

Abstract
Manganese superoxide dismutase (MnSOD) is considered a critical component of the antioxidant systems that protect against oxidative damage. We are interested in the role of oxidative stress in bladder detrusor smooth muscle (SM) in different disease states. In this study, we generated an inducible, SM-specific Sod2−/− mouse model to investigate the effects of MnSOD depletion on the function of the bladder. We crossbred floxed Sod2 (Sod2lox/lox) mice with mice containing heterozygous knock-in of a gene encoding a tamoxifen-activated Cre recombinase in the SM22α promoter locus [SM-CreERT2(ki)Cre/+]. We obtained Sod2lox/lox,SM-CreERT2(ki)Cre/+ mice and injected 8-wk-old males with 4-hydroxytamoxifen to induce Cre-mediated excision of the floxed Sod2 allele. Twelve weeks later, SM-specific deletion of Sod2 and depletion of MnSOD were confirmed by polymerase chain reaction, immunoblotting, and immunohistochemistry. SM-specific Sod2−/− mice exhibited normal growth with no gross abnormalities. A significant increase in nitrotyrosine concentration was found in bladder SM tissue of SM-specific Sod2−/− mice compared with both wild-type mice and Sod2+/+, SM-CreERT2(ki)Cre/+ mice treated with 4-hydroxytamoxifen. Assessment of 24-h micturition in SM-specific Sod2−/− mice revealed significantly higher voiding frequency compared with both wild-type and SM-specific Cre controls. Conscious cystometry revealed significantly shorter intercontraction intervals and lower functional bladder capacity in SM-specific Sod2−/− mice compared with wild-type mice. This novel model can be used for exploring the mechanistic role of oxidative stress in organs rich in SM in different pathological conditions.
Keywords: oxidative stress, inducible cre-loxp recombination
reactive oxygen species (ROS) are normal byproducts produced at low levels during normal physiological conditions and scavenged by endogenous antioxidant defense systems, including superoxide dismutase (SOD), glutathione peroxidase, glutathione reductase, catalase, and some vitamins such as C and E (41). Oxidative stress (OS) can result from an imbalance between ROS generation and the scavenging capacity of antioxidant enzymes, namely overproduction of ROS and/or impaired ROS removal due to diminished antioxidant activities (35). OS has emerged as a potentially important pathogenic factor in the development of bladder dysfunction in animal models of diabetes (3), bladder outlet obstruction (16), and ischemic overactive bladder (2). Induction of OS in the bladder with intravesical hydrogen peroxide (H2O2) can induce bladder overactivity through activation of afferent C fibers (27). Although effects of OS on bladder innervation have been demonstrated, the role of OS in bladder smooth muscle (SM) function in bladder disorders has not been elucidated.
SODs are important antioxidant enzymes that catalyze the conversion of superoxide anions to H2O2 and O2. H2O2 is further reduced to water by catalase and glutathione peroxidases. Three SOD isoforms are expressed in mammalian cells. Copper/zinc SOD (Cu/ZnSOD) is found mainly in the cytoplasm, manganese superoxide dismutase (MnSOD) is localized in the mitochondrial matrix, and extracellular SOD is usually present in the extracellular space (29). Although all three SOD isoforms play crucial roles in cellular antioxidant defense mechanisms, MnSOD appears to be the most important one, since mitochondria are not only a major source of ROS production from respiratory chains but also represent a major target of ROS-induced cellular injury.
Because MnSOD is responsible for protecting the mitochondrial compartment from oxidative damage, it has been an area of interest for several investigators who have used methods including overexpression via transgenic mice or downregulation via knockout mice (26) to see if manipulation of MnSOD can change the level of OS and alter tissue functions or the impact of pathological conditions. Previous studies showed that global deletion of Sod2 resulted in neonatal lethality, underscoring the vital cytoprotective role of MnSOD (13, 15). However, several groups have generated viable knockout mice by deleting Sod2 selectively in different tissues, with various effects on OS in and functions of the tissues [for review, see Marecki et al. (26)].
To investigate the mechanistic role of OS in bladder dysfunction, this study was designed to generate conditional SM-specific Sod2−/− mice using an inducible Cre-loxP recombination strategy, and characterize the bladder function in this model. The generated mouse model can be a valuable tool for investigating the pathological consequences of increased OS in SM-related disorders.
MATERIALS AND METHODS
Generation of Sod2lox/lox,SM-CreERT2(ki)Cre/+ mice.
Inducible SM-specific Sod2−/− mice were generated by crossbreeding two established transgenic mouse lines. One line contains a heterozygous knock-in, into the SM22α promoter locus, of a gene encoding a fusion protein of Cre recombinase and a mutated human estrogen receptor ligand-binding domain [SM-CreERT2(ki)Cre/+ mice, kindly provided by Dr. Robert Feil, University of Tübingen, Germany] (12). This transgenic mouse expresses the CreERT2 fusion protein specifically in SM cells under control of the SM22α promotor. Cre recombinase activity of the fusion protein is induced by 4-hydroxytamoxifen (OHT), but not by natural 17β-estradiol (8). In the second transgenic mouse line, exon 3 of both Sod2 alleles is flanked by LoxP sites in introns 2 and 3 (Sod2lox/loxmice, kindly provided by Dr. Takuji Shirasawa, Tokyo Metropolitan Institute of Gerontology, Tokyo, Japan) (10). Both mouse lines are on a C57BL/6 background. After initial matings of SM-CreERT2(ki)Cre/+ mice with Sod2lox/lox mice, double heterozygous [Sod2lox/+,SM-CreERT2(ki)Cre/+] offspring were identified by genotyping and were crossed with each other to obtain Sod2lox/lox,SM-CreERT2(ki)Cre/+ mice. Treatment of Sod2lox/lox,SM-CreERT2(ki)Cre/+ mice with OHT activates Cre-mediated excision of Sod2 exon 3 specifically in SM cells, yielding a gene encoding a truncated peptide (Fig. 1). SM-specific Cre [Sod2+/+,SM-CreERT2(ki)Cre/+], homozygous floxed Sod2 (Sod2lox/lox,SM22α+/+), and wild-type mice were also obtained from the matings of double heterozygous mice to serve as controls.
Fig. 1.
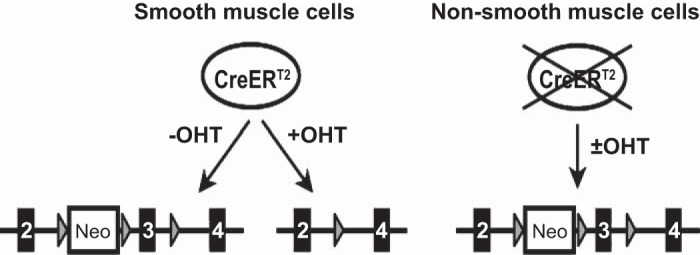
Schema of conditional, smooth muscle (SM)-specific excision of superoxide dismutase (Sod) 2 exon 3. The circled CreERT2 indicates the CreERT2 protein, expressed exclusively in SM cells. The lines at the bottom show the Sod2 gene locus. Exons 2–4, loxP sequences, and the neomycin resistance gene are indicated by the numbered black boxes, gray triangles, and boxed Neo, respectively. Floxed exon 3 is excised upon 4-hydroxytamoxifen (OHT) activation of CreERT2 in SM cells.
Mice were housed in cages at a constant room temperature (25 ± 2°C) and relative humidity (55 ± 5%) under a 12:12-h light-dark cycle with free access to food and water. Mice were maintained according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. All of the animal protocols were approved by the Institutional Animal Care and Use Committee of Case Western Reserve University.
Genotype analysis.
Genomic DNA was extracted using a commercially available RED Extract-N-Amp Tissue PCR Kit (Sigma-Aldrich, St. Louis, MO) from either tail biopsy of 4-wk-old offsprings or from different harvested organs after killing the mice at the end of the experimental protocol (the latter was performed to assess the tissue specificity of Sod2 exon 3 deletion).
Genotyping of the CreERT2 and floxed Sod2 genes was performed by PCR analysis according to the published protocols (10, 12). The wild-type SM22α allele was detected using sense primer RF67 (5′-CTCAGAGTGGAAGGCCTGCTT-3′) near the 3′-end of intron 1 and antisense primer RF90 (5′-CACACCATTCTTCAGCCACA-3′) at the 3′-end of exon 2, which amplify a 276-bp product. The SM-CreERT2(ki) transgene was detected using primer RF67 and antisense primer SC135 (5′-GGCGATCCCTGAACATGTCC-3′) on the 5′-side of the Cre gene, which amplify a 220-bp product.
