

Abstract
Glycosphingolipids (GSLs) play a role in insulin resistance and diabetes, but their role in diabetic nephropathy (DN) has received limited attention. We used 9- and 17-wk-old nondiabetic db/m and diabetic db/db mice to examine the role of GSLs in DN. Cerebrosides or monoglycosylated GSLs [hexosylceramides (HexCers); glucosyl- and galactosylceramides] and lactosylceramide (LacCers) were elevated in db/db mouse kidney cortices, specifically in glomeruli, and also in urine. In our recent paper (25), we observed that the kidneys exhibited glomerular hypertrophy and proximal tubular vacuolization and increased fibrosis markers at these time points. Mesangial cells contribute to hyperglycemia-induced glomerular hypertrophy in DN. Hyperglycemic culture conditions, similar to that present in diabetes, were sufficient to elevate mesangial cell HexCers and increase markers of fibrosis, extracellular matrix proteins, and cellular hypertrophy. Inhibition of glucosylceramide synthase or lowering glucose levels decreased markers of fibrosis and extracellular matrix proteins and reversed mesangial cell hypertrophy. Hyperglycemia increased phosphorylated (p)SMAD3 and pAkt levels and reduced phosphatase and tensin homolog levels, which were reversed with glucosylceramide synthase inhibition. These data suggest that inhibition of glucosylceramide synthase reversed mesangial cell hypertrophy through decreased pAkt and pSmad3 and increased pathways responsible for protein degradation. Importantly, urinary GSL levels were higher in patients with DN compared with healthy control subjects, implicating a role for these lipids in human DN. Thus, hyperglycemia in type II diabetes leads to renal dysfunction at least in part by inducing accumulation of HexCers and LacCers in mesangial cells, resulting in fibrosis, extracellular matrix production, and hypertrophy.
Keywords: glycosphingolipids, kidney, diabetic nephropathy, hypertrophy, mesangial cells
diabetic nephropathy (DN) is major complication of diabetes mellitus and is the most prevalent cause of chronic renal failure and end-stage renal disease in the Western world (13). DN is clinically characterized by an initial rise in the glomerular filtration rate followed later by an increase in urinary albumin excretion. Without intervention, the glomerular filtration rate decreases over time.
DN is thought to arise from hemodynamic and metabolic factors that act independently and in concert with one another (14, 23). Hemodynamic factors include alterations in intraglomerular vascular pressure as well as vasoactive hormone pathways such as the renin-angiotensin aldosterone system and endothelin (14, 23). Chronic hyperglycemia has been reported to induce an initial phase of increased mesangial cell proliferation that is followed by mesangial matrix expansion and renal hypertrophy (17, 23, 29, 62). In addition, there is an induction of inflammatory signaling pathways within the glomerulus that results in the activation of macrophages and infiltration of immune cells (20). The mechanisms by which chronic hyperglycemia induces these changes in the glomeruli are not completely understood.
Glycosphingolipids (GSLs) are a heterogenous class of lipids in the sphingolipid family. One of the most simple subclass of GSLs is glucosylceramide, which is generated from ceramides synthesized in the endoplasmic reticulum that are transported to the Golgi, where UDP-Glc:ceramide glucosyltransferase [glucosylceramide synthase (GCS)] catalyzes the addition of glucose (35). Once formed, a galactose (from UDP-galactose) can be added to glucosylceramide to form lactosylceramides (LacCer). LacCer is the central hub for the synthesis of complex GSLs, including other neutral GSLs, such as globotriaosylceramide (Gb3), and acidic GSLs, such as ganglioside GM3.
GSLs regulate a number of cellular processes, including cell proliferation and inflammation, and have been implicated in metabolic syndrome and diabetes (7, 12, 26, 27, 35, 38, 55, 63). Inhibition of GCS by d,l-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol hydrochloride (PDMP), d,l-threo-1-phenyl-2-palmitoylamino-3-morpholino-1-propanol (PPMP), deoxynojirimycin, and miglustat protect from various disease conditions in rodent models; in particular, inhibition of GCS prevents and reverses insulin resistance and hepatic steatosis in rodent models of diabetes (1, 59, 65). GCS inhibitors, such as miglustat and eliglustat, are beneficial in the treatment of Gaucher disease in humans (9, 28, 36, 43, 52).
GSLs are highly abundant in the kidney, including podocytes, mesangial cells, and tubular epithelial cells (35). Defects in GSL metabolism that either result in their accumulation or enhanced degradation occur in Fabry's kidney disease, polycystic kidney disease, and renal cell carcinoma (35). Renal GSLs have been shown to play a role in streptozotocin (STZ)-induced type I DN in rats, and their inhibition with PDMP reversed glomerular hypertrophy (61). However, their role in nephropathy associated with type II diabetes has not been determined.
We hypothesized that chronic hyperglycemia in type II diabetes elevates levels of kidney GSLs, which lead to glomerular pathology. We tested this in vivo in the db/db mouse model of type II diabetes and in vitro in cultured mesangial cells. We observed elevation of GSLs in the kidney of diabetic db/db mice and that inhibition of the synthesis of these lipids reversed hyperglycemia-induced mesangial cell hypertrophy through decreased phosphorylated (p)Smad3 and pAkt signaling and enhanced p-phosphatase and tensin homolog (pPTEN)-mediated protein degradation pathways. The present work indicates a novel role for GSLs in the induction of glomerular hypertrophy in response to hyperglycemia in DN.
MATERIALS AND METHODS
Materials.
DMEM (low and high glucose), trypsin-EDTA solution, HEPES, FBS, and penicillin-streptomycin solution were from GIBCO/Invitrogen. F-12 HAM's supplement was purchased from Hyclone. The BCA protein assay kit was from Pierce (catalog nos. 23223 and 23224). We used an inhibitor of glucosylceramide synthase, d-threo-1-ethylendioxyphenyl-2-decanoylamino-3-pyrrolidino-propanol, the 10-carbon analog of eliglustat (C10) (33). Concentration determined by glucosylceramide synthase activity assays verified that 48 h after a single treatment of 0.15 μM C10, the activity of glucosylceramide synthase was significantly reduced (decreased by 90%) and did not decrease viability (data not shown).
Mice.
Female diabetic db/db mice (BKS.Cg-m +/+ Leprdb/J; related genotype: a/a+Leprdb/+ Leprdb) and female nondiabetic db/m mice (BKS.Cg-m +/+ Leprdb/J; related genotype: a/a+Dock7m +/+ Leprdb) were purchased from Jackson Laboratory (stock no. 000642, Bar Harbor, ME) and housed in temperature-controlled conditions under a light-dark cycle with food and water supplied ad libitum. At 9 wk (n = 11 mice/group) and 17 wk (n = 6 mice/group) of age, db/m and db/db mice were placed in metabolism cages for 24 h and then euthanized as previously described (25), and body weight, serum glucose, proinflammatory markers, fibrosis, creatinine clearance, and urinary protein excretion were evaluated (25).
Isolation of glomeruli from db/db and db/m mice.
Db/db and db/m mice were euthanized by cervical dislocation, and kidneys were dissected. The kidneys of three mice were pooled (6 total). The kidney cortex was dissected, and the cortices were minced with a razor in sterile ice-cold PBS. Kidney cortex homogenate was filtered through three consecutive nylon sieves arranged largest pore size on top to smallest pore size on bottom (pore sizes: 180, 106, and 53 μm). The end of a plunger from a 10-ml syringe was used to press the homogenate through the top sieve. The contents of the final 53-μm sieve were washed into a beaker using ice-cold PBS and centrifuged for 5 min at 500 g at 4°C. The resulting pellet was resuspended in collagenase solution that was prewarmed at 37°C (3 ml of 5 mg/ml collagenase type II, catalog no. 17101-015, GIBCO) and incubated at 37°C for 30 min with gentle vortexing every 10 min. After the collagenase digestion, 5 ml ice-cold PBS was added, and the homogenate was then centrifuged at 500 g for 5 min. The resulting pellet was washed twice by resuspending in ice-cold PBS and centrifugation at 500 g for 5 min. The final pellet was snap frozen and stored at −80°C until analysis.
