consistent with prior reports showing a spectrum of elastic fiber abnormalities in TNX haploinsufficient samples (15). Immunohistochemical staining using an antifibrillin-1 antibody showed disrupted fibrillin-1 organization in CAH-X CH-1 and CAH-X CH-2 dermal biopsies compared with normal controls (Figure 3D).
CAH-X CH-2 disrupts the fibrinogen domain's ability to bind TGF-β1
We hypothesized that CAH-X CH-2 patients may not fully bind TGF-β1 as a consequence of having a mutated fibrinogen-like domain. We coimmunoprecipitated a TNX fragment at the molecular weight of the fibrinogen domain using a TGF-β1 antibody in a CAH-X CH-2 patient's whole-cell lysate compared with CAH controls (16). Serum TNX exists as the full-length form, as well as multiple functional fragments, which is also likely reflected at the tissue level (6, 16, 27). Less TNX fragment was pulled down by the TGF-β1 antibody in the CAH-X CH-2 patients compared with CAH controls by Western blot, suggesting a disrupted interaction with TGF-β1 (Figure 3E).
Discussion
In this study, we describe increased CAH-X syndrome prevalence relative to our previous report (8) in a large cohort of patients with CAH due to 21-hydroxylase deficiency. Through detailed genetic studies, we identify a pseudogene-derived variant c.12174C>G (p.C4058W), representing a novel TNXA/TNXB chimera that does not involve a 120-bp deletion in exon 35. Patients with this novel variant have cardinal EDS features, such as joint hypermobility and chronic joint pain as well as midline defects and skin hyperextensibility in a patient subset (8). All patients with CAH-X syndrome described to date have a similar phenotype, disrupted extracellular matrix, and an altered TGF-β pathway, although we describe here mutation-specific effects on TNX protein expression (8).
We previously identified CAH-X in 7% of CAH patients; however, the genetic cause underlying the CAH-X phenotype in a patient subgroup was unknown. The lack of unequivocally distinguishable sites between intron 8 of CYP21 and intron 35 of TNX made identification of chimeric gene junction sites challenging. Due to the homologous sequence in this region, an in-depth understanding of the sequence differences between the active genes and pseudogenes is required. Unlike CYP21A2 and its pseudogene CYP21A1P, which have been extensively studied and well characterized (28), TNXB and TNXA sequences are still largely unknown. A reference sequence for TNXA (NR_001284.2) exists; however, the variations within this pseudogene have never been investigated in detail. In our current study, we used the TNXA-specific sequence present in the previously identified TNXA/TNXB chimera (120-bp deletion in exon 35) to compare with allelic sequences in unaffected controls. TNXA and TNXB variation profiling, and a variant comparison between them in a proper sample size in the general population is essential for future genetic studies of CAH-X and EDS. Canturk et al (28) showed that CYP21A1P-specific variants present with various allele frequencies within CYP21A1P. Similarly, our data suggest that the c.12174C>G variant is present in every copy of TNXA, whereas the other three identified TNXA-specific variants (c.12218G>A, c.12514G>A, and c.12524G>A) may have a lower allele frequency.
The TNX protein is known to regulate collagen deposition and cross-linking (4, 15, 29), although the mechanism is largely unknown. Recent studies with recombinant TNX constructs and the discovery of a link between TNX and the TGF-β signaling pathway is beginning to elucidate this mechanism. The highly conserved C4058W variant found in this study is located in the fibrinogen-like domain and is likely disulfide bonded and upon disruption would potentially lead to at least partial protein misfolding and a functional impact. Prior studies of CAH-X and EDS patients carrying the previously identified TNXA/TNXB CH-1 gene showed reduced dermal and serum TNX expression compared with controls, supporting a haploinsufficient mechanism (7, 8). However, TNX expression was unchanged in our CAH-X patients carrying the TNXA/TNXB CH-2 or TNXB variants, suggesting a dominant-negative mechanism. Previous studies showed disrupted elastin fibers and reduced fibrillin-1 staining in patients with autosomal recessive complete TNX deficiency (30) and, to some extent, patients with partial deficiency due to TNX haploinsufficiency (15). Our data revealed reduced elastin staining in dermal biopsies from CAH-X CH-2 patients compared with controls, whereas the reduction in elastin staining in CAH-X CH-1 samples was less dramatic. Similarly, variable effects of TNX haploinsufficiency on elastin have been reported (15). In our study, fibrillin-1 organization was highly disrupted in all CAH-X patients. Although fibrillin-1 is a part of the elastic fiber and microfibrillar network, it also exists independently and is believed to interact with TNX indirectly to stabilize elastic fiber assembly (30).
