

Abstract
Recent studies of myocardial infarction in secreted Frizzled-related protein 2 (sFRP2) knockout mice and our hamster heart failure therapy based on sFRP2 blockade have established sFRP2 as a key profibrotic cytokine in the heart. The failing hamster heart is marked by prominent fibrosis and calcification with elevated expression of sFRP2. Noting the involvement of tissue-nonspecific alkaline phosphatase (TNAP) in bone mineralization and vascular calcification, we determined whether sFRP2 might be an upstream regulator of TNAP. Biochemical assays revealed an approximately twofold increase in the activity of TNAP and elevated levels of inorganic phosphate (Pi) in the failing heart compared with the normal heart. Neither was this change detected in the liver or hamstring muscle nor was it associated with systemic hyperphosphatemia. TNAP was readily cloned from the hamster heart and upon overexpression increased the level of extracellular but not intracellular Pi, which is consistent with the cell surface location of the ectoenzyme. In line with the previous demonstration that sFRP2 blockade attenuated fibrosis, we show here that the therapy downregulated TNAP. This in vivo finding is corroborated by the in vitro study showing that cultured cardiac fibroblasts treated with recombinant sFRP2 protein exhibited progressive increase in the expression and activity of TNAP, which was completely abrogated by cycloheximide or tunicamycin. Induction of TNAP by sFRP2 is restricted to cardiac fibroblasts among the multiple cell types examined, and was not observed with sFRP4. The current work indicates that sFRP2 may promote cardiac fibrocalcification through coordinate activation of tolloid-like metalloproteinases and TNAP.
Keywords: sFRP2, TNAP, fibrocalcification, heart failure
the family of secreted Frizzled-related proteins (sFRPs) are known to have multiple roles in development and disease (6, 41, 45). A novel postnatal role of sFRP2 in regulating extracellular matrix (ECM) remodeling was first revealed by Kobayashi et al. (25), demonstrating that sFRP2-null mice exhibited reduced collagen deposition and significantly improved cardiac function after myocardial infarction. This study reveals a profibrotic function of sFRP2 in the adult mouse heart, and is consistent with the findings that heart failure is associated with increased expression of sFRP2, sFRP3, and sFRP4 (3, 37, 47). Since excessive collagen deposition leads to scar formation, distorted tissue architecture, and ultimately organ failure (15, 55), intervention of sFRP2-mediated fibrogenic pathways may represent a novel approach to restoring tissue function. Indeed, we recently documented an antibody-based sFRP2 blockade strategy, which coordinately reduced myocardial fibrosis, increased angiogenesis and improved cardiac function in the failing hamster heart (37). These therapeutic benefits are comparable to those achieved by regenerative therapy based on mesenchymal stem cells (36, 48) or vascular endothelial growth factor (57, 58).
sFRP2 is known to function in Wnt-dependent and -independent mechanisms that are influenced by developmental stage, protein concentration, and cell/tissue specificity (41). Although sFRP2 has traditionally been viewed as a Wnt inhibitor due to the presence of a Frizzled (Fzd) domain and absence of a transmembrane domain, we and others have demonstrated its Wnt-activating function in multiple organ systems (27, 37, 51). This Wnt-activating effect of sFRP2 is noteworthy because excessive activation of Wnt signaling is associated with pulmonary, renal, cardiac, and skeletal muscle fibrosis (7, 12, 23, 52). On the other hand, the profibrotic role of sFRP2 as proposed by Kobayashi et al. (25) appears to operate in a Wnt-independent manner through serving as enhancer of procollagen C proteinase activity of tolloid-like metalloproteinases. Given the complex nature of the fibrogenic machinery, additional targets of sFRP2 likely exist in the dynamic multistage cascade of pathogenic fibrosis.
The ECM serves crucial roles as a site for sequestration and controlled release of cytokines and matrix-modifying enzymes, which can promote adverse ECM remodeling in disease states (53). The secreted enzyme lysyl oxidase catalyzes formation of aldehydes from lysine residues in collagen and elastin, resulting in insoluble and stiff ECM fibrils (18). Tissue transglutaminase (TG2) mediates extensive cross-linking of collagen fibers via isopeptide bond linkages (21). Both lysyl oxidase and TG2 have been recognized as key fibrosis markers (34, 42). Another matrix-modifying player is the phosphate-splitting enzyme termed tissue nonspecific alkaline phosphatase (TNAP), which has been reported as a marker of pulmonary fibrosis in chronic interstitial disorders (9). TNAP is normally induced during the early stage of osteogenesis and participates in collagen calcification during bone formation (8, 39). Recent studies on vascular calcification have revealed TNAP as a pathogenic player in the abnormal mineralization process (14). Although clinical test of circulating levels of alkaline phosphatase is routinely performed to check for a wide range of disease conditions, the relation between TNAP dysregulation and cardiac pathology has not been examined. The current work identified TNAP as a novel enzyme target activated by sFRP2 in myocardial fibrocalcification.1
MATERIALS AND METHODS
Animal use.
The TO2 strain cardiomyopathic hamsters (4-mo-old males) and age-matched normal controls (F1B strain) were obtained from Bio Breeders (Watertown, MA). All procedures and protocols conformed to institutional guidelines for the care and use of animals in research as documented (37) and were approved by the Institutional Animal Care and Use Committee of University at Buffalo. Normal and ischemic porcine heart tissue samples, as described previously (35), were kindly provided by Dr. Gen Suzuki (Department of Medicine, University at Buffalo).
Cell culture.
Cardiac fibroblasts were isolated from the heart of 3- to 5-mo-old Balb/c mice using an established protocol (37). 3T3 fibroblasts, HEK293, C2C12 myoblasts, and HL1 cardiomyocytes were maintained in minimal essential medium (MEM) containing 10% fetal bovine serum (FBS), 50 μg/ml gentamycin, and 0.125 μg/ml amphotericin B (Fungizone). Cardiac fibroblasts receiving less than two trypsin passages were used for experiments. Cells plated on 6- or 12-well plates at 1 to 2 × 105 cells per well were grown to confluency prior to treatment. Cells were then switched to MEM containing 5% FBS and treated with recombinant proteins for 1–3 days, following which cells were rinsed with normal saline and harvested for Western blotting and enzyme assays.
Histology.
Paraffin-embedded tissue sections were used for histological staining. Von Kossa staining for calcified lesions was performed by immersing deparaffinized sections in 2% silver nitrate in a glass Coplin jar, which was then exposed to UV light for ∼3 h in a UV crosslinker (model FB-UVXL-1000, Fisher Scientific). Sections were rinsed with dH2O briefly, immersed in 2% sodium thiosulfate for 1 min, and rinsed with dH2O. The sections were then further processed for staining of fibrosis areas. Masson Trichrome staining for fibrotic areas and quantification are as described previously (49). Analysis of fibrotic areas was performed by Photoshop-aided quantification of image pixels as described previously (49). In brief, the blue color range was selected to represent fibrotic areas. At least 15 random fields at ×200 magnification were assessed for each slide. White space from images was subtracted for calculation of total tissue area. The ratio of fibrotic area to total tissue area was calculated as % fibrotic area.