Mesangial cell culture and treatment.
Mouse mesangial cells were obtained from the American Type Culture Collection (CRL 1927). Cells were cultured in a 3:1 (vol/vol) mixture of high-glucose DMEM, Ham's F-12 medium, and 14 mM HEPES and supplemented with 5% FBS and 1% penicillin-streptomycin solution. The total glucose concentration of this media composition was 18.8 mM. Once we received this cell line, one set of cells was switched to normal glucose conditions (cells were grown for 25 days in normal glucose and split when cells were confluent; this set was frozen and used for further experiments, which used a total glucose concentration of 5.5 mM). Glucose concentrations were controlled by altering the glucose level of DMEM to achieve either a high (18.8 mM) or normal (5.5 mM) total glucose concentration in the culture media. As an osmotic control, mannitol was used at an equal concentration as glucose. In all cases, media were filter sterilized before use. Cells were grown at 37°C in a humidified 5% CO2 incubator not exceeding seven passages.
Sphingolipidomic mass spectrometry.
For kidney measurements, the cortex of the kidney was homogenized using a Tissue-Tearor (Biospec Products) in 20 mM Tris·HCl buffer. A Complete Protease Inhibitor Cocktail Tablet was added to the buffer (Sigma, St. Louis, MO). Protein was quantified using a BCA Protein Determination Kit. From each kidney cortex homogenate, 1 mg of protein was sent for sphingolipid analysis. For measurement of urine GSLs, 300 μl mouse or 100 μl human urine was centrifuged, and the supernatant was transferred to a glass tube. To obtain enough material for the quantification of hexosylceramides (HexCers), purified glomeruli from the kidneys of three mice were combined into a single sample. For cultured mesangial cells, ∼3 × 106 cells were sent for sphingolipid analysis. All samples were snap frozen and submitted for sphingolipid analysis performed at the Medical University of South Carolina (Lipidomics Core) on a Thermo Finnigan TSQ 7000, triple-stage quadrupole mass spectrometer operating in a multiple reaction monitoring positive ionization mode, as previously described (3). Data were normalized to total lipid phosphate, and urine sample data were normalized to creatinine.
Mesangial cell proliferation by counting.
Mouse mesangial cells grown in normal or high-glucose medium were plated in six-well plates at a density of 1.6 × 105 cells/well (in 2 ml of complete growth medium). At 24 or 48 h, cells were washed with PBS, trypsinized, and resuspended in growth media. Cell counts were determined using a Bio-Rad TC20 automated cell counter.
Mesangial cell proliferation by Alamar blue assay.
Mouse mesangial cells grown in normal or high-glucose medium were plated in 96-well plates at a density of 3 × 103 cells/well (in 200 μl of complete growth medium) for 24, 48, and 72 h, at which point 10% of Alamar blue reagent from Invitrogen (catalogue no. DAL1100) was added to each well. Plates were then incubated, and the fluorescence of the Alamar blue reduction was determined on a SPECTRAmax Gemini EM plate reader every hour until untreated wells were midlinear (∼4,000 arbitrary units, 540-nm excitation and 594-nm emission).
Mesangial cell proliferation by bromodeoxyuridine/7-aminoactinomycin D flow cytometry.
Mouse mesangial cells grown in normal or high-glucose medium were plated in 10-cm2 dishes at a density of 0.5 × 106 cells/plate (in 2 ml of complete growth medium). At 24 or 48 h, cells were washed with PBS, trypsinized, and resuspended in growth media. At 45 min before trypsinization, bromodeoxyuridine (BrdU; catalog no. 552598, BD Pharmingen) was added at a final concentration of 10 μM. Cells were fixed, permeabilized, and washed according to the manufacturer's instructions. BrdU and 7-aminoactinomycin D (7-AAD) were analyzed via flow cytometry (FACS scan, DxP multicolor upgrades powered by Cytek Development).
Mesangial cell proliferation by BrdU ELISA.
Mouse mesangial cells grown in normal or high-glucose medium were plated in 96-well plates at a density of 3 × 103 cells/well (in 200 μl of complete growth medium). At the indicated time points, 20 μl of 1× BrdU solution was added and incubated for 24 h, and cells were then fixed. BrdU measurements were performed according to the manufacturer's instructions (catalog no. 2750, Millipore). The absorbance was then read at 450 nm.
Mesangial cell thickness by confocal microscopy.
Mouse mesangial cells grown in normal or high-glucose medium were plated in 35-mm diameter confocal dishes at a density of 1.6 × 105 cells/plate (in 2 ml of complete growth medium). Where indicated, cells were treated with GCS inhibitor (0.15 μM C10) at the time of plating. After 48 h, cells were fixed with 4% formaldehyde for 10 min at room temperature (24°C). After plates were washed twice with PBS for 5 min each, cells were permeabilized with blocking buffer (2 ml, 1× PBS, 5% FBS, and 0.3% Triton X-100) for 10 min at room temperature. Blocking buffer was aspirated, and cells were stained with the F-actin stain phalloidin (diluted 1:1,000 in PBS, Alexa fluor568, catalog no. 12380, Life Technologies) at room temperature for 10 min. After cells had been washed three times for 5 min each with PBS, they were stained with the nuclear stain DRAQ (catalog no. 4084, Cell Signalling) at room temperature for 10 min. Cells were then washed three times for 10 min each at room temperature. PBS remained on the cells to avoid drying until they were imaged on a Nikon (NIS-Elements, version 4.00) confocal microscope. The microscope was setup to take z-stacks every 0.5 μm from several micrometers above the cells until the bottom of the cells at the surface of the slide. The average number of z-stacks required to transverse the cells (where we saw F-actin stain from the top to bottom of the cells) was defined the average thickness of the cells. z-Stacks were performed using the ×60 objective on samples prepared from three independent experiments. Measurements of at least 10 fields of view were taken per experiment.
Immunoblot analysis.
Mouse mesangial cells grown in normal or high-glucose medium were plated in 10-cm2 dishes at a density of 0.5 × 106 cells/plate (in 10 ml of complete medium). Where indicated, cells were treated with GCS inhibitor (0.15 μM C10) at the time of plating. After 48 h, media were removed, and cells were washed with ice-cold PBS and scraped with 1 ml PBS. Cells were centrifuged, and the supernatant was removed. Cells were resuspended with Nonidet P-40 lysis buffer containing Pierce Halt protease and phosphatase inhibitor. Processing of Western blot samples was performed as previously described by Qureshi et al. (45). Protein (30 μg) was used for Western blot analysis. Membranes were then incubated with primary antibodies in 1:2,000 dilutions and incubated overnight with continuous shaking at 4°C. The primary antibodies were as follows: Akt, pAkt (Ser473), PTEN, pPTEN (Ser380/Thr382/383), and Smad3 (catalog nos. 4691S, 4060S, 9552S, 9549S, and 9513S, respectively, Cell Signaling); pSmad3 (Ser423/Ser425, catalog no. 600-401-919, Rockland); and fibronectin and β-actin (catalog nos. F3648 and A5441, respectively, Sigma). After extensive washes, membranes were incubated with peroxidase-conjugated donkey anti-rabbit (1:10,000, catalog no. SC-2313, Santa Cruz Biotechnology). Signals were visualized using ECL or SuperSignal west Dura (Thermo Scientific).
Data and statistical analysis.
Data are expressed as means ± SD for all experiments. Multiple comparisons of normally distributed data were analyzed by one-way ANOVA, as appropriate, and group means were compared using the Student-Newman-Keuls post hoc test. Single comparisons were analyzed by Student's t-test where appropriate.
Animal usage.
All animal and treatment protocols were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the Medical University of South Carolina.
Human urine samples.
