

fibrosis of the heart, caused by overproduction of extracellular matrix (ECM) proteins by fibroblasts, has diverse negative functional consequences. For example, fibrosis increases the passive stiffness of the myocardium, resulting in diastolic dysfunction (9), and disrupts electrical conduction in the heart, causing arrhythmias (3). Additionally, the quantity of fibrosis in the heart is predictive of adverse cardiac outcomes (4). Despite the well-accepted role of fibrosis in the pathogenesis of numerous cardiac and noncardiac diseases, there is only one FDA-approved antifibrotic therapy, pirfenidone, for the treatment of idiopathic pulmonary fibrosis. While encouraging, unfortunately the molecular target(s) of pirfenidone are unknown, and the efficacy of the drug is limited, with no impact on quality of life, symptoms, or mortality (6). This highlights the need to more extensively define the molecular mechanisms that control fibrosis, with the goal of revealing novel therapeutic targets that can impact hard outcomes.
Lee and colleagues provide evidence that tissue-nonspecific alkaline phosphatase (TNAP) serves a novel role in the activation of cardiac fibroblasts, and thus may contribute to cardiac fibrosis (8). This new finding stemmed from the group's work on secreted frizzled-related protein 2 (sFRP2), which physically associates with Wnt ligands and prevents them from transducing intracellular signals via frizzled transmembrane receptors. Wnt-dependent and -independent roles for sFRP2 in cardiac fibrosis have previously been suggested. Through a Wnt-independent mechanism, sFRP2 was shown to enhance tolloid-like metalloproteinases to cleave procollagen into collagen and promote fibrosis of the heart, and mice lacking sFRP2 were found to be protected from adverse remodeling post-myocardial infarction (MI) (7). Consistent with a profibrotic role for sFRP2, the Lee group previously demonstrated that a blocking antibody against sFRP2 repressed cardiac fibrosis and improved systolic function in the TO2 δ-sarcoglycan-null cardiomyopathic hamster model (10). However, blockade of sFRP2 was associated with reduced Wnt signaling in this model, suggesting that sFRP2 also stimulates cardiac fibrosis through a Wnt-dependent mechanism. Adding further complexity was the demonstration that intracardiac injection of recombinant sFRP2 paradoxically reduces cardiac fibrosis in a rat MI model (5). It has been proposed that these contradictory findings (stimulation and inhibition of fibrosis by sFRP2) are related to the ability of sFRP2 to enhance or inhibit Wnt signaling depending on dosage, with low and high sFRP2 concentrations stimulating and inhibiting Wnt, respectively (10).
In the current study, the Lee group noted that TO2 hamster hearts have concomitant fibrosis and calcification; fibrotic plaques can become calcified through a process known as fibrocalcification. Based on this finding, the authors began assessing the possible connection to TNAP, an ectoenzyme that promotes calcification by hydrolyzing inorganic pyrophosphate (PPi), a potent mineralization inhibitor, into inorganic phosphate (Pi) (Fig. 1). In addition to its function in normal bone development, TNAP has been implicated in the pathogenesis of vascular calcification. The authors found that TNAP expression and activity were elevated in TO2 hearts and in pigs with ischemic cardiomyopathy due to stenosis of the left anterior descending and left circumflex coronary arteries. TNAP activity was reduced in hearts of TO2 hamsters treated with sFRP2 blocking antibody, while treatment of cultured mouse cardiac fibroblasts with recombinant sFRP2 led to increased TNAP expression and activity. Together, these findings suggest that sFRP2 enhances cardiac fibrosis, and possibly fibrocalcification of the heart, by stimulating TNAP activity.
Fig. 1.
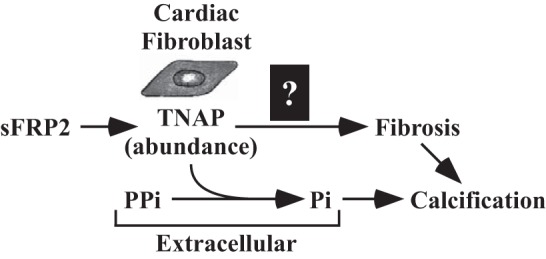
Model for the regulation of cardiac fibrosis and calcification by tissue-nonspecific alkaline phosphatase (TNAP). Secreted frizzled-related protein 2 (sFRP2) stimulates expression of TNAP in cardiac fibroblasts through an undefined mechanism. TNAP is an ectoenzyme that promotes calcification by hydrolyzing inorganic pyrophosphate (PPi), a mineralization inhibitor, into inorganic phosphate (Pi). The current study also suggests a role for TNAP in cardiac fibrosis.