The wild-type Sod2 allele was detected by using sense primer P1 (5′-CGAGGGGCATCTAGTGGAGAAG-3′) in intron 2 and antisense primer P2 (5′-TTAGGGCTCAGGTTTGTCCAGAA-3′) in exon 3, which amplify a 500-bp product. The floxed Sod2 allele was detected by using primer P1 with antisense primer P4 (5′-AGCTTGGCTGGACGTAA-3′) in the loxP-flanked neomycin resistance-exon 3 cassette, which amplify a 358-bp product. A third antisense primer, P3 (5′-CTAGTGAGATGGCTCAGC-3′) in intron 3, was used with P1 to amplify a 400-bp product corresponding to the deleted Sod2 allele.
The PCR conditions were as follows: 95°C for 3 min and then 35 cycles of 94°C for 30 s, 59°C for 1 min, and 72°C for 45 s, and finally 72°C for 2 min.
Preparation of OHT.
OHT (10 mg; Sigma-Aldrich) was suspended in ethanol at a concentration of 10 mg/100 μl and then further diluted to 10 mg/ml by addition of autoclaved sunflower oil. The solution was sonicated for 30 min in a Branson ultrasonicator (model 2510), and aliquots were stored at −20°C for no more than 4 wk (28). Aliquots were sonicated briefly just before injection into mice to activate CreERT2 in cells expressing the protein.
Experimental design.
Through successive interbreeding and selections as described above, we obtained Sod2lox/lox,SM-CreERT2(ki)Cre/+ offspring, as well as the control genotypes (wild-type, homozygous floxed Sod2, and SM-specific Cre mice). Not all of the possible control groups with or without OHT treatment were used in all experiments. However, all experiments examining phenotypic effects of Sod2 deletion included SM-specific Cre mice [Sod2+/+,SM-CreERT2(ki)Cre/+], treated with OHT to activate CreERT2 transiently, to control for potential Cre-mediated effects independent of floxed Sod2 (40).
Only male mice were used in this study, to exclude possible effects of estrous cycle hormones in female mice on CreERT2. At 8 wk of age, male Sod2lox/lox,SM-CreERT2(ki)Cre/+ mice and age-matched controls were injected with OHT (40 mg·kg body wt−1·day−1 ip) or sunflower oil vehicle for five consecutive days (28). Eight weeks after the final OHT injection, body weights were recorded and tissues were harvested for RT-PCR, histology, and immunoblotting of MnSOD and Cu/ZnSOD. In other mice, 12 wk after the final OHT injection, bladder function was assessed by measuring 24-h micturition and conscious cystometry (CMG). Bladders were harvested from additional mice 12 wk after the final OHT injection for immunoblotting of nitrotyrosine and MnSOD.
Tissue collection.
Before death, organs were harvested from mice under isoflurane anesthesia. Whole organs analyzed included liver, lung, heart, thoracic aorta, urethra, left ureter, and skeletal muscle from the left hind leg. Small intestine specimens were cut open, and the epithelium was removed by rubbing with a cotton swab, leaving primarily intestinal SM for analysis. Bladders were washed with ice-cooled PBS and then dried on filter paper and weighed. For RT-PCR, immunoblotting, and SOD activity assay, harvested bladders were carefully cut open to expose the urothelial lining and pinned to a Sylgard-coated dish containing ice-cooled PBS. The urothelium was removed under a surgical microscope, and both urothelium and detrusor SM were snap-frozen in liquid nitrogen and stored at −80°C for further immunoblotting analysis and biochemical assays.
Immunoblot analyses.
Frozen tissues were homogenized in ice-cooled RIPA buffer containing Complete Mini Protease Inhibitor Cocktail (1 tablet/10 ml; Roche Diagnostics, Mannheim, Germany), using a PowerGen 125 homogenizer (Fisher Scientific, Waltham, MA). Protein concentrations were determined using the DC protein assay reagent (Bio-Rad Laboratories, Hercules, CA). Proteins were separated by SDS-PAGE and then transferred to polyvinylidene difluoride membranes (Immobilon-P; EMD Millipore, Billerica, MA). Membranes were blocked with 5% skim milk for 1 h at room temperature and then incubated at 4°C overnight in blocking buffer containing one of the following antibodies: SOD-1 (for detection of Cu/ZnSOD, sc11407, 1:1,000 dilution; Santa Cruz, Dallas, TX), SOD-2 (for detection of MnSOD, sc30080, 1:500 dilution; Santa Cruz), GAPDH (no. 2118, 1:1,000 dilution; Cell Signaling, Boston, MA), and nitrotyrosine (sc-65385, dilution 1:1,000; Santa Cruz). Blots were incubated at room temperature for 1 h with the appropriate secondary antibody. The membranes were then treated with ECL reagent (Amersham Biosciences, Piscataway, NJ) and exposed to light-sensitive film.
For quantification of nitrotyrosine and MnSOD, after being developed, the membranes were stripped using Restore Western blot stripping buffer (Thermo Scientific, Rockford, IL) for 15 min and then reprobed with β-actin antibody (sc-47778, dilution 1:10,000) to confirm equal protein loading. The bands were quantified using Image J software and then divided to the corresponding β-actin bands.
Histology and immunohistochemistry.
For histological examinations, bladders of SM-specific Sod2−/− mice and wild-type control mice were fixed in 10% neutral buffered formalin and embedded in paraffin. Bladder cross sections of 5 μm were cut at the equatorial midline and stained with Masson's trichrome solution. Stained sections were analyzed with Image-Pro Plus 6 image analysis software (Media Cybernetics, Silver Spring, MD), which can distinguish regions stained with different colors and accurately measure such areas. The tissue areas occupied by SM (red), collagen (blue), and urothelium (pink) were determined and expressed as percentages of the total tissue area as we described previously (19). Also, the ratio of collagen to SM area was calculated. In all cases image processing was done by one investigator blinded to treatment group assignments.
For immunohistochemistry (IHC), paraffin sections (5 μm) were deparaffinized with xylene and rehydrated through graded ethanol. Antigen retrieval was performed using heat-induced epitope retrieval with citrate buffer (Dako, Carpinteria, CA). After the slides were incubated in blocking buffer, the primary antibody (rabbit polyclonal anti-MnSOD antibody, ab13534, 1:2,000 dilution; Abcam, Cambridge, MA) in PBS with 1% BSA was applied overnight at 4°C. After being rinsed three times for 5 min, the sections were incubated with secondary antibody (biotinylated anti-rabbit IgG H+L, BA-1000, 1:1,000; Vector Laboratories, Burlingame, CA) in PBS with 1% BSA for 2 h at room temperature. Avidin-labeled peroxidase was applied for 30 min at room temperature, and then avidin-biotinylated antibody complexes were developed with diaminobenzidine for 10–30 min at room temperature. The sections were counterstained with hematoxylin, dehydrated, cleared, and mounted for light microscopy examination. Negative controls were performed by parallel incubation without the primary antibody.
SOD activity.
Detrusor SM tissues were homogenized in cold 20 mM HEPES buffer, pH 7.2, containing 1 mM EGTA, 210 mM mannitol, and 70 mM sucrose and then centrifuged at 1,500 g for 15 min at 4°C. The supernatant was stored at −80°C. SOD activity was measured using an SOD assay kit (Cayman Chemical, Ann Arbor, MI) that utilizes a tetrazolium salt for detection of superoxide radicals generated by xanthine oxidase and hypoxanthine, following the manufacturer's instructions. To measure MnSOD activity, potassium cyanide was added to the supernatant at a concentration of 1 mM to inhibit Cu/ZnSOD activity. Cu/ZnSOD activity was then determined by subtracting MnSOD activity from the total SOD activity. One unit of SOD activity is defined as the amount of enzyme needed to exhibit 50% dismutation of the superoxide radical. Results were expressed as units of SOD activity per milligram protein.
Measurement of 24-h fluid consumed and micturition.