Clean catch urine samples for the LacCer analyses were obtained from frozen, centrifuged, and aliquoted samples that were collected for an unrelated urine biomarker study. All volunteers gave documented informed consent to participate in Institutional Review Board-approved protocols. A 100-μl aliquot of urine was submitted to the Medical University of South Carolina Lipidomics Core facility for the quantification of LacCers. In human urine, LacCers can be easily quantified, but HexCers are often below the detectable limit (data not shown). This may reflect differences in their stability in solution and/or their rates of shedding into the urine. LacCers were normalized to milligram of urine creatinine using a creatinine assay kit (BioAssay Systems, Hayward, CA) as per the manufacturer's instructions.
RESULTS
GSLs are elevated in the kidney cortex and urine of diabetic db/db mice.
We have previously reported that the db/db mouse model of type II diabetes has elevated markers of inflammation and fibrosis in the kidney as early 9 wk. A decline in kidney function and pathological changes typical of DN, such as mesangial matrix expansion, thickening of the glomerular basement membrane, and vacuolization of the proximal tubular cells, were observed at 17 wk of age (25). Thus, we defined 9 and 17 wk of age as early and late DN in these mice. Using the same db/db diabetic and db/m nondiabetic control mice from these two time points (25), we measured HexCers and LacCers in kidney cortex homogenates. HexCers are synthesized from ceramides, which vary in the chain length of the N-linked fatty acyl chain. In the diabetic kidney, several major species of HexCers and LacCers were elevated in the kidney cortex at both 9 and 17 wk in db/db mice compared with age-matched db/m control mice (Fig. 1, A and B). Very-long-chain species of HexCers (C22- to C26-HexCers) were elevated to a larger extent than long-chain species of C16- and C18-HexCers (Fig. 1A). However, the pattern for LacCers was the opposite, with C16-, C18-, and C20-LacCers increased to a larger extent than very-long-chain species (C24-, C24:1-, and C26-LacCers; Fig. 1B).
Fig. 1.

Glycosphingolipids (GSLs) are elevated in the kidney cortex and urine of diabetic db/db mice. GSLs [hexosylceramides (HexCers) and lactosylceramides (LacCers)] were quantified by HPLC-tandem mass spectroscopy (HPLC-MS/MS). HexCers (A) and LacCers (B) were quantified in 1 mg of kidney cortex homogenate obtained from either db/m or db/db mice at 9 or 17 wk of age. Lipids in A and B were normalized to total lipid phophate. C: total HexCers and LacCers in a 24-h urine sample collected from nondiabetic db/m or diabetic db/db mice at 9 wk of age. Lipids were normalized to creatinine. Data in A–C are means ± SD; n = 5. *P ≤ 0.01 compared with age-matched nondiabetic control mice.
Because GSLs are elevated in several kidney diseases and can be secreted in the urine, we determined if GSLs were higher in the urine of db/db diabetic mice. Total GSLs were elevated in the urine of 9-wk-old db/db mice compared with db/m control mice (Fig. 1C). These data demonstrate that renal and urinary HexCers and LacCers are elevated early in DN and represent multiple species with very-long-chain species of HexCers and long-chain species of LacCers being elevated.
HexCer levels are higher in glomeruli obtained from db/db mice, and hyperglycemia induces their generation in mesangial cells in vitro.
Because DN is characterized in part by glomerular changes, we determined if GSLs were elevated in glomeruli. Glomeruli-enriched fractions were obtained from kidney cortices of 9-wk-old db/db diabetic and db/m control mice, and HexCers were quantified. HexCers were higher in glomeruli obtained from db/db mice [long-chain (C16, C18, and C20) HexCer but not-very-long chain (C22, C22:1, and C24:1) HexCer; Fig. 2A]. Kidney GSLs were elevated after STZ-induced type I DN in rats, and administration of insulin to these rats reduced kidney GSLs, suggesting that they accumulate as a result of hyperglycemia (61). Thus, we hypothesized that hyperglycemia in diabetes is sufficient to induce the generation of HexCers. To test this, we used mouse mesangial cells grown in culture and determined if hyperglycemic conditions, similar to that present in DN, increase HexCers. HexCers were measured in cells cultured in normal glucose (5.5 mM supplemented with 13.3 mM mannitol to control for changes in osmolarity) or high-glucose (18.8 mM) media for 48 h. High-glucose media led to the accumulation of long-chain and very-long-chain HexCers (C16-, C18-, C20-, and C24-HexCer), with the largest changes being long-chain-species (C16- and C20-HexCers; Fig. 2B). The pattern of accumulated HexCers in mesangial cells cultured in hyperglycemic conditions was similar to those that accumulated in vivo in glomerular-enriched fractions (Fig. 2A). These data reveal that elevated glucose is sufficient to increase GSLs in mesangial cells.
Fig. 2.

HexCer levels are higher in glomerular cells obtained from db/db mice and mesangial cells grown in elevated glucose media in vitro. A: HexCers were elevated in glomerular enriched fractions obtained from the kidneys of 9-wk-old diabetic db/db mice compared with age-matched nondiabetic db/m control mice. Cells were purified as described in materials and methods from the kidney cortex. Kidneys from two mice were combined to create a single sample of three million cells, and three separate samples were analyzed. B: elevated glucose induces an accumulation of HexCers in cultured mesangial cells. Mouse mesangial cells were cultured for 48 h in normal or high-glucose medium. Sample of three million cells and three separate samples were analyzed. HexCers were measured by HPLC-MS/MS, normalized to total lipid phosphate, and expressed as fold changes over db/m control mice (A) and mannitol-treated control cells (B). Data are means ± SD; n = 3. *P ≤ 0.01 (as determined by a Student's t-test).
High glucose induces mesangial cell hypertrophy, which is reversed by glucosylceramide synthase inhibition.
Glomerular hypertrophy occurs in DN, and in the db/db model of type II DN, glomerular hypertrophy is observed at 17 wk of age (25). Hyperglycemia-induced glomerular hypertrophy is thought to occur in part by an initial transient phase of increased mesangial cell proliferation, which is followed by mesangial cell expansion, extracellular matrix accumulation, podocyte hypertrophy, and thickening of the glomerular basement membrane; hyperglycemic culture conditions have been shown to be sufficient to induce many of these changes (2, 30, 32, 34, 42, 49, 51, 57, 58, 62). We cultured mesangial cells in high-glucose media, and cell size was measured by determining the average cell thickness using confocal microscopy. Cells grown in normal glucose media had an average cell thickness of 7.7 ± 0.3 μm (mean ± SD; Fig. 3, A and D). In contrast, the cell thickness of mesangial cells grown in high-glucose media increased to 12.3 ± 0.8 μm (mean ± SD; Fig. 3, B and D). As mesangial cell GSLs were elevated in high-glucose media (Fig. 2B), we determined if pharmacological inhibition of glucosylceramide synthase was sufficient to reduce mesangial cell thickness in high-glucose media. Mesangial cells grown in high-glucose media and treated with an inhibitor of glucosylceramide synthase, d-threo-1-ethylendioxyphenyl-2-decanoylamino-3-pyrrolidino-propanol, the 10-carbon analog of eliglustat (C10), for 48 h exhibited decreased cell thickness to 8.0 ± 0.7 μm (mean ± SD; Fig. 3, C and D), similar to cells grown in normal glucose medium.
Fig. 3.


High glucose induces mesangial cell hypertrophy, which is reversed by glucosylceramide synthase inhibition. Mesangial cells were grown in normal glucose (NG; A), high glucose (HG; B), or HG supplemented with 0.15 μM C10 (C). At 48 h, cells were fixed, permeabilized, and stained with the F-actin marker phalloidin and the nuclear stain DRAQ. Serial 0.5-μm-thick z-sections were obtained using confocal microscopy. D: average thickness of mesangial cells calculated by the distance from the top to the bottom of the F-actin staining. Data are means ± SD; n ≥ 5. ***P ≤ 0.0001, HG compared with NG; $$$P ≤ 0.0001, HG + C10 compared with HG (as determined by one-way ANOVA). Scale bars = 50 μm.
Chronic high glucose does not increase mesangial cell proliferation.