TGF-β is known to regulate collagen (31), and disrupted TGF-β signaling is implicated in a number of connective tissue disorders (32–36), including CAH-X (17). We previously reported elevated TGF-β biomarkers in both dermal fibroblasts and tissue as well as in circulation and secreted from cells in patients with CAH-X due to the TNXA/TNXB CH-1 chimera (17). We hypothesized that TGF-β pathway disruption could also be responsible for some of the connective tissue phenotypes in our CAH-X CH-2 patients, although likely through a different mechanism. Alcaraz et al (16) showed that the TNX C-terminal fibrinogen-like domain activated the TGF-β pathway by directly interacting with the latent TGF-β complex and inducing a conformational change, causing the release of active TGF-β1 and subsequent downstream phosphorylated Smad signaling. Similarly, we found that less TNX fibrinogen domain fragment was bound by TGF-β1 in dermal fibroblast whole-cell lysate from CAH-X CH-2 patients compared with controls, suggesting a disrupted interaction between TNX and TGF-β1. Further studies are needed to analyze this interaction in CAH-X CH-2 patients.
The clinical impact of carrying a TNXB defect includes the risk of chronic joint pain in adulthood, joint subluxations, hernias, and/or organ prolapse as well as developmental cardiac defects. Unlike previously described patients with CAH-X, several patients with CAH-X due to the novel TNXA/TNXB CH-2 had skin laxity with normal healing, reflecting a phenotypic spectrum that may or may not be variant specific. Most hypermobile EDS cases are unexplained (37). Novel ways in which TNX is disrupted, including the novel TNXA/TNXB chimera described here, represent potential genetic causes of hypermobile EDS of unknown etiology and requires further study.
A strength of our study is the large cohort size with extensive phenotyping. However, limited sequence information on the TNXA and TNXB genes remains a challenge. Also, the lack of an appropriate CAH-X mouse model is one limitation to fully dissecting the mechanism behind TNX deficiency, although the development of in vitro TNX constructs could address this issue.
Our study shows that multiple TNXA/TNXB chimeric genes can lead to CAH-X syndrome, and likely further chimeras remain to be discovered. TNX deficiency effects can be seen through a disrupted ECM and TGF-β signaling pathway in CAH-X patients, although the mechanism behind these effects is likely specific to the nature of the TNXB variant. This study reveals novel ways in which connective tissue features can manifest in CAH patients; therefore, more patients may be affected by CAH-X syndrome than previously recognized. We can now estimate that approximately 9% of CAH patients have CAH-X syndrome, expanding the phenotypic spectrum of CAH. Therefore, evaluation for a connective tissue dysplasia is warranted in CAH patients, and further studies of TNXB may provide insight into the pathogenesis of EDS.
Acknowledgments
We are grateful to the patients for their participation in this study.
This study had a Clinical Trial registration identifier number of NCT00250159 (clinicaltrials.gov).
This work was supported by the Intramural Research Programs of the National Institutes of Health Clinical Center, the Eunice Kennedy Shriver National Institute of Child Health and Human Development, the National Cancer Institute, and the National Institute on Aging. D.P.M. is a commissioned officer in the US Public Health Service.
Disclosure Summary: D.P.M. received research funds from Diurnal Limited, Ltd. The other authors have nothing to disclose.
Footnotes
- CAH
- congenital adrenal hyperplasia
- CAH-X
- contiguous gene deletion syndrome
- ECM
- extracellular matrix
- EDS
- Ehlers Danlos syndrome
- OMIM
- online Mendelian inheritance in man
- TNX
- tenascin-X.
References
- 1. Merke DP, Bornstein SR. Congenital adrenal hyperplasia. Lancet. 2005;365:2125–2136. [DOI] [PubMed] [Google Scholar]
- 2. Speiser PW, Azziz R, Baskin LS, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95:4133–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Egging D, van Vlijmen-Willems I, van Tongeren T, Schalkwijk J, Peeters A. Wound healing in tenascin-X deficient mice suggests that tenascin-X is involved in matrix maturation rather than matrix deposition. Connect Tissue Res. 2007;48:93–98. [DOI] [PubMed] [Google Scholar]
- 4. Mao JR, Taylor G, Dean WB, et al. Tenascin-X deficiency mimics Ehlers-Danlos syndrome in mice through alteration of collagen deposition. Nat Genet. 2002;30:421–425. [DOI] [PubMed] [Google Scholar]
- 5. Burch GH, Gong Y, Liu W, et al. Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nat Genet. 1997;17:104–108. [DOI] [PubMed] [Google Scholar]
- 6. Schalkwijk J, Zweers MC, Steijlen PM, et al. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N Engl J Med. 2001;345:1167–1175. [DOI] [PubMed] [Google Scholar]
- 7. Zweers MC, Bristow J, Steijlen PM, et al. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet. 2003;73:214–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Merke DP, Chen W, Morissette R, et al. Tenascin-X haploinsufficiency associated with Ehlers-Danlos syndrome in patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2013;98:E379–E387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vrzalova Z, Hruba Z, Hrabincova ES, et al. Chimeric CYP21A1P/CYP21A2 genes identified in Czech patients with congenital adrenal hyperplasia. Eur J Med Genet. 2011;54:112–117. [DOI] [PubMed] [Google Scholar]