Tissue homogenization.
Each snap frozen tissue sample (∼30 mg) was homogenized in 1 ml of an ice-cold lysis solution containing 0.1% TX-100 and 2 mM EDTA. Homogenates were briefly clarified by a 5-s, 5,000-rpm spin to remove tissue debris. Clarified homogenates were further centrifuged at 13,000 rpm at 4°C for 5 min to separate the membrane and soluble fractions. The membrane fraction was resuspended in the lysis solution. Protein concentrations were determined using the Protein Assay Kit/DC from Biomedical Research Service Center (University at Buffalo). Trichloroacetic acid (TCA) deproteinization was carried out by homogenizing tissue (∼100 mg) in 1 ml of an ice-cold 10% TCA solution. Homogenates were incubated on ice for 30 min, following which homogenates were centrifuged at 13,000 rpm for 5 min. The supernatant was extracted with 0.5 ml of water-saturated ether three times to remove TCA.
Western blotting.
Protein extracts (1 mg/ml) prepared in SDS-PAGE sample buffer were treated with 1% β-mercaptoethanol and boiled for 10 min. Proteins (40 μg)were loaded in each lane and fractionated by 10% SDS-PAGE. Proteins were electrotransferred to Immobilon-P membrane, which was incubated with a 1,000-fold diluted primary antibody solution overnight at 4°C. Washed membrane was probed with a horseradish peroxidase-conjugated secondary antibody at ∼10 ng/ml. Signals were developed using the SuperSignal chemiluminescent substrate from Pierce Biotechnology and digitally imaged. The TNAP bands and Coomassie Blue-stained bands were quantified by densitometry, and TNAP was normalized to the Coomassie Blue stain as shown in the figure. TNAP antibody (no. sc-30203) and GAPDH antibody (no. sc-20357) were purchased from Santa Cruz Biotechnology.
Enzyme assays.
Colorimetric TNAP and GAPDH assay kits were obtained from University at Buffalo Biomedical Research Service Center. Typically 10−20 μg protein extracts were used for the enzyme assays. Assay of TNAP activity is based on conversion of p-nitrophenol phosphate to nitrophenol in an alkaline buffer. TNAP activity was measured at 405 nm and calculated using a molar extinction coefficient of 18.75 mM/cm. Assay of GAPDH activity is based on reduction of the tetrazolium salt INT in a NADH-coupled enzymatic reaction to formazan, which exhibits an absorption maximum at 492 nm. GAPDH activity was calculated using a molar extinction coefficient of 18 mM/cm. For β-galactosidase (LacZ) assay, 10 μl sample was incubated with 100 μl ONPG substrate (0.1 M sodium phosphate pH 7.4, 1.5 mg/ml o-nitrophenyl-β-d-galactopyranoside, 1 mM MgCl2, and 10 mM β-mercaptoethanol) at 30°C for 60 min. Reaction was terminated by addition of 50 μl 0.5 M Na2CO3. Enzyme activity was measured by absorbance at 414 nm and calculated using a molar extinction coefficient of 4.5 mM/cm.
In-gel TNAP activity staining.
Protein extracts for TNAP activity staining were not treated with β-mercaptoethanol nor boiled prior to SDS-PAGE. After electrophoresis, gel was washed in a buffer containing 50 mM Tris-Cl pH 8, 200 mM NaCl, 5 mM CaCl2, 5 μM ZnSO4, and 2.5% TX-100 three times (15 min each), following which the gel was finally washed in the buffer without TX-100 for another 15 min. The washed gel was incubated in a TNAP substrate solution containing 100 mM Tris-Cl pH 10.5, 2 mM MgCl2, 1 mg/ml 5-bromo-4-chloro-3-indoxyl phosphate, and 0.1 mg/ml 3-[4,5-dimethylthiazol-2-yl]2,5-diphenyltetrazolium bromide at 37°C overnight.
Metabolite assays.
Phosphate and l-lactate assay kits were obtained from University at Buffalo Biomedical Research Service Center. Conditioned culture medium was mixed with an equal volume of ice-cold 10% TCA solution and incubated on ice for 30 min. Tissues were homogenized in 10% TCA as described above. Both assays were set up in 96-well plates. Twenty microliters of each TCA extract (after a 5-fold dilution with dH2O) was mixed with 100 μl of the ammonium molybdate-based phosphate assay solution and 50 μl of the lactate dehydrogenase-based lactate assay solution. Phosphate assay was incubated at room temperature for 10 min and lactate assay was incubated at 37°C for 30 min. A set of NaH2PO4 and lactate standards were used as reference. Absorbance was measured at 620 nm and 492 nm for phosphate and lactate assays, respectively. Sample metabolite concentrations were derived by comparison to a standard curve performed at the same time.
RT-PCR.
RNA extraction was performed using Qiagen's RNA isolation kits. Hamster TNAP cDNA was PCR amplified from hamster heart RNA using primers CACCATGATCTCGCCATTTTTA and CTGGGCCCTCAGAACAGGGTGC. DNA was excised from agarose gel, inserted into pCR2.1-TOPO, and sequenced in both orientations (GenBank accession no. KF934412). DNA sequencing was performed by RPCI DNA sequencing core facility. qRT-PCR was performed to quantify expression of TNAP using the SYBR green kit on MyIQ (Bio-Rad) as previously described (31). β2-Microglobulin was used as the reference gene for data calculation based on the 2ΔΔCT method. β2-Microglobulin and TNAP primers for qRT-PCR are 5′-TCTCTTGGCTCACAGGGAGT/3′-ATGTCTCGTTCCCAGGTGAC and 5′-ACAACCTGACTGACCCTTCG/3′-GCCTTTGAGGTTTTTGGTCA, respectively. Oligonucleotides were synthesized by Midland Oligo.
Replication-deficient TNAP adenovirus.
Construction of replication-deficient recombinant adenovirus was as described previously (30). The TNAP cDNA was inserted into NotI and HindIII sites of the adenoviral shuttle vector pShuttle-CMV. The recombinant shuttle DNA and pAdEasy-1 DNA were recombined in HEK293 cells. The virus was amplified in HEK293 cells to yield ∼109 viral particles/ml. Virus was confirmed by PCR and Western blotting. For gene expression analysis, viral lysates were diluted 10-fold with MEM and added to adherent cells for 3 h with occasional agitation, following which cells were rinsed with HBSS twice and maintained in MEM containing 10% FBS, 50 μg/ml gentamycin, and 0.125 μg/ml Fungizone.
