

This study assesses effects of aerobic exercise training on the release of microparticles from endothelial cells and corroborates these findings using an in vitro experimental exercise stimulant, laminar shear stress. Furthermore, this study demonstrated that shear stress-induced mitochondrial biogenesis mediates these effects against endothelial cell activation and injury.
Keywords: shear stress, exercise, mitochondrial biogenesis, endothelial microparticle
Abstract
The concept of enhancing structural integrity of mitochondria has emerged as a novel therapeutic option for cardiovascular disease. Flow-induced increase in laminar shear stress is a potent physiological stimulant associated with exercise, which exerts atheroprotective effects in the vasculature. However, the effect of laminar shear stress on mitochondrial remodeling within the vascular endothelium and its related functional consequences remain largely unknown. Using in vitro and in vivo complementary studies, here, we report that aerobic exercise alleviates the release of endothelial microparticles in prehypertensive individuals and that these salutary effects are, in part, mediated by shear stress-induced mitochondrial biogenesis. Circulating levels of total (CD31+/CD42a−) and activated (CD62E+) microparticles released by endothelial cells were significantly decreased (∼40% for both) after a 6-mo supervised aerobic exercise training program in individuals with prehypertension. In cultured human endothelial cells, laminar shear stress reduced the release of endothelial microparticles, which was accompanied by an increase in mitochondrial biogenesis through a sirtuin 1 (SIRT1)-dependent mechanism. Resveratrol, a SIRT1 activator, treatment showed similar effects. SIRT1 knockdown using small-interfering RNA completely abolished the protective effect of shear stress. Disruption of mitochondrial integrity by either antimycin A or peroxisome proliferator-activated receptor-γ coactivator-1α small-interfering RNA significantly increased the number of total, and activated, released endothelial microparticles, and shear stress restored these back to basal levels. Collectively, these data demonstrate a critical role of endothelial mitochondrial integrity in preserving endothelial homeostasis. Moreover, prolonged laminar shear stress, which is systemically elevated during aerobic exercise in the vessel wall, mitigates endothelial dysfunction by promoting mitochondrial biogenesis.
NEW & NOTEWORTHY
This study assesses effects of aerobic exercise training on the release of microparticles from endothelial cells and corroborates these findings using an in vitro experimental exercise stimulant, laminar shear stress. Furthermore, this study demonstrated that shear stress-induced mitochondrial biogenesis mediates these effects against endothelial cell activation and injury.
in endothelial cells (ECs), mitochondria occupy a relatively small compartment of cytoplasmic volume (∼2–6%) than other energy demanding cell-types such as cardiomyocytes (32). The endothelial mitochondria are, however, thought to play a key regulatory role in cell signaling (39), calcium handling (44), and cell survival (49). Indeed, an impaired mitochondrial network has been associated with dysfunctional endothelium associated with cardiovascular diseases (CVDs) (21, 22). Therefore, promoting enhanced mitochondrial networks and their function has emerged as a potential therapy for preventing and reversing endothelial dysfunction.
Mitochondrial biogenesis is a complex process involving replication of mitochondrial DNA (mtDNA) and expression of nuclear- and mitochondrial-encoded genes through which new mitochondria are formed (35). Studies have shown that peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) plays a crucial role in mitochondrial biogenesis by activating nuclear respiratory factor-1 (NRF-1), which regulates the expression of nuclear genes required for mitochondrial biogenesis (35). PGC-1α also activates mitochondrial transcription factor A (TFAM), which is required for mtDNA transcription and mtDNA replication, through coactivation of the NRF-1. The activity of PGC-1α is controlled by multiple factors, including nitric oxide (NO) and sirtuin 1 (SIRT1)-mediated deacetylation (29, 30). NO and SIRT1 synergistically increase mitochondrial biogenesis in ECs, which may involve shear-sensitive SIRT1 induction and SIRT1-mediated endothelial NO synthase (eNOS) deacetylation (12).
Besides its well-known effects on CVD risk reduction, aerobic exercise directly improves vascular health not only by increasing NO-dependent vasodilation but possibly by decreasing EC activation and apoptosis in the arterial wall (48). To this end, long-term exercise intervention has been shown to reduce the severity of atherosclerosis (31), to stabilize vascular lesion (36), and to lessen post-injury neointima growth (38). The mechanism underlying these adaptive changes is believed to be governed by high wall shear rates occurring during exercise. Aerobic exercise enhances the level of unidirectional laminar flow and diminishes disturbed or turbulent flow occurring in certain arterial geometries such as bifurcations, branch points, and curvatures (13, 45). In cultured ECs, laminar shear stress (LSS) modulates endothelial activation by suppressing expression of inflammatory adhesion molecules (1). LSS also prevents EC apoptosis (17), which is known feature of atherosclerosis. However, limited information is available regarding the mechanistic relationship of mitochondrial biogenesis and the release of endothelial microparticles (EMP) under enhanced LSS.
The aim of this study was to elucidate the mechanism underlying the effect of exercise on reducing the release of EMP. Microparticles (MP) are submicron membrane vesicles formed from plasma membrane and released from virtually all cells into the plasma (15). The surface of MP contains most of the cell membrane-associated proteins of their parent cells (27). It is interesting to note that ECs release membrane-derived MP into the circulation when they are activated or are undergoing apoptosis (5). Previous studies showed that apoptosis-induced EMP expresses the constitutive EC marker PECAM (CD31), whereas activation-induced EMP increases their expression of inducible endothelial markers such as E-selectin (CD62E). Because CD31 is also expressed by platelet derived MP; EMP is ensured by the CD31+/CD42a− (platelet glycoprotein GP9) phenotype (14). Therefore, elevated EMP production is thought to be an emerging biomarker of endothelial dysfunction or injuries such as those associated with atherosclerosis, hypertension, coronary artery disease, and metabolic syndrome (7). Using complementary in vivo and in vitro studies, we have tested a hypothesis that LSS would decrease the release of activated (CD62E+)- and total (CD31+/CD42a−) MP from ECs through LSS-mediated mitochondrial biogenesis.