Twenty four-hour voiding frequency and volume per void were measured in real time in a 12:12-h light-dark cycle using mouse micturition chambers custom designed and built by Med Associates (St. Albans, VT), as we described previously (17). The mice were put in the micturition chambers that included a wire mesh bottom. Directly below the bottom opening was a balance with a data port connected to a data acquisition system. Changes in the weight of the collection were recorded at a sampling speed of 10 times/s (Origin 7.5; OriginLab, Northampton, MA). Twenty-four hours before micturition measurement, solid food was removed from the cages and replaced with lactose-free milk to reduce the frequency and weight of feces generated during testing, thereby preventing distortion of the urine collection (20). The volume of milk remaining at the end of the 24-h micturition measurement period was subtracted from the volume at the start of the period and recorded.
Suprapubic bladder catheter implantation and CMG.
Two days before CMG, suprapubic implantation of a catheter (PE-10 tubing with a flared tip) in the bladder was performed as we described previously (6). On the day of CMG, mice were placed individually in specially modified metabolic cages as we described previously (6). Briefly, the implanted bladder catheter was attached via a stopcock to both a pressure transducer (BP-100; CB Sciences, Dover, NH) and a flow pump (Kent Scientific, Torrington, CT). The bladder was emptied through the catheter using a syringe and then filled with room temperature 0.9% saline (1 ml/h) while bladder pressure was recorded. Urine was collected in a beaker on a force transducer (FT-03 D; Grass Instrument, Quincy, MA) placed beneath each cage. The pressure and force transducers were connected to an amplifier (ETH-400; CB Sciences), and multiport controller software (MED-CMG; Med Associates and Catamount Research and Development, Saint Albans, VT) was used for data recording through a computer. After an initial 0.5- to 1-h stabilization period, the data on 9–12 representative micturition cycles were collected, and the mean values were used for analysis of the cystometric parameters. The functional bladder capacity was calculated by multiplying the time from infusion to the first void by the infusion rate. Voided volume is the volume expelled at micturition. The residual urine volume was calculated by subtracting the voided volume from the functional bladder capacity. Peak voiding pressure was measured at the peak of the detrusor contraction. The intercontraction interval between two successive voiding contractions was calculated in each micturition cycle. The bladder compliance was calculated by dividing the functional bladder capacity by the difference between the threshold pressure (immediately before the start of a voiding contraction) and the basal pressure (immediately before the start of bladder filling).
Statistical analysis.
Statistical analysis was performed using GraphPad Prism 6 (GraphPad Software, La Jolla, CA). All data are expressed as means ± SE. Comparisons of total SOD, MnSOD, and Cu/ZnSOD activities between two groups (Fig. 6) were done by multiple t-test analysis, using the Holm-Sidak method to correct for multiple comparisons. For multiple group comparisons (nitrotyrosine and MnSOD in Fig. 7; Tables 1–4), one-way ANOVA was used, followed by Tukey's posttest of pairwise comparisons. For parameters in Tables 1–4 where no significant differences were found, posttests to detect linear trends from wild-type to SM-specific Cre to SM-specific Sod2−/− mice were performed. P < 0.05 was considered statistically significant.
Fig. 6.
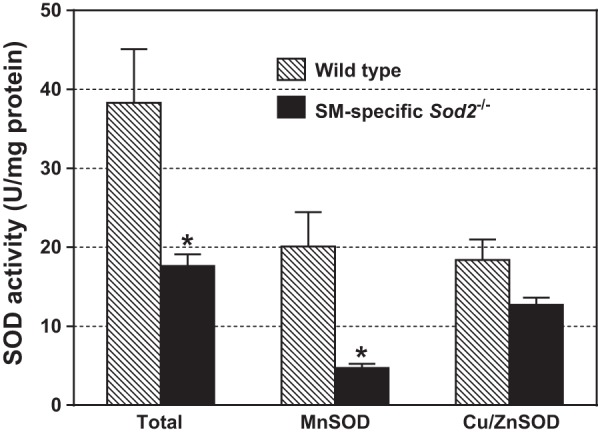
Enzymatic activities of total SOD, MnSOD, and Cu/ZnSOD in detrusor SM from 17-wk-old wild-type and Sod2lox/lox,SMCreERT2(ki)Cre/+ mice treated with OHT at 8 wk of age. Bars with error bars indicate means ± SE (n = 6 experiments/group). *Significantly different from wild-type group at P < 0.005.
Fig. 7.

Representative immunoblot results and quantitative analysis of nitrotyrosine (A and B) and MnSOD (C and D) levels relative to β-actin in detrusor SM. Wild-type mice (WT), SM-specific Cre mice (Cre), and Sod2lox/lox,SMCreERT2(ki)Cre/+ mice (KO) were treated with OHT at 8 wk of age. Later (12 wk), detrusor SM was harvested from 4 mice/group for immunoblot analyses. Bars with error bars represent means ± SE. *Significantly different from WT and KO groups at P < 0.0001. €Significantly different from KO group at P < 0.0001. #Significantly different from WT and Cre groups at P < 0.0001.
Table 1.
Body weight, bladder weight, and bladder weight-to-body weight ratio in wild-type, SM-specific Cre, and SM-specific Sod2−/− mice
| Groups | n | Body Wt, g | Bladder Wt, mg | Bladder Wt/Body Wt, mg/g |
|---|---|---|---|---|
| Wild type | 12 | 30.1 ± 0.80 | 29.6 ± 1.26 | 0.99 ± 0.04 |
| SM-specific Cre | 7 | 31.3 ± 1.30 | 27.5 ± 1.36 | 0.91 ± 0.04 |
| SM-specific Sod2−/− | 18 | 28.3 ± 0.89 | 27.9 ± 1.09 | 0.97 ± 0.03 |
Data are expressed as means ± SE; n, no. of mice. SM, smooth muscle; SOD, superoxide dismutase. All mice were treated with 4-hydroxytamoxifen (OHT) at 8 wk of age and assessed 8 wk after the final OHT injection.
Table 4.
Area percentage of SM, collagen, and urothelium, as well as collagen:SM ratio of bladders of wild-type, SM-specific Cre, and SM-specific Sod2−/− mice
| Groups | SM, % | Collagen, % | Urothelium, % | Collagen-to-SM Ratio |
|---|---|---|---|---|
| Wild type | 69.05 ± 2.95 | 22.38 ± 2.48 | 8.58 ± 1.30 | 0.33 ± 0.05 |
| SM-specific Cre | 65.90 ± 5.80 | 24.42 ± 4.42 | 9.70 ± 2.14 | 0.40 ± 0.09 |
| SM-specific Sod2−/− | 60.46 ± 1.90 | 27.18 ± 2.39 | 12.36 ± 1.29 | 0.46 ± 0.05 |
Data are expressed as means ± SE; n = 5 mice/group. All mice were treated with OHT at 8 wk of age and assessed 8 wk after the final OHT injection.
RESULTS
Generation of inducible SM-specific Sod2 deletion mice.
After initial crosses of homozygous floxed Sod2 (Sod2lox/lox) mice with heterozygous SM-specific Cre [SM-CreERT2(ki)Cre/+] mice and identification of double heterozygous [Sod2lox/+,SM-CreERT2(ki)Cre/+] offspring by PCR, matings of double heterozygous mice to each other yielded the four genotypes used in our experiments (Fig. 2). Genomic DNA from tail biopsies was amplified by PCR with the primer pairs described in materials and methods. As shown in Fig. 2A, a representative wild-type mouse yielded a 500-bp product corresponding to the wild-type Sod2 gene (lane 1) and a 267-bp product indicating the wild-type SM22α gene (lane 3), but no bands corresponding to floxed Sod2 or SM-CreERT2 knock-in alleles (lanes 2 and 4, respectively). Mice homozygous for floxed Sod2 yielded a 358-bp product for Sod2lox (Fig. 2B, lane 2) and a 267-bp product for the wild-type SM22α gene (Fig. 2B, lane 3). SM-specific Cre mice [SM-CreERT2(ki)Cre/+] yielded a 220-bp product indicating the SM-CreERT2(ki) transgene (Fig. 2C, lane 4), as well as a 267-bp product for the wild-type SM22α allele (Fig. 2C, lane 3) and a 500-bp product for the wild-type Sod2 gene (Fig. 2C, lane 1). Genotype analysis of a representative inducible SM-specific Sod2 deletion mouse [Sod2lox/lox,SM-CreERT2(ki)Cre/+] is shown in Fig. 2D. In addition to the 220- and 267-bp products for the SM-CreERT2(ki) transgene and the wild-type SM22α allele (lanes 4 and 3, respectively), a 358-bp product for floxed Sod2 was generated (lane 2), while no product corresponding to wild-type Sod2 was observed.