Recombinant proteins.
Commercially available recombinant sFRP2 and sFRP4 proteins were purchased from R&D. To produce bacterially expressed sFRP2, the sFRP2 DNA was excised from pET3d-sFRP2 (37) using NcoI and BamHI and inserted into the glutathione S-transferase (GST) fusion vector pGEX (24). Expression of GST and GST-sFRP2 fusion protein in Escherichia coli (strain BL21) was induced by adding 0.5 mM isopropyl thiogalactopyranoside (IPTG) to a log phase culture for a 3-h induction. GST and GST-sFRP2 were purified by glutathione-agarose chromatography as described previously (24). Purified GST-sFRP2 was cleaved with ∼10 units of thrombin in the presence of 2.5 mM CaCl2 at room temperature for 2 h, following which thrombin and GST were removed by incubation with p-aminobenzamidine and glutathione agarose beads. Proteins were dialyzed in normal phosphate-buffered saline at 4°C and filter sterilized for cell treatment.
Statistical analysis.
Comparisons between two and multiple experimental groups were made with Student's t-test and one-way ANOVA, respectively. A value of P < 0.05 was considered significant. Data are expressed as means ± SD.
RESULTS
Fibrocalcification as a prominent feature in the failing hamster heart.
The δ-sarcoglycan-null TO2 strain hamster develops congestive heart failure and fibrosis resembling that seen in the general population of heart failure patients (44). To determine whether the failing hamster heart exhibits concurrent fibrosis and calcification, we used Trichrome staining and von Kossa staining to locate areas of fibrosis and calcification, respectively. Digital quantification of the histological images showed that the failing heart exhibited ∼12% fibrotic area in comparison to ∼1% fibrotic area in the age-matched F1B strain normal heart (Fig. 1). While no calcification was observed in the F1B heart, prominent calcium deposits (black staining) were found to be associated with the fibrotic areas (blue staining) of the TO2 heart at 5 mo of age (Fig. 1), indicating that cardiac fibrocalcification is a prominent pathology in the failing hamster heart.
Fig. 1.
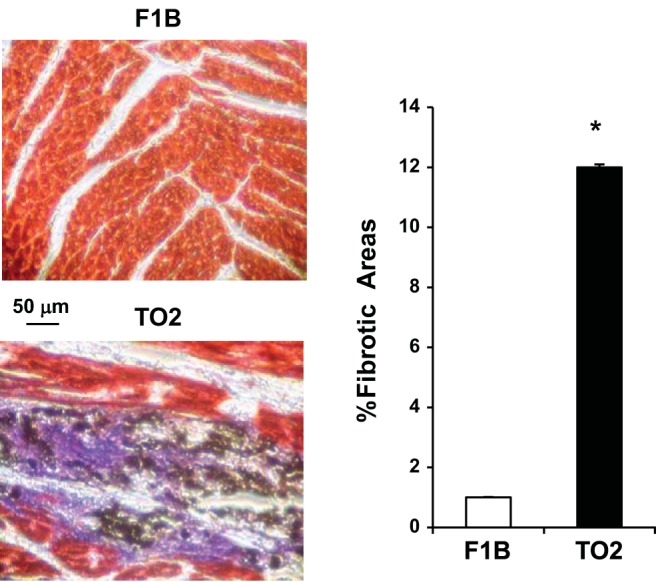
Trichrome and von Kossa double staining of the F1B and TO2 hamster heart. Deparaffinized ventricular tissue sections from 5-mo F1B (normal control) and TO2 (cardiomyopathic) hearts were sequentially processed by von Kossa staining and Trichrome staining. Black, blue, and red staining represents calcium deposits, fibrous tissue, and cardiomyocytes, respectively. Representative images are shown (scale bar, 50 μm). Quantification of fibrotic areas was described previously (49); n = 5 per group (*P < 0.05).
Upregulation of cardiac alkaline phosphatase.
Recognizing that TNAP is responsible for collagen calcification during bone formation and is also involved in vascular calcification pathology (8, 14, 39), we investigated whether TNAP might also have a role in cardiac fibrocalcification. We measured the activity of TNAP present in heart tissue homogenates, which showed a significant approximately twofold increase in the TO2 heart TNAP activity (Fig. 2A). Notably, TNAP activities of the liver and hamstring homogenates were similar between F1B and TO2 hamsters (Fig. 2A). As an additional assay control, GAPDH enzyme activity was measured and found to be similar in all of the tissue samples (Fig. 2B). We further examined whether altered TNAP activity might also be associated with cardiac ischemia. Since we previously characterized the ischemic porcine myocardium (35), we analyzed TNAP activity of the normal and ischemic porcine heart tissue extracts. The assay revealed that cardiac ischemia mildly but significantly stimulated TNAP activity in the porcine heart (Fig. 2C). GAPDH activity was again not altered (Fig. 2D).
Fig. 2.

Assays of tissue-nonspecific alkaline phosphatase (TNAP) and GAPDH enzyme activity. TNAP (A) and GAPDH (B) enzyme activity was determined for the hearts, livers, and hamstrings of F1B and TO2 hamsters. Normal and ischemic porcine heart tissues were also analyzed (C and D). Enzyme activity in units per μg protein is shown; n = 3 per group (*P < 0.05).
Since TNAP dimer represents the catalytically active form (46), we used an in-gel activity staining method to further confirm the observed difference in cardiac TNAP activity. Figure 3A shows an activity-stained protein band of ∼120 kDa corresponding to TNAP dimer. Densitometric analysis revealed again an approximately twofold increase in the TO2 heart TNAP activity (Fig. 3B), corroborating the activity assay presented in Fig. 2A.
Fig. 3.
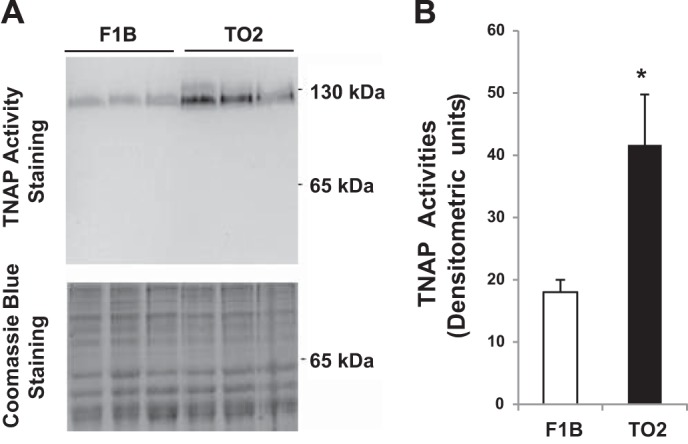
In-gel activity staining of hamster heart TNAP. A: hamster heart protein extracts (30 μg) were loaded in each lane for SDS-PAGE. Top: the gel was processed for TNAP activity staining. Two protein molecular mass markers are illustrated. Bottom: the gel was stained by Coomassie Blue as loading reference. B: densitometric quantification of the stained TNAP dimer shown in A; n = 3 per group (*P < 0.05 TO2 vs. F1B).