METHODS
Study subjects and aerobic exercise intervention.
The study protocol was approved by and conducted in accordance with the Temple University Institutional Review Board. All subjects provided their written, informed consent during their first laboratory visit. Twenty-one male and female subjects were recruited in this study who met the following inclusion criteria: sedentary life style (regular aerobic exercise <2 days/wk), nonsmoking, not on lipid-lowering medication, blood pressure <160/100 mmHg, body mass index <40 kg/m2, and no history of diabetes, cardiovascular, liver, kidney, or lung disease. Under the supervision of the study physician, qualified participants who were using only one antihypertensive medication were tapered off the medication and remained off for the remainder of the study. Subjects using two or more anti-hypertensive medications were excluded from the study. Following a successful clinical and cardiopulmonary examination by a study physician, qualified subjects participated in a supervised aerobic exercise training (AEXT)session 3 days/wk for 6 mo. Each exercise session consisted of 40 min of aerobic exercise at an intensity level of 65% of individually predicted maximal heart rate. Adherence was assessed by examining individual training logs for frequency data. Only data from participants that achieved 80% adherence were included in the analyses.
Cell culture and shear stress experiments.
Seven separate human umbilical vein EC (HUVEC) lines from Lonza were grown in M199 medium supplemented with 20% fetal bovine serum and EC growth supplement (Sigma-Aldrich no. E2759) and maintained at 37°C in a 5% CO2 atmosphere. All experiments with HUVECs were conducted between passages 3 and 5. For some experiments, cells were treated with trans-3,4′,-5-trihydroxystilebene [resveratrol (RSV)] at indicated concentrations, and DMSO was used for the control. For shear experiments, HUVECs, grown at 90–100% confluence, were replaced with shear media and exposed to the physiological levels of shear stress for various times using a cone and plate viscometer (0.5°Cone angle), which was placed inside of a CO2 cell incubator as previously described (33). Shear media was M199 supplemented with 2% fetal bovine serum and the EC growth supplement. All shear experiments were conducted under sterile conditions. For some experiment cells were treated with antimycin A (10 μM, 12 h) before shear exposure.
EMP analysis.
Level of EMP production was determined by flow cytometry, as previously described (6). Briefly, samples were thawed at room temperature and centrifuged at 1,500 g for 20 min at room temperature to obtain platelet poor plasma. The top two-thirds of platelet poor plasma were further centrifuged at 1,500 g for 20 min at room temperature to obtain cell free plasma. The supernatant was used for EMP analysis. A volume of 100 μl of sample was incubated with the different fluorochrome-labeled antibodies for 20 min at room temperature in the dark on a shaker (200 rpm). The two-color combination of phycoerythrin (PE)-labeled anti-CD31 (BD Biosciences no. 555446) and fluorescein isothiocyanate (FITC)-labeled anti-CD42a (BD Biosciences no. 558818) was used. Samples were diluted with 500 ml of 0.22 μm double filtered phosphate buffered saline before analysis. Samples were measured using a BD LSRII flow cytometer and BD FACSDIVA software. Total and activated MP from ECs were defined as CD62E+ and CD31+/CD42a−, respectively, and as vesicles sizes of <1.0 μm. A logarithmic scale was implemented for forward scatter signal, side scatter signal, and each fluorescent channel. Size calibration was done with 0.9 μm beads. Nonstained samples were used to discriminate true events from noise and to increase the specificity for EMP detection for each sample. The flow rate was set on medium, and all samples were run for 180 s. The mean sample processing rate of 101 μl/180 s was calculated using calibration beads. For the human study, EMP counts per μl plasma were determined, and for the cell culture study, EMP counts per μL media per μg cell protein were determined. The inter- and intra-assay CVs were 8% and 6%, respectively.
Detection of surface marker on ECs.
After trypsinization, 1.0 × 106 cells in a single cell suspension were collected in 100 μl FACS buffer, which includes PBS, 10% FBS, and 0.1% NaN3. Antibody (20 μl) was added, and cells were incubated for 30 min at 4°C in the dark. The antibody combinations for this experiment were CD62E-PE/CD31-FITC. Then, 1 ml FACS buffer was added and the cells were washed three times by centrifugation at 1,500 rpm for 5 min. Cells were resuspended in 500 μl ice-cold FACS buffer with 2% formaldehyde (Polysciences no. 04018). Cells were kept in the dark on ice until flow cytometry analysis was performed. Percent CD62E-positive ECs were measured using a BD LSRII flow cytometer and BD FACSDIVA software.
NO measures.
NO levels were indirectly measured by determining NO end-products, nitrate and nitrite, using an Enzo Life Sciences/Assay Designs assay kit as described previously (18). Absorbance was read at 540 nm. The inter-assay CV was 8%, and the intra-assay CV was 11%.
Western blotting.
Antibodies were from the following sources: rabbit polyclonal anti-SIRT1 (Cell signaling no. 2496), rabbit polyclonal anti-PGC-1α, (NOVUS no. NBP1-04676), mouse polyclonal anti-TFAM (NOVUS no. H00007019-B01P), and mouse monoclonal anti-VDAC1/Porin (Abcam no. ab14734). Mouse monoclonal anti-α-Tubulin (Sigma no. T9026) was used as the internal control. ECs were homogenized in ice-cold RIPA lysis buffer with protease inhibitor (Roche no. 11836153001). Proteins from cell lysates were resolved by Tris-glycine SDS-PAGE and transferred to Immobilon-P PVDF membrane for standard electrochemiluminescence Western blot analysis. Total protein expression was detected by chemiluminescence. Band densiometry analyses were completed using ImageJ software, National Institute of Health, to direct comparisons between experimental sets. The values calculated by ImageJ are arbitrary units. The densities of the selected protein bands were expressed relative to the densities of the internal control gene, α-Tubulin.
MitoTracker staining.