Fig. 2.
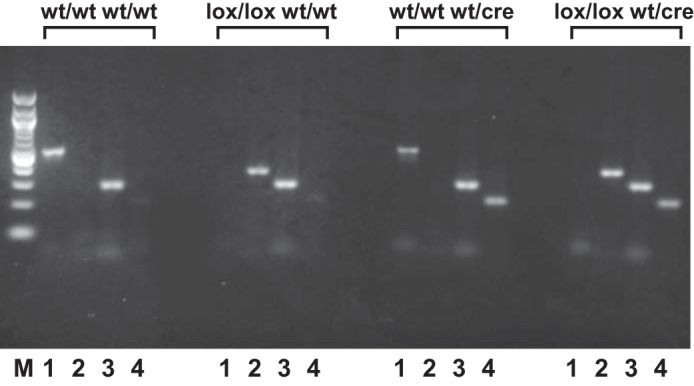
Representative PCR products amplified from genomic DNA isolated from tail biopsies of mice with different genotypes. wt/wt wt/wt, wild-type mouse; lox/lox wt/wt, mouse with homozygous floxed Sod2 and wild-type SM22α genes (Sod2lox/lox); wt/wt wt/cre, mouse with homozygous wild-type Sod2 and heterozygous CreERT2 knock-in [SM-CreERT2(ki)Cre/+ or SM-specific Cre]; lox/lox wt/cre, inducible SM-specific Sod2 deletion mouse [Sod2lox/lox,SM-CreERT2(ki)Cre/+]. The primer pairs in the PCR reactions in A–D were as follows: lane 1, primers P1 and P2, which amplify a 500-bp product from the wild-type Sod2 allele; lane 2, primers P1 and P4, which amplify a 358-bp product from the floxed Sod2 allele; lane 3, primers RF67 and RF90, which amplify a 276-bp product from the wild-type SM22α allele; and lane 4, primers RF67 and SC135, which amplify a 220-bp product from the SM-CreERT2(ki) transgene; lane M, 100-bp DNA ladder (New England Biolabs, Ipswich, MA).
To assess the tissue specificity of Sod2 exon 3 excision by activated CreERT2 (Fig. 1), Sod2lox/lox,SM-CreERT2(ki)Cre/+ mice were treated with OHT, and different organs were harvested 8 wk after the final OHT injection. DNA was isolated from the tissues, and PCR was performed using primers P1 and P3 to amplify a 400-bp fragment from Sod2 alleles with exon 3 excised. As shown in Fig. 3, strong bands of 400 bp were generated from bladder SM (denuded of urothelium) and aorta, the two tissues consisting predominantly of SM, confirming Cre-mediated excision of the floxed Sod2 allele in those tissues. Weaker bands were generated from urethra, ureter, bladder urothelium, skeletal muscle, liver, and heart, presumably due to excision of Sod2 exon 3 in the SM component of the urethra and in vascular SM infiltrating those tissues.
Fig. 3.
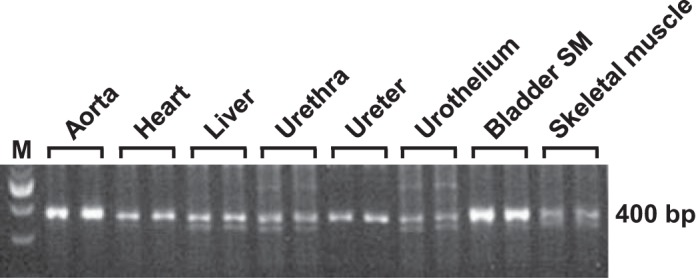
Representative results of PCR amplification with primers P1 and P3 from genomic DNA isolated from different tissues of a Sod2lox/lox,SM-CreERT2(ki)Cre/+ mouse treated with OHT. Sense primer P1 and antisense primer P3 corresponding to sequences in Sod2 exons 2 and 3, respectively, delineate a 400-bp region in Sod2 alleles with floxed exon 3 excised. Lane M, 100-bp DNA ladder.
Depletion of MnSOD in SM in OHT-treated Sod2lox/lox,SM-CreERT2(ki)Cre/+ mice.
Immunoblot analysis revealed that injection of OHT into Sod2lox/lox,SM-CreERT2(ki)Cre/+ mice resulted in a dramatic depletion of MnSOD protein in bladder detrusor SM, whereas no effect of OHT on detrusor MnSOD content was observed in wild-type mice, mice with homozygous floxed Sod2 only (Sod2lox/lox), or mice with heterozygous CreERT2 knock-in only [SM-CreERT2(ki)Cre/+] (Fig. 4A). Conversely, the protein expression of Cu/ZnSOD was similar in detrusor SM in all four genotypes, with or without OHT injection.
Fig. 4.
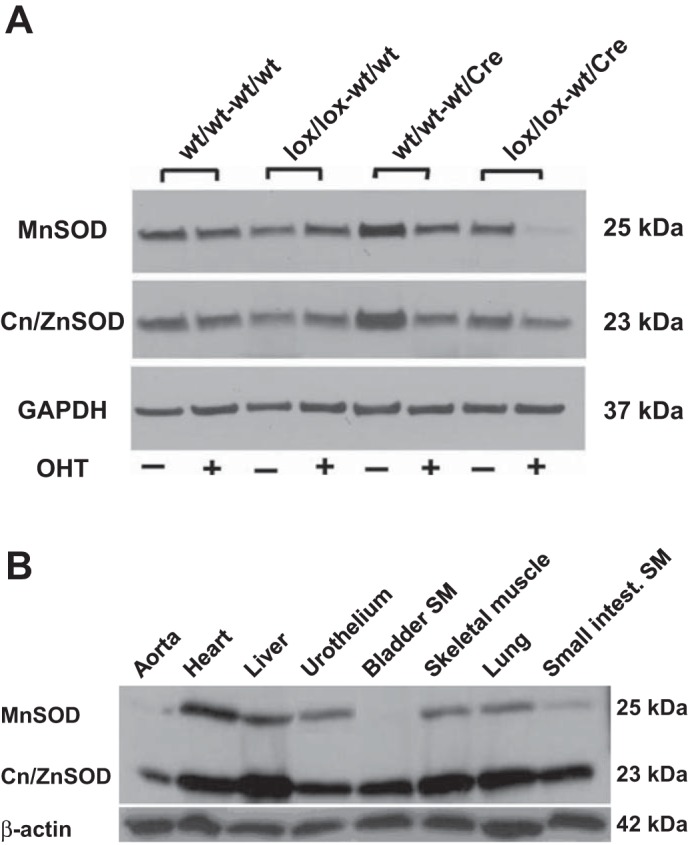
A: representative immunoblots of manganese superoxide dismutase (MnSOD), copper/zinc superoxide dismutase (Cu/ZnSOD), and GAPDH in bladder SM obtained from mice with different genotypes with (+) or without (−) OHT injection. B: representative immunoblots of MnSOD, Cu/ZnSOD, and β-actin in different tissues of OHT-treated Sod2lox/lox,SM-CreERT2(ki)Cre/+ mice.
To confirm that the Cre-mediated deletion of Sod2 was restricted to SM-containing tissues, the expression of MnSOD protein was examined by immunoblot analysis in different tissues obtained from OHT-treated Sod2lox/lox,SM-CreERT2(ki)Cre/+ mice (Fig. 4B). A marked decline of MnSOD protein expression was observed in detrusor SM and other predominantly SM tissues (aorta and small intestinal SM), whereas MnSOD was expressed in heart, liver, lung, skeletal muscle, and urothelium. On the other hand, Cu/ZnSOD protein was abundantly expressed in all of the examined tissues from OHT-treated Sod2lox/lox,SM-CreERT2(ki)Cre/+ mice.
Immunohistochemical evidence of MnSOD depletion in SM-specific Sod2−/− mice.
Positive immunohistochemical staining of MnSOD was observed in both detrusor SM and urothelium in wild-type mice without or with OHT treatment (Fig. 5, A and B, respectively), as well as in Sod2lox/lox,SM-CreERT2(ki)Cre/+ mice without OHT treatment (Fig. 5C). OHT treatment of Sod2lox/lox,SM-CreERT2(ki)Cre/+ mice resulted in a loss of MnSOD immunoreactivity in detrusor SM, without affecting the MnSOD level in the urothelium (Fig. 5D).
Fig. 5.