Elevated inorganic phosphate in the fibrocalcification state.
Although the physiological substrates of TNAP remain to be definitively determined, elevated activity of TNAP, which is a glycosyl-phosphatidylinositol (GPI)-anchored ectoenzyme (39), is expected to increase the tissue level of inorganic phosphate (Pi) through its robust phosphate-splitting activity. F1B and TO2 hamster tissue samples were extracted and analyzed by the Malachite green-based phosphate assay. The TO2 heart extract was found to contain a significantly elevated Pi level compared with the F1B heart (Fig. 4). This increase in Pi is likely mediated by the increased TNAP activity in the TO2 heart because neither the TO2 liver nor hamstring tissue extracts exhibited an elevation in Pi levels (Fig. 4). In addition, the F1B and TO2 sera exhibited similar Pi levels (Fig. 4), indicating that the detected Pi increase in the TO2 heart, which would promote calcium phosphate crystal deposition in the myocardium, is not secondary to systemic hyperphosphatemia (10).
Fig. 4.
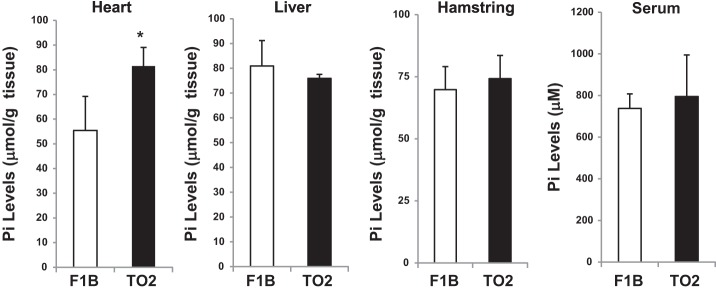
Measurement of hamster tissue inorganic phosphate (Pi). Trichloroacetic acid (TCA)-extracted F1B and TO2 hamster tissues were analyzed by the ammonium molybdate-based phosphate assay method. The Pi concentration is shown as μmol/g tissue or μM for serum samples; n = 3 per group (*P < 0.05 TO2 vs. F1B).
Cloning and expression of hamster heart TNAP.
To confirm the identity and activity of TNAP in cardiac fibrocalcification, we cloned by RT-PCR the full-length TNAP cDNA from the hamster heart (GenBank accession no. KF934412). Sequence analysis reveals that the human and hamster TNAP enzymes are highly conserved, both being 524 amino acids in length and exhibiting a 92% amino-acid sequence homology. In addition, the five N-linked glycosylation sites (Asn-X-Ser/Thr) are all conserved. Since cardiac fibroblasts are the most abundant cell type in the heart and mediate fibrosis (29), we used cultured mouse fibroblasts to further characterize the cloned TNAP. We generated a replication-deficient recombinant TNAP adenovirus and used the LacZ adenovirus as control (30). As expected, cells expressing the LacZ reporter and the TNAP exhibited prominent β-galactosidase (Fig. 5A) and alkaline phosphatase (Fig. 5B) activity, respectively. In-gel alkaline phosphatase activity staining illustrated that cardiac fibroblasts exhibited a basal level phosphatase activity conferred by TNAP dimer, which was further amplified by adenoviral expression of TNAP (Fig. 5C).
Fig. 5.

Overexpression of cloned TNAP in isolated cardiac fibroblasts. Cardiac fibroblasts were infected with LacZ and TNAP adenovirus and cultured for 4 days prior to analysis. A and B: cell lysates were analyzed by β-galactosidase (A) and TNAP (B) activity assays. *P < 0.05 TNAP vs. LacZ. C: cell lysates were processed for in-gel TNAP activity staining as illustrated in Fig. 3. D: cells were extracted with TCA and analyzed by the phosphate assay. The result represents the intracellular Pi levels. E: fibroblast-conditioned medium was extracted with TCA and analyzed by the phosphate assay. The result represents the extracellular Pi levels. *P < 0.05 TNAP vs. LacZ. F: fibroblast-conditioned medium was extracted with TCA and analyzed by the lactate assay; n = 3 per group.
Modulation of extracellular Pi by TNAP.
The LacZ- and TNAP-expressing fibroblasts along with the conditioned medium were harvested and deproteinized. Phosphate assays show that the intracellular Pi levels were indistinguishable between the LacZ and TNAP expressors (Fig. 5D). In contrast, the extracellular Pi present in the conditioned medium was significantly elevated by forced expression of TNAP (Fig. 5E). As a medium assay control, we show that there was no difference in medium lactate levels between the LacZ and TNAP expressors (Fig. 5F). The finding that the extracellular but not intracellular Pi is affected by TNAP expression is consistent with the cell surface location of TNAP. It also suggests that the detected Pi increase in the TO2 heart (Fig. 4) is likely of extracellular origin.
TNAP as a target of sFRP2 in vivo.
Upregulation of TNAP in the fibrotic heart prompted us to determine whether TNAP might be a downstream target of profibrotic cytokines. We recently showed that the fibrotic heart exhibits a higher expression level of sFRP2 and that administration of sFRP2 blocking antibody attenuated myocardial fibrosis and improved function in the TO2 heart (37). Heart tissues from the study were homogenized to assess the effects of the antifibrotic therapy on the TNAP axis. Phosphatase enzyme activity assays reveal that the beneficial effects of sFRP2 blockade were associated with a significant reduction in the TO2 heart TNAP activity (Fig. 6A). Pi concentration assays reveal a concurrent reduction in the level of Pi, but not lactate, after sFRP2 blockade (Fig. 6, B and C), although the decrease in Pi did not reach statistical significance. These findings led us to hypothesize that sFRP2 may possess a TNAP-inducing activity through which the profibrotic cytokine may promote calcification in the fibrotic heart.
Fig. 6.