Live HUVECs were incubated with 200 nM prewarmed MitoTracker Green FM or MitoTracker Red CMXRos (Molecular Probes) solution at 37°C for 30 min. After removal of the incubation solution, cells were washed three times with prewarmed PBS and then mounted in HBSS. For quantitative analyses, more than 100 images per each group were acquired using an epifluorescence upright microscope with a 63× objective oil lens. For MitoTracker Green FM staining, excitation/emission wavelengths were set at 470/525 nm (FL filter Set 38; Zeiss), and for MitoTracker Red CMXRos staining, excitation/emission wavelengths were set at 587/647 nm (FL filter Set 64HE). Images were initially acquired using an AxioCam MRm and AxioVision image processing system (Zeiss), and the fluorescence intensities were assessed using ImageJ software.
mtDNA quantification.
Total genomic DNAs were isolated by using the DNeasy kit (QIAGEN) and mtDNA contents were assessed by semi-quantitative PCR. The relative ratio between mtDNA [Cytochrome c oxidase subunits I or II (COX I or COX II, respectively) and nuclear DNA (18s rRNA) amount was measured. Primer sequences were as follows: COX I: sense, 5′-CATAGGAGGCTTCATTCACTG-3′ and antisense, 5′-CAGGTTTATGGAGGGTTCTTC-3′; COX II: sense, 5′-CCATAGGGCACCAATGATACTG-3′ and antisense, 5′-AGTCGGCCTGGGATGGCATC-3′; and 18s rRNA: sense, 5′-CTTAGAGGGACAAGTGGCGTTC-3′ and antisense, 5′-CGCTGAGCCAGTCAGTGTAG-3′.
Transfection.
Cultured ECs, reached 60–80% confluence, were incubated with a mixture containing either SIRT1 small-interfering RNA (siRNA) (Qiagen no. SI00098434) or control siRNA (Qiagen no. 1027281) and FuGene 6 transfection reagent (Promega no. E2691) in Opti-MEM serum-free medium (Life Tech no. 31985).
Statistical analysis.
Data are presented as percentages or means ± SE. For all in vivo data, pre- and post-AEXT variables were compared using the paired samples t-test or the paired samples Wilcoxon signed ranks test, as appropriate. For all in vitro data, differences across experimental conditions were assessed by analysis of variance followed by post hoc testing with the Fisher's least significant difference test. Pearson's correlation analysis was used to assess the relationship between the expression level of mitochondrial content and EMP production. P < 0.05 was considered statistically significant for all analyses.
RESULTS
Aerobic exercise training and LSS reduce EMP release.
Table 1 shows clinical parameters of the participants before and after 6 mo of AEXT. Except the reduction of triglycerides, other serum measurements, blood pressure, and body mass index remain unchanged. Maximum oxygen consumption (VO2max) was significantly increased following AEXT, indicating increased aerobic capacity. Following AEXT, there were significant reductions in CD62E+ EMP (−41%) and CD31+/CD42a− EMP (−38%) (Fig. 1A). In contrast, plasma NOx concentration was significantly increased (+53%) after AEXT (Fig. 1B). Previous anti-hypertensive medication use, menopausal status, or gender did not influence these outcomes. EMP levels were also assessed in cell culture media of HUVEC preparations from seven different donors upon distinct LSS stimulations. When compared with low (5 dyne/cm2) physiological levels of LSS condition, CD62E+ EMP level was significantly lower (−44%) after high (20 dyne/cm2) physiological levels of LSS (Fig. 1C). NOx levels in the cell culture media were significantly greater (+37%) under the high LSS compared with the low LSS condition (Fig. 1D).
Table 1.
Clinical characteristics pre- and post-AEXT
| Pre-AEXT | Post-AEXT | Percent Change | N | |
|---|---|---|---|---|
| Age, yr | 52.1 ± 1.4 | 21 | ||
| Total cholesterol, mg/dl | 190.9 ± 6.1 | 184.8 ± 6.5 | −2.8 | 18 |
| Triglycerides, mg/dl | 85.7 ± 9.0 | 69.7 ± 5.0* | −12.3 | 19 |
| Lipoprotein cholesterol, mg/dl | ||||
| High density | 63.8 ± 3.8 | 63.0 ± 2.9 | 0.5 | 19 |
| Low density | 112.2 ± 5.6 | 110.2 ± 6.1 | −1.7 | 19 |
| Fasting glucose, mg/dl | 92.9 ± 2.6 | 89.0 ± 2.7 | −3.7 | 17 |
| Blood pressure, mmHg | ||||
| Systolic | 122.3 ± 2.3 | 121.1 ± 2.6 | −0.8 | 21 |
| Diastolic | 77.5 ± 1.3 | 78.5 ± 1.8 | 1.4 | 21 |
| Body mass index, kg/m2 | 29.8 ± 1.1 | 29.1 ± 0.9 | −2.1 | 21 |
| Maximal oxygen consumption, ml·kg−1·min−1 | 25.7 ± 1.0 | 29.0 ± 1.4** | 12.6 | 21 |
Values are means ± SE; N, usable sample for variables. AEXT, aerobic exercise training.
P < 0.05 and
P < 0.01, significant differences baseline vs. final.
Fig. 1.

Effects of aerobic exercise training (AEXT) and shear stress on endothelial cell microparticle and nitric oxide production. A and B: plasma CD62+ endothelial cell microparticle (EMP), CD31+/CD42a− EMP, and nitric oxide (NOx) levels from human subjects at before (pre) and after (post) 6-mo AEXT (n = 21). C and D: 7 separate human umbilical vein endothelial cell (HUVEC) cell lines from 7 different donors were exposed to either low (5 dyne/cm2) or high (20 dyne/cm2) laminar shear stress (LSS) for 24 h. Accumulated CD62+EMP and NOx were measured from the cell culture media. Each column represents means ± SE. *P < 0.05; **P < 0.01.
LSS reduces EMP release through SIRT1-mediated mitochondrial biogenesis.