Several studies have reported that glomerular hypertrophy in DN is associated with an initial phase of enhanced mesangial cell proliferation (23, 62). High-glucose media in vitro has also been reported to increase cell proliferation of mesangial cells (5, 39, 60, 62). In addition, GSLs have been reported to play a role in cell proliferation (6). To test if cell proliferation played a role in this system, we compared mesangial cells chronically cultured in high-glucose conditions with those chronically cultured in normal glucose conditions. Cell proliferation was examined using multiple approaches, including counting cell numbers (Fig. 4A), Alamar blue fluorescence (Fig. 4B), and BrdU/7-AAD staining and flow cytometry (Fig. 4C). These results show that chronic high-glucose culture conditions do not alter mesangial cell proliferation. In addition, we determined the effects of a switching normal glucose to high-glucose media and high-glucose to normal glucose complete growth media for 24 or 48 h on cell proliferation by measuring BrdU incorporation (Fig. 4D). There were no changes in cell proliferation under any condition (Fig. 4D). Therefore, the data shown in Fig. 3 provide evidence that hyperglycemia-induced cell hypertrophy is not accompanied by enhanced cellular proliferation.
Fig. 4.

Chronic HG does not increase mesangial cell proliferation. Cells were plated in NG or HG, and cell proliferation determined by counting cells (A), determining relative cell numbers with Alamar blue fluorescence (B), measuring incorporation of bromodeoxyuridine (BrdU) at the indicated time points by flowcytometry (C), and measuring the incorporation of BrdU at the indicated time points by ELISA after plating (D). Data are means ± SD; n = 3.
Chronic high glucose increases signaling pathways associated with enhanced cell growth, extracellular matrix production, and fibrosis, which are reduced with glucosylceramide synthase inhibitors.
To decipher the mechanism by which inhibition of glucosylceramide synthase reverses hyperglycemia-induced mesangial cell hypertrophy, we measured the expression of key molecules that regulate cell growth, protein degradation, and extracellular matrix protein fibronectin. It has been shown that growth stimulatory signals and inhibition of protein degradation pathways can cause cell hypertrophy (10, 32, 64). Mesangial cells in high-glucose media had elevated cell growth signals with increased pAkt (Ser473) with no change in total Akt (Fig. 5, A and B) and decreased levels of the protein degradation marker pPTEN (Fig. 5D) even though total PTEN was increased (Fig. 5C). Our results indicate that hyperglycemia increases growth signals in association with decreased protein degradation by decreasing PTEN phosphorylation in mesangial cells.
Fig. 5.

Chronic HG alters protein expression. Mesangial cells were grown in NG or HG. At 48 h, cells were collected and immunobloted for cell growth signaling pathways (A and B), protein degradation markers (C and D), and fibrosis and extracellular matrix (ECM) markers (E–G). p, Phosphorylated; PTEN, phosphatase and tensin homolog. The blots shown are from one experiment and were reprobed for β-actin, which was used as an internal control. Results are representative of at least 3 independent experiments. Data are means ± SD. *P ≤ 0.05; ***P ≤ 0.001; ****P ≤ 0.0001 (as determined by a Student's t-test).
Transforming growth factor (TGF)-β has been implicated in renal hypertrophy and the production of extracellular matrix in DN (22). Because TGF-β mediates phosphorylation of Smad3, we analyzed Smad3 and pSmad3 in mesangial cells in the presence of normal and high glucose. High-glucose culture conditions increased levels of Smad3 and pSmad3 in mesangial cells, suggesting enhanced TGF-β signaling (Fig. 5, E and F). TGF-β production is upstream of and regulates extracellular matrix production. It has been established that mesangial extracellular matrix buildup is a characteristic feature of DN (37). High-glucose media have also been reported to induce expression of cell-associated fibronectin in mesangial cells (37). High glucose enhanced levels of total fibronectin associated with mesangial cells (Fig. 5G). In conclusion, high-glucose culture conditions enhanced pSmad3 and pAkt, decreased pPTEN, and increased production of the extracellular matrix protein fibronectin via Smad3 and pAkt induced by TGF-β.
To determine the role of GSLs in high-glucose mesangial cell signaling, we treated mesangial cells grown in hyperglycemic conditions with the C10 inhibitor of GCS and analyzed the above signaling pathways. Cells were treated with a 0.15 μM concentration of C10 for 48 h. GCS inhibition did not alter total Akt (Fig. 6A) but decreased Akt phosphorylation (Fig. 6B), suggesting reduced Akt signaling. Similarly, C10 did not alter total PTEN levels (Fig. 6C) but increased levels of pPTEN (Fig. 6D), suggesting enhanced protein degradation pathways. To determine if GCS inhibition alters signaling through TGF-β, we analyzed levels of pSmad3 and total Smad3. C10 reduced levels of total Smad3 and pSmad3 (Fig. 6, E and F) in cells grown in high glucose, suggesting inhibition of TGF-β signaling pathways. Likewise, C10 reduced levels of fibronectin (Fig. 6G), suggesting reduced levels of extracellular matrix proteins. The data shown in Fig. 6 suggest that inhibition of GCS reverses mesangial cell hypertrophy by reducing Akt and Smad3 signaling and reducing extracellular matrix proteins by enhanced protein degradation pathways.
Fig. 6.

Inhibition of glucosylceramide synthase reduces mesangial hypertrophy by reducing pAkt and pSmad3 and increasing pPTEN. Mesangial cells were grown in HG, plated, and supplemented with DMSO or 0.15 μM C10. At 48 h, cells were collected and immunobloted for cell growth signaling pathways (A and B), protein degradation markers (C and D), and fibrosis and ECM marker (E–G). The blots shown are from one experiment and were reprobed for β-actin, which was used as an internal control. Results are representative of at least 3 independent experiments. Data are means ± SD. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.005; ****P ≤ 0.0001 (as determined by a Student's t-test).
Urinary LacCers are elevated in humans with DN.
The data presented thus far indicate that in the db/db mouse model of type II DN, GSLs are elevated in the kidney and urine and play a role in mediating DN and the functional role of GSLs on mesangial hypertrophy. We hypothesized that GSLs would also be elevated in the urine of patients with DN. GSLs were quantified in urine obtained from either patients diagnosed with DN or from healthy control subjects. Total urine LacCers were increased ∼12-fold in patients with DN compared with levels detected in healthy control subjects (2.9 ± 4.1 vs. 35.1 ± 14 pmol LacCer/mg creatinine; Fig. 7). These data suggest a possible role for GSLs in human DN.
Fig. 7.
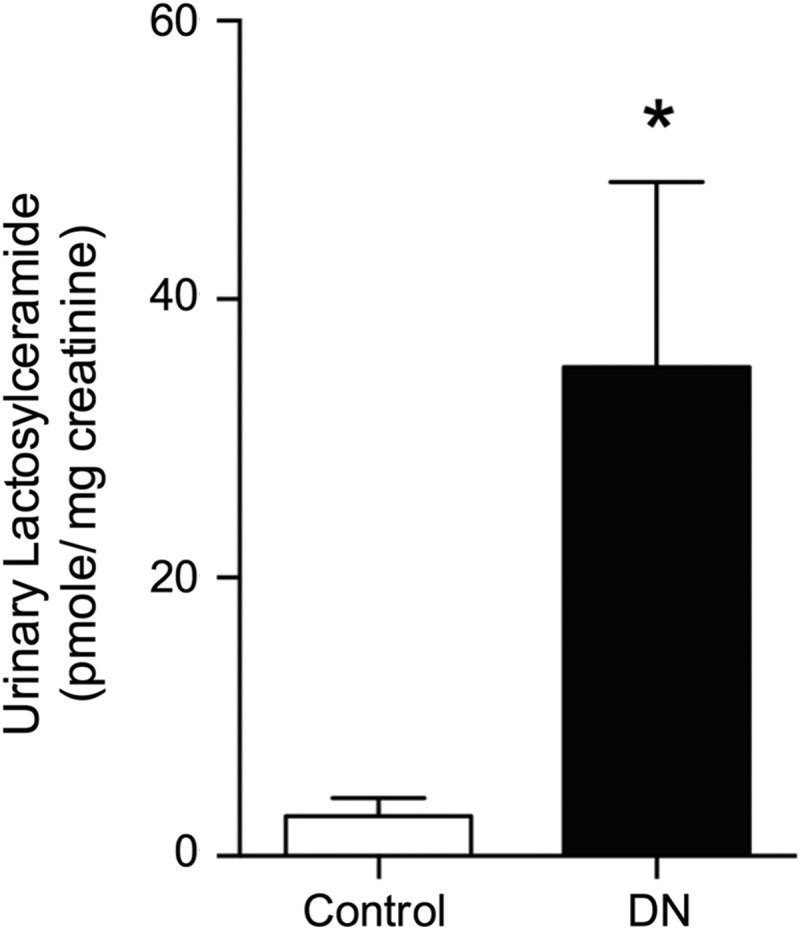
Urinary LacCers are elevated in humans with diabetic nephropathy (DN). LacCers were quantified in the urine of control subjects without nephropathy or from patients with confirmed DN via HPLC-MS/MS. Lipids were normalized to urine creatinine. Data are means ± SD; n = 10. *P ≤ 0.01 compared with control subjects (as determined by a Student's t-test).