Representative photomicrographs showing immunohistochemical staining of MnSOD in transverse sections of bladder specimens from wild-type and Sod2lox/lox,SM-CreERT2(ki)Cre/+ mice, with or without OHT treatment, confirming that OHT treatment depleted MnSOD in detrusor SM of inducible SM-specific Sod2 deletion mice (modification: ×40). Bladder sections from a wild-type mouse without OHT injection (A), an OHT-treated wild-type mouse (B), and a Sod2lox/lox,SM-CreERT2(ki)Cre/+ mouse without OHT injection (C) show positive staining of MnSOD in detrusor SM and urothelium. Conversely, a bladder section from an OHT-treated Sod2lox/lox,SMCreERT2(ki)Cre/+ mouse (SM-specific Sod2−/−) (D) shows positive staining of MnSOD in urothelium, but not in detrusor SM. U, urothelium.
SOD activity.
As shown in Fig. 6, total SOD activity was markedly decreased in detrusor homogenates of SM-specific Sod2−/− mice compared with wild-type mice due entirely to a decline of MnSOD activity (by ∼76%) without a significant reduction in Cu/ZnSOD activity.
General characteristics of SM-specific Sod2−/− mice.
In the following experiments, SM-specific Sod2−/− mice [OHT-treated Sod2lox/lox,SM-CreERT2(ki)Cre/+ mice] were compared with age-matched wild-type and SM-specific Cre [OHT-treated Sod2+/+,SM-CreERT2(ki)Cre/+] control mice, with all mice treated with OHT at 8 wk of age. Eight weeks after the final OHT injection, there were no significant differences in body weight, bladder weight, or bladder weight-to-body weight ratio among those three genotypes (Table 1). The SM-specific Sod2−/− mice appeared phenotypically normal and showed no significant differences in growth or survival compared with the control mice when monitored out to 21 wk of age.
Increased OS in bladder detrusor of SM-specific Sod2−/−.
In the remaining experiments described below, OHT-treated SM-specific Sod2−/− mice, SM-specific Cre mice, and wild-type mice were assessed 12 wk after the final OHT injection. Nitration of protein tyrosine residues by peroxynitrite, the product of the reaction of nitric oxide with superoxide, is a recognized marker of severe or prolonged OS (38). Protein nitrotyrosination was measured in detrusor SM extracted from OHT-treated SM-specific Sod2−/− and control mice by immunoblotting. Major bands were detected by the nitrotyrosine antibody at about 26 kDa and in the 50- to 75-kDa range, the latter likely representing multiple nitrotyrosine-containing proteins. The intensities of all of those bands were quantified in each lane and normalized to the corresponding actin signal. SM-specific Sod2−/− mice exhibited a significantly higher mean level of the major nitrotyrosine-containing proteins, by 1.9-fold (Fig. 7, A and B), along with 80% reduction in MnSOD protein in bladder SM (Fig. 7, C and D) compared with wild-type mice.
Surprisingly, the mean nitrotyrosine levels in detrusor SM from SM-specific Cre mice were significantly higher than in wild-type mice (Fig. 7, A and B), indicating an effect of transiently activated CreERT2 on development of OS in the absence of MnSOD depletion (Fig. 7, C and D).
Higher voiding frequency in SM-specific Sod2−/− mice.
The 24-h micturition parameters and fluid consumed by wild-type, SM-specific Cre, and SM-specific Sod2−/− mice are summarized in Table 2. There were no significant differences in 24-h fluid consumed or total urine output among the three groups. However, the mean voiding frequency in SM-specific Sod2−/− mice was significantly higher compared with both wild-type and SM-specific Cre mice. The mean volume per void was significantly lower in SM-specific Sod2−/− mice compared with wild-type mice, but not compared with the SM-specific Cre mice.
Table 2.
24-h fluid consumed and voiding parameters in wild-type, SM-specific Cre, and SM-specific Sod2−/− mice
| 24-h Voiding Parameters |
|||||
|---|---|---|---|---|---|
| Groups | n | 24-h Fluid Consumed, ml | Voiding events | Mean void volume, ml/event | Total urine, ml |
| Wild type | 9 | 33.0 ± 0.94 | 53 ± 3.12 | 0.49 ± 0.036 | 25.0 ± 0.91 |
| SM-specific Cre | 6 | 28.5 ± 1.09 | 52 ± 5.16 | 0.45 ± 0.043 | 23.5 ± 0.83 |
| SM-specific Sod2−/− | 13 | 31.2 ± 1.12 | 74 ± 4.70* | 0.37 ± 0.024# | 26.3 ± 1.17 |
Data are expressed as means ± SE; n, no. of mice. All mice were treated with OHT at 8 wk of age and assessed 12 wk after the final OHT injection.
Significantly higher than wild type (P = 0.006) and SM-specific Cre (P = 0.011).
Significantly lower than wild type (P = 0.026).
Evidence for overactive bladder revealed by conscious CMG.
The results of conscious CMG revealed some differences between SM-specific Sod2−/− mice and controls suggestive of bladder overactivity in the knockout mice (Table 3). Functional bladder capacity was significantly lower and the intercontraction interval between successive voiding reflexes was significantly shorter in SM-specific Sod2−/− mice compared with wild-type mice, although not compared with the SM-specific Cre mice. There were no significant differences between the SM-specific Cre and wild-type control groups in any of the CMG parameters. However, there were significant linear trends from the wild-type to the SM-specific Cre to the SM-specific Sod2−/− group toward lower voiding volume (P = 0.025) and intercontraction interval (P = 0.016).
Table 3.
CMG parameters in wild-type, SM-specific Cre, and SM-specific Sod2−/− mice
| Parameter | Wild Type (n = 6) | SM-Specific Cre (n = 6) | SM-Specific Sod2−/− (n = 8) |
|---|---|---|---|
| Peak voiding pressure, cmH2O | 31.81 ± 1.55 | 32.57 ± 1.53 | 36.95 ± 3.10 |
| Functional bladder capacity, ml | 0.24 ± 0.03 | 0.15 ± 0.01 | 0.13 ± 0.03* |
| Voiding volume, ml | 0.12 ± 0.015 | 0.081 ± 0.009 | 0.070 ± 0.017 |
| Residual volume, ml | 0.11 ± 0.032 | 0.066 ± 0.020 | 0.049 ± 0.029 |
| Compliance, ml/cmH2O | 0.052 ± 0.013 | 0.039 ± 0.008 | 0.023 ± 0.009 |
| Intercontraction interval, min | 14.2 ± 1.93 | 8.77 ± 0.82 | 7.41 ± 2.04* |
Data are expressed as means ± SE; n, no. of mice. CMG, cystometry. All mice were treated with OHT at 8 wk of age and assessed 12 wk after the final OHT injection.
Significantly lower than wild type (P < 0.05).
Similar gross bladder morphology in SM-specific Sod2−/− mice and controls.
Histological examination of Masson's trichrome-stained cross-sectional areas of bladder specimens from wild-type, SM-specific Cre, and SM-specific Sod2−/− mice revealed no significant morphological differences. In all three groups, the detrusor muscle bundles were regularly arranged and closely packed, the urothelial cells were tightly packed, and the collagen fibers, mainly localized in the lamina propria and between the muscle bundles, were dense (Fig. 8).
Fig. 8.