To examine the effect of shear stress on mitochondrial biogenesis, confluent monolayers of human ECs were subjected to different levels of LSS (5–20 dyne/cm2). The morphology of HUVECs presented an elongation shape after exposure to 36 h LSS (Fig. 2A). Likewise, the key regulators of mitochondrial biogenesis, SIRT1, PGC-1α, and TFAM, were gradually increased by LSS in a magnitude-dependent manner (Fig. 2B). Accordingly, mitochondrial content determined by abundance of mitochondrial outer membrane protein, porin (VDAC), was gradually increased by LSS (Fig. 2C). The increased mitochondrial content in the ECs was confirmed by MitoTracker Green FM staining and mtDNA copy number assay (Fig. 2, D and E). Then, a potential relationship between mitochondrial biogenesis and EMP release was examined in shear-conditioned cells. Likewise, LSS (20 dyne/cm2, for 36 h) significantly increased mitochondrial content (Fig. 2C). In contrast, CD62E+ EMP and CD31+/CD42a− EMP levels were decreased in LSS-exposed cells (Fig. 3A). In addition, Pearson's correlation analysis indicated that mitochondrial content was inversely correlated with either CD62E+ EMP or CD31+/CD42a− EMP (Fig. 3B). Moreover, the number of ECs expressing CD62E surface marker was decreased in LSS-exposed cells (Fig. 3, C and D). RSV is a potent stimulus of mitochondrial biogenesis through SIRT1 pathway (22, 23). RSV increased SIRT1 and PGC-1α levels in a time- and a dose-dependent manner, which peaked at 24 h with 20 μM RSV (Fig. 4, A and B). LSS and RSV demonstrated additive effects on mitochondrial content (Fig. 4C). RSV or the combination with LSS reduced CD62E+ EMP and CD31+/CD42a− EMP production, but a nonstatistically greater reduction was observed with the combination (Fig. 4D). Because SIRT1-dependent pathway has been shown to be involved in mitochondrial biogenic processes in ECs (11), role of SIRT1 in the shear stress-induced adaptive responses was investigated. SIRT1 silencing by a specific siRNA completely abolished flow-induced mitochondrial biogenesis (Fig. 5, A and B) and the reduction of EMP release (Fig. 5C).
Fig. 2.

Shear stress induces mitochondrial biogenesis in human endothelial cells. A: phase-contrast images of HUVECs exposed to LSS at 5 to 20 dyne/cm2 for 36 h; 0 dyne/cm2 indicates static control (STT). Arrows indicate the direction of flow. Scale bars indicate 100 μm. B: cell lysates were analyzed by Western blotting using anti-SIRT1, anti-peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), and anti-transcription factor A (TFAM) antibodies. The bar graphs are results of densitometry analyses. C: mitochondrial content determined by abundance of porin. For both Western blots in B and C, α-Tubulin was used as a loading control. D: representative fluorescence microscopic images of MitoTracker Green FM staining of HUVECs under static or LSS (20 dyne/cm2 for 36 h). Nu, nucleus. The bar graphs are results of densitometry analysis. E: mitochondrial DNA copy number calculated by relative mitochondrial DNA (mtDNA)-encoded genes (COX I or COX II) copy number normalized by 18S. Bar graphs in all panels are means ± SE from 3–5 independent experiments. *P < 0.05; **P < 0.01 vs. static control.
Fig. 3.

EMP release inversely correlates with LSS-induced mitochondrial biogenesis. Confluent monolayers of HUVECs were subjected to LSS (20 dyne/cm2) for 36 h. A: activated- (CD62E+) or total- (CD31+/CD42a−) EMP released from HUVECs under STT or LSS. B: Pearson's correlation plots between mitochondrial content vs. CD62E+ EMP or CD31+/CD42a− EMP. C and D: cell surface expression of CD62E determined by flow cytometry. Bar graphs in all panels are means ± SE from 3–5 independent experiments. *P < 0.05; **P < 0.01 vs. static control.
Fig. 4.

EMP release inversely correlates with mitochondrial enrichment. Confluent monolayers of HUVECs were subjected to various conditions of resveratrol (RSV; 20 μM) or RSV + LSS (20 dyne/cm2) combination treatment for the indicated times. A and B: HUVECs were incubated with RSV at various concentrations for 24 h or at 20 μM for various times. Cell lysates were analyzed by Western blotting with specific antibodies. C: mitochondrial content. D: activated- (CD62E+) or total- (CD31+/CD42a−) EMPs. Bar graphs in all panels are means ± SE from 3–5 independent experiments. *P < 0.05; **P < 0.01 vs. static control. α-Tub, α-Tubulin; SIRT, sirtuin 1.
Fig. 5.

LSS reduces EMP release by SIRT1-mediated mitochondrial biogenesis. HUVECs were transiently transfected with either scrambled or SIRT1 small-interfering RNA (siRNA) and maintained in STT or LSS (20 dyne/cm2 for 24 h). A: SIRT1, PGC-1α, and TFAM protein expressions. α-Tubulin was used as a loading control (CTR). The bar graphs are results of densitometry analyses. B: mitochondrial content. C: activated- (CD62E+) or total- (CD31+/CD42a−) EMP. Each column represents means ± SE from 3–5 independent experiments. *P < 0.05; **P < 0.01. N.S., not significant.
Shear stress decreases EMP release in ECs with defective mitochondria.
To investigate the effect of LSS on restoring the mitochondrial content and EMP release, mitochondrial complex III inhibitor, antimycin A, was used in HUVECs. Treatment with antimycin A (10 μM for 12 h) decreased mitochondrial content by 80% compared with the untreated cells. In contrast, release of CD62E+ EMP (180%) and CD31+/CD42a− (220%) EMPs were significantly increased with the treatment. LSS partially restored mitochondrial content in the antimycin A-preconditioned HUVECs, and the CD62E+ and CD31+/CD42a− EMP production was significantly decreased by LSS (Fig. 6, A and B). Figure 7 shows the schematics of the proposed mechanism.
Fig. 6.