DISCUSSION
GSLs have been documented to play a role in metabolic syndrome, insulin resistance, and diabetes. In particular, inhibition of GCS prevents and reverses insulin resistance in a variety of rodent models of diabetes (1). Furthermore, GSL accumulation has been shown to play a role in nephropathy resulting from STZ-induced type I diabetes in rats, and inhibition of their production using the glucosylceramide synthase inhibitor PDMP reversed renal hypertrophy (61). In that study (61), GSLs were measured by thin-layer chromatography, which can separate glucosylceramide and galactosylceramide using borate-impregnated plates but does not separate individual species of GSLs based on fatty acyl groups. The mass spectroscopy methods used in the present study do not distinguish between glucosylceramide and galactosylceramide. However, given that UDP-glucose and UDP-galactose are interconvertable through a ubiquitously expressed epimerase, it is likely that total HexCer content reflects changes in glucosylceramide. In addition, the location within the kidney in which GSLs were generated and their role in nephropathy were also not determined (61). Our data reveal that GSLs accumulated in the kidney cortex in glomeruli suggest that they are mediators of type II DN via the induction of glomerular hypertrophy. Our in vitro data indicate that GSLs play a role in mesangial cell hypertrophy. There are many cell types in addition to mesangial cells that are present and play a role in glomerular hypertrophy in DN. Future work will be aimed at determining the specific glomerular cell type in which GSLs accumulate during DN.
Db/db mice become very obese and develop insulin resistance and hyperglycemia at a very young age. Changes in the kidney such as inflammation, immune cell infiltration, and fibrosis occur early (9 wk), whereas changes such as altered creatinine clearance occur later (17 wk) (25). Mice in this model develop glomerular hypertrophy and proximal tubule vacuolization similar to human disease (25). Here, we documented renal cortical accumulation of the GSLs HexCers and LacCers early in DN (9 wk) and continuing at later stages of DN (17 wk; Fig. 1, A and B). We have recently reported (41) increased GSL levels in the kidneys and urine of lupus-prone mice. Similarly, urinary hexosyl and LacCers were increased in db/db mice at 9 wk of age (Fig. 1C). As we previously reported in lupus nephritis (41), GSLs in db/db mice were elevated at least in part in the glomeruli. We have recently reported that GSL levels in hematopoietic and leukemia cells are in part determined by glucose availability (47). Similarly, culture of mesangial cells in hyperglycemic conditions, similar to those present in the diabetic kidney, were sufficient to induce the accumulation of GSLs.
The mechanism by which elevated glucose leads to increased levels of HexCers is unproven. In vivo data provide evidence that blood glucose levels regulate at least in part kidney GSL concentrations; administration of insulin to rats with insulin-dependent diabetes as a result of STZ treatment significantly decreased blood glucose levels as well as kidney GSLs (61). Shayman and coauthors (61) have suggested that UDP-glucose/UDP-galactose are required substrates for the synthesis of glucosylceramide and LacCer and that the levels of UDP-glucose/UDP-galactose in the kidney are lower than the Km of glucsosylceramide synthase and thus are rate limiting. These substrates are generated in the glycogen synthesis pathway. In addition, NADPH produced in the pentose phosphate pathway is rate limiting for the synthesis of fatty acids, the building blocks of all lipids, including sphingolipids. UTP generated in the pentose phosphate pathway feeds into the glycogen synthesis pathway for the generation of UDP-glucose and UDP-galactose. It is possible that the enhanced flux of glucose into these otherwise minor pathways in the kidney during diabetes leads to an enhanced production of rate-limiting substrates required for a glucosylceramide synthesis. Indeed, our recent data suggest that this is the case (47). Cells made to be highly glycolytic have elevated HexCers, which are depleted upon glucose withdrawal. In addition, inhibitors of glycolysis or the pentose phosphate pathway significantly decreased HexCers (47). These changes in HexCers were likely substrate driven, as they were independent of glucosylceramide synthase expression or activity.
In addition to hyperglycemia, ROS generation in the diabetic kidney could also play a role in GSL production. Grove et al. (16) previously reported that the ROS inhibitor pyridoxamine, when given to endothelial nitric oxide synthase-deficient C57BLKS db/db DN mice upon the development of hyperglycemia (∼6 wk of age), prevented the accumulation in glomeruli and/or tubules of specific phospho- and glycolipid species from four different classes, such as gangliosides, sulfoglycosphingolipids, lysophospholipids, and phosphatidylethanolamines, and prevented nephropathy. Pyridoxamine is also efficacious when used in several other animal models of diabetes (11, 48, 50) and in clinical trials in patients with early stages of the disease. Indeed, treatment of mesangial cells with the oxidant H2O2 increases HexCer levels (data not shown). Thus, in addition to hyperglycemia, increased ROS in DN could further enhance synthesis of GSLs.
Hyperglycemia is a prerequisite for the development of DN and mediates early glomerular pathological changes. Consistent with the literature (8, 18, 21, 32, 34, 44, 49), our data indicate that high-glucose media induce hypertrophy of mesangial cells. Transgenic mice overexpressing glucose transporter (GLUT)1 in glomeruli develop glomerular hypertrophy, glomerulosclerosis, and decreased renal function (54); these data reveal that hyperglycemia induces many pathological changes in glomeruli even in the absence of diabetes and hypertension. Overexpression of GLUT1 in mesangial cells in culture is sufficient to induce hypertrophy (19). We showed that mesangial cells grown in high-glucose media are 1.6-fold greater in size than those grown in normal glucose media (Fig. 3, B and D). High glucose induced an accumulation of GSLs in mesangial cells (Fig. 2B), and when GSL synthesis was inhibited pharmacologically with C10, mesangial cell thickness was reduced to the size of cells grown in normal glucose (Fig. 3, C and D). Our data are consistent with those reported in the STZ-induced model of type I DN in which PDMP administration to diabetic rats reversed glomerular hypertrophy (61). In summary, these data indicate that GSLs mediate hyperglycemia-induced mesangial cell hypertrophy.
To explore the molecular mechanism by which GCS inhibition reverses hyperglycemia-induced mesangial cell hypertrophy, we evaluated the expression of key signaling molecules that mediate this process. Chronic hyperglycemia induced enhanced Akt activation and reduced PTEN expression (Fig. 5, B and D). A recent report (64) has indicated that activated Akt and a reduction in PTEN levels are crucially involved in vivo in db/db mouse model DN and in vitro in hyperglycemia-induced hypertrophy. Overexpression of wild-type PTEN reduces Akt phosphorylation and prevents hyperglycemia-induced mesangial cell hypertrophy, whereas overexpression of dominant negative PTEN is sufficient to induce hypertrophy in mesangial cells even when grown in normal glucose conditions (32). Our data suggest that GSLs and glucosylceramide synthase act upstream of PTEN and Akt (Fig. 6, B and D) as inhibition of their generation was sufficient to reduce pAkt and enhance pPTEN even in hyperglycemic culture conditions. Other studies also have shown that inhibition of GCS is protective in a variety of diseases, such as type I diabetes, Parkinson disease, polycystic kidney disease, and cancer, and that this protection is mediated at least in part by inhibition of Akt signaling (1, 40, 46).