Given the intriguing findings of the Lee group, and the recent demonstration that TNAP promotes cardiomyocyte hypertrophy (2), efforts to further understand the roles of TNAP in pathological cardiac remodeling are justified, and the next steps should be relatively straightforward. First, the generalizability of the current findings should be addressed by quantifying TNAP activity in additional preclinical models of heart failure (e.g., pressure overload and chronic angiotensin signaling) and, more crucially, TNAP activity should be measured in explanted hearts from humans with heart failure. The importance of this additional work is underscored by the fact that overt interstitial calcification within the myocardium, as shown in TO2 hamsters, is not a prominent feature of chronic heart failure in humans. Therefore, it will be critical to rule out the possibility that TNAP-driven cardiac fibrosis is limited to the cardiomyopathic hamster model; the findings with pig hearts are encouraging but need to be expanded. Second, the impact of fibroblast-specific genetic knockdown/knockout of TNAP on ECM gene expression and fibrosis should be determined. Finally, the availability of small molecule TNAP inhibitors, which are being developed for vascular calcification (1), provides an excellent opportunity to perform preclinical pharmacology studies to address the translational potential of inhibiting TNAP catalytic activity for the treatment of heart failure. Together, these follow-up “target validation” studies should enable a rapid Go-/No-Go decision on whether to extend investigations of TNAP in the pathogenesis of heart failure, or whether to relegate this target to a power nap.
GRANTS
K. B. Schuetze received funding from a National Institutes of Health (NIH) T32 training grant (5T32HL007822-12) and T. A. McKinsey was supported by grants from the NIH (R01HL116848, R21AG043822, R01HL127240) and American Heart Association (13GRNT14510001).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
K.B.S. drafted manuscript; T.A.M. edited and revised manuscript.
REFERENCES
- 1.Debray J, Chang L, Marques S, Pellet-Rostaing S, Le DD, Mebarek S, Buchet R, Magne D, Popowycz F, Lemaire M. Inhibitors of tissue-nonspecific alkaline phosphatase: design, synthesis, kinetics, biomineralization and cellular tests. Bioorg Med Chem 21: 7981–7987, 2013. [DOI] [PubMed] [Google Scholar]
- 2.Gan XT, Taniai S, Zhao G, Huang CX, Velenosi TJ, Xue J, Urquhart BL, Karmazyn M. CD73-TNAP crosstalk regulates the hypertrophic response and cardiomyocyte calcification due to alpha1 adrenoceptor activation. Mol Cell Biochem 394: 237–246, 2014. [DOI] [PubMed] [Google Scholar]
- 3.Grand T, Salvarani N, Jousset F, Rohr S. Aggravation of cardiac myofibroblast arrhythmogeneicity by mechanical stress. Cardiovasc Res 104: 489–500, 2014. [DOI] [PubMed] [Google Scholar]
- 4.Gulati A, Jabbour A, Ismail TF, Guha K, Khwaja J, Raza S, Morarji K, Brown TD, Ismail NA, Dweck MR, Di PE, Roughton M, Wage R, Daryani Y, O'Hanlon R, Sheppard MN, Alpendurada F, Lyon AR, Cook SA, Cowie MR, Assomull RG, Pennell DJ, Prasad SK. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA 309: 896–908, 2013. [DOI] [PubMed] [Google Scholar]
- 5.He W, Zhang L, Ni A, Zhang Z, Mirotsou M, Mao L, Pratt RE, Dzau VJ. Exogenously administered secreted frizzled related protein 2 (Sfrp2) reduces fibrosis and improves cardiac function in a rat model of myocardial infarction. Proc Natl Acad Sci USA 107: 21110–21115, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, Lederer DJ, Nathan SD, Pereira CA, Sahn SA, Sussman R, Swigris JJ, Noble PW. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 370: 2083–2092, 2014. [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi K, Luo M, Zhang Y, Wilkes DC, Ge G, Grieskamp T, Yamada C, Liu TC, Huang G, Basson CT, Kispert A, Greenspan DS, Sato TN. Secreted Frizzled-related protein 2 is a procollagen C proteinase enhancer with a role in fibrosis associated with myocardial infarction. Nat Cell Biol 11: 46–55, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin S, Lin H, Ejimadu C, Lee T. Tissue-nonspecific alkaline phosphatase as a target of sFRP2 in cardiac fibroblasts. Am J Physiol Cell Physiol (May 13, 2015). doi: 10.1152/ajpcell.00009.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martos R, Baugh J, Ledwidge M, O'Loughlin C, Conlon C, Patle A, Donnelly SC, McDonald K. Diastolic heart failure: evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation 115: 888–895, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Mastri M, Shah Z, Hsieh K, Wang X, Wooldridge B, Martin S, Suzuki G, Lee T. Secreted Frizzled-related protein 2 as a target in antifibrotic therapeutic intervention. Am J Physiol Cell Physiol 306: C531–C539, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]