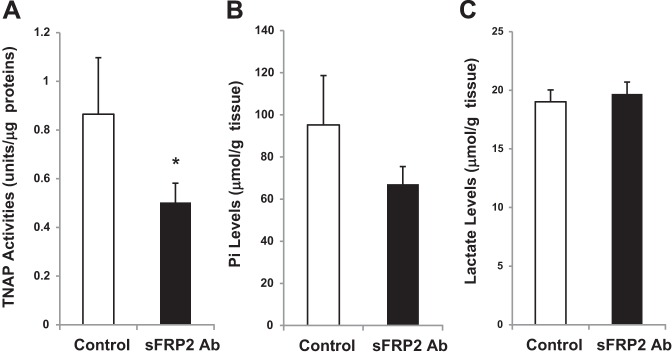
Effect of secreted Frizzled-related protein 2 (sFRP2) blockade on TNAP in the failing heart. TO2 hamster heart tissues from the previous sFRP2 blockade study (37) were processed for measurement of TNAP activity (A), Pi concentration (B), and lactate concentration (C). The antibody control and sFRP2 antibody groups were compared. TNAP activities of the membrane fractions are presented; n = 3 per group (*P < 0.05 sFRP2 antibody vs. control IgG).
Induction of TNAP by sFRP2 in vitro.
To provide evidence for the hypothesis, we treated cultured cardiac fibroblasts with commercially available sFRP2 and sFRP4 proteins for 1–3 days as described previously (37), following which TNAP activity was measured. We found that only sFRP2 treatment steadily increased TNAP activity during the 3-day period, reaching significant activation on day 2 and day 3 (Fig. 7A). Increasing the concentration of sFRP2 and sFRP4 by up to 10-fold (30 nM) did not cause further stimulation (data not shown). To rule out the possibility of cytokine contamination in the commercial sFRP2 preparation, we expressed sFRP2 in E. coli bacteria as a GST fusion protein and purified the recombinant protein using glutathione agarose affinity chromatography as previously described (24). We found that GST-sFRP2 induced a more robust activation of TNAP at the same protein concentration (3 nM), achieving a two- to threefold increase in TNAP activity (Fig. 7B). Analysis of the conditioned medium, however, failed to reveal a concurrent increase in extracellular Pi after sFRP2-mediated induction of TNAP (data not shown). Since the Pi increase shown in Fig. 5E was mediated by a ∼30-fold increase in TNAP activity after adenoviral infection, it is possible that the two- to threefold induction of TNAP by sFRP2 is insufficient to cause a detectable Pi increase in the culture medium, which has a high Pi background (∼1 mM Pi). To rule out the possibility that the more potent TNAP-activating effect of GST-sFRP2 might be due to the presence of the GST domain, we cleaved the fusion protein with thrombin to remove the GST domain. The cleaved protein (c-sFRP2) was found to retain its robust TNAP-activating effect (Fig. 7B). These findings thus demonstrate for the first time that sFRP2 is an inducer of TNAP in the fibrotic heart and isolated cardiac fibroblasts.
Fig. 7.

Effect of sFRP2 treatment on TNAP in the isolated cardiac fibroblast (CaFb). A: cardiac fibroblasts were treated with 3 nM commercial sFRP2 or sFRP4 for 1–3 days, following which TNAP activities were measured (n = 3 per group; *P < 0.05 sFRP2 vs. control on day 2; #P < 0.05 sFRP2 vs. control on day 3). B: cardiac fibroblasts were treated with 3 nM glutathione S-transferase (GST), GST-sFRP2, or c-sFRP2 (sFRP2 derived from cleavage of the GST-sFRP2 fusion protein) for 3 days, following which TNAP activities were measured (n = 3 per group; %P < 0.05 GST-sFRP2 vs. control; ^P < 0.05 c-sFRP2 vs. control).
De novo protein synthesis and cell specificity in TNAP induction.
We performed qRT-PCR analysis to determine whether sFRP2-induced TNAP activity might be mediated by increased expression of TNAP. Figure 8A shows an approximately twofold increase in TNAP gene expression after GST-sFRP2 treatment. This increase is associated with a corresponding increase in TNAP protein as assessed by Western blot analysis (Fig. 8B). Dependence of TNAP activation on de novo gene induction and protein synthesis is further revealed by the finding that inhibition of protein synthesis by cycloheximide completely abolished sFRP2-mediated induction of TNAP activity (Fig. 8C). Since TNAP is a glycoprotein and its catalytic property has been shown to be modulated by N-linked glycosylation (22, 39), we performed GST-sFRP2 treatment in the presence of tunicamycin to determine whether the induced TNAP requires N-linked protein glycosylation. Figure 8B shows that tunicamycin also completely abolished TNAP induction. In contrast, treatment with thiamet G, a potent inhibitor of β-N-acetylglucosaminidase (56), had no effect on TNAP induction (Fig. 8C). Of note, the TNAP-activating effect of sFRP2 was not observed in 3T3 fibroblasts (Fig. 8D), HL1 cardiomyocytes (Fig. 8E), HEK293 cells, C2C12 myoblasts, and bone marrow mesenchymal stem cells (data not shown), indicating a high degree of cell specificity in sFRP2 regulation of TNAP. Further, we found that the induction mechanism requires cell confluency (data not shown). Taken together, the in vivo sFRP2 blockade study showing downregulation of TNAP (Fig. 6) and the in vitro sFRP2 treatment study showing upregulation of TNAP in cardiac fibroblasts (Fig. 7 and 8) establish a novel role of sFRP2 in ECM regulation through coupling cardiac fibrosis and calcification.
Fig. 8.

Characterization of sFRP2-mediated induction of TNAP. A: qRT-PCR analysis of cardiac fibroblasts treated with GST or GST-sFRP2 for 3 days. β2-Microglobulin was used as the internal reference gene for analysis of TNAP expression (*P < 0.05; n = 4). B: Western blot analysis of cardiac fibroblasts treated with GST or GST-sFRP2 for 3 days. Blots were probed with TNAP antibody (top) and GAPDH antibody (bottom). The detected protein bands are TNAP monomer (∼60 kDa) and GAPDH (∼37 kDa). C: cardiac fibroblasts were treated with 3 nM GST or GST-sFRP2 in the presence of 3.5 μM cycloheximide (Cyc), 2.5 μM tunicamycin (Tun), or 0.1 μM thiamet G (Thi) for 3 days, following which TNAP activities were measured (*P < 0.05 GST-sFRP2 vs. GST; #P < 0.05 cycloheximide vs. no drug; ^P < 0.05 tunicamycin vs. no drug). D and E: 3T3 fibroblasts (D) and HL1 cardiomyocytes (E) were treated with 3 nM GST or GST-sFRP2 for 3 days, following which TNAP activities were measured; n = 3 per group.