In addition to pAkt and pPTEN, TGF-β enhances activation of Smad3, which stimulates the expression of extracellular matrix proteins, including fibronectin (22). Indeed, mesangial matrix expansion is halted in Smad3 knockout diabetic mice (15, 53). Inhibition of glucosylceramide synthase reduced Smad3, pSmad3, and fibronectin even under hyperglycemic culture conditions (Fig. 6, E–G). Our data indicate that GSLs act upstream of TGF-β, pSmad3, and extracellular matrix production. Kato et al. (24) has previously proposed a signaling pathway demonstrating a mechanism for Akt activation that is downstream of PTEN downregulation as a result of TGF-β upregulation. Our data support a model (Fig. 8) in which inhibition of GCS under hyperglycemic conditions reduces hypertrophy and extracellular matrix expression at least in part by decreasing pSmad3, maintaining levels of activated PTEN, inhibiting activation of Akt, and reducing production of fibronectin.
Fig. 8.

Schematic representation of the glucosylceramide-induced signaling cascade of the DN process. Excessive glucose enhances GSL production by increasing the availability of substrate ceramide and UDP-glucose. GSLs activate signaling through Smad3 and downregulate pPTEN phosphorylation, which increases Akt phosphorylation, leading to hypertrophy and ECM accumulation in diabetic mesangial cells. Inhibition of GSL by C10 improves the pathology by reversing the signaling pathway.
To determine if the above data have translational relevance, we analyzed urine samples collected from 10 diabetic nephropathy patients and 10 healthy individuals (Fig. 7). Total LacCer levels increased 12-fold in the urine of DN patients (Fig. 7) compared with healthy individuals (HexCer levels were below the detectable limit). Levels of GSLs in the urine of diabetic patients were consistent with those observed for other kidney diseases in which GSLs play a role. For example, we recently reported elevated levels of GSLs in the urine of patients with lupus nephritis (41). In addition, levels and activity of the enzyme neuraminidase were also increased in the urine of lupus nephritis patients (41). Proteomics analysis by Caseiro et al. (4) identified 219 unique proteins in the urine of 15 individuals that have type I diabetes for 15 years compared with healthy individuals; many proteins involved in ganglioside metabolism and GSL catabolic pathways were detected in diabetic urine. Whitfield et al. (56) showed that the GSL Gb3 excreted in the urine was elevated in Fabry disease patients. In Fabry disease, there is a lysosomal accumulation of Gb3 as a result of a deficiency of α-galactosidase A. Thus, the level and species of GSL present in the different kidney cell types and urine may be specific for particular cellular stresses and stimuli as well as different kidney diseases and may be a potential diagnostic tool.
Inhibition of glucosylceramide synthase may be a viable therapeutic option for mediating the pathological impact of hyperglycemia on the kidney during DN. Future studies using GCS inhibitors and GCS inducible knockout mice in models of type II DN will be important for determining if modulation of this pathway prevents DN or inhibits the progression of DN to renal failure. GCS inhibitors reverse insulin resistance in rodents (1, 27, 65). Thus, dissecting the indirect and direct effects of these pharmacological glucosylceramide synthase inhibitors on DN will be difficult. Rather, in vivo studies using kidney cell type-specific glucosylceramide synthase inducible knockout mice will be highly valuable in determining if GSLs are indeed direct mediators of DN. Pharmacological inhibitors of glucosylceramide synthase, such as eliglustat, are currently being used in humans for the treatment of Gaucher disease and improve platelet and hemoglobin levels as well as reduce spleen and liver size (28, 31). These inhibitors are also well tolerated for long-term use. Thus, glucosylceramide synthase inhibitors may have therapeutic potential for the treatment of DN.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants P20-RR-17677 (COBRE in Lipidomics and Pathobiology pilot project), R01-DK-093462 (to L. J. Siskind), R01-DK-101034 (to J. M. Arthur), and R01-GM-084147 (to R. G. Schnellmann), Veterans Administration Merit Review Grant BX-000851 (to R. G. Schnellmann), Veterans Affairs Research Enhancement Award Program pilot project funding (to L. J. Siskind, J. M. Arthur, and R. G. Schnellmann), and the Lipidomics Shared Resource of the Hollings Cancer Center at the Medical University of South Carolina, supported by NIH Grant P30-CA-138313.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
DISCLAIMER
The contents of this publication do not represent the views of the Department of Veterans Affairs or the United States Government or any of the other funding entities.
AUTHOR CONTRIBUTIONS
Author contributions: M.S., M.K., J.M.A., J.A.S., R.G.S., and L.J.S. conception and design of research; M.S., M.K., L.A.H., and L.J.S. performed experiments; M.S., L.A.H., and L.J.S. analyzed data; M.S., R.G.S., and L.J.S. interpreted results of experiments; M.S. and L.J.S. prepared figures; M.S. and L.J.S. drafted manuscript; M.S., J.A.S., R.G.S., and L.J.S. edited and revised manuscript; M.S., R.G.S., and L.J.S. approved final version of manuscript.
REFERENCES
- 1.Aerts JM, Ottenhoff R, Powlson AS, Grefhorst A, van Eijk M, Dubbelhuis PF, Aten J, Kuipers F, Serlie MJ, Wennekes T, Sethi JK, O'Rahilly S, Overkleeft HS. Pharmacological inhibition of glucosylceramide synthase enhances insulin sensitivity. Diabetes 56: 1341–1349, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baccora MH, Cortes P, Hassett C, Taube DW, Yee J. Effects of long-term elevated glucose on collagen formation by mesangial cells. Kidney Int 72: 1216–1225, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Bielawski J, Szulc ZM, Hannun YA, Bielawska A. Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods 39: 82–91, 2006. [DOI] [PubMed] [Google Scholar]
- 4.Caseiro A, Barros A, Ferreira R, Padrao A, Aroso M, Quintaneiro C, Pereira A, Marinheiro R, Vitorino R, Amado F. Pursuing type 1 diabetes mellitus and related complications through urinary proteomics. Transl Res 163: 188–199, 2014. [DOI] [PubMed] [Google Scholar]
- 5.Chae YM, Park KK, Magae J, Lee IS, Kim CH, Kim HC, Hong S, Lee JG, Choi IJ, Kim HS, Min KS, Lee IK, Chang YC. Sp1-decoy oligodeoxynucleotide inhibits high glucose-induced mesangial cell proliferation. Biochem Biophys Res Commun 319: 550–555, 2004. [DOI] [PubMed] [Google Scholar]
- 6.Chatterjee S, Shi WY, Wilson P, Mazumdar A. Role of lactosylceramide and MAP kinase in the proliferation of proximal tubular cells in human polycystic kidney disease. J Lipid Res 37: 1334–1344, 1996. [PubMed] [Google Scholar]
- 7.Chatterjee S, Wei H. Roles of glycosphingolipids in cell signaling: adhesion, migration, and proliferation. Methods Enzymol 363: 300–312, 2003. [DOI] [PubMed] [Google Scholar]