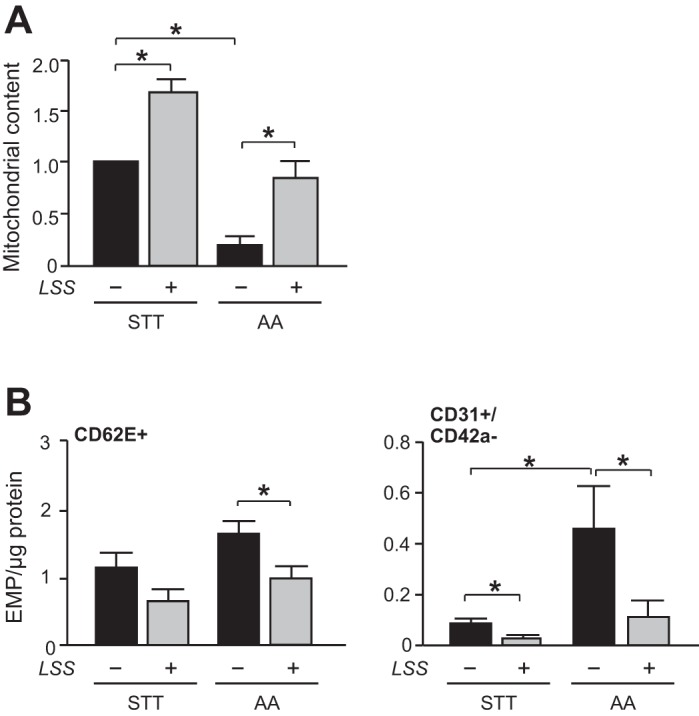
LSS normalizes increased EMP production in endothelial cells with defective mitochondria. HUVECs were treated with antimycin A (AA; 10 μM) or LSS (20 dyne/cm2) for 12 h. Antimycin A-preconditioned HUVECs were subjected to STT or LSS (20 dyne/cm2) for succeeding 24 h. A: mitochondrial content. B: activated- (CD62E+) or total- (CD31+/CD42a−) EMP. Each column represents means ± SE from 3–5 independent experiments. *P < 0.05.
Fig. 7.
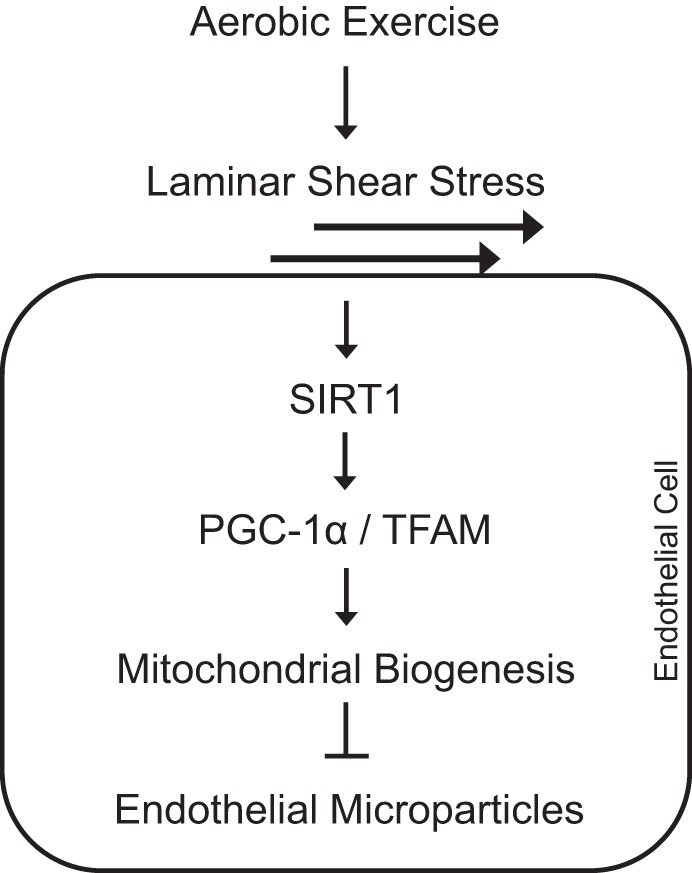
Schematics of the proposed mechanism.
DISCUSSION
Numerous studies have shown that physiological levels of LSS improve EC phenotype. Although several mechanisms through which LSS exerts a beneficial effect on the endothelium have been described, roles of mitochondria and its potential relationship with EMP biogenesis have not been explored. The present study is the first to assess the effects of AEXT on the release of total and activated MP from ECs and to corroborate these effects using an in vitro experimental exercise stimulant, shear stress. Furthermore, the present study demonstrated that LSS-induced mitochondrial biogenesis plays a protective role against the endothelial activation and injury.
We demonstrate, here, that long-term endurance exercise training reduced CD62E+ and CD31+/CD42a− EMP release. This effect is likely attributed to the repeated exposure of endothelium to high laminar flow by aerobic endurance exercise regimen. These findings are consistent with previous studies demonstrating that LSS inhibited leukocyte adhesion and EC apoptosis in response to oxidant stress or growth factor withdrawal (1). In cultured ECs, oscillatory shear stress exaggerates the expression of EC adhesion molecules, whereas high LSS decreases these proinflammatory processes (2, 9, 11). A recent study also demonstrated that disturbed flow acutely increased activation and apoptosis of the human vascular endothelium (20). Moreover, a strong inverse relationship between arterial shear stress and circulating EMP levels has been shown in hemodialysis patients (4). In addition, except the reduction in serum triglyceride, other metabolic parameters and blood pressure remain unchanged in our exercised individuals. Therefore, enhanced LSS by exercise and the subsequent improvement in systemic EC function may directly combat against EC activation in the pre- and mild hypertensive individuals rather indirectly through improvement of their chronic metabolic or hemodynamic status.