- 10. New MI, Abraham M, Gonzalez B, et al. Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc Natl Acad Sci USA. 2013;110:2611–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen W, Xu Z, Sullivan A, et al. Junction site analysis of chimeric CYP21A1P/CYP21A2 genes in 21-hydroxylase deficiency. Clin Chem. 2012;58:421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tucker RP, Drabikowski K, Hess JF, Ferralli J, Chiquet-Ehrismann R, Adams JC. Phylogenetic analysis of the tenascin gene family: evidence of origin early in the chordate lineage. BMC Evol Biol. 2006;6:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gbadegesin RA, Brophy PD, Adeyemo A, et al. TNXB mutations can cause vesicoureteral reflux. J Am Soc Nephrol. 2013;24:1313–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Penisson-Besnier I, Allamand V, Beurrier P, et al. Compound heterozygous mutations of the TNXB gene cause primary myopathy. Neuromusc Disord. 2013;23:664–669. [DOI] [PubMed] [Google Scholar]
- 15. Zweers MC, Dean WB, van Kuppevelt TH, Bristow J, Schalkwijk J. Elastic fiber abnormalities in hypermobility type Ehlers-Danlos syndrome patients with tenascin-X mutations. Clin Genet. 2005;67:330–334. [DOI] [PubMed] [Google Scholar]
- 16. Alcaraz LB, Exposito JY, Chuvin N, et al. Tenascin-X promotes epithelial-to-mesenchymal transition by activating latent TGF-β. J Cell Biol. 2014;205:409–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morissette R, Merke DP, McDonnell NB. Transforming growth factor-β (TGF-β) pathway abnormalities in tenascin-X deficiency associated with CAH-X syndrome. Eur J Med Genet. 2014;57:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Finkielstain GP, Chen W, Mehta SP, et al. Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2011;96:E161–E172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998;77:31–37. [DOI] [PubMed] [Google Scholar]
- 20. Ignotz RA, Kelly B, Davis RJ, Massague J. Biologically active precursor for transforming growth factor type α, released by retrovirally transformed cells. Proc Natl Acad Sci USA. 1986;83:6307–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim DE, Chivian D, Malmstrom L, Baker D. Automated prediction of domain boundaries in CASP6 targets using Ginzu and Rosetta DOM. Proteins. 2005;61(suppl 7):193–200. [DOI] [PubMed] [Google Scholar]
- 22. Simons KT, Kooperberg C, Huang E, Baker D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J Mol Biol. 1997;268:209–225. [DOI] [PubMed] [Google Scholar]
- 23. Rohl CA, Strauss CE, Chivian D, Baker D. Modeling structurally variable regions in homologous proteins with rosetta. Proteins. 2004;55:656–677. [DOI] [PubMed] [Google Scholar]
- 24. Ceroni A, Passerini A, Vullo A, Frasconi P. DISULFIND: a disulfide bonding state and cysteine connectivity prediction server. Nucleic Acids Res. 2006;34:W177–W181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yaseen A, Li Y. Dinosolve: a protein disulfide bonding prediction server using context-based features to enhance prediction accuracy. BMC Bioinformatics. 2013;14(suppl 13):S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Garlatti V, Martin L, Gout E, et al. Structural basis for innate immune sensing by M-ficolin and its control by a pH-dependent conformational switch. J Biol Chem. 2007;282:35814–35820. [DOI] [PubMed] [Google Scholar]
- 27. Egging DF, Peeters AC, Grebenchtchikov N, et al. Identification and characterization of multiple species of tenascin-X in human serum. FEBS J. 2007;274:1280–1289. [DOI] [PubMed] [Google Scholar]
- 28. Canturk C, Baade U, Salazar R, Storm N, Portner R, Hoppner W. Sequence analysis of CYP21A1P in a German population to aid in the molecular biological diagnosis of congenital adrenal hyperplasia. Clin Chem. 2011;57:511–517. [DOI] [PubMed] [Google Scholar]
- 29. Valcourt U, Alcaraz LB, Exposito JY, Lethias C, Bartholin L. Tenascin-X: beyond the architectural function. Cell Adhesion Migration. 2015;9:154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zweers MC, van Vlijmen-Willems IM, van Kuppevelt TH, et al. Deficiency of tenascin-X causes abnormalities in dermal elastic fiber morphology. J Invest Dermatol. 2004;122:885–891. [DOI] [PubMed] [Google Scholar]
- 31. Ignotz RA, Massague J. Transforming growth factor-β stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986;261:4337–4345. [PubMed] [Google Scholar]
- 32. Morissette R, Schoenhoff F, Xu Z, et al. Transforming growth factor-β and inflammation in vascular (type IV) Ehlers-Danlos syndrome. Circ Cardiovasc Genet. 2014;7:80–88. [DOI] [PubMed] [Google Scholar]
- 33. Doyle AJ, Doyle JJ, Bessling SL, et al. Mutations in the TGF-β repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat Genet. 2012;44:1249–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lindsay ME, Schepers D, Bolar NA, et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. 2012;44:922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. [DOI] [PubMed] [Google Scholar]
- 37. Colombi M, Dordoni C, Chiarelli N, Ritelli M. Differential diagnosis and diagnostic flow chart of joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type compared to other heritable connective tissue disorders. Am J Med Genet Part C Semin Med Genet. 2015;169:6–22. [DOI] [PubMed] [Google Scholar]