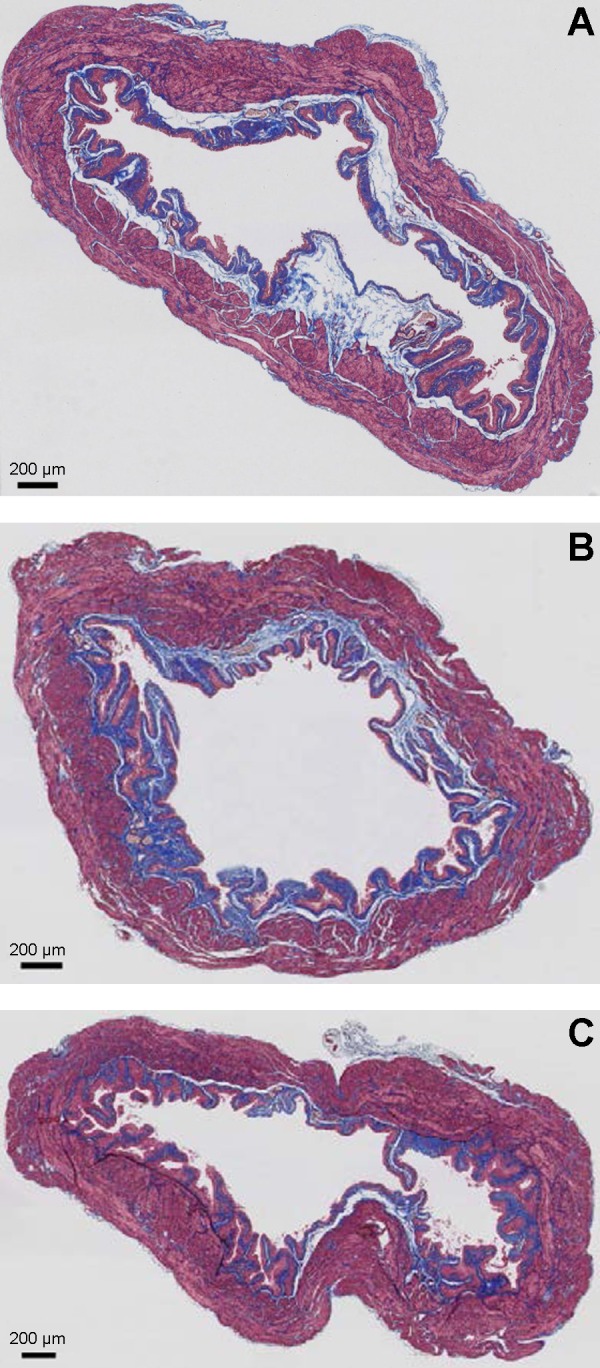
Representative images of Masson's trichrome staining of equatorial bladder sections of age-matched wild-type mice (A), SM-specific Cre mice (B), and SM-specific Sod2−/− mice (C) revealing SM (outer magenta), collagen (blue), and urothelium (inner light magenta). Scale bar indicates 200 μm.
The three major bladder components, detrusor SM, urothelium, and collagen, were quantified and expressed as percentages of the total tissue area (Table 4). From the wild-type to SM-specific Cre to SM-specific Sod2−/− groups, possible linear trends toward increased collagen and urothelium relative to detrusor SM were observed, but were not significant.
DISCUSSION
Many lines of experimental and clinical evidence suggest that OS plays a key role in the pathogenesis of bladder dysfunction in different pathological conditions, such as diabetes (3), bladder outlet obstruction (16), and ischemic overactive bladder (2). Investigating the pathological role of OS in bladder dysfunction is of great clinical interest and may encourage evaluation of the efficiency of antioxidant therapy in ameliorating bladder dysfunction.
Although there are three SOD isoforms in mammals, MnSOD is considered to be the first and most important line of defense against OS. This is mainly because the mitochondrial electron transport is the major source of superoxide anions. It has been reported that low expression or decreased enzymatic activity of MnSOD can contribute to excessive production of superoxide radicals and other toxic oxidants, resulting in cellular damage through oxidation of proteins and lipids, which in turn can lead to disruptions in protein function and membrane symmetry (37). Global homozygous Sod2 knockout mice have been developed, but those mice died with severe dilated cardiomyopathy and neurodegeneration within a few days after birth, revealing a vital role for MnSOD in development and/or survival (13, 15). The availability of several different transgenic mouse lines expressing Cre in a tissue-specific manner, along with mice containing floxed Sod2 alleles, has led to the development of multiple viable tissue-specific Sod2 knockout mice. The floxed Sod2 transgenic mouse line used in our study, which allows for Cre-mediated excision of essential exon 3, has been used previously for selective deletion of Sod2 from tissues including liver (10), cardiac/skeletal muscle (31), brain (30), skeletal muscle (22), and kidney (34).
Our interest in the role of OS in bladder dysfunction, in particular through its effect on detrusor SM, led us to generate an inducible SM-specific Sod2−/− mouse model to investigate the effects of MnSOD depletion in SM. Three different promoters have been reported to direct the expression of Cre recombinase in SM cells in mice, including SM myosin heavy chain (23), SM α-actin (39), and SM22α (transgelin) (1). Although the SM α-actin promoter appears to be the strongest of those three, it has yielded substantial expression in bone and heart in several founders (24). SM22α is a calponin-related protein that is expressed specifically in SM cells of adult mice. The SM-CreERT2(ki) transgenic mouse used in our study provides the additional advantage of allowing inducible activation of the CreERT2 fusion protein by OHT in adult mice (12). The dose and schedule of OHT administration used in this study was previously shown to be nontoxic (12), and we did not detect any deleterious effects of the drug in the mice in our study.
The efficiency and specificity of OHT-induced, Cre-mediated excision of Sod2 exon 3 and depletion of MnSOD in Sod2lox/lox,SM-CreERT2(ki)Cre/+ mice were evaluated by PCR, immunoblotting, IHC, and SOD activity assay. Amplification of the 400-bp P1–P3 product indicating excision of exon 3 was more robust with genomic DNA isolated from bladder SM and SM-rich thoracic aorta than from the other tissues in OHT-treated Sod2lox/lox,SM-CreERT2(ki)Cre/+ mice shown in Fig. 3. Most of the tissues yielding weaker 400-bp bands in Fig. 3 (urothelium, heart, liver, skeletal muscle) lack SM other than in blood vessels, although the urethral and ureteral walls contain thin layers of SM.
Confirmation of SM-specific depletion of MnSOD was provided by immunoblot analysis of tissues harvested from Sod2lox/lox,SM-CreERT2(ki)Cre/+ mice 8 wk after OHT injection, which showed suppression of MnSOD protein expression in SM-containing tissues, to very low levels in aorta and small intestinal SM and to a nearly undetectable levels in bladder SM (Fig. 4B). The reduction of MnSOD enzymatic activity in detrusor SM of SM-specific Sod2−/− mice by 76% (Fig. 6) correlates well with the 80% reduction of MnSOD protein shown by immunoblot analysis (Fig. 7, C and D). More complete elimination of MnSOD in the detrusor tissue was likely precluded by MnSOD-expressing endothelial, nerve, and other cells infiltrating the tissue. Our results concur with previous studies that reported 60–82% reductions in MnSOD activity in other tissue-specific Sod2 knockout mouse models (10, 22, 34). The absence of an effect of Sod2 deletion on the expression or enzymatic activity of Cu/ZnSOD in detrusor SM (Figs. 4A and 6) indicates that regulation of Cu/ZnSOD is independent of MnSOD expression.
The dependence of Cre-mediated depletion of MnSOD on OHT injection in Sod2lox/lox,SM-CreERT2(ki)Cre/+ mice was shown by immunoblot and IHC analyses (Figs. 4A and 6, respectively). These results confirmed that Cre-ERT2 is a tightly regulated recombinase that displays undetectable activity in the absence of OHT and can be effectively activated by OHT treatment. Thus, MnSOD depletion could be induced at a specific time point in adult mice. SM-specific Sod2−/− mice that had been treated with OHT at 8 wk of age displayed normal growth patterns and behavior and survived as well as wild-type mice until killed 12 wk later.
A significant increase in expression of nitrotyrosine, a commonly used OS marker (38), was observed in detrusor SM in SM-specific Sod2−/− mice compared with OHT-treated wild-type and SM-specific Cre (Fig. 7, A and B), illuminating the important role of MnSOD in protection against free radicals produced during normal physiological conditions. Surprisingly, OHT treatment of SM-specific Cre mice containing two wild-type Sod2 alleles also resulted in a significant increase in nitrotyrosine level in detrusor SM tissue compared with wild-type mice (Fig. 7, A and B). Although it was originally assumed that Cre activation in mammalian cells does not interfere with cellular functions, a growing body of literature has reported some adverse effects of activated Cre recombinase, especially at high expression levels, in the absence of introduced loxP sequences. These effects include growth inhibition and DNA damage in mammalian cell lines (21), as well as gastric epithelial atrophy (9), glucose intolerance (14), and dilated cardiomyopathy (4) in transgenic mice expressing Cre globally, in pancreatic β-cells, and in cardiac muscle, respectively. Earlier studies have shown that Cre recombinase can identify and catalyze the excision between cryptic LoxP sites, naturally present in mammalian genome, which can eventually trigger several stress pathways (36). More relevant to our study is a recent report indicating that the expression of Cre in Sertoli cells in the testes of mice was associated with enhanced lipid peroxidation and OS, with more pronounced effects observed in homozygous Cre transgenic mice than in heterozygous mice (40). In our study, heterozygous expression of CreERT2 and activation by OHT in SM-specific Cre mice resulted in OS (Fig. 7, A and B), without affecting MnSOD expression (Fig. 7, C and D), in bladder SM. This result underscores the importance of including control mice that express active Cre in the absence of introduced loxP sites for an appropriate time when using the Cre-loxP recombination system.