Protection from EC dysfunction by LSS and its mechanisms such as eNOS activation have been extensively studied. Our data presented here add new insight into the signal transduction pathway where EC mitochondria seem to be a master regulator. To support this notion, mitochondrial dysfunction has been implicated in the initiation and progression of various CVDs. For instance, excessive mitochondrial reactive oxygen species production has been associated with hypertension, whereas administration of mitochondrial-targeted superoxide scavengers decreased blood pressure (16, 47). Genetic ablation of a mitochondrial antioxidant enzyme [i.e., MnSOD (3) or thioredoxin-2 (50)] in ApoE-knockout mice accelerates the progression of atherosclerosis. Our study showed that LSS-induced mitochondrial biogenesis was significantly associated with reduction in the release of EMP. These data highlight the crucial role of endothelial mitochondria in maintaining vascular homeostasis.
Although the precise mechanisms underlying mitochondrial life cycle and their quality control remain largely unknown, recent studies suggest that mitochondrial content and their functional integrity are maintained primarily by mitochondrial biogenesis (35). Mitochondrial biogenesis requires the expansion of mitochondrial content by transcription of nuclear and mtDNA-encoded genes. PGC-1α is involved in transactivation of the vast majority of the mitochondrial proteins and those involved in mitochondrial transcription such as TFAM, mitochondrial TFBM1 and 2, and the mitochondrial polymerases (43). SIRT1 physically interacts with, and deacetylates, PGC-1α at multiple lysine residues, consequently increasing PGC-1α activity (40). In agreement with a previous report (12), we demonstrate that EC dysfunction as assessed by EMP generation was induced when mitochondrial biogenesis was retarded with a siRNA against SIRT1. Conversely, LSS rescued EC dysfunction by induction of mitochondrial biogenesis. Furthermore, it is important to note that NO is an important inducer of mitochondrial biogenesis in vascular tissues through direct activation of PGC-1α (30). A recent study showed that eNOS inhibition attenuated mitochondrial adaptation to an exercise intervention in the aorta (26). Therefore, it is possible that LSS-induced SIRT1 activation contributes to the preservation of EC function via activation of a SIRT1/eNOS/PGC-1α signaling cascade.
TFAM, a key activator of mitochondrial transcription and replication, is known to be important for mitochondrial biogenesis pathway. The expression of TFAM is coregulated by PGC-1α and NRF-1. In this study, we observed that SIRT-1 knockdown does not change in PGC-1α and TFAM expression level under static flow condition (Fig. 5A). This result is consistent with a previous finding showing that SIRT1 knockdown alone does not affect expression levels of NRF1 or mitochondrial content in ECs under basal condition (12). This might be attributed to a signal redundancy. For example, our group previously reported that tumor suppressor p53 also transcriptionally regulates TFAM expression (34). Because SIRT1 downregulates p53 activity (41), SIRT1 knockdown condition may activate the alternative p53 pathway. Future studies are warranted to elucidate a precise mechanism for induction of TFAM expression in ECs.
Recent studies suggest that SIRT1 and PGC-1α modulate endothelial function in a manner that is independent of mitochondrial biogenesis. EC-specific overexpression of PGC-1α alleviates angiotensin II-induced hypertension through activation of eNOS (24). Endothelial PGC-1α inhibits angiogenesis, and this was shown to be mediated by promoting notch signaling (42). In addition, SIRT1 depletion in ECs exaggerated fibrotic response and atherogenesis via downregulation of matrix metalloproteinase-14 (46). Future studies of these genetic models in endothelial mitochondrial biology are warranted.
The activation of AMP-activated protein kinase (AMPK), a key enzyme for the cellular energy metabolism, is known to be regulated by SIRT1 (37). AMPK was found to increase SIRT1 activity by increasing intracellular level of NAD+ under conditions of energy deprivation, such as exercise (10). Under exercise, activated AMPK increases the level of intracellular NAD+ and induces further enhancement of NAD+-dependent deacetylase SIRT1(19). Activation of AMPK is known to alleviate apoptosis in vascular ECs (25, 28). Together, the crosstalk in the pathway between SIRT1 and AMPK can be another possible mechanism for the reduction of endothelial activation/apoptosis under aerobic exercise.
Stimuli implicated in MP formation from ECs include reactive oxygen species, high glucose, angiotensin II, uremic toxins, and pro-inflammatory and pro-coagulant factors (7). We previously demonstrated that MP release from ECs was increased in response to TNF-α and was attenuated with superoxide dismutase (6). Therefore, the level of EMP may reflect the inflammatory and oxidative stress of the ECs in vitro and in vivo as proposed recently (8). Furthermore, in the present study, our data suggest that mitochondrial dysfunction in ECs may be a critical determinant of EMP production associated with several CVDs including hypertension. However, further research should be done to determine the precise mechanism underlying mitochondrial regulation of MP biogenesis.
A large body of literature has demonstrated that regular practice of physical activity improves endothelial function in animals and humans with and without CVD risk factors. Studying cellular mechanisms of the adaptive response may provide valuable insights for therapeutic strategies against endothelial dysfunction. Because EMP is emerging as a biomarker of EC health status, the results of the present study support the concept that EMP may provide a reliable assessment of the changes in cellular status of the endothelium resulting from interventions. However, an inordinate amount of work is ahead to determine whether the level of EMP is a marker of the natural history of a disease and correlates longitudinally with known clinical indices and whether the level depicts the effects of a therapeutic intervention.
GRANTS
This work was supported by Temple University's University Fellowships (to J. Kim, B. Kim, and H. Lee), American Heart Association Grants 12SDG12070327 (to J. Park) and Predoctoral Fellowship 12PRE11960049 (to B. Kim), and National Heart, Blood, and Lung Institute Grants R01-HL085497 (to M. Brown) and R01-HL126952 (to J. Park).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.-S.K., B.K., H.L., S.T., and D.M.B. performed experiments; J.-S.K., B.K., H.L., S.T., D.M.B., M.D.B., and J.-Y.P. analyzed data; J.-S.K., B.K., and J.-Y.P. prepared figures; J.-S.K. drafted manuscript; J.-S.K., B.K., H.L., S.T., D.M.B., S.E., M.D.B., and J.-Y.P. approved final version of manuscript; S.E., M.D.B., and J.-Y.P. interpreted results of experiments; S.E., M.D.B., and J.-Y.P. edited and revised manuscript; M.D.B. and J.-Y.P. conception and design of research.