DISCUSSION
The study by Kobayashi et al. (25) demonstrating reduced fibrosis and improved function in sFRP2-knockout mice after myocardial infarction and that by us showing the same benefits in the failing hamster heart after sFRP2 blockade (37) established sFRP2 as a key profibrotic cytokine in cardiac pathology. The proposed profibrotic role of sFRP2 targets collagen metabolism through enhancing the procollagen C proteinase activity of tolloid-like metalloproteinases (25), which promotes collagen maturation and ECM thickening. The current study discovered that sFRP2 possesses the ability to further modify the fibrotic milieu through TNAP-mediated ECM mineralization. Clinical studies have demonstrated myocardial calcification in the settings of post myocardial infarction, myocarditis, and congenital heart disease (4, 43, 50) and human pericardial pathology often exhibits fibrocalcification (32). The term porcelain heart was previously coined to describe a case of massive myocardial calcification (2). However, the molecular underpinning of concurrent fibrosis and calcification remains unclear. Our findings here suggest that sFRP2 may promote cardiac fibrocalcification through coordinate activation of tolloid-like metalloproteinases and TNAP. In addition, the finding that bacterially produced sFRP2 is functional suggests that protein glycosylation, which is required for the function of Wnt signaling proteins (26, 28), is dispensable for the TNAP-activating function of sFRP2.
TNAP is normally induced during the early stage of osteogenesis and participates in collagen calcification during bone formation (8, 39). It has been proposed that the role of TNAP in the bone matrix is to generate the Pi needed for hydroxyapatite crystallization. Extracellular Pi serves not only as a substrate for hydroxyapatite formation but also as an inducer of the gene expression program that promotes calcification (19, 20). Extracellular Pi for instance is a key regulator of cementoblast during tooth development, signaling a marked increase in osteopontin gene expression (16). Indeed, upregulation of osteopontin gene expression has been observed in the failing hamster heart (40). TNAP knockout mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia (17), which is an inborn error of TNAP, leading to severe rickets and osteomalacia (54). On the other hand, aberrantly elevated circulating levels of alkaline phosphatase can also be associated with a wide range of disease conditions. Although clinical test of circulating levels of alkaline phosphatase is routinely performed, the abnormal increase of TNAP identified in the current study is confined to the failing heart and is not accompanied by systemic hyperphosphatemia.
Vascular calcification is prevalent in aging and many pathological conditions, and is recognized as a strong predictor of cardiovascular events in the general population as well as diabetic and end-stage renal disease patients (20). In chronic kidney disease, vascular calcification is associated with hyperphosphatemia. However, vascular calcification is a multifaceted process that involves not only the gain of inducers of calcification but also the loss of calcification suppressors. Recent studies on vascular calcification have revealed TNAP as a key player in the pathological mineralization process (14). In the study of sheep aortic valve interstitial cells in vitro, transforming growth factor-β1 (TGF-β1) was found to induce progressive calcification and maximal levels of TNAP (13). Since TGF-β1 is a well-established profibrotic cytokine (55), sFRP2 and TGF-β1 may share a similar role in promoting tissue fibrocalcification. However, given that sFRP2-mediated induction of TNAP exhibits cell specificity as observed in our study, whether sFRP2 may also be involved in vascular calcification remains to be determined.
Wnt/β-catenin signaling appears to be more active in the fibrotic hamster heart (38). Consistent with this demonstration, postnatal activation of Wnt signaling is known to promote fibrosis (7), and is required for TGF-β1-mediated fibrosis (1). For instance, Wnt3a activates MAPK p38, resulting in the stimulation of TNAP and matrix mineralization in C3H10T1/2 mesenchymal cells (11). Excessive Wnt/β-catenin signaling may thus have a role in the initiation and/or maintenance of cardiac fibrocalcification. Along this line, we have previously shown that sFRP2 activates Wnt signaling in cardiac fibroblasts (37), which likely mediates the induction of TNAP. Of note, TNAP induction can also be mediated at the posttranslational level independent of gene induction (5, 33). Although protein glycosylation is known to influence the biological activities of Wnt regulators (26, 28), our study indicates that it is not involved in the sFRP2-TNAP axis because the bacterially expressed sFRP2 appears to retain its full TNAP regulatory activity.
Although heart failure has been shown to be associated with increased expression of sFRP2, sFRP3, and sFRP4 (3, 37, 47), the current result based on the use of a commercial source of sFRP4 protein shows that sFRP2 but not sFRP4 mediates TNAP induction. Further work is needed to obtain sFRP4 protein from bacterial overexpression so as to validate this finding. If confirmed, it would indicate a high degree of molecular specificity in the mechanism underlying TNAP induction that also exhibits cell density and cell type dependence. In particular, the induction by sFRP2 has so far only been observed in cardiac fibroblasts among the multiple cell types examined. Given that activation of TNAP has also emerged as a marker of pulmonary fibrosis (9), identification of TNAP as a target of sFRP2 in cardiac fibrosis uncovers a disease pathway amenable to therapeutic intervention.
GRANTS
This work was supported in part by National Institutes of Health (National Heart, Lung, and Blood Institute Grant R01 HL-84590) and University at Buffalo Biomedical Research Service Center.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S.M., C.E., and T.L. conception and design of research; S.M., H.L., and C.E. performed experiments; S.M., H.L., C.E., and T.L. analyzed data; S.M., H.L., C.E., and T.L. interpreted results of experiments; S.M., H.L., C.E., and T.L. prepared figures; T.L. drafted manuscript; T.L. edited and revised manuscript; T.L. approved final version of manuscript.
Footnotes
This article is the topic of an Editorial Focus by Katherine B. Schuetze and Timothy A. McKinsey (46a).