The effect of conditional SM-specific depletion of MnSOD on bladder function was assessed by 24-h urine output analysis and conscious CMG. The SM-specific Sod2−/− mice displayed higher micturition frequency than both the wild-type and SM-specific Cre mice, and lower mean void volume than the wild-type group (Table 2). The CMG data revealed a significant decrease in functional bladder capacity and shorter intercontraction intervals in SM-specific Sod2−/− mice compared with wild-type mice (Table 3). Those data indicate bladder overactivity in the SM-specific Sod2−/− mice. However, the peak voiding pressure and residual volume did not differ significantly in the SM-specific Sod2−/− mice compared with the wild mice, suggesting that bladder voiding efficiency was largely unaffected by 12 wk of Sod2 depletion. Furthermore, an apparent trend toward lower bladder compliance from wild-type to SM-specific Cre to SM-specific Sod2−/− mice did not reach significance, suggesting that the tissue composition of the bladder was relatively unchanged. That deduction is supported by the absence of significant differences in the percentages of detrusor SM, collagen, and urothelium areas (Table 4), despite apparent, but nonsignificant, trends from wild-type to SM-specific Cre to SM-specific Sod2−/− mice toward increased collagen and urothelium relative to SM.
The enhanced OS in bladder SM in SM-specific Sod2−/− mice may cause bladder overactivity through its effects on the detrusor SM tissue itself or by affecting the nerve fibers innervating the detrusor, which are essential for normal micturition. Recently, it has been shown that OS was correlated with increased collagen and reduced bladder capacity in a rat model of chronic bladder ischemia, which was further supported by melatonin treatment that restored bladder capacity and improved bladder functions, through its antioxidative properties (32). Masuda et al. demonstrated that OS induced by H2O2 activated C-fiber afferent pathways, leading to detrusor overactivity in the bladders of anesthetized rats (27). Moreover, it was shown that OS was associated with bladder hyperactivity (evidenced by a decrease in the voiding interval) in a rat model of atherosclerosis-induced chronic bladder ischemia (33).
Although SM-specific Cre mice had an increased level of nitrotyrosine in detrusor SM, between the levels in wild-type and SM-specific Sod2−/− mice, there were no significant differences between wild-type and SM-specific Cre mice in any of the micturition or CMG parameters. Thus, the level of OS in SM-specific Cre mice appears to have been insufficient to cause bladder overactivity. However, the significant trend toward lower voiding volume from wild-type to SM-specific Cre to SM-specific Sod2−/− mice suggests that the observed effect of transiently active Cre on OS independent of MnSOD may have contributed to the bladder overactivity caused by Sod2 deletion.
Although some reports (5, 25) have indicated that OS is associated with impaired detrusor contractility, we have postulated that the amount and duration of ROS generation are important factors that affect detrusor contractility (7, 18). A previous study has shown that the adverse effects of intravesicular administration of H2O2 on bladder functions were dose dependent (27). Low concentrations of H2O2 (0.003–0.3%) induced detrusor hyperactivity, associated with an increased peak voiding pressure. However, a high dose of H2O2 (3%) significantly reduced the peak voiding pressure and impaired detrusor contractility (27). Excessive free radicals can induce destructive changes in cellular and subcellular components of detrusor muscle cells, including lipids, proteins, and DNA, and ultimately result in cellular degeneration and activation of apoptotic pathways, which in turn contribute to impairment of detrusor function (11). Therefore, the effects of longer periods of SM-specific Sod2 depletion on ROS accumulation and bladder function and morphology are pertinent areas for future study.
In conclusion, we successfully created and characterized a novel mouse model with inducible SM-specific deletion of the Sod2 gene. SM-specific deletion of Sod2 in 8-wk-old male mice resulted in normal growth, behavior, and survival, at least up to the experimental endpoint of 21 wk of age. No gross morphological changes were detected in bladders of SM-specific Sod2−/− mice; however, the depletion of MnSOD caused OS in the bladder and affected the bladder function. This mouse model can be used to investigate the role of OS in different pathological conditions in the bladder and other SM-rich tissues, and may be useful in evaluating therapeutic interventions.
GRANTS
This study was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants U01-DK-076162, P20-DK-090871, and R01-DK-083733. R. Elrashidy was supported by a fund from the Egyptian Cultural Affairs and Missions Sector, within the joint supervision scholarship.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: G.L. and F.D. conception and design of research; G.L., R.A.E., N.X., M.K., Y.H., and M.T. performed experiments; G.L., R.A.E., E.K., and G.S. analyzed data; G.L. and R.A.E. interpreted results of experiments; G.L., R.A.E., and C.T.P. prepared figures; G.L. and R.A.E. drafted manuscript; G.L., R.A.E., C.T.P., and H.E.M. edited and revised manuscript; G.L., R.A.E., N.X., M.K., Y.H., M.T., C.T.P., E.K., G.S., H.E.M., and F.D. approved final version of manuscript.
REFERENCES
- 1.Akyurek LM, Yang ZY, Aoki K, San H, Nabel GJ, Parmacek MS, Nabel EG. SM22alpha promoter targets gene expression to vascular smooth muscle cells in vitro and in vivo. Mol Med 6: 983–991, 2000. [PMC free article] [PubMed] [Google Scholar]
- 2.Azadzoi KM, Radisavljevic ZM, Golabek T, Yalla SV, Siroky MB. Oxidative modification of mitochondrial integrity and nerve fiber density in the ischemic overactive bladder. J Urol 183: 362–369, 2010. [DOI] [PubMed] [Google Scholar]
- 3.Beshay E, Carrier S. Oxidative stress plays a role in diabetes-induced bladder dysfunction in a rat model. Urology 64: 1062–1067, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Buerger A, Rozhitskaya O, Sherwood MC, Dorfman AL, Bisping E, Abel ED, Pu WT, Izumo S, Jay PY. Dilated cardiomyopathy resulting from high-level myocardial expression of Cre-recombinase. J Card Fail 12: 392–398, 2006. [DOI] [PubMed] [Google Scholar]
- 5.Changolkar AK, Hypolite JA, Disanto M, Oates PJ, Wein AJ, Chacko S. Diabetes induced decrease in detrusor smooth muscle force is associated with oxidative stress and overactivity of aldose reductase. J Urol 173: 309–313, 2005. [DOI] [PubMed] [Google Scholar]
- 6.Daneshgari F, Huang X, Liu G, Bena J, Saffore L, Powell CT. Temporal differences in bladder dysfunction caused by diabetes, diuresis, and treated diabetes in mice. Am J Physiol Regul Integr Comp Physiol 290: R1728–R1735, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Daneshgari F, Liu G, Birder L, Hanna-Mitchell AT, Chacko S. Diabetic bladder dysfunction: current translational knowledge. J Urol 182: S18–S26, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feil R, Wagner J, Metzger D, Chambon P. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Commun 237: 752–757, 1997. [DOI] [PubMed] [Google Scholar]
- 9.Huh WJ, Mysorekar IU, Mills JC. Inducible activation of Cre recombinase in adult mice causes gastric epithelial atrophy, metaplasia, and regenerative changes in the absence of “floxed” alleles. Am J Physiol Gastrointest Liver Physiol 299: G368–G380, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ikegami T, Suzuki Y, Shimizu T, Isono K, Koseki H, Shirasawa T. Model mice for tissue-specific deletion of the manganese superoxide dismutase (MnSOD) gene. Biochem Biophys Res Commun 296: 729–736, 2002. [DOI] [PubMed] [Google Scholar]