REFERENCES
- 1.Ando J, Tsuboi H, Korenaga R, Takada Y, Toyama-Sorimachi N, Miyasaka M, Kamiya A. Shear stress inhibits adhesion of cultured mouse endothelial cells to lymphocytes by downregulating VCAM-1 expression. Am J Physiol Cell Physiol 267: C679–C687, 1994. [DOI] [PubMed] [Google Scholar]
- 2.Ando J, Yamamoto K. Effects of shear stress and stretch on endothelial function. Antioxid Redox Signal 15: 1389–1403, 2011. [DOI] [PubMed] [Google Scholar]
- 3.Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA, Hu Z, Reuf J, Horaist C, Lebovitz R, Hunter GC, McIntyre K, Runge MS. Mitochondrial integrity and function in atherogenesis. Circulation 106: 544–549, 2002. [DOI] [PubMed] [Google Scholar]
- 4.Boulanger CM, Amabile N, Guerin AP, Pannier B, Leroyer AS, Mallat CN, Tedgui A, London GM. In vivo shear stress determines circulating levels of endothelial microparticles in end-stage renal disease. Hypertension 49: 902–908, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Boulanger CM, Amabile N, Tedgui A. Circulating microparticles: a potential prognostic marker for atherosclerotic vascular disease. Hypertension 48: 180–186, 2006. [DOI] [PubMed] [Google Scholar]
- 6.Brown MD, Feairheller DL, Thakkar S, Veerabhadrappa P, Park JY. Racial differences in tumor necrosis factor-alpha-induced endothelial microparticles and interleukin-6 production. Vasc Health Risk Manag 7: 541–550, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burger D, Schock S, Thompson CS, Montezano AC, Hakim AM, Touyz RM. Microparticles: biomarkers and beyond. Clin Sci (Lond) 124: 423–441, 2013. [DOI] [PubMed] [Google Scholar]
- 8.Burger D, Touyz RM. Cellular biomarkers of endothelial health: microparticles, endothelial progenitor cells, and circulating endothelial cells. J Am Soc Hypertens 6: 85–99, 2012. [DOI] [PubMed] [Google Scholar]
- 9.Burns MP, DePaola N. Flow-conditioned HUVECs support clustered leukocyte adhesion by coexpressing ICAM-1 and E-selectin. Am J Physiol Heart Circ Physiol 288: H194–H204, 2005. [DOI] [PubMed] [Google Scholar]
- 10.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458: 1056–1060, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chappell DC, Varner SE, Nerem RM, Medford RM, Alexander RW. Oscillatory shear stress stimulates adhesion molecule expression in cultured human endothelium. Circ Res 82: 532–539, 1998. [DOI] [PubMed] [Google Scholar]
- 12.Chen Z, Peng IC, Cui X, Li YS, Chien S, Shyy JY. Shear stress, SIRT1, and vascular homeostasis. Proc Natl Acd Sci USA 107: 10268–10273, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng C. Abdominal aortic hemodynamic conditions in healthy subjects aged 50–70 at rest and during lower limb exercise: in vivo quantification using MRI. Atherosclerosis 168: 323–331, 2003. [DOI] [PubMed] [Google Scholar]
- 14.Chironi GN, Boulanger CM, Simon A, Dignat-George F, Freyssinet JM, Tedgui A. Endothelial microparticles in diseases. Cell Tissue Res 335: 143–151, 2009. [DOI] [PubMed] [Google Scholar]
- 15.Dignat-George F, Boulanger CM. The many faces of endothelial microparticles. Arterioscler Thromb Vasc Biol 31: 27–33, 2011. [DOI] [PubMed] [Google Scholar]
- 16.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res 107: 106–116, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dimmeler S, Haendeler J, Rippmann V, Nehls M, Zeiher AM. Shear stress inhibits apoptosis of human endothelial cells. FEBS Lett 399: 71–74, 1996. [DOI] [PubMed] [Google Scholar]
- 18.Feairheller DL, Park JY, Rizzo V, Kim B, Brown MD. Racial differences in the responses to shear stress in human umbilical vein endothelial cells. Vasc Health Risk Manag 7: 425–431, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol 5: 253–295, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jenkins NT, Padilla J, Boyle LJ, Credeur DP, Laughlin MH, Fadel PJ. Disturbed blood flow acutely induces activation and apoptosis of the human vascular endothelium. Hypertension 61: 615–621, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kizhakekuttu TJ, Wang J, Dharmashankar K, Ying R, Gutterman DD, Vita JA, Widlansky ME. Adverse alterations in mitochondrial function contribute to type 2 diabetes mellitus-related endothelial dysfunction in humans. Arterioscler Thromb Vasc Biol 32: 2531–2539, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ Res 112: 1171–1188, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127: 1109–1122, 2006. [DOI] [PubMed] [Google Scholar]
- 24.Li C, Cai S, Keaney JFJ. Mitochondrial regulation of endothelial function. Circulation 126: A16000, 2012. [Google Scholar]
- 25.Liu C, Liang B, Wang Q, Wu J, Zou MH. Activation of AMP-activated protein kinase alpha1 alleviates endothelial cell apoptosis by increasing the expression of anti-apoptotic proteins Bcl-2 and survivin. J Biol Chem 285: 15346–15355, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Miller MW, Knaub LA, Olivera-Fragoso LF, Keller AC, Balasubramaniam V, Watson PA, Reusch JE. Nitric oxide regulates vascular adaptive mitochondrial dynamics. Am J Physiol Heart Circ Physiol 304: H1624–H1633, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morel O, Toti F, Hugel B, Bakouboula B, Camoin-Jau L, Dignat-George F, Freyssinet JM. Procoagulant microparticles: disrupting the vascular homeostasis equation? Arterioscler Thromb Vasc Biol 26: 2594–2604, 2006. [DOI] [PubMed] [Google Scholar]