- 8.Covington MD, Schnellmann RG. Chronic high glucose downregulates mitochondrial calpain 10 and contributes to renal cell death and diabetes-induced renal injury. Kidney Int 81: 391–400, 2012. [DOI] [PubMed] [Google Scholar]
- 9.Cox T, Lachmann R, Hollak C, Aerts J, van Weely S, Hrebicek M, Platt F, Butters T, Dwek R, Moyses C, Gow I, Elstein D, Zimran A. Novel oral treatment of Gaucher's disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet 355: 1481–1485, 2000. [DOI] [PubMed] [Google Scholar]
- 10.Das F, Dey N, Venkatesan B, Kasinath BS, Ghosh-Choudhury N, Choudhury GG. High glucose upregulation of early-onset Parkinson's disease protein DJ-1 integrates the PRAS40/TORC1 axis to mesangial cell hypertrophy. Cell Signal 23: 1311–1319, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Degenhardt TP, Alderson NL, Arrington DD, Beattie RJ, Basgen JM, Steffes MW, Thorpe SR, Baynes JW. Pyridoxamine inhibits early renal disease and dyslipidemia in the streptozotocin-diabetic rat. Kidney Int 61: 939–950, 2002. [DOI] [PubMed] [Google Scholar]
- 12.Degroote S, Wolthoorn J, van Meer G. The cell biology of glycosphingolipids. Semin Cell Dev Biol 15: 375–387, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Foley RN, Collins AJ. The growing economic burden of diabetic kidney disease. Curr Diabetes Rep 9: 460–465, 2009. [DOI] [PubMed] [Google Scholar]
- 14.Forbes JM, Fukami K, Cooper ME. Diabetic nephropathy: where hemodynamics meets metabolism. Exp Clin Endocrinol Diabetes 115: 69–84, 2007. [DOI] [PubMed] [Google Scholar]
- 15.Fujimoto M, Maezawa Y, Yokote K, Joh K, Kobayashi K, Kawamura H, Nishimura M, Roberts AB, Saito Y, Mori S. Mice lacking Smad3 are protected against streptozotocin-induced diabetic glomerulopathy. Biochem Biophys Res Commun 305: 1002–1007, 2003. [DOI] [PubMed] [Google Scholar]
- 16.Grove KJ, Voziyan PA, Spraggins JM, Wang S, Paueksakon P, Harris RC, Hudson BG, Caprioli RM. Diabetic nephropathy induces alterations in the glomerular and tubule lipid profiles. J Lipid Res 55: 1375–1385, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gruden G, Perin PC, Camussi G. Insight on the pathogenesis of diabetic nephropathy from the study of podocyte and mesangial cell biology. Curr Diabetes Rev 1: 27–40, 2005. [DOI] [PubMed] [Google Scholar]
- 18.Han HJ, Lee YJ, Park SH, Lee JH, Taub M. High glucose-induced oxidative stress inhibits Na+/glucose cotransporter activity in renal proximal tubule cells. Am J Physiol Renal Physiol 288: F988–F996, 2005. [DOI] [PubMed] [Google Scholar]
- 19.Heilig CW, Concepcion LA, Riser BL, Freytag SO, Zhu M, Cortes P. Overexpression of glucose transporters in rat mesangial cells cultured in a normal glucose milieu mimics the diabetic phenotype. J Clin Invest 96: 1802–1814, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hickey FB, Martin F. Diabetic kidney disease and immune modulation. Curr Opin Pharmacol 13: 602–612, 2013. [DOI] [PubMed] [Google Scholar]
- 21.Higgins GC, Coughlan MT. Mitochondrial dysfunction and mitophagy: the beginning and end to diabetic nephropathy? Br J Pharmacol 171: 1917–1942, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Isono M, Chen S, Hong SW, Iglesias-de la Cruz MC, Ziyadeh FN. Smad pathway is activated in the diabetic mouse kidney and Smad3 mediates TGF-β-induced fibronectin in mesangial cells. Biochem Biophys Res Commun 296: 1356–1365, 2002. [DOI] [PubMed] [Google Scholar]
- 23.Kanwar YS, Wada J, Sun L, Xie P, Wallner EI, Chen S, Chugh S, Danesh FR. Diabetic nephropathy: mechanisms of renal disease progression. Exp Biol Med 233: 4–11, 2008. [DOI] [PubMed] [Google Scholar]
- 24.Kato M, Putta S, Wang M, Yuan H, Lanting L, Nair I, Gunn A, Nakagawa Y, Shimano H, Todorov I, Rossi JJ, Natarajan R. TGF-β activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat Cell Biol 11: 881–889, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korrapati MC, Howell LH, Shaner BE, Megyesi JK, Siskind LJ, Schnellmann RG. Suramin: a potential therapy for diabetic nephropathy. PLos One 8: e73655, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lahiri S, Futerman AH. The metabolism and function of sphingolipids and glycosphingolipids. Cell Mol Life Sci 64: 2270–2284, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langeveld M, Aerts JM. Glycosphingolipids and insulin resistance. Prog Lipid Res 48: 196–205, 2009. [DOI] [PubMed] [Google Scholar]
- 28.Lee L, Abe A, Shayman JA. Improved inhibitors of glucosylceramide synthase. J Biol Chem 274: 14662–14669, 1999. [DOI] [PubMed] [Google Scholar]
- 29.Lehmann R, Schleicher ED. Molecular mechanism of diabetic nephropathy. Clin Chim Acta 297: 135–144, 2000. [DOI] [PubMed] [Google Scholar]
- 30.Liu L, Hu X, Cai GY, Lv Y, Zhuo L, Gao JJ, Cui SY, Feng Z, Fu B, Chen XM. High glucose-induced hypertrophy of mesangial cells is reversed by connexin43 overexpression via PTEN/Akt/mTOR signaling. Nephrol Dial Transplant 27: 90–100, 2012. [DOI] [PubMed] [Google Scholar]
- 31.Lukina E, Watman N, Dragosky M, Pastores GM, Arreguin EA, Rosenbaum H, Zimran A, Angell J, Ross L, Puga AC, Peterschmitt JM. Eliglustat, an investigational oral therapy for Gaucher disease type 1: Phase 2 trial results after 4 years of treatment. Blood Cells Mol Dis 53: 274–276, 2014. [DOI] [PubMed] [Google Scholar]
- 32.Mahimainathan L, Das F, Venkatesan B, Choudhury GG. Mesangial cell hypertrophy by high glucose is mediated by downregulation of the tumor suppressor PTEN. Diabetes 55: 2115–2125, 2006. [DOI] [PubMed] [Google Scholar]
- 33.Marshall J, Ashe KM, Bangari D, McEachern K, Chuang WL, Pacheco J, Copeland DP, Desnick RJ, Shayman JA, Scheule RK, Cheng SH. Substrate reduction augments the efficacy of enzyme therapy in a mouse model of Fabry disease. PLos One 5: e15033, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Masson E, Lagarde M, Wiernsperger N, El Bawab S. Hyperglycemia and glucosamine-induced mesangial cell cycle arrest and hypertrophy: Common or independent mechanisms? IUBMB Life 58: 381–388, 2006. [DOI] [PubMed] [Google Scholar]
- 35.Mather AR, Siskind LJ. Glycosphingolipids and kidney disease. Adv Exp Med Biol 721: 121–138, 2011. [DOI] [PubMed] [Google Scholar]
- 36.McEachern KA, Fung J, Komarnitsky S, Siegel CS, Chuang WL, Hutto E, Shayman JA, Grabowski GA, Aerts JM, Cheng SH, Copeland DP, Marshall J. A specific and potent inhibitor of glucosylceramide synthase for substrate inhibition therapy of Gaucher disease. Mol Genet Metab 91: 259–267, 2007. [DOI] [PubMed] [Google Scholar]
- 37.Miller CG, Pozzi A, Zent R, Schwarzbauer JE. Effects of high glucose on integrin activity and fibronectin matrix assembly by mesangial cells. Mol Biol Cell 25: 2342–2350, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morales A, Colell A, Mari M, Garcia-Ruiz C, Fernandez-Checa JC. Glycosphingolipids and mitochondria: role in apoptosis and disease. Glycoconj J 20: 579–588, 2004. [DOI] [PubMed] [Google Scholar]
- 39.Nahman NS Jr, Leonhart KL, Cosio FG, Hebert CL. Effects of high glucose on cellular proliferation and fibronectin production by cultured human mesangial cells. Kidney Int 41: 396–402, 1992. [DOI] [PubMed] [Google Scholar]