- 11.Kanika ND, Chang J, Tong Y, Tiplitsky S, Lin J, Yohannes E, Tar M, Chance M, Christ GJ, Melman A, Davies KD. Oxidative stress status accompanying diabetic bladder cystopathy results in the activation of protein degradation pathways. BJU Int 107: 1676–1684, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuhbandner S, Brummer S, Metzger D, Chambon P, Hofmann F, Feil R. Temporally controlled somatic mutagenesis in smooth muscle. Genesis 28: 15–22, 2000. [DOI] [PubMed] [Google Scholar]
- 13.Lebovitz RM, Zhang H, Vogel H, Cartwright J Jr, Dionne L, Lu N, Huang S, Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci USA 93: 9782–9787, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JY, Ristow M, Lin X, White MF, Magnuson MA, Hennighausen L. RIP-Cre revisited, evidence for impairments of pancreatic beta-cell function. J Biol Chem 281: 2649–2653, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet 11: 376–381, 1995. [DOI] [PubMed] [Google Scholar]
- 16.Lin WY, Guven A, Juan YS, Neuman P, Whitbeck C, Chichester P, Kogan B, Levin RM, Mannikarottu A. Free radical damage as a biomarker of bladder dysfunction after partial outlet obstruction and reversal. BJU Int 101: 621–626, 2008. [DOI] [PubMed] [Google Scholar]
- 17.Lin YH, Liu G, Kavran M, Altuntas CZ, Gasbarro G, Tuohy VK, Daneshgari F. Lower urinary tract phenotype of experimental autoimmune cystitis in mouse: a potential animal model for interstitial cystitis. BJU Int 102: 1724–1730, 2008. [DOI] [PubMed] [Google Scholar]
- 18.Liu G, Daneshgari F. Diabetic bladder dysfunction. Chinese Med J 127: 1357–1364, 2014. [PMC free article] [PubMed] [Google Scholar]
- 19.Liu G, Lin YH, Li M, Xiao N, Daneshgari F. Temporal morphological and functional impact of complete urinary diversion on the bladder: a model of bladder disuse in rats. J Urol 184: 2179–2185, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, Zhao Y, Pawlyk B, Damaser M, Li T. Failure of elastic fiber homeostasis leads to pelvic floor disorders. Am J Pathol 168: 519–528, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loonstra A, Vooijs M, Beverloo HB, Allak BA, van Drunen E, Kanaar R, Berns A, Jonkers J. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proc Natl Acad Sci USA 98: 9209–9214, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lustgarten MS, Jang YC, Liu Y, Muller FL, Qi W, Steinhelper M, Brooks SV, Larkin L, Shimizu T, Shirasawa T, McManus LM, Bhattacharya A, Richardson A, Van Remmen H. Conditional knockout of Mn-SOD targeted to type IIB skeletal muscle fibers increases oxidative stress and is sufficient to alter aerobic exercise capacity. Am J Physiol Cell Physiol 297: C1520–C1532, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Madsen CS, Regan CP, Hungerford JE, White SL, Manabe I, Owens GK. Smooth muscle-specific expression of the smooth muscle myosin heavy chain gene in transgenic mice requires 5′-flanking and first intronic DNA sequence. Circ Res 82: 908–917, 1998. [DOI] [PubMed] [Google Scholar]
- 24.Maeda S, Sutliff RL, Qian J, Lorenz JN, Wang J, Tang H, Nakayama T, Weber C, Witte D, Strauch AR, Paul RJ, Fagin JA, Clemens TL. Targeted overexpression of parathyroid hormone-related protein (PTHrP) to vascular smooth muscle in transgenic mice lowers blood pressure and alters vascular contractility. Endocrinology 140: 1815–1825, 1999. [DOI] [PubMed] [Google Scholar]
- 25.Mannikarottu AS, Changolkar AK, Disanto ME, Wein AJ, Chacko S. Over expression of smooth muscle thin filament associated proteins in the bladder wall of diabetics. J Urol 174: 360–364, 2005. [DOI] [PubMed] [Google Scholar]
- 26.Marecki JC, Parajuli N, Crow JP, MacMillan-Crow LA. The use of the Cre/loxP system to study oxidative stress in tissue-specific manganese superoxide dismutase knockout models. Antioxid Redox Signal 20: 1655–1670, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masuda H, Kihara K, Saito K, Matsuoka Y, Yoshida S, Chancellor MB, de Groat WC, Yoshimura N. Reactive oxygen species mediate detrusor overactivity via sensitization of afferent pathway in the bladder of anaesthetized rats. BJU Int 101: 775–780, 2008. [DOI] [PubMed] [Google Scholar]
- 28.Metzger D, Chambon P. Site- and time-specific gene targeting in the mouse. Methods 24: 71–80, 2001. [DOI] [PubMed] [Google Scholar]
- 29.Miao L, St. Clair DK. Regulation of superoxide dismutase genes: implications in disease. Free Rad Biol Med 47: 344–356, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Misawa H, Nakata K, Matsuura J, Moriwaki Y, Kawashima K, Shimizu T, Shirasawa T, Takahashi R. Conditional knockout of Mn superoxide dismutase in postnatal motor neurons reveals resistance to mitochondrial generated superoxide radicals. Neurobiol Dis 23: 169–177, 2006. [DOI] [PubMed] [Google Scholar]
- 31.Nojiri H, Shimizu T, Funakoshi M, Yamaguchi O, Zhou H, Kawakami S, Ohta Y, Sami M, Tachibana T, Ishikawa H, Kurosawa H, Kahn RC, Otsu K, Shirasawa T. Oxidative stress causes heart failure with impaired mitochondrial respiration. J Biol Chem 281: 33789–33801, 2006. [DOI] [PubMed] [Google Scholar]
- 32.Nomiya M, Burmeister DM, Sawada N, Campeau L, Zarifpour M, Yamaguchi O, Andersson KE. Effect of melatonin on chronic bladder-ischaemia-associated changes in rat bladder function. BJU Int 112: E221–E230, 2013. [DOI] [PubMed] [Google Scholar]
- 33.Nomiya M, Sagawa K, Yazaki J, Takahashi N, Kushida N, Haga N, Aikawa K, Matsui T, Oka M, Fukui T, Andersson KE, Yamaguchi O. Increased bladder activity is associated with elevated oxidative stress markers and proinflammatory cytokines in a rat model of atherosclerosis-induced chronic bladder ischemia. Neurourol Urodynamics 31: 185–189, 2012. [DOI] [PubMed] [Google Scholar]
- 34.Parajuli N, Marine A, Simmons S, Saba H, Mitchell T, Shimizu T, Shirasawa T, Macmillan-Crow LA. Generation and characterization of a novel kidney-specific manganese superoxide dismutase knockout mouse. Free Rad Biol Med 51: 406–416, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shimizu T, Nojiri H, Kawakami S, Uchiyama S, Shirasawa T. Model mice for tissue-specific deletion of the manganese superoxide dismutase gene. Geriatr Gerontol Int 10, Suppl 1: S70–S79, 2010. [DOI] [PubMed] [Google Scholar]
- 36.Thyagarajan B, Guimaraes MJ, Groth AC, Calos MP. Mammalian genomes contain active recombinase recognition sites. Gene 244: 47–54, 2000. [DOI] [PubMed] [Google Scholar]
- 37.Vaziri ND, Dicus M, Ho ND, Boroujerdi-Rad L, Sindhu RK. Oxidative stress and dysregulation of superoxide dismutase and NADPH oxidase in renal insufficiency. Kidney Int 63: 179–185, 2003. [DOI] [PubMed] [Google Scholar]
- 38.Viappiani S, Schulz R. Detection of specific nitrotyrosine-modified proteins as a marker of oxidative stress in cardiovascular disease. Am J Physiol Heart Circ Physiol 290: H2167–H2168, 2006. [DOI] [PubMed] [Google Scholar]
- 39.Wang J, Niu W, Witte DP, Chernausek SD, Nikiforov YE, Clemens TL, Sharifi B, Strauch AR, Fagin JA. Overexpression of insulin-like growth factor-binding protein-4 (IGFBP-4) in smooth muscle cells of transgenic mice through a smooth muscle alpha-actin-IGFBP-4 fusion gene induces smooth muscle hypoplasia. Endocrinology 139: 2605–2614, 1998. [DOI] [PubMed] [Google Scholar]
- 40.Xiao Y, Karnati S, Qian G, Nenicu A, Fan W, Tchatalbachev S, Holand A, Hossain H, Guillou F, Luers GH, Baumgart-Vogt E. Cre-mediated stress affects sirtuin expression levels, peroxisome biogenesis and metabolism, antioxidant and proinflammatory signaling pathways. PLoS One 7: e41097, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zorov DB, Bannikova SY, Belousov VV, Vyssokikh MY, Zorova LD, Isaev NK, Krasnikov BF, Plotnikov EY. Reactive oxygen and nitrogen species: friends or foes? Biochemistry (Mosc) 70: 215–221, 2005. [DOI] [PubMed] [Google Scholar]