REFERENCES
- 1.Akhmetshina A, Palumbo K, Dees C, Bergmann C, Venalis P, Zerr P, Horn A, Kireva T, Beyer C, Zwerina J, Schneider H, Sadowski A, Riener MO, MacDougald OA, Distler O, Schett G, Distler JH. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat Commun 3: 735, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aras D, Topaloglu S, Demirkan B, Deveci B, Ozeke O, Korkmaz S. Porcelain heart: a case of massive myocardial calcification. Int J Cardiovasc Imaging 22: 123–126, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Askevold ET, Aukrust P, Nymo SH, Lunde IG, Kaasboll OJ, Aakhus S, Florholmen G, Ohm IK, Strand ME, Attramadal H, Fiane A, Dahl CP, Finsen AV, Vinge LE, Christensen G, Yndestad A, Gullestad L, Latini R, Masson S, Tavazzi L, Ueland T. The cardiokine secreted Frizzled-related protein 3, a modulator of Wnt signalling, in clinical and experimental heart failure. J Intern Med 275: 621–630, 2014. [DOI] [PubMed] [Google Scholar]
- 4.Barnard DC, Pape L, Missri J, Dalen JE. Massive myocardial calcification and normal coronary arteries. Tex Heart Inst J 12: 363–365, 1985. [PMC free article] [PubMed] [Google Scholar]
- 5.Baudry A, Bitard J, Mouillet-Richard S, Locker M, Poliard A, Launay JM, Kellermann O. Serotonergic 5-HT(2B) receptor controls tissue-nonspecific alkaline phosphatase activity in osteoblasts via eicosanoids and phosphatidylinositol-specific phospholipase C. J Biol Chem 285: 26066–26073, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bovolenta P, Esteve P, Ruiz JM, Cisneros E, Lopez-Rios J. Beyond Wnt inhibition: new functions of secreted Frizzled-related proteins in development and disease. J Cell Sci 121: 737–746, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, Rando TA. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 317: 807–810, 2007. [DOI] [PubMed] [Google Scholar]
- 8.Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, McDonald JM, Chen Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor runx-2 by akt signaling. J Biol Chem; 283: 15319–15327, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Capelli A, Lusuardi M, Cerutti CG, Donner CF. Lung alkaline phosphatase as a marker of fibrosis in chronic interstitial disorders. Am J Respir Crit Care Med 155: 249–253, 1997. [DOI] [PubMed] [Google Scholar]
- 10.Caudarella R, Vescini F, Buffa A, Francucci CM. Hyperphosphatemia: effects on bone metabolism and cardiovascular risk. J Endocrinol Invest 30: 29–34, 2007. [PubMed] [Google Scholar]
- 11.Caverzasio J, Manen D. Essential role of Wnt3a-mediated activation of mitogen-activated protein kinase p38 for the stimulation of alkaline phosphatase activity and matrix mineralization in C3H10T1/2 mesenchymal cells. Endocrinology 148: 5323–5330, 2007. [DOI] [PubMed] [Google Scholar]
- 12.Chilosi M, Poletti V, Zamo A, Lestani M, Montagna L, Piccoli P, Pedron S, Bertaso M, Scarpa A, Murer B, Cancellieri A, Maestro R, Semenzato G, Doglioni C. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am J Pathol 162: 1495–1502, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clark-Greuel JN, Connolly JM, Sorichillo E, Narula NR, Rapoport HS, Mohler ER 3rd, Gorman JH 3rd, Gorman RC, Levy RJ. Transforming growth factor-beta1 mechanisms in aortic valve calcification: increased alkaline phosphatase and related events. Ann Thorac Surg 83: 946–953, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Demer LL, Tintut Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler Thromb Vasc Biol 34: 715–723, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dobaczewski M, Frangogiannis NG. Chemokines and cardiac fibrosis. Front Biosci 1: 391–405, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fatherazi S, Matsa-Dunn D, Foster BL, Rutherford RB, Somerman MJ, Presland RB. Phosphate regulates osteopontin gene transcription. J Dent Res 88: 39–44, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fedde KN, Blair L, Silverstein J, Coburn SP, Ryan LM, Weinstein RS, Waymire K, Narisawa S, Millan JL, MacGregor GR, Whyte MP. Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia. J Bone Miner Res 14: 2015–2026, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gacheru SN, Trackman PC, Shah MA, O'Gara CY, Spacciapoli P, Greenaway FT, Kagan HM. Structural and catalytic properties of copper in lysyl oxidase. J Biol Chem 265: 19022–19027, 1990. [PubMed] [Google Scholar]
- 19.Giachelli CM. The emerging role of phosphate in vascular calcification. Kidney Int 75: 890–897, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giachelli CM, Speer MY, Li X, Rajachar RM, Yang H. Regulation of vascular calcification: roles of phosphate and osteopontin. Circ Res 96: 717–722, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Griffin M, Casadio R, Bergamini CM. Transglutaminases: nature's biological glues. Biochem J 368: 377–396, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halling Linder C, Narisawa S, Millan JL, Magnusson P. Glycosylation differences contribute to distinct catalytic properties among bone alkaline phosphatase isoforms. Bone 45: 987–993, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He W, Dai C, Li Y, Zeng G, Monga SP, Liu Y. Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J Am Soc Nephrol 20: 765–776, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalenik JL, Chen D, Bradley ME, Chen SJ, Lee TC. Yeast two-hybrid cloning of a novel zinc finger protein that interacts with the multifunctional transcription factor YY1. Nucleic Acids Res 25: 843–849, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kobayashi K, Luo M, Zhang Y, Wilkes DC, Ge G, Grieskamp T, Yamada C, Liu TC, Huang G, Basson CT, Kispert A, Greenspan DS, Sato TN. Secreted Frizzled-related protein 2 is a procollagen C proteinase enhancer with a role in fibrosis associated with myocardial infarction. Nat Cell Biol 11: 46–55, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Komekado H, Yamamoto H, Chiba T, Kikuchi A. Glycosylation and palmitoylation of Wnt-3a are coupled to produce an active form of Wnt-3a. Genes Cells 12: 521–534, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Kress E, Rezza A, Nadjar J, Samarut J, Plateroti M. The frizzled-related sFRP2 gene is a target of thyroid hormone receptor alpha1 and activates beta-catenin signaling in mouse intestine. J Biol Chem 284: 1234–1241, 2009. [DOI] [PubMed] [Google Scholar]
- 28.Kurayoshi M, Yamamoto H, Izumi S, Kikuchi A. Post-translational palmitoylation and glycosylation of Wnt-5a are necessary for its signalling. Biochem J 402: 515–523, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leask A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res 106: 1675–1680, 2010. [DOI] [PubMed] [Google Scholar]
- 30.Lin H, McGrath J, Wang P, Lee T. Cellular toxicity induced by SRF-mediated transcriptional squelching. Toxicol Sci 96: 83–91, 2007. [DOI] [PubMed] [Google Scholar]
- 31.Lin H, Shabbir A, Molnar M, Yang J, Marion S, Canty JM Jr, Lee T. Adenoviral expression of vascular endothelial growth factor splice variants differentially regulate bone marrow-derived mesenchymal stem cells. J Cell Physiol 216: 458–468, 2008. [DOI] [PubMed] [Google Scholar]