- 40.Natoli TA, Smith LA, Rogers KA, Wang B, Komarnitsky S, Budman Y, Belenky A, Bukanov NO, Dackowski WR, Husson H, Russo RJ, Shayman JA, Ledbetter SR, Leonard JP, Ibraghimov-Beskrovnaya O. Inhibition of glucosylceramide accumulation results in effective blockade of polycystic kidney disease in mouse models. Nat Med 16: 788–792, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nowling TK, Mather AR, Thiyagarajan T, Hernandez-Corbacho MJ, Powers TW, Jones EE, Snider AJ, Oates JC, Drake RR, Siskind LJ. Renal glycosphingolipid metabolism is dysfunctional in lupus nephritis. J Am Soc Nephrol 26: 1402–1413, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petermann AT, Pippin J, Durvasula R, Pichler R, Hiromura K, Monkawa T, Couser WG, Shankland SJ. Mechanical stretch induces podocyte hypertrophy in vitro. Kidney Int 67: 157–166, 2005. [DOI] [PubMed] [Google Scholar]
- 43.Peterschmitt MJ, Burke A, Blankstein L, Smith SE, Puga AC, Kramer WG, Harris JA, Mathews D, Bonate PL. Safety, tolerability, and pharmacokinetics of eliglustat tartrate (Genz-112638) after single doses, multiple doses, and food in healthy volunteers. J Clin Pharmacol 51: 695–705, 2011. [DOI] [PubMed] [Google Scholar]
- 44.Phillips AO, Baboolal K, Riley S, Grone H, Janssen U, Steadman R, Williams J, Floege J. Association of prolonged hyperglycemia with glomerular hypertrophy and renal basement membrane thickening in the Goto Kakizaki model of non-insulin-dependent diabetes mellitus. Am J Kidney Dis 37: 400–410, 2001. [DOI] [PubMed] [Google Scholar]
- 45.Qureshi A, Subathra M, Grey A, Schey K, Del Poeta M, Luberto C. Role of sphingomyelin synthase in controlling the antimicrobial activity of neutrophils against Cryptococcus neoformans. PLos One 5: e15587, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen W, Henry AG, Paumier KL, Li L, Mou K, Dunlop J, Berger Z, Hirst WD. Inhibition of glucosylceramide synthase stimulates autophagy flux in neurons. J Neurochem 129: 884–894, 2014. [DOI] [PubMed] [Google Scholar]
- 47.Stathem M, Marimuthu S, O'Neal J, Rathmell JC, Chesney JA, Beverly LJ, Siskind LJ. Glucose availability and glycolytic metabolism dictate glycosphingolipid levels. J Cell Biochem 116: 67–80, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sugimoto H, Grahovac G, Zeisberg M, Kalluri R. Renal fibrosis and glomerulosclerosis in a new mouse model of diabetic nephropathy and its regression by bone morphogenic protein-7 and advanced glycation end product inhibitors. Diabetes 56: 1825–1833, 2007. [DOI] [PubMed] [Google Scholar]
- 49.Tahara A, Tsukada J, Tomura Y, Yatsu T, Shibasaki M. Effects of high glucose on AVP-induced hyperplasia, hypertrophy, and type IV collagen synthesis in cultured rat mesangial cells. Endocr Res 37: 216–227, 2012. [DOI] [PubMed] [Google Scholar]
- 50.Tanimoto M, Gohda T, Kaneko S, Hagiwara S, Murakoshi M, Aoki T, Yamada K, Ito T, Matsumoto M, Horikoshi S, Tomino Y. Effect of pyridoxamine (K-163), an inhibitor of advanced glycation end products, on type 2 diabetic nephropathy in KK-A(y)/Ta mice. Metabolism 56: 160–167, 2007. [DOI] [PubMed] [Google Scholar]
- 51.Uttarwar L, Gao B, Ingram AJ, Krepinsky JC. SREBP-1 activation by glucose mediates TGF-β upregulation in mesangial cells. Am J Physiol Renal Physiol 302: F329–F341, 2012. [DOI] [PubMed] [Google Scholar]
- 52.Vunnam RR, Radin NS. Analogs of ceramide that inhibit glucocerebroside synthetase in mouse brain. Chem Phys Lipids 26: 265–278, 1980. [DOI] [PubMed] [Google Scholar]
- 53.Wang A, Ziyadeh FN, Lee EY, Pyagay PE, Sung SH, Sheardown SA, Laping NJ, Chen S. Interference with TGF-β signaling by Smad3-knockout in mice limits diabetic glomerulosclerosis without affecting albuminuria. Am J Physiol Renal Physiol 293: F1657–F1665, 2007. [DOI] [PubMed] [Google Scholar]
- 54.Wang Y, Heilig K, Saunders T, Minto A, Deb DK, Chang A, Brosius F, Monteiro C, Heilig CW. Transgenic overexpression of GLUT1 in mouse glomeruli produces renal disease resembling diabetic glomerulosclerosis. Am J Physiol Renal Physiol 299: F99–F111, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wennekes T, van den Berg RJ, Boot RG, van der Marel GA, Overkleeft HS, Aerts JM. Glycosphingolipids–nature, function, and pharmacological modulation. Angew Chem Int Ed Engl 48: 8848–8869, 2009. [DOI] [PubMed] [Google Scholar]
- 56.Whitfield PD, Calvin J, Hogg S, O'Driscoll E, Halsall D, Burling K, Maguire G, Wright N, Cox TM, Meikle PJ, Deegan PB. Monitoring enzyme replacement therapy in Fabry disease–role of urine globotriaosylceramide. J Inherit Metab Dis 28: 21–33, 2005. [DOI] [PubMed] [Google Scholar]
- 57.Wu D, Peng F, Zhang B, Ingram AJ, Kelly DJ, Gilbert RE, Gao B, Krepinsky JC. PKC-β1 mediates glucose-induced Akt activation and TGF-β1 upregulation in mesangial cells. J Am Soc Nephrol 20: 554–566, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu D, Peng F, Zhang B, Ingram AJ, Kelly DJ, Gilbert RE, Gao B, Kumar S, Krepinsky JC. EGFR-PLCγ1 signaling mediates high glucose-induced PKCβ1-Akt activation and collagen I upregulation in mesangial cells. Am J Physiol Renal Physiol 297: F822–F834, 2009. [DOI] [PubMed] [Google Scholar]
- 59.Yamashita T, Hashiramoto A, Haluzik M, Mizukami H, Beck S, Norton A, Kono M, Tsuji S, Daniotti JL, Werth N, Sandhoff R, Sandhoff K, Proia RL. Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc Natl Acad Sci USA 100: 3445–3449, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yuan P, Xue H, Zhou L, Qu L, Li C, Wang Z, Ni J, Yu C, Yao T, Huang Y, Wang R, Lu L. Rescue of mesangial cells from high glucose-induced over-proliferation and extracellular matrix secretion by hydrogen sulfide. Nephrol Dial Transplant 26: 2119–2126, 2011. [DOI] [PubMed] [Google Scholar]
- 61.Zador IZ, Deshmukh GD, Kunkel R, Johnson K, Radin NS, Shayman JA. A role for glycosphingolipid accumulation in the renal hypertrophy of streptozotocin-induced diabetes mellitus. J Clin Invest 91: 797–803, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang L, Pang S, Deng B, Qian L, Chen J, Zou J, Zheng J, Yang L, Zhang C, Chen X, Liu Z, Le Y. High glucose induces renal mesangial cell proliferation and fibronectin expression through JNK/NF-κB/NADPH oxidase/ROS pathway, which is inhibited by resveratrol. Int J Biochem Cell Biol 44: 629–638, 2012. [DOI] [PubMed] [Google Scholar]
- 63.Zhang X, Kiechle FL. Review: Glycosphingolipids in health and disease. Ann Clin Lab Sci 34: 3–13, 2004. [PubMed] [Google Scholar]
- 64.Zhang Z, Peng H, Chen J, Chen X, Han F, Xu X, He X, Yan N. MicroRNA-21 protects from mesangial cell proliferation induced by diabetic nephropathy in db/db mice. FEBS Lett 583: 2009–2014, 2009. [DOI] [PubMed] [Google Scholar]
- 65.Zhao H, Przybylska M, Wu IH, Zhang J, Siegel C, Komarnitsky S, Yew NS, Cheng SH. Inhibiting glycosphingolipid synthesis improves glycemic control and insulin sensitivity in animal models of type 2 diabetes. Diabetes 56: 1210–1218, 2007. [DOI] [PubMed] [Google Scholar]