- 28.Nagata D, Kiyosue A, Takahashi M, Satonaka H, Tanaka K, Sata M, Nagano T, Nagai R, Hirata Y. A new constitutively active mutant of AMP-activated protein kinase inhibits anoxia-induced apoptosis of vascular endothelial cell. Hypertens Res 32: 133–139, 2009. [DOI] [PubMed] [Google Scholar]
- 29.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. J Biol Chem 280: 16456–16460, 2005. [DOI] [PubMed] [Google Scholar]
- 30.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 299: 896–899, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Okabe TA, Shimada K, Hattori M, Murayama T, Yokode M, Kita T, Kishimoto C. Swimming reduces the severity of atherosclerosis in apolipoprotein E deficient mice by antioxidant effects. Cardiovasc Res 74: 537–545, 2007. [DOI] [PubMed] [Google Scholar]
- 32.Oldendorf WH, Cornford ME, Brown WJ. The large apparent work capability of the blood-brain barrier: a study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann Neurol 1: 409–417, 1977. [DOI] [PubMed] [Google Scholar]
- 33.Park JY, Farrance IK, Fenty NM, Hagberg JM, Roth SM, Mosser DM, Wang MQ, Jo H, Okazaki T, Brant SR, Brown MD. NFKB1 promoter variation implicates shear-induced NOS3 gene expression and endothelial function in prehypertensives and stage I hypertensives. Am J Physiol Heart Circ Physiol 293: H2320–H2327, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park JY, Wang PY, Matsumoto T, Sung HJ, Ma W, Choi JW, Anderson SA, Leary SC, Balaban RS, Kang JG, Hwang PM. p53 improves aerobic exercise capacity and augments skeletal muscle mitochondrial DNA content. Circ Res 105: 705–712, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patten IS, Arany Z. PGC-1 coactivators in the cardiovascular system. Trends Endocrinol Metab 223: 90–97, 2012. [DOI] [PubMed] [Google Scholar]
- 36.Pellegrin M, Miguet-Alfonsi C, Bouzourene K, Aubert JF, Deckert V, Berthelot A, Mazzolai L, Laurant P. Long-term exercise stabilizes atherosclerotic plaque in ApoE knockout mice. Med Sci Sports Exerc 41: 2128–2135, 2009. [DOI] [PubMed] [Google Scholar]
- 37.Price NL, Gomes AP, Ling AJ, Duarte FV, Martin-Montalvo A, North BJ, Agarwal B, Ye L, Ramadori G, Teodoro JS, Hubbard BP, Varela AT, Davis JG, Varamini B, Hafner A, Moaddel R, Rolo AP, Coppari R, Palmeira CM, de Cabo R, Baur JA, Sinclair DA. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab 15: 675–690, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pynn M, Schafer K, Konstantinides S, Halle M. Exercise training reduces neointimal growth and stabilizes vascular lesions developing after injury in apolipoprotein e-deficient mice. Circulation 109: 386–392, 2004. [DOI] [PubMed] [Google Scholar]
- 39.Quintero M, Colombo SL, Godfrey A, Moncada S. Mitochondria as signaling organelles in the vascular endothelium. Proc Natl Acd Sci USA 103: 5379–5384, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434: 113–118, 2005. [DOI] [PubMed] [Google Scholar]
- 41.Sasca D, Hahnel PS, Szybinski J, Khawaja K, Kriege O, Pante SV, Bullinger L, Strand S, Strand D, Theobald M, Kindler T. SIRT1 prevents genotoxic stress-induced p53 activation in acute myeloid leukemia. Blood 124: 121–133, 2014. [DOI] [PubMed] [Google Scholar]
- 42.Sawada N, Jiang A, Takizawa F, Safdar A, Manika A, Tesmenitsky Y, Kang KT, Bischoff J, Kalwa H, Sartoretto JL, Kamei Y, Benjamin LE, Watada H, Ogawa Y, Higashikuni Y, Kessinger CW, Jaffer FA, Michel T, Sata M, Croce K, Tanaka R, Arany Z. Endothelial PGC-1alpha mediates vascular dysfunction in diabetes. Cell Metab 19: 246–258, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 88: 611–638, 2008. [DOI] [PubMed] [Google Scholar]
- 44.Szabadkai G, Duchen MR. Mitochondria: the hub of cellular Ca2+ signaling. Physiology (Bethesda) 23: 84–94, 2008. [DOI] [PubMed] [Google Scholar]
- 45.Taylor CA, Hughes TJ, Zarins CK. Effect of exercise on hemodynamic conditions in the abdominal aorta. J Vasc Surg 29: 1077–1089, 1999. [DOI] [PubMed] [Google Scholar]
- 46.Vasko R, Xavier S, Chen J, Lin CH, Ratliff B, Rabadi M, Maizel J, Tanokuchi R, Zhang F, Cao J, Goligorsky MS. Endothelial sirtuin 1 deficiency perpetrates nephrosclerosis through downregulation of matrix metalloproteinase-14: relevance to fibrosis of vascular senescence. J Am Soc Nephrol 25: 276–291, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Widlansky ME, Gutterman DD. Regulation of endothelial function by mitochondrial reactive oxygen species. Antioxid Redox Signal 15: 1517–1530, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilund KR. Is the anti-inflammatory effect of regular exercise responsible for reduced cardiovascular disease? Clin Sci (Lond) 112: 543–555, 2007. [DOI] [PubMed] [Google Scholar]
- 49.Yu E, Mercer J, Bennett M. Mitochondria in vascular disease. Cardiovasc Res 95: 173–182, 2012. [DOI] [PubMed] [Google Scholar]
- 50.Zhang H, Luo Y, Zhang W, He Y, Dai S, Zhang R, Huang Y, Bernatchez P, Giordano FJ, Shadel G, Sessa WC, Min W. Endothelial-specific expression of mitochondrial thioredoxin improves endothelial cell function and reduces atherosclerotic lesions. Am J Pathol 170: 1108–1120, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]