- 32.Liu X, Tan M, Gong D, Han L, Lu F, Huang S, Xu Z. Characteristics of pericardial interstitial cells and their implications in pericardial fibrocalcification. J Mol Cell Cardiol 53: 780–789, 2012. [DOI] [PubMed] [Google Scholar]
- 33.Lopez-Posadas R, Gonzalez R, Ballester I, Martinez-Moya P, Romero-Calvo I, Suarez MD, Zarzuelo A, Martinez-Augustin O, Sanchez de Medina F. Tissue-nonspecific alkaline phosphatase is activated in enterocytes by oxidative stress via changes in glycosylation. Inflamm Bowel Dis 17: 543–556, 2011. [DOI] [PubMed] [Google Scholar]
- 34.Lopez B, Gonzalez A, Lindner D, Westermann D, Ravassa S, Beaumont J, Gallego I, Zudaire A, Brugnolaro C, Querejeta R, Larman M, Tschope C, Diez J. Osteopontin-mediated myocardial fibrosis in heart failure: a role for lysyl oxidase? Cardiovasc Res 99: 111–120, 2013. [DOI] [PubMed] [Google Scholar]
- 35.Lynch P, Lee T, Fallavollita JA, Canty JM Jr, Suzuki G. Intracoronary administration of AdvFGF-5 (fibroblast growth factor-5) ameliorates left ventricular dysfunction and prevents myocyte loss in swine with developing collaterals and ischemic cardiomyopathy. Circulation 116: I71–I76, 2007. [DOI] [PubMed] [Google Scholar]
- 36.Mastri M, Shah Z, McLaughlin T, Greene CJ, Baum L, Suzuki G, Lee T. Activation of Toll-like receptor 3 amplifies mesenchymal stem cell trophic factors and enhances therapeutic potency. Am J Physiol Cell Physiol 303: C1021–C1033, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mastri M, Shah Z, Wang W, Hsieh K, Wooldridge B, Suzuki G, Lee T. Secreted Frizzled-related protein 2 as a target in antifibrotic intervention. Am J Physiol Cell Physiol 306: C531–C539, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Masuelli L, Bei R, Sacchetti P, Scappaticci I, Francalanci P, Albonici L, Coletti A, Palumbo C, Minieri M, Fiaccavento R, Carotenuto F, Fantini C, Carosella L, Modesti A, Di Nardo P. Beta-catenin accumulates in intercalated disks of hypertrophic cardiomyopathic hearts. Cardiovasc Res 60: 376–387, 2003. [DOI] [PubMed] [Google Scholar]
- 39.Millan JL. The role of phosphatases in the initiation of skeletal mineralization. Calcif Tissue Int 93: 299–306, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Missihoun C, Zisa D, Shabbir A, Lin H, Lee T. Myocardial oxidative stress, osteogenic phenotype, and energy metabolism are differentially involved in the initiation and early progression of delta-sarcoglycan-null cardiomyopathy. Mol Cell Biochem 321: 45–52, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nathan E, Tzahor E. sFRPs: a declaration of (Wnt) independence. Nat Cell Biol 11: 13, 2009. [DOI] [PubMed] [Google Scholar]
- 42.Olsen KC, Epa AP, Kulkarni AA, Kottmann RM, McCarthy CE, Johnson GV, Thatcher TH, Phipps RP, Sime PJ. Inhibition of transglutaminase 2, a novel target for pulmonary fibrosis, by two small electrophilic molecules. Am J Respir Cell Mol Biol 50: 737–747, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roubille C, Brunel AS, Gahide G, Vernhet Kovacsik H, Le Quellec A. Cytomegalovirus (CMV) and acute myocarditis in an immunocompetent patient. Intern Med 49: 131–133, 2010. [DOI] [PubMed] [Google Scholar]
- 44.Ryoke T, Gu Y, Mao L, Hongo M, Clark RG, Peterson KL, Ross J Jr. Progressive cardiac dysfunction and fibrosis in the cardiomyopathic hamster and effects of growth hormone and angiotensin-converting enzyme inhibition. Circulation 100: 1734–1743, 1999. [DOI] [PubMed] [Google Scholar]
- 45.Satoh W, Gotoh T, Tsunematsu Y, Aizawa S, Shimono A. Sfrp1 and Sfrp2 regulate anteroposterior axis elongation and somite segmentation during mouse embryogenesis. Development 133: 989–999, 2006. [DOI] [PubMed] [Google Scholar]
- 46.Satou Y, Al-Shawafi HA, Sultana S, Makita S, Sohda M, Oda K. Disulfide bonds are critical for tissue-nonspecific alkaline phosphatase function revealed by analysis of mutant proteins bearing a C(201)-Y or C(489)-S substitution associated with severe hypophosphatasia. Biochim Biophys Acta 1822: 581–588, 2012. [DOI] [PubMed] [Google Scholar]
- 46a.Schuetze KB, McKinsey TA. TNAP: A new player in cardiac fibrosis? Focus on “Tissue-nonspecific alkaline phosphatase as a target of sFRP2 in cardiac fibroblasts.” Am J Physiol Cell Physiol (June 24, 2015). doi: 10.1152/ajpcell.00167.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schumann H, Holtz J, Zerkowski HR, Hatzfeld M. Expression of secreted frizzled related proteins 3 and 4 in human ventricular myocardium correlates with apoptosis related gene expression. Cardiovasc Res 45: 720–728, 2000. [DOI] [PubMed] [Google Scholar]
- 48.Shabbir A, Zisa D, Lin H, Mastri M, Roloff G, Suzuki G, Lee T. Activation of host tissue trophic factors through JAK/STAT3 signaling: a mechanism of mesenchymal stem cell-mediated cardiac repair. Am J Physiol Heart Circ Physiol 299: H1428–H1438, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shabbir A, Zisa D, Suzuki G, Lee T. Heart failure therapy mediated by the trophic activities of bone marrow mesenchymal stem cells: a non-invasive therapeutic regimen. Am J Physiol Heart Circ Physiol 296: H1888–H1897, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Topaz O. Myocardial calcifications in infants with congenital heart disease. Pediatr Cardiol 7: 75–78, 1986. [DOI] [PubMed] [Google Scholar]
- 51.von Marschall Z, Fisher LW. Secreted Frizzled-related protein-2 (sFRP2) augments canonical Wnt3a-induced signaling. Biochem Biophys Res Commun 400: 299–304, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X, Gerdes AM. Chronic pressure overload cardiac hypertrophy and failure in guinea pigs: III. Intercalated disc remodeling. J Mol Cell Cardiol 31: 333–343, 1999. [DOI] [PubMed] [Google Scholar]
- 53.Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol 10: 15–26, 2013. [DOI] [PubMed] [Google Scholar]
- 54.Whyte MP. Physiological role of alkaline phosphatase explored in hypophosphatasia. Ann NY Acad Sci 1192: 190–200, 2010. [DOI] [PubMed] [Google Scholar]
- 55.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 18: 1028–1040, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yuzwa SA, Macauley MS, Heinonen JE, Shan X, Dennis RJ, He Y, Whitworth GE, Stubbs KA, McEachern EJ, Davies GJ, Vocadlo DJ. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat Chem Biol 4: 483–490, 2008. [DOI] [PubMed] [Google Scholar]
- 57.Zisa D, Shabbir A, Mastri M, Suzuki G, Lee T. Intramuscular VEGF repairs the failing heart: role of host-derived growth factors and mobilization of progenitor cells. Am J Physiol Regul Integr Comp Physiol 297: R1503–R1515, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zisa D, Shabbir A, Mastri M, Taylor T, Aleksic I, McDaniel M, Suzuki G, Lee T. Intramuscular VEGF activates an SDF1-dependent progenitor cell cascade and an SDF1-independent muscle paracrine cascade for cardiac repair. Am J Physiol Heart Circ Physiol 301: H2422–H